
Abstract
Background
Despite multiple attempts, there is still no effective HIV-1 vaccine available. The HIV-1 polymerase (pol) gene is highly conserved and encodes cytotoxic T-lymphocyte (CTL) epitopes. The aim of the study was to characterise HIV-1 Pol CTL epitopes in mostly sample pairs obtained during early and chronic stages of infection.
Methods
Illumina deep sequencing was performed for all samples while Sanger sequencing was only performed on baseline samples. Codons under immune selection pressure were assessed by computing nonsynonymous to synonymous mutation ratios using MEGA. Minority CTL epitope variants occurring at \(\ge\) 5% were detected using low-frequency variant tool in CLC Genomics. Los Alamos HIV database was used for mapping mutations to known HIV-1 CTL epitopes.
Results
Fifty-two participants were enrolled in the study. Their median age was 28 years (interquartile range: 24–32 years) and majority of participants (92.3%) were female. Illumina minority variant analysis identified a significantly higher number of CTL epitopes (n = 65) compared to epitopes (n = 8) identified through Sanger sequencing. Most of the identified epitopes mapped to reverse transcriptase (RT) and integrase (IN) regardless of sequencing method. There was a significantly higher proportion of minority variant epitopes in RT (n = 39, 60.0%) compared to IN (n = 17, 26.2%) and PR (n = 9, 13.8%), p = 0.002 and < 0.0001, respectively. However, no significant difference was observed between the proportion of minority variant epitopes in IN versus PR, p = 0.06. Some epitopes were detected in either early or chronic HIV-1 infection whereas others were detected in both stages. Different distribution patterns of minority variant epitopes were observed in sample pairs; with some increasing or decreasing over time, while others remained constant. Some of the identified epitopes have not been previously reported for HIV-1 subtype C. There were also variants that could not be mapped to reported CTL epitopes in the Los Alamos HIV database.
Conclusion
Deep sequencing revealed many Pol CTL epitopes, including some not previously reported for HIV-1 subtype C. The findings of this study support the inclusion of RT and IN epitopes in HIV-1 vaccine candidates as these proteins harbour many CTL epitopes.
Similar content being viewed by others


Introduction
The HIV/AIDS pandemic has been a global crisis for four decades [1, 2]. At the end of 2020, there were 37.6 million people living with HIV globally, 1.5 million new HIV-1 infections, and 690,000 AIDS-related deaths [3]. South Africa has 7.9 million people living with HIV-1 (PLWH), making it the country with the highest number of infections in the world [4, 5]. This highlights the need to better understand HIV-1 natural immune responses in order to bolster the efforts of developing effective HIV-1 vaccines.
During early HIV-1 infection, effective cytotoxic T-lymphocyte (CTL) immune responses play an important role in the control of viraemia, by contributing to suppression of the viral load (VL) to a set-point [6]. CTL responses target and kill virus-infected cells, via recognition of viral peptide epitopes and releasing cytokines and cytotoxic granules that facilitate cell killing [7,8,9]. Viral peptide epitopes are presented on the surface of infected cells by the human leukocyte antigen type I (HLA I) trans-membrane proteins encoded by HLA-A, B and C alleles [10,11,12]. The targeted epitopes undergo immune selection pressure, which leads to the emergence of viral escape mutations [13, 14]. This results in evasion of the immune system, loss of immune control and ultimately progression to AIDS [7, 10]. The majority of epitopes that play a role in the control of viraemia have been extensively studied and reported for HIV-1 Gag [15, 16]. However, some studies show that HIV-1 Pol also harbours important CTL epitopes that contribute to the control of HIV-1 viraemia [17,18,19], hence pol is included in some HIV-1 vaccine candidates [20,21,22,23,24,25]. There are limited data on the characterisation of HIV-1 Pol CTL epitopes in early and chronic HIV-1 disease stages. The aim of this study was to characterise and assess the evolution of CTL epitopes encoded by HIV-1 pol during early and chronic stages of HIV-1 infection, using deep sequencing methods.
Materials and methods
Study population
This was a retrospective study that used stored plasma samples that were collected from individuals in the Tshwane district of South Africa. Participants enrolled in this study had a confirmed diagnosis of early or chronic HIV-1 infection and were antiretroviral therapy (ART) naïve. They were identified in a study that screened for early HIV-1 infection in individuals who had a negative rapid test at point-of-care facilities, and diagnosis was confirmed through HIV-1 VL, antibody (enzyme-linked immunoassay), p24 antigen, Western blot and avidity testing [26]. Most of the participants were followed up, and thus samples at two time-points were available (at baseline and follow-up) for analysis.
Nucleic acid extraction and amplification of HIV-1 pol
Total nucleic acids were manually extracted from plasma samples using the QIAamp UltraSens Virus kit (Qiagen, Hilden, Germany). Samples with a VL > 1000 copies/ml were extracted from a plasma input volume of 500 µl, which was adjusted to 1 ml using phosphate-buffered saline (PBS). Samples with a VL ≤ 1000 copies/ml were extracted directly from 1 ml plasma input volume to increase nucleic acid yield. Extraction was performed according to the manufacturer’s instructions except for the first centrifugation step, which was optimized to 800 relative centrifugal force (RCF) instead of the recommended 1200 RCF. This modification facilitated a more efficient dissolution of the pellet. Nucleic acids were eluted in a volume of 60 µl and all the eluates were stored at − 70 °C immediately after extraction.
The complete HIV-1 pol gene was amplified in all samples using an in-house nested PCR method, employing the SuperScript™ III One-Step RT-PCR System with Platinum™ Taq High Fidelity DNA Polymerase (Invitrogen, Carlsbad, California, USA). Each PCR reaction was performed in a 50 µl reaction, which included 5 µl of extracted RNA / DNA template, 2X reaction mix containing 2.4 mM magnesium sulphate (MgSO4), 0.4 mM of each deoxynucleotide triphosphate (dNTPs), 10 mM sense primer, 10 mM anti-sense primer, 5 units (U) of enzyme mix for (first round) or 1U of Platinum Taq polymerase (for second round), and nuclease-free water. The first-round amplicon was used as template for the second-round amplification (www.lifetechnologies.com) (Additional file 1: Table S1). One or two extracted HIV-1-negative controls were included in each experiment to assess potential contamination.
The following sets of primers were used for amplification of the complete pol gene: SM-F1 (outer forward) 5′-GCG GCT ACA TTA GAA GAA ATG ATG-3′ (HXB2 1807–1830) and SM-R1 (outer reverse) 5′-GCC AAG TAT TGT AGA GAT CCT ACC T-3′ (HXB2 5462–5488), SM-F3 (inner forward) 5′-AGA TTG TTA AAT GTT TCA ACT GTG G-3′ (HXB2 1952–1976) and SM-R3 (inner reverse) 5′-CTC CTG TAT GCA AAC CCC AAT A-3′ (HXB2 5245–5266). The nested primers amplified a fragment size of 3268 bp.
Sanger sequencing and sequence analysis
Sanger sequencing was performed on PCR amplicons, but only for baseline samples. Sequencing was performed commercially in six overlapping regions covering the entire pol ORF (Inqaba Biotechnical Industries, Tshwane, South Africa) (Additional file 1: Table S1). Sequences were edited in CLC Main Workbench 21 software (Qiagen, Hilden, Germany) and consensus sequences were generated, viewed in BioEdit 7.2.5 (https://bioedit.software.informer.com/download/) and aligned using the online version of the MAFFT program (https://mafft.cbrc.jp/alignment/server/). The HIV-1 HXB2 reference sequence (K03455.1) was used for nucleotide numbering. Phylogenetic analysis was performed on the multiple alignment and the ratio of nonsynonymous to synonymous (dN-dS) mutations within the HIV-1 Pol region was computed using MEGA software (https://www.megasoftware.net/). A dN-dS ratio \(\ge\) 5 was used to identify codons under high selection pressure and these sites were subsequently mapped to CTL epitopes documented in the Los Alamos HIV database [27].
Illumina sequencing and analysis of consensus sequences
Sequencing of PCR amplicons was performed at Inqaba Biotechnical Industries (Pretoria, South Africa), a commercial next generation sequencing (NGS) service provider. Briefly paired-end libraries (2 × 300 bp) were prepared using the NEBNext® Ultra™ II DNA library prep kit for Illumina® (New England Biolabs, USA) and sequencing was performed on an Illumina MiSeq instrument (Illumina, USA).
Some samples (n = 12) that were viewed to have poor quality scores were sent for repeat sequencing to the National Institute for Communicable Diseases (NICD), Sequencing Core Facility, Johannesburg, South Africa. Multiplexed paired-end libraries (2 × 300 bp) were prepared using the Nextera™ XT DNA Library Sample Preparation kit (Illumina Inc., California, United States) according to the manufacturer’s instructions and sequencing was carried out on an Illumina MiSeq instrument (Illumina Inc., California, United States).
Illumina sequencing data was analysed in the PASeq program (https://paseq.org/) using the feature that excludes APOBEC-induced hypermutations, and consensus sequences were generated. The quality of the Illumina reads was assessed in PASeq and CLC Genomics Workbench 21 (Qiagen, Hilden, Germany). PASeq-generated Illumina consensus sequences were viewed in BioEdit 7.2.5 software and aligned to HIV-1 reference sequences obtained from the GenBank database, in the MAFFT program available online. Sanger and Illumina consensus sequences obtained from baseline samples were compared through phylogenetic analysis in MEGA software and were observed to correctly cluster together (Additional file 4: Figure S1).
Analysis of Illumina sequencing reads for minority variants
Deep sequencing reads were mapped to an HIV-1 subtype C reference (AY162225.1) in CLC Genomics. Minority variants were analysed using a low-frequency variant detection tool in CLC Genomics, setting the cut-off for significance at 1% and the minimum frequency for variants at 5%. Minority variants were assessed in all participants and only nonsynonymous variants were mapped to HIV-1 CTL epitopes in the Los Alamos HIV database, to identify the epitope they fall within. Thus, in this study, minority variant epitopes were defined as epitopes existing at proportions above 5% but less than 20%. The Los Alamos HIV database was also used to predict human leukocyte antigen (HLA) allotypes that may be involved in presenting the identified epitopes to CTLs. Evolution within Pol CTL epitopes was assessed by comparing the proportion of minority variants between baseline and follow-up samples, and in early and chronic HIV-1 disease stages.
Data analysis
Descriptive statistics was used to present median values and interquartile range (IQR) for age, HIV-1 VL and sequencing depth. Fisher’s exact test was used to assess if there was an association between the distribution of mutations and HIV-1 disease stage for epitopes identified through Sanger sequencing. Two sample t-test was used for comparing the proportions of minority variant epitopes among the different regions of the Pol protein. A p-value of ≤ 0.05 was considered statistically significant. All statistical tests were performed on the STATA 16.0 software package (StataCorp LP, College Station, TX, USA). The statistical analyses of dN–dS ratios were computed on the MEGA programme using the HyPhy test for selection. Due to the small sample size, codons with dN–dS ratios of ≥ 5 were considered for assessment of epitopes under immune selection pressure. GraphPad Prism v.9.1.2 (GraphPad Software, San Diego, California, USA) was used to prepare graphs for the frequently recognised minority variant epitopes among the different regions of the Pol protein, and also for showing the dynamics of these epitopes in sample pairs obtained during early and chronic HIV-1 infection stages.
Results
Demographics
This study enrolled 52 participants with HIV-1 infection, 34 of whom had paired plasma samples. Their median age was 28 years (IQR: 24–32 years), and majority were females (92.3%). There were 15 participants (28.9%) with early HIV-1 infection and 37 (71.1%) with chronic HIV-1 infection. The median HIV-1 VL at baseline was 2.8 × \({10}^{4}\) copies/mL (IQR: 8.6 × \({10}^{3}\)–9.1 × \({10}^{4}\) copies/mL). The median interval between baseline and follow-up sampling was 4 weeks (IQR: 3–8 weeks) (Table 1).
Amplification of HIV-1 pol and phylogenetic analysis
The complete HIV-1 pol gene was successfully amplified in all project samples using the in-house nested PCR method. Phylogenetic analysis of Illumina consensus sequences showed that most study sequences clustered with HIV-1 subtype C strains. All baseline and follow-up sequences (OK127761 – OK127810 and OK275500 – OK275537, respectively) for each participant correctly clustered together (Fig. 1), showing accurate amplification and sequencing of viral genes from the same participant.

Neighbour-joining phylogenetic tree of all samples including sample pairs. Group O strain of HIV-1 was used for rooting. Only bootstrap values above 70% are shown. Baseline and follow-up (fu) sequences of the same participant correctly clustered
Pol CTL epitopes based on Sanger consensus sequences
Eight CTL epitopes with amino acid residues under high selection pressure were identified. The majority of the identified epitopes were located within RT. The distribution of escape mutations was comparable between early and chronic HIV-1 disease stages (Table 2). However, these escape mutations were identified mostly in participants with chronic HIV-1 infection (Additional file 2: Table S2). One or more escape mutations were observed in each epitope. Two other codons were identified to be under high selection pressure but could not be mapped to the reported CTL epitopes in the Los Alamos HIV database (Table 2).
Pol CTL epitopes based on analysis of minority variants
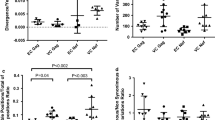
The median sequencing depth (for baseline and follow-up) was 23052X (IQR: 13,011–49753X) (Table 1). Minority variant analysis identified 65 frequently targeted Pol CTL epitopes. There was a significantly higher proportion of epitopes in RT (n = 39, 60.0%) compared to IN (n = 17, 26.2%) and PR (n = 9, 13.8%), p = 0.002 and < 0.0001, respectively. However, no significant difference was observed between the proportion of minority epitopes in IN versus PR, p = 0.06. The majority of these epitopes were identified in participants with chronic HIV-1 infection compared to those with early HIV-1 infection. Some epitopes were only identified in either early or chronic HIV-1 infection, whereas others were identified in both stages of disease (Fig. 2). Different distribution patterns of minority variant epitopes were observed. Some variant epitopes increased or decreased between baseline and follow-up, while others remained constant between these two time-points (Fig. 3, Additional file 3: Table S3). There were minority variants (2 in early and 26 in chronic HIV-1 infection) that could not be mapped to the reported CTL epitopes in the Los Alamos HIV database (Additional file 3: Table S3).

Frequently recognized Pol CTL epitopes in early and chronic HIV infection. These are epitopes that were recognized by more than one participant. A Epitopes within the protease (PR) region. B Epitopes within the reverse transcriptase (RT) region. C Epitopes within the integrase (IN) region. Many epitopes were located within RT and IN as compared to PR, and most were found in chronic HIV infection. CTL = cytotoxic T-lymphocytes

Dynamics of minority variants in sample pairs during A early HIV infection and B chronic HIV infection. (i) Some variants either increased in proportion between baseline and follow-up or remained at / above the detection limit of 5% (black solid line). (ii) Other variants decreased in proportion over time and were detected below 5% at follow-up. (iii) There were variants that increased in proportion over time to become majority variants and were thus maintained above 20% (black dotted line). (iv) Some minority variants that were detected at follow-up existed as majority variants at baseline. The median interval between baseline and follow-up sampling was 4 weeks (IQR: 3–8 weeks)
Pol CTL epitopes and predicted HLA class I presentation
HLA class I allotypes that possibly present the identified epitopes were predicted from the Los Alamos HIV database. Some participants recognised more epitopes than others. This was observed for participants 8047 and 6743 who each recognised seven epitopes during early HIV-1 infection. The most commonly targeted epitope during early HIV-1 infection was SAAVKAACW (IN 123–131), located in IN, and possibly presented by HLA-B*58:01 [27]. The same trend was noticed in chronic HIV-1 infection where the most frequently targeted epitope, MASDFNLPPIV (IN 22–31), was also located in IN and possibly presented by HLA-A*02 [27]. Some of the frequently targeted epitopes identified in this study have never been reported for HIV-1 subtype C. HLA class I allotypes that possibly present the identified epitopes in this study have been reported previously in the South African population [11, 12, 28,29,30,31,32]. Some epitopes were predicted to be recognised by multiple HLA class I allotypes. There were also frequently recognised epitopes that seemed to have similar HLA class I restriction such as ALQDSGLEV (RT 485–494) and RAMASDFNL (IN 20–28) in early HIV-1 infection (Table 3).
Discussion
To our knowledge, this is the first study to assess the evolution of HIV-1 Pol CTL epitopes in samples obtained during early and chronic disease stages in a setting where HIV-1 subtype C is predominant. We identified epitopes within all three regions of Pol, unlike previous studies that have only characterised epitopes within RT and PR [16, 29, 33]. This highlights the advantage of using a PCR method that amplifies the complete pol gene. Baseline and follow-up sequences for each participant correctly clustered together, indicating accurate amplification and sequencing of viral genes belonging to the same participant. It was expected that the majority of sequences would cluster with HIV-1 subtype C as this is the most common subtype in the southern African region [34, 35]. A small number of non-subtype C strains was detected, which could have been introduced through migration, and this has also been reported in previous South African studies [36, 37].
Many epitopes were identified through the analysis of minority variants obtained by deep sequencing compared to majority variants obtained by Sanger sequencing. This shows the advantage of using deep sequencing for characterisation of Pol CTL epitopes as this method can detect variants that exist below 20% of the total virus population, which are not readily detected by Sanger sequencing [38, 39]. Some identified minority variant epitopes have never been reported for HIV-1 subtype C and were possibly missed by previous studies that employed Sanger sequencing. The use of Sanger sequencing in most studies has most likely underestimated the presence of CTL epitopes in Pol. Other studies have also shown increased detection of CTL epitopes when using NGS methods [40, 41]. This shows an advantage in the use of NGS for characterising HIV-1 CTL epitopes in future research studies.
In this study, CTL epitopes were identified in all regions of Pol, but mostly in RT and IN. Some of these epitopes such as GKKAIGTVL (PR 68–76) and IAMESIVIW (RT 375–383) (Table 3) have been previously reported to induce CTL responses [42, 43]. These data indicate that all three regions of Pol are immunogenic and may play a role in the control of viraemia and the establishment of a viral set-point during early HIV-1 infection, in agreement with previous studies that assessed CTL responses against the HIV-1 proteome [42,43,44]. In a study that assessed CTL responses during acute HIV-1 infection, Kim et al.showed that the larger proportion of targeted epitopes were within Pol as compared to other regions of the HIV-1 proteome, and some of these epitopes stimulated strong CTL responses [42]. Ojwach et al.reported that CTL responses towards epitopes in RT and IN during acute and chronic HIV-1 infection induced mutations that decreased viral replicative fitness, indicating that RT and IN harbour crucial CTL epitopes [45]. Fewer epitopes were identified in PR especially during early HIV-1 infection and this could indicate that this protein harbours more subdominant CTL epitopes that are mostly recognised later during the course of infection [46]. Thus, PR might have a limited role in control of viraemia during early HIV-1 infection. CTL epitopes encoded by pol have also been reported to play a role in the control of viraemia in long-term non-progressors (LTNPs) and elite controllers (ECs) [47, 48].
HLA class I allotypes play an important role in the control of HIV-1 viraemia [29]. Several HLA class I alleles that encode allotypes predicted to present epitopes identified in this study are known to be present in the South African population, including HLA-B*58:01 and HLA-A*02 that present SAAVKAACW (IN 123–131) and MASDFNLPPIV (IN 22–31), respectively [28, 30, 49]. Past gene association studies showed that alleles within the HLA-B group correlate with greater viral control than alleles in the HLA-A and HLA-C groups [28, 29, 50]. Payne et al.found that HLA-B*58:01 was associated with slower HIV-1 disease progression and was protective against HIV-1 infection [51], which could explain why some participants such as 8047 and 6743 recognised more epitopes, as they also recognised an epitope reported to be presented by HLA-B*58:01. A hierarchy in epitope presentation may be an explanation for recognition of more epitopes during chronic HIV-1 infection. Immunodominant epitopes could be preferentially presented earlier during the course of infection, followed by presentation of subdominant epitopes later [52]. Immunodominant epitopes are often located in variable domains whereas subdominant epitopes usually map to more conserved regions of the HIV-1 proteome [18, 53]. The HIV-1 pol gene is more conserved than gag and env [54] and therefore harbours more subdominant epitopes [18]. Examples of subdominant responses being more effective in controlling viraemia have been shown in past studies [55, 56].
The evolution of Pol CTL epitopes was monitored by assessing minority variants in sample pairs. Minority variant epitopes that increased in proportion from baseline to follow-up may have replaced the wild-type epitopes over time and facilitated immune escape [57]. Fitness cost is the likely explanation for minority variant epitopes that decreased in proportion over time, and these may have been out-competed by new variants [57]. The proportion of some minority variant epitopes remained constant between the two time-points, and these could also represent variants that do not replicate efficiently and are hence maintained at lower levels within the HIV-1 quasispecies [40, 57]. Some variant sites were located outside of reported epitopes (Additional file 3: Table S3), and these may represent new unreported epitopes or could highlight important adjacent sites that may affect epitope processing and presentation [13, 58]. Viral evolution was also assessed by comparing sequences from early and chronic HIV-1 stages. Data from this study showed that evolution within CTL epitopes began quite early following infection, as epitopes with escape mutations were detected in participants who had early HIV-1 infection [59]. Many epitopes with escape mutations were detected in participants in the chronic HIV-1 stage, indicating that viral evolution of HIV-1 results from immune selection pressure that occurs throughout the course of infection and diverse viral populations evolve that constantly evade immune responses [57, 60].
All three expressed proteins (PR, RT and IN) of Pol harbour CTL epitopes and this highlights the importance of including pol in the design of a vaccine candidate [2, 20]. Some researchers have included RT and IN in the design of HIV-1 vaccines [22, 23, 53]; this is supported by the findings of our study as the majority of epitopes mapped to these two proteins. Conserved epitopes have been shown to provide cross-clade protection against HIV-1 infection [2, 18]. Some of the conserved Pol epitopes such as IETVEPVKL (RT 5–12) and SVPLDEGFRK (RT 117–126) (Table 3), that were previously observed in a study that looked at using a vaccine candidate with conserved immunogens were also identified in our study [18]. The immune responses directed towards epitopes in conserved regions are usually subdominant but may provide protection against multiple HIV-1 subtypes [17, 53]. Many studies that included Pol epitopes in vaccine candidates showed stimulation of effective CTL responses [18, 56, 61, 62]. Ahmed et al., showed control of HIV-1 replication in vitro through stimulation of Pol CTL responses by a vaccine that contained subdominant epitopes [56].
Limitations of this study include the small sample size and fewer participants with early HIV-1 infection. The duration of infection in participants with chronic HIV-1 infection was not known. HIV-1 VL was not measured in the participants at follow-up, therefore no correlation between VL and the number of minority variant epitopes detected could be made. Functional CTL responses against identified epitopes were not determined and genomic DNA samples were unavailable to conduct HLA class I genotyping. Longitudinal samples were only obtained at two time-points for most of the participants at a median interval of 4 weeks and this might not be sufficient for assessing viral evolution.
Conclusions
Deep sequencing revealed many HIV-1 Pol CTL epitopes, including some which have never been reported for HIV-1 subtype C. Immune selection pressure on Pol CTL epitopes was observed during early HIV-1 infection highlighting the possibility that these epitopes might contribute to the control of viraemia and the establishment of a VL set-point. Variable patterns of distribution of epitopes in sample pairs reflect the ongoing generation of escape mutants capable of evading the immune responses throughout the course of infection. The findings of this study support the inclusion of RT and IN in potential HIV-1 vaccine candidates, as these regions harbour the majority of CTL epitopes. Variants that could not be mapped to known epitopes in the Los Alamos HIV database might represent new unreported epitopes. Future research, using deep sequencing, is needed for better characterisation of Pol epitopes as this protein contains highly conserved epitopes that have the potential to provide HIV-1 cross-clade protection.
Availability of data and materials
All data generated or analysed during this study are included in this manuscript and its supplementary information files.
Change history
05 May 2022
A Correction to this paper has been published: https://doi.org/10.1186/s12985-022-01803-4
Abbreviations
- HIV-1:
-
Human immunodeficiency virus type 1
- PLWH:
-
People living with HIV
- CTL:
-
Cytotoxic T-lymphocyte
- VL:
-
Viral load
- HLA I:
-
Human leukocyte antigen type 1
- ART:
-
Antiretroviral therapy
- RCF:
-
Relative centrifugal force
- RT-PCR:
-
Reverse transcriptase polymerase chain reaction
- RNA:
-
Ribonucleic acid
- DNA:
-
Deoxyribonucleic acid
- dNTPs:
-
Deoxynucleotide triphosphate
- ORF:
-
Open reading frame
- NGS:
-
Next generation sequencing
- IQR:
-
Interquartile range
- Gag:
-
Group-specific antigen
- Pol:
-
Polymerase
- Env:
-
Envelope
- PR:
-
Protease
- RT:
-
Reverse transcriptase
- IN:
-
Integrase
- LTNP:
-
Long-term non-progressors
- EC:
-
Elite controllers
References
Greene WC. A history of AIDS: looking back to see ahead. Eur J Immunol. 2008. https://doi.org/10.1002/eji.200737441.
Fauci AS, Marston HD. Ending AIDS-is an HIV vaccine necessary? N Engl J Med. 2014;370:495–8. https://doi.org/10.1056/NEJMp1313771.
UNAIDS [Internet]. Fact Sheet 2021 - Preliminary epidemiological estimates. 2021 [accessed July 2021]. Available from: https://www.unaids.org/sites/default/files/media_asset/UNAIDS_FactSheet_en.pdf.
UNAIDS [Internet]. Global 2019 HIV/AIDS Statistics. 2020 [accessed May 2021]. Available from: https://www.unaids.org/en/resources/documents/2020/unaids-data.
HSRC. The fifth South African national HIV prevalence, incidence, behaviour and communication survey, 2017: HIV impact assessment summary report. HSRC Press Cape Town; 2018.
Tiemessen CT, Martinson N. Elite controllers: understanding natural suppressive control of HIV-1 infection. CMEJ. 2012;30:282–5.
Gulzar N, Copeland KF. CD8+ T-cells: function and response to HIV infection. Curr HIV Res. 2004;2:23–37. https://doi.org/10.2174/1570162043485077.
Collins DR, Gaiha GD, Walker BD. CD8+ T cells in HIV control, cure and prevention. Nat Rev Immunol. 2020;20:471–82. https://doi.org/10.1038/s41577-020-0274-9.
Perdomo-Celis F, Taborda NA, Rugeles MT. CD8(+) T-cell response to HIV infection in the era of antiretroviral therapy. Front Immunol. 2019;10:1896. https://doi.org/10.3389/fimmu.2019.01896.
McMichael AJ, Borrow P, Tomaras GD, Goonetilleke N, Haynes BF. The immune response during acute HIV-1 infection: clues for vaccine development. Nat Rev Immunol. 2010;10:11–23. https://doi.org/10.1038/nri2674.
Tshabalala M, Mellet J, Pepper MS. Human Leukocyte Antigen diversity: A Southern African perspective. J Immunol Res. 2015;2015: 746151. https://doi.org/10.1155/2015/746151.
Hammond M, Anley D, editors. Tamil from Natal province South Africa. 13th International Histocompatibility Workshop; 2006; Seattle. Washington, USA: International Histocompatibility Working Group Press.
Allen TM, Altfeld M, Yu XG, O’Sullivan KM, Lichterfeld M, Le Gall S, et al. Selection, transmission, and reversion of an antigen-processing cytotoxic T-lymphocyte escape mutation in human immunodeficiency virus type 1 infection. J Virol. 2004;78:7069–78. https://doi.org/10.1128/jvi.78.13.7069-7078.2004.
Draenert R, Le Gall S, Pfafferott KJ, Leslie AJ, Chetty P, Brander C, et al. Immune selection for altered antigen processing leads to cytotoxic T lymphocyte escape in chronic HIV-1 infection. J Exp Med. 2004;199:905–15. https://doi.org/10.1084/jem.20031982.
Kiepiela P, Ngumbela K, Thobakgale C, Ramduth D, Honeyborne I, Moodley E, et al. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat Med. 2007;13:46–53. https://doi.org/10.1038/nm1520.
Masemola A, Mashishi T, Khoury G, Mohube P, Mokgotho P, Vardas E, et al. Hierarchical targeting of subtype C human immunodeficiency virus type 1 proteins by CD8+ T cells: correlation with viral load. J Virol. 2004;78(7):3233–43. https://doi.org/10.1128/jvi.78.7.3233-3243.2004.
Kunwar P, Hawkins N, Dinges WL, Liu Y, Gabriel EE, Swan DA, et al. Superior control of HIV-1 replication by CD8+ T cells targeting conserved epitopes: implications for HIV vaccine design. PLoS ONE. 2013;8: e64405. https://doi.org/10.1371/journal.pone.0064405.
Borthwick N, Lin Z, Akahoshi T, Llano A, Silva-Arrieta S, Ahmed T, et al. Novel, in-natural-infection subdominant HIV-1 CD8+ T-cell epitopes revealed in human recipients of conserved-region T-cell vaccines. PLoS ONE. 2017;12:e0176418. https://doi.org/10.1371/journal.pone.0176418.
Acevedo-Sáenz L, Ochoa R, Rugeles MT, Olaya-García P, Velilla-Hernández PA, Diaz FJ. Selection pressure in CD8+ T-cell epitopes in the pol gene of HIV-1 infected individuals in Colombia. A bioinformatic approach. Viruses. 2015;7:1313–31. https://doi.org/10.3390/v7031313.
Hsu DC, O’Connell RJ. Progress in HIV vaccine development. Hum Vaccin Immunother. 2017;13:1018–30. https://doi.org/10.1080/21645515.2016.1276138.
Harmon TM, Fisher KA, McGlynn MG, Stover J, Warren MJ, Teng Y, et al. Exploring the potential health impact and cost-effectiveness of AIDS Vaccine within a comprehensive HIV/AIDS response in low- and middle-income countries. PLoS ONE. 2016;11:e0146387. https://doi.org/10.1371/journal.pone.0146387.
Buchbinder SP, Mehrotra DV, Duerr A, Fitzgerald DW, Mogg R, Li D, et al. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): a double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet. 2008;372:1881–93. https://doi.org/10.1016/s0140-6736(08)61591-3.
Gray GE, Allen M, Moodie Z, Churchyard G, Bekker LG, Nchabeleng M, et al. Safety and efficacy of the HVTN 503/Phambili study of a clade-B-based HIV-1 vaccine in South Africa: a double-blind, randomised, placebo-controlled test-of-concept phase 2b study. Lancet Infect Dis. 2011;11:507–15. https://doi.org/10.1016/s1473-3099(11)70098-6.
Priddy FH, Brown D, Kublin J, Monahan K, Wright DP, Lalezari J, et al. Safety and immunogenicity of a replication-incompetent adenovirus type 5 HIV-1 clade B gag/pol/nef vaccine in healthy adults. Clin Infect Dis. 2008;46:1769–81. https://doi.org/10.1086/587993.
Stephenson KE, Wagh K, Korber B, Barouch DH. Vaccines and broadly neutralizing antibodies for HIV-1 prevention. Annu Rev Immunol. 2020;38:673–703. https://doi.org/10.1146/annurev-immunol-080219-023629.
Mayaphi SH. Detection and characterisation of primary (acute and early) HIV-1 infections in an HIV hyper-endemic area [Thesis]. Pretoria: University of Pretoria; 2018.
LANL [Internet]. HIV molecular immunology database. Los Alamos National Laboratory (NIH); 2021. Available from: https://www.hiv.lanl.gov/content/immunology/tables/ctl_summary.html.
Leslie A, Matthews PC, Listgarten J, Carlson JM, Kadie C, Ndung’u T, et al. Additive contribution of HLA class I alleles in the immune control of HIV-1 infection. J Virol. 2010;84:9879–88. https://doi.org/10.1128/JVI.00320-10.
Carlson JM, Listgarten J, Pfeifer N, Tan V, Kadie C, Walker BD, et al. Widespread impact of HLA restriction on immune control and escape pathways of HIV-1. J Virol. 2012;86:5230–43. https://doi.org/10.1128/JVI.06728-11.
Loubser S, Paximadis M, Gentle NL, Puren A, Tiemessen CT. Human leukocyte antigen class I (A, B, C) and class II (DPB1, DQB1, DRB1) allele and haplotype variation in Black South African individuals. Hum Immunol. 2020;81:6–7. https://doi.org/10.1016/j.humimm.2019.12.003.
Paximadis M, Mathebula TY, Gentle NL, Vardas E, Colvin M, Gray CM, et al. Human Leukocyte Antigen class I (A, B, C) and II (DRB1) diversity in the black and Caucasian South African population. Hum Immunol. 2012;73:80–92. https://doi.org/10.1016/j.humimm.2011.10.013.
Coetzee V, Barrett L, Greeff JM, Henzi SP, Perrett DI, Wadee AA. Common HLA alleles associated with health, but not with facial attractiveness. PLoS ONE. 2007;2: e640. https://doi.org/10.1371/journal.pone.0000640.
Brumme ZL, Brumme CJ, Carlson J, Streeck H, John M, Eichbaum Q, et al. Marked epitope-and allele-specific differences in rates of mutation in human immunodeficiency type 1 (HIV-1) gag, pol, and nef cytotoxic T-lymphocyte epitopes in acute/early HIV-1 infection. J Virol. 2008;82:9216–27. https://doi.org/10.1128/JVI.01041-08.
Bbosa N, Kaleebu P, Ssemwanga D. HIV subtype diversity worldwide. Curr Opin HIV AIDS. 2019;14:153–60. https://doi.org/10.1097/coh.0000000000000534.
Buonaguro L, Tornesello M, Buonaguro F. Human immunodeficiency virus type 1 subtype distribution in the worldwide epidemic: pathogenetic and therapeutic implications. J Virol. 2007;81:10209–19. https://doi.org/10.1128/JVI.00872-07.
Jacobs GB, Wilkinson E, Isaacs S, Spies G, De Oliveira T, Seedat S, et al. HIV-1 subtypes B and C unique recombinant forms (URFs) and transmitted drug resistance identified in the Western Cape Province, South Africa. PLoS ONE. 2014;9:e90845. https://doi.org/10.1371/journal.pone.0090845.
Heipertz RA Jr, Ayemoba O, Sanders-Buell E, Poltavee K, Pham P, Kijak GH, et al. Significant contribution of subtype G to HIV-1 genetic complexity in Nigeria identified by a newly developed subtyping assay specific for subtype G and CRF02_AG. Medicine. 2016;95(32):e4346.
Kumar KR, Cowley MJ, Davis RL. Next-generation sequencing and emerging technologies. Semin Thromb Hemost. 2019;45(7):661–73. https://doi.org/10.1055/s-0039-1688446.
Pareek CS, Smoczynski R, Tretyn A. Sequencing technologies and genome sequencing. J Appl Genet. 2011;52(4):413–35. https://doi.org/10.1007/s13353-011-0057-x.
Caetano DG, Côrtes FH, Bello G, Teixeira SLM, Hoagland B, Grinsztejn B, et al. Next-generation sequencing analyses of the emergence and maintenance of mutations in CTL epitopes in HIV controllers with differential viremia control. BMC Retrovirol. 2018;15(1):62. https://doi.org/10.1186/s12977-018-0444-z.
Tumiotto C, Riviere L, Bellecave P, Recordon-Pinson P, Vilain-Parce A, Guidicelli GL, et al. Sanger and Next-Generation Sequencing data for characterization of CTL epitopes in archived HIV-1 proviral DNA. PLoS ONE. 2017;12(9): e0185211. https://doi.org/10.1371/journal.pone.0185211.
Kim J, De La Cruz J, Lam K, Ng H, Daar ES, Balamurugan A, et al. CD8(+) cytotoxic T lymphocyte responses and viral epitope escape in acute HIV-1 infection. Viral Immunol. 2018;31(7):525–36. https://doi.org/10.1089/vim.2018.0040.
Li F, Finnefrock AC, Dubey SA, Korber BT, Szinger J, Cole S, et al. Mapping HIV-1 vaccine induced T-cell responses: bias towards less-conserved regions and potential impact on vaccine efficacy in the Step study. PLoS ONE. 2011;6: e20479. https://doi.org/10.1371/journal.pone.0020479.
Goonetilleke N, Liu MK, Salazar-Gonzalez JF, Ferrari G, Giorgi E, Ganusov VV, et al. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J Exp Med. 2009;206:1253–72. https://doi.org/10.1084/jem.20090365.
Ojwach DBA, MacMillan D, Reddy T, Novitsky V, Brumme ZL, Brockman MA, et al. Pol-driven replicative capacity impacts disease progression in HIV-1 subtype C infection. J Virol. 2018. https://doi.org/10.1128/jvi.00811-18.
Rhee SY, Sankaran K, Varghese V, Winters MA, Hurt CB, Eron JJ, et al. HIV-1 protease, reverse transcriptase, and integrase variation. J Virol. 2016;90(13):6058–70. https://doi.org/10.1128/jvi.00495-16.
Miura T, Brumme ZL, Brockman MA, Rosato P, Sela J, Brumme CJ, et al. Impaired replication capacity of acute/early viruses in persons who become HIV controllers. J Virol. 2010;84(15):7581–91. https://doi.org/10.1128/jvi.00286-10.
Pernas M, Casado C, Arcones C, Llano A, Sánchez-Merino V, Mothe B, et al. Low-replicating viruses and strong anti-viral immune response associated with prolonged disease control in a superinfected HIV-1 LTNP elite controller. PLoS ONE. 2012;7(2):e31928. https://doi.org/10.1371/journal.pone.0031928.
Mellet J, Tshabalala M, Agbedare O, Meyer P, Gray CM, Pepper MS. Human leukocyte antigen (HLA) diversity and clinical applications in South Africa. S Afr Med J. 2019;109:29–34. https://doi.org/10.7196/SAMJ.2019.v109i8b.13825.
Goulder PJ, Walker BD. HIV and HLA class I: an evolving relationship. Immunity. 2012;37:426–40. https://doi.org/10.1016/j.immuni.2012.09.005.
Payne R, Muenchhoff M, Mann J, Roberts HE, Matthews P, Adland E, et al. Impact of HLA-driven HIV adaptation on virulence in populations of high HIV seroprevalence. Proc Natl Acad Sci USA. 2014;111(50):E5393–400. https://doi.org/10.1073/pnas.1413339111.
Adland E, Hill M, Lavandier N, Csala A, Edwards A, Chen F, et al. Differential Immunodominance Hierarchy of CD8(+) T-cell responses in HLA-B*27:05- and -B*27:02-mediated control of HIV-1 infection. J Virol. 2018. https://doi.org/10.1128/jvi.01685-17.
Liu Y, McNevin J, Rolland M, Zhao H, Deng W, Maenza J, et al. Conserved HIV-1 epitopes continuously elicit subdominant cytotoxic T-lymphocyte responses. J Infect Dis. 2009;200(12):1825–33. https://doi.org/10.1086/648401.
Tebit DM, Arts EJ. Tracking a century of global expansion and evolution of HIV to drive understanding and to combat disease. Lancet Infect Dis. 2011;11:45–56. https://doi.org/10.1016/S1473-3099(10)70186-9.
Friedrich TC, Valentine LE, Yant LJ, Rakasz EG, Piaskowski SM, Furlott JR, et al. Subdominant CD8+ T-cell responses are involved in durable control of AIDS virus replication. J Virol. 2007;81(7):3465–76. https://doi.org/10.1128/jvi.02392-06.
Ahmed T, Borthwick NJ, Gilmour J, Hayes P, Dorrell L, Hanke T. Control of HIV-1 replication in vitro by vaccine-induced human CD8(+) T cells through conserved subdominant Pol epitopes. Vaccine. 2016;34(9):1215–24. https://doi.org/10.1016/j.vaccine.2015.12.021.
Liu Y, McNevin J, Zhao H, Tebit DM, Troyer RM, McSweyn M, et al. Evolution of human immunodeficiency virus type 1 cytotoxic T-lymphocyte epitopes: fitness-balanced escape. J Virol. 2007;81:12179–88. https://doi.org/10.1128/jvi.01277-07.
Yokomaku Y, Miura H, Tomiyama H, Kawana-Tachikawa A, Takiguchi M, Kojima A, et al. Impaired processing and presentation of cytotoxic-T-lymphocyte (CTL) epitopes are major escape mechanisms from CTL immune pressure in human immunodeficiency virus type 1 infection. J Virol. 2004;78(3):1324–32. https://doi.org/10.1128/jvi.78.3.1324-1332.2004.
Sun J, Zhao Y, Peng Y, Han Z, Liu G, Qin L, et al. Multiple T-cell responses are associated with better control of acute HIV-1 infection: An observational study. Medicine. 2016;95: e4429. https://doi.org/10.1097/md.0000000000004429.
Koibuchi T, Allen TM, Lichterfeld M, Mui SK, O’Sullivan KM, Trocha A, et al. Limited sequence evolution within persistently targeted CD8 epitopes in chronic human immunodeficiency virus type 1 infection. J Virol. 2005;79(13):8171–81. https://doi.org/10.1128/jvi.79.13.8171-8181.2005.
Mohamed YS, Borthwick NJ, Moyo N, Murakoshi H, Akahoshi T, Siliquini F, et al. Specificity of CD8+ T-Cell responses following vaccination with conserved regions of HIV-1 in Nairobi. Kenya Vaccine. 2020;8:260. https://doi.org/10.3390/vaccines8020260.
Zou C, Murakoshi H, Kuse N, Akahoshi T, Chikata T, Gatanaga H, et al. Effective suppression of HIV-1 replication by cytotoxic T lymphocytes specific for pol epitopes in conserved mosaic vaccine immunogens. J Virol. 2019. https://doi.org/10.1128/jvi.02142-18.
Acknowledgements
We thank Prof. Marietjie Venter and Dr. Adriano Mendes for granting us access to their CLC Genomics software program.
Funding
PN was funded by the National Research Foundation (NRF) and Poliomyelitis Research Foundation (PRF grant number: 20/22). SHM received funding from Discovery Foundation, National Health Laboratory Service Research Trust (NHLS-RT), South African Medical Research Council Self-Initiated Research (MRC-SIR), and University of Pretoria Faculty of Health Sciences Research Committee. CTT receives funding through the South African Research Chairs Initiative of the Department of Science and Innovation and National Research Foundation of South Africa (84177). TCQ and ADR—The Division of Intramural Research, NIAID, NIH.
Author information
Authors and Affiliations
Contributions
Conceptualization: SHM. Methodology: PN, SHM. Funding acquisition: PN, SHM. Investigation: PN, SHM. Supervision: SHM, CTT. Project administration: PN, SHM. Data analysis: PN, SHM, CTT, TCQ, SL, AR, AI. Writing—original draft: PN. Writing—review & editing: PN, SHM, CTT, TCQ, SL, AR, AI. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethics approval was obtained for this study from the Faculty of Health Sciences Research Ethics Committee, University of Pretoria, South Africa: Reference number 282/2020. Written consent was obtained from all participants in the parent study (Ethics reference: 295/2015) [26].
Consent for publication
Not applicable.
Competing interests
The authors declared no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised: the epitopes from Table 3 have been corrected.
Supplementary Information
Additional file 1. Supplementary Table 1:
The in-house complete HIV pol nested PCR conditions and Sanger sequencing primers.
Additional file 2. Supplementary Table 2:
Pol CTL epitopes identified through Sanger sequencing and comparison by stage of infection
Additional file 3. Supplementary Table 3:
Minority variant proportions within Pol CTL epitopes by stage of infection.
Additional file 4. Supplementary Figure 1:
Neighbour-joining phylogenetic analysis of Illumina consensus and Sanger consensus sequences for baseline samples. Sanger and Illumina consensus of the same sample significantly clustered together. Majority of the sequences clustered with HIV-1 subtype C references. HIV group O was used for rooting the tree and a bootstrap value of 1000 was used for analysis. S = Sanger sequencing; D = Deep sequencing.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Nkone, P., Loubser, S., Quinn, T.C. et al. Deep sequencing of the HIV-1 polymerase gene for characterisation of cytotoxic T-lymphocyte epitopes during early and chronic disease stages. Virol J 19, 56 (2022). https://doi.org/10.1186/s12985-022-01772-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-022-01772-8