
Abstract
Background
Aphids are important vectors of numerous plant viruses. Besides plant viruses, a number of insect specific viruses (ISVs), such as nege/nege-like viruses, have been recently discovered in aphids of the genera Aphis, Rhopalosiphum, and Sitobion.
Findings
In this study, the complete genome sequence of a novel nege-like virus, tentatively named “Indomegoura nege-like virus 1” (INLV1), was identified in aphids of the genus Indomegoura. INLV1 possessed a single positive-stranded RNA genome with 8945 nucleotides, which was predicted to contain three typical open reading frames (ORFs) of negeviruses (including ORF1, ORF2, and ORF3), a 44-nt 5′ untranslated region (UTR) and a 98-nt 3′ UTR. Five conserved domains were predicted for INLV1, including an Alphavirus-like methyltransferase domain, a RNA virus helicase core domain, and a RNA-dependent RNA polymerase domain (RdRP) in ORF1, a DISB-ORF2_chro domain in ORF2, and a SP24 domain in ORF3. According to the maximum likelihood phylogenetic tree based on RdRP, INLV1 was grouped with barley aphid RNA virus 1 and Hubei virga-like virus 4, together with another two invertebrate viruses, which formed a distinct clade in the proposed group Centivirus. The alignment of RdRP domains for INLV1 and other nege/kita-like viruses suggested that RdRP of INLV1 contained the permuted C (GDD)- A [DX(4–5)D] –B [GX(2–3)TX(3)N] motifs, which were conserved in the Centivirus and Sandewavirus groups. Furthermore, the high abundance and typical characteristics of INLV1 derived small interfering RNAs clearly showed the active replication of INLV1 in the aphid Indomegoura.
Conclusion
INLV1 is the first nege-like virus infecting aphids of the genus Indomegoura. As far as we know, it is also the first ISV revealed in this aphid genus.
Similar content being viewed by others


Background
Aphids, which belong to the order Hemiptera, family Aphididae, are one of the most serious pests of agricultural and horticultural crops. Aphids will cause damage through direct feeding or by means of vectors of many important plant viruses [1]. Apart from plant viruses, recent studies have indicated that aphids are known to harbor numerous novel insect specific viruses (ISVs) belonging to the families Dicistroviridae and Iflaviridae, and others from various unclassified taxa [2,3,4,5,6,7]. Negeviruses are a newly proposed group of ISVs well-known for their wide geographic distribution and broad host range [8,9,10]. The genome of negeviruses is a single positive-sense RNA with the size of 9–10 kb, which encodes three open reading frames (ORFs). Negeviruses are currently classified into two distinct phylogenetic clades called Nelorpivirus and Sandewavirus, and they are also closely related to plant viruses in the family Kitaviridae [11,12,13].
A few nege/nege-like viruses have hitherto been reported in aphids. The first aphid nege-like virus was discovered in soybean aphid (Aphis glycines), which was obtained from Ohio State University using Next Generation Sequencing [14]. More recently, a number of nege/kita-like viruses have been discovered in the other two genera of aphids (Rhopalosiphum and Sitobion). According to phylogenetic analysis, the nege/kita-like viruses infecting these two aphid genera can be classified into two new distinct clades, tentatively designated as Centivirus and Aphiglyvirus, respectively [13].
Main text
In this study, a novel nege-like virus was discovered in aphids of the genus Indomegoura. The aphids were harvested from the host plant Hemerocallis fulva at Ningbo University, Ningbo, China in 2020. Then, we used TRIzol reagent (Invitrogen, MA, USA) to extract total RNA from a pool of ten aphids. The Nano Drop spectrophotometer (Thermo Scientific, MA, USA) was used to determine the RNA content. Paired-end (150 bp) sequencing of the RNA library was performed using the Illumina HiSeq 4000 sequencer (Novogene, Tianjin, China). Afterwards, the 22,045,205 pairs of raw reads generated were subjected to quality trimming and de novo assembly by adopting Trinity (version 2.8.5) with the default parameters [15]. To determine the accurate aphid species, all the 63,158 assembled contigs were compared with cytochrome oxidase subunit 1 (COI) records derived from the Barcode of Life Data (BOLD) Systems (http://www.boldsystems.org/), and later the potential aphid COI sequence was extracted. The aphid COI sequence was then compared with the nucleotide (nt) database in NCBI, which showed high homology (97% similarities) to the COI of Indomegoura indica (Accession number: NC_045897.1), confidentially indicating that the collected aphid species were highly similar to I. indica and belonged to the genus Indomegoura. The COI sequence of aphids Indomegoura was further confirmed by Sanger sequencing and stored in GenBank under the accession number MW533423 (Additional file 1: File S1).
To identify the potential viral-like contigs in the transcriptome, the assembled contigs were searched against the local generated virus database with the sequences retrieved from NCBI viral reference database (https://www.ncbi.nlm.nih.gov/genome/viruses). As a result, a confidently nege-like viral contig was discovered in aphids, which represented almost the complete viral genome with the length of 8876 nt. To investigate the transcript abundance and coverage of the contig, the adaptor- and quality-trimmed reads from the transcriptome were mapped back to this contig using Bowtie2 and Samtools. As a result, high coverage (290X) was confirmed for this nege-like viral contig. Thereafter, the identified viral contig was further compared with the entire NCBI nucleotide (NT) and non-redundant (NR) protein database to avoid false positive results (Additional file 3: Table S1). Then, the viral contig was confirmed with reverse transcription-PCR (RT-PCR), followed by Sanger sequencing. Furthermore, the full genome of the nege-like virus was successfully achieved by the rapid amplification of cDNA ends (RACE) with SMARTer® RACE 5′/3′ kit (Takara, Dalian, China). The primers used for RT-PCR and RACE are listed in Additional file 4: Table S2. The novel nege-like virus from aphids of genus Indomegoura was temporarily named “Indomegoura nege-like virus 1” (INLV1), and its full genome sequence was deposited in GenBank with the accession number MW285725 (Additional file 2: File S2).
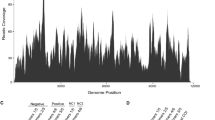
The RT-PCR and Sanger sequencing results confirmed the sequences of the assembled viral-like contig (with a few corrections of the nucleotides). Furthermore, the complete 5′ and 3′ untranslated region (UTR) were obtained using RACE technology followed by Sanger sequencing, and the full genome sequences of INLV1 was successfully reconstructed. INLV1 had a genome size of 8945 nt (excluding polyA), which was the most homologous to Hubei virga-like virus 4 (HVLV-4) (accession number APG77770.1) and barley aphid RNA virus 1 (BARV-1) (accession number BBV14745.1), with the amino acid (aa) sequence identities of 59.00% and 58.47%, respectively. In terms of the genome organization, INLV1 contained three typical negevirus ORFs (ORF1, ORF2, and ORF3) predicted using the Expasy online server (https://web.expasy.org/translate/), a 44-nt 5′ UTR and a 98-nt 3′ UTR (nucleotide position in the genome: 8848–8945 nt) (Fig. 1a). Additionally, the conserved domains predicted using InterProScan (https://www.ebi.ac.uk/interpro) suggested that the long ORF1 (nucleotide position in the genome: 45-6908 nt) consisted of an Alphavirus-like methyltransferase domain (vMet, IPR002588), a RNA virus helicase core domain (HEL, PF01443), and a RNA-dependent RNA polymerase domain (RdRP, PF00978). In addition, RNA ribosomal methyltransferase domain (FstJ), which was demonstrated to be present or absent in various negeviruses [10, 12, 13], was not detected in the ORF1 of INLV1, indicating that FstJ might not be well-conserved in the taxon Negevirus. ORF2 and ORF3 of INLV1 possessed the conserved domains of DiSB-ORF2_chro (a putative virion glycoprotein, PF16506) and SP24 (a putative virion membrane protein, PF16504), respectively (Fig. 1a), which were similar to another negevirus isolated from Aedes vexans mosquitoes in Finland [16]. According to previous studies, overlaps between different ORFs of negeviruses are common [8, 10]. In our study, an overlap between ORF1 and ORF2 by 263-nt was also found with different frames in INLV1 (Fig. 1a). To further understand the abundance and coverage of sequenced reads derived from INLV1, we realigned the RNA-seq reads to the confirmed full genome of INLV1. Noteworthily, viral reads were apparently accumulated within the 3′ region of the genome, especially in ORF3 (Fig. 1a), consistent with the recently reported negeviruses discovered in a dungfly [10]. In addition, the transmembrane domains of INLV1 ORF3 were predicted by the TMHMM server v. 2.0 (http://www.cbs.dtu.dk/services/TMHMM/). As a result, the four transmembrane domains were evidently present in the ORF3 of INLV1 (Additional file 6: Figure S1), indicating that SP24 was probably an integral membrane protein of INLV1, conforming to previous report [17].

a Genome organization and transcriptome raw read coverage of Indomegoura nege-like virus 1 (INLV1). vMet, Alphavirus-like methyltransferase domain; HEL, RNA virus helicase core domain; RdRp, RNA-dependent RNA polymerase domain; UTR, untranslated region; IR, intergenic region. b Maximum likelihood phylogenetic tree based on the RdRp domain of INLV1, previously reported representative nege/nege-like viruses, and plant viruses in the families Kitaviridae and Virgaviridae
To further evaluate the taxonomical status of INLV1, we aligned the conserved RdRP domain of INLV1 and the previously reported nege/nege-like viruses by MAFFT (version 7.450), and further trimmed the gaps by Gblock [18]. Besides, the substitution model was evaluated by ModelTest-NG, and a maximum likelihood (ML) tree was constructed using IQ-tree with 1000 bootstrap replications [19, 20]. Two plant viruses in the family Virgaviridae, Tobacco mosaic virus (NP_597746.1) and Cucumber green mottle mosaic virus (NP_044577.1), were used as outgroup. According to recent phylogenetic study on aphid nege/kita-like viruses, it is proposed that the two newly identified groups, Centivirus and Aphiglyvirus, together with the Negevirus subgroups (Nelorpivirus and Sandewavirus), can be classified into a novel viral family or assigned to the family Kitaviridae [13]. In this study, the reconstructed phylogenetic ML tree based on the viral RdRP domain sequences indicated that INLV1 was clearly grouped with BARV-1 and HVLV-4, together with another two invertebrate viruses, which formed a distinct group in the clade Centivirus closely related to Aphiglyvirus (Fig. 1b).Using MegAlign (version 7.1.0) and BioEdit Sequence Alignment Editor (version 7.1.11) [21], we aligned INLV1 and the related viruses based on the predicted RdRP protein/nucleotide sequences of INLV1 and the previously reported nege/kita-like viruses, so as to determine the homology of INLV1 with the related viruses. Previous study indicates that the RdRP of nege/kita-like viruses contains three conserved motifs, namely, motif A [DX(4–5)D], motif B [GX(2–3)TX(3)N], and motif C (GDD), in the canonical order A-B-C or the permuted order C-A-B [22]. In our study, these two motif types of nege/kita-like viruses were also observed, and the RdRP domain of INLV1 showed the clear permuted C-A-B motif pattern (Additional file 7: Figure S2). More interestingly, the canonical A-B-C type of RdRPs exclusively belonged to the groups Nelorpivirus and Aphiglyvirus, as well as the plant virus of families Kitaviridae and Virgaviridae, whereas the permuted C-A-B pattern was observed in the groups Centivirus and Sandewavirus (Additional file 7: Figure S2), consistent with the taxonomical status of each group in the phylogenic tree (Fig. 1b). Furthermore, we compared the aa/nt identity of INLV1 RdRP sequences with other reported nege/kita-like viruses. As a result, INLV1 was the most closely related to BARV-1 and HVLV-4 in the group Centivirus, with the aa (nt) identities of 77.2% (69.2%) and 76.3% (69.0%), respectively (Table 1). For the phylogenetically related aphiglyviruses (Fig. 1b), INLV1 shared 31.4–32.9% (aa) and 47.5–50.3% (nt) identities (Table 1).
To explore small interfering RNA (siRNA) based anti-viral immunity in aphid host, small RNAs (sRNA) of the aphids were sequenced and virus derived siRNAs (vsiRNAs) were comprehensively characterized. In brief, a sRNA library was prepared using the Illumina TruSeq small RNA sample preparation kit (Illumina, San Diego, CA, USA), and sRNA sequencing was performed by Novogene on an Illumina HiSeq 2500 platform. The sRNA reads were pretreated (removal of adapters, low quality, and junk sequences) and sRNAs with the length of 18-nt to 30-nt were extracted. The processed sRNA reads were mapped back to the full viral genome sequence of INLV1 using Bowtie with zero mismatches. vsiRNAs were further analyzed using the custom perl scripts and the Linux bash scripts. As a result, a total of 13,203 (4,181 unique) vsiRNAs perfectly mapped to INLV1 genome were identified, accounting for 0.06% (0.48% unique) of the sRNA library. The vsiRNAs were mostly 22-nt long (accounting for 69.0% and 48.1% of total and unique vsiRNAs, respectively), and they were equally derived from the sense and antisense strands of the viral genome (Fig. 2a, b). Besides, equal distribution alongside the viral genome and a strong A/U bias in the 5′-terminal nucleotide of vsiRNAs was also observed (Fig. 2c, d), which have been characterized for vsiRNAs derived from various organisms, including insects [23]. These typical characteristics of INLV1 derived siRNAs strongly suggested that the active involvement of RNA interference antiviral pathway in the aphid genus Indomegoura.

Analysis of virus derived small interfering RNAs (vsiRNAs) of INLV1. The size distribution of INLV1-derived siRNAs of total reads (a) or unique reads (b). c Distribution of INLV1-derived siRNA on the viral genome. d 5′ terminal nucleotide preference of siRNAs derived from INLV1. Black represents siRNAs derived from the sense genomic strand (Plus), and red represents small RNAs derived from the antisense genomic strand (Minus)
Conclusions
In addition to the recent discoveries of nege/nege-like viruses in various aphid genera including Aphis, Rhopalosiphum, and Sitobion, INLV1 provides the first report on a novel nege-like virus in another aphid genus Indomegoura. Our results imply that the actual diversity of nege/nege-like viruses in aphids may still be largely undetermined, and the associations between different aphid species and nege/nege-like viruses will be of great interest in future investigation. More intriguingly, it is necessary to further explore the effects of these nege/nege-like viruses (such as INLV1) on aphid competence, and to evaluate whether they can be used as the biological agents to control aphid-borne plant viruses in the field.
Availability of data and materials
The COI sequence of aphids used in this study was stored in GenBank under the accession number MW533423. The full genome sequence of Indomegoura nege-like virus 1 was deposited in GenBank with the accession number MW285725.
Abbreviations
- ISV:
-
Insect specific virus
- INLV1:
-
Indomegoura nege-like virus 1
- ORF:
-
Open reading frame
- UTR:
-
Untranslated region
- RdRP:
-
RNA-dependent RNA polymerase
- COI:
-
Cytochrome oxidase subunit 1
- RACE:
-
Rapid amplification of cDNA ends
- ML:
-
Maximum likelihood
- siRNA:
-
Small interfering RNA
References
Carr JP, Tungadi T, Donnelly R, Bravo-Cazar A, Rhee SJ, Watt LG, Mutuku JM, Wamonje FO, Murphy AM, Arinaitwe W, Pate AE, Cunniffe NJ, Gilligan CA. Modelling and manipulation of aphid-mediated spread of non-persistently transmitted viruses. Virus Res. 2020;277:197845.
Moon JS, Domier LL, McCoppin NK, D’Arcy CJ, Jin H. Nucleotide sequence analysis shows that Rhopalosiphum padi virus is a member of a novel group of insect-infecting RNA viruses. Virology. 1998;243:54–65.
Ryabov EV. A novel virus isolated from the aphid Brevicoryne brassicae with similarity to Hymenoptera picorna-like viruses. J Gen Virol. 2007;88:2590–5.
Shi M, Lin XD, Vasilakis N, Tian JH, Li CX, Chen LJ, Eastwood G, Diao XN, Chen MH, Chen X, Qin XC, Widen SG, Wood TG, Tesh RB, Xu J, Holmes EC, Zhang YZ. Divergent viruses discovered in arthropods and vertebrates revise the evolutionary history of the Flaviviridae and related viruses. J Virol. 2016;90:659–69.
Teixeira M, Sela N, Ng J, Casteel CL, Peng HC, Bekal S, Girke T, Ghanim M, Kaloshian I. A novel virus from Macrosiphum euphorbiae with similarities to members of the family Flaviviridae. J Gen Virol. 2016;97:1261–71.
Teixeira MA, Sela N, Atamian HS, Bao E, Chaudhary R, MacWilliams J, He J, Mantelin S, Girke T, Kaloshian I. Sequence analysis of the potato aphid Macrosiphum euphorbiae transcriptome identified two new viruses. PLoS ONE. 2018;13:e0193239.
van Munster M, Dullemans AM, Verbeek M, van den Heuvel J, Clerivet A, van der Wilk F. Sequence analysis and genomic organization of Aphid lethal paralysis virus: a new member of the family Dicistroviridae. J Gen Virol. 2002;83:3131–8.
Vasilakis N, Forrester NL, Palacios G, Nasar F, Savji N, Rossi SL, Guzman H, Wood TG, Popov V, Gorchakov R, Gonzalez AV, Haddow AD, Watts DM, da Rosa AP, Weaver SC, Lipkin WI, Tesh RB. Negevirus: a proposed new taxon of insect-specific viruses with wide geographic distribution. J Virol. 2013;87:2475–88.
Vasilakis N, Tesh RB. Insect-specific viruses and their potential impact on arbovirus transmission. Curr Opin Virol. 2015;15:69–74.
Lu G, Ye ZX, He YJ, Zhang Y, Wang X, Huang HJ, Zhuo JC, Sun ZT, Yan F, Chen JP, Zhang CX, Li JM. Discovery of two novel negeviruses in a dungfly collected from the arctic. Viruses. 2020;12:692.
Kallies R, Kopp A, Zirkel F, Estrada A, Gillespie TR, Drosten C, Junglen S. Genetic characterization of goutanap virus, a novel virus related to negeviruses, cileviruses and higreviruses. Viruses. 2014;6:4346–57.
Nunes MRT, Contreras-Gutierrez MA, Guzman H, Martins LC, Barbirato MF, Savit C, Balta V, Uribe S, Vivero R, Suaza JD, Oliveira H, NunesNeto JP, Carvalho VL, da Silva SP, Cardoso JF, de Oliveira RS, da Silva LP, Wood TG, Widen SG, Vasconcelos PFC, Fish D, Vasilakis N, Tesh RB. Genetic characterization, molecular epidemiology, and phylogenetic relationships of insect-specific viruses in the taxon Negevirus. Virology. 2017;504:152–67.
Kondo H, Fujita M, Hisano H, Hyodo K, Andika IB, Suzuki N. Virome analysis of aphid populations that infest the barley field: the discovery of two novel groups of nege/kita-like viruses and other novel RNA viruses. Front Microbiol. 2020;11:509.
Feng Y, Krueger EN, Liu S, Dorman K, Bonning BC, Miller WA. Discovery of known and novel viral genomes in soybean aphid by deep sequencing. Phytobiomes PBIOMES. 2017;11:16–10.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–52.
Suvanto MT, Truong Nguyen P, Uusitalo R, Korhonen EM, Faolotto G, Vapalahti O, Huhtamo E, Smura T. A novel negevirus isolated from Aedes vexans mosquitoes in Finland. Arch Virol. 2020;165:2989–92.
Kuchibhatla DB, Sherman WA, Chung BY, Cook S, Schneider G, Eisenhaber B, Karlin DG. Powerful sequence similarity search methods and in-depth manual analyses can identify remote homologs in many apparently “orphan” viral proteins. J Virol. 2014;88:10–20.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74.
Darriba D, Posada D, Kozlov AM, Stamatakis A, Morel B, Flouri T. ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol Biol Evol. 2020;37:291–4.
Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucl Acids Symp Ser. 1999;41:95–8.
Nabeshima T, Inoue S, Okamoto K, Posadas-Herrera G, Yu F, Uchida L, Ichinose A, Sakaguchi M, Sunahara T, Buerano CC, Tadena FP, Orbita IB, Natividad FF, Morita K. Tanay virus, a new species of virus isolated from mosquitoes in the Philippines. J Gen Virol. 2014;95:1390–5.
Li JM, Andika IB, Shen JF, Lv YD, Ji YQ, Sun LY, Chen JP. Characterization of rice black-streaked dwarf virus- and rice stripe virus-derived siRNAs in singly and doubly infected insect vector Laodelphax striatellus. PLoS ONE. 2013;8:e66007.
Funding
This work was supported by the National Natural Science Foundation of China (U20A2036), the Natural Science Foundation of Zhejiang Province (LQ20C140003), and the Ningbo Science and Technology Innovation 2025 Major Project (2019B10004). This work was sponsored by the K. C. Wong Magna Fund in Ningbo University.
Author information
Authors and Affiliations
Contributions
J-PC, C-XZ, and J-ML conceived the study and designed the project. Y-HQ, L-YX, JZ, Z-XY, and GL performed the experiment, analyzed the data, and drafted the manuscript. J-PC, C-XZ, and J-ML helped to revise the manuscript. All authors contributed to the article and approved the submitted version. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
File S1: COI sequence of the aphid (Genus Indomegoura) used in this study.
Additional file 2.
File S2: Full genome sequence of Indomegoura nege-like Virus 1.
Additional file 3.
Table S1: The BLAST results of INLV1 compare to the NCBI NT and NR database.
Additional file 4.
Table S2: Primers used in this study.
Additional file 5.
Table S3: Abbreviations of virus names and GenBank accession numbers used in this study.
Additional file 6.
Figure S1: Prediction of transmembrane domains (TM) in the ORF3 of INLV1. TM 1:68-87 aa; TM 2:107-126 aa; TM 3:138-157 aa; TM 4:177-196 aa.
Additional file 7.
Figure S2: Alignment of RdRp amino acid sequences of INLV1, previously reported representative nege/kita-like viruses, and plant viruses in the families Kitaviridae and Virgaviridae. Red boxes indicate the position of the motifs - A: DX(4-5)D, B: GX(2-3)TX(3)N, and C: GDD. Virus names and GenBank accessions numbers are listed in Supplementary Table S3.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Qi, YH., Xu, LY., Zhai, J. et al. Complete genome sequence of a novel nege-like virus in aphids (genus Indomegoura). Virol J 18, 76 (2021). https://doi.org/10.1186/s12985-021-01552-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-021-01552-w