
Abstract
Background
The generation of accurate and reproducible viral sequence data is necessary to understand the diversity present in populations of RNA viruses isolated from clinical samples. While various sequencing methods are available, they often require high quality templates and high viral titer to ensure reliable data.
Methods
We modified a multiplex PCR and sequencing approach to characterize populations of simian immunodeficiency virus (SIV) isolated from nonhuman primates. We chose this approach with the aim of reducing the number of required input templates while maintaining fidelity and sensitivity. We conducted replicate sequencing experiments using different numbers of quantified viral RNA (vRNA) or viral cDNA as input material. We performed assays with clonal SIVmac239 to detect false positives, and we mixed SIVmac239 and a variant with 24 point mutations (SIVmac239-24X) to measure variant detection sensitivity.
Results
We found that utilizing a starting material of quantified viral cDNA templates had a lower rate of false positives and increased reproducibility when compared to that of quantified vRNA templates. This study identifies the importance of rigorously validating deep sequencing methods and including replicate samples when using a new method to characterize low frequency variants in a population with a small number of templates.
Conclusions
Because the need to generate reproducible and accurate sequencing data from diverse viruses from low titer samples, we modified a multiplex PCR and sequencing approach to characterize SIV from populations from non-human primates. We found that increasing starting template numbers increased the reproducibility and decreased the number of false positives identified, and this was further seen when cDNA was used as a starting material. Ultimately, we highlight the importance of vigorously validating methods to prevent overinterpretation of low frequency variants in a sample.
Similar content being viewed by others

Introduction
Characterizing the sequence diversity of RNA virus populations is an essential component of studying viral pathogenesis and transmission in individuals [1, 2]. This sequence data can be used to identify antiviral drug resistance mutations [3], understand how viruses evolve [4, 5], and track virus transmission during epidemics [6], such as the Ebola virus outbreak in West Africa in 2014, the Zika virus outbreak in Brazil in 2015 [7, 8], and the current SARS-CoV-2 outbreak [9, 10].
The accumulation of mutations in RNA viruses can impact their pathogenesis [11]. While many mutations can be deleterious or neutral, some are beneficial for virus proliferation, survival, or transmission [4, 12]. Naturally elicited host immune responses that fail to eliminate replicating viruses select for variants that avoid immune detection [4]. Drug resistance mutations can also accumulate when antiretroviral therapy does not fully suppress virus replication [13]. Accurate detection of these variants in RNA virus populations can help determine whether therapeutic interventions eliminate or exacerbate mutations from the replicating virus population.
Sequencing RNA viruses requires the generation of viral cDNA, followed by amplification of either long (> 1000 bp) or short (< 400 bp) DNA segments. Long amplicons are used to study distantly linked nucleotides on the same virus templates using Pacific Biosciences (Brese et al. 2018) or Oxford Nanopore instruments [14, 15], In contrast, Illumina technology can generate sequence data from shorter viral segments with higher throughput, better fidelity, and improved efficiency [16]. While each approach has advantages and disadvantages, the desire to acquire sequence data with newer assays often trumps taking the time to perform experiments required to validate their sensitivity and reproducibility.
Our goal was to implement a multiplex PCR approach, similar to those used for Ebola [17], ZIKV [8], and SARS-CoV-2 [10], to improve the reproducibility and sensitivity of sequencing SIV derived from plasma with low virus titers or cell-associated vRNA isolated from different tissues. SIV dynamics are frequently studied in nonhuman primates [18,19,20], but the samples collected from animals with interesting biological phenotypes often have a low virus titer. With an ongoing emphasis on understanding the dynamics of SIV replication in nonhuman primates [21], we aimed to determine if the multiplex method could be applied to SIV to improve the characterization of virus populations with improved sensitivity and reproducibility.
We developed a multiplex PCR approach to amplify and sequence SIV. To validate this method, we sequenced different numbers of vRNA and viral cDNA templates of clonal SIVmac239, as well as variable ratios of two clonal SIV strains differing at 24 nucleotide positions. We found improved sensitivity and reproducibility of variant calling when normalizing to the number of viral cDNA templates added to the reaction when compared to the number of vRNA templates added to the reaction. By validating the SIV multiplex sequencing method here, we identify the strengths and limitations of this method, which are essential for defining the usability of any new technique.
Results
Design of a multiplex PCR assay for SIV
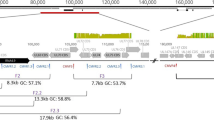
Candidate multiplex primers for SIV were designed in Primal Scheme, a tool developed by Quick et al. [8]. Each primer set was tested individually and then pooled such that the amplicon products would not overlap with each other Fig. 1a, Table 1. The most 5′ primer binds just upstream of the start codon for gag, and has an identical sequence to the 3′ LTR, so it is not necessary to amplify the 5′ LTR as well. Primers generated by PrimalScheme are selected based on Primer3 software, as described in Quick et al. [8]. Primer pools were tested to verify that individual primer pairs would generate amplicons spanning the entire viral genome when combined. Final primer pair concentrations, corresponding sequences, and positions relative to SIVmac239 (Accession: M33262) reference can be found in Table 1.

a SIV multiplex primer scheme. Two non-overlapping pools corresponding to even (red) and odd (blue) primer sets were designed using Primal Scheme to generate small amplicons spanning the entire SIVmac239 genome. Primer pairs were pooled at varying concentrations described in Table 1. b Locations of SNPs in SIVmac239-24x. SNPs denoted with a red X. SNPs are present across the entire SIVmac239 genome and are present in all genes
We first isolated vRNA from a stock of clonal SIVmac239. For Method 1, we quantified the vRNA stock and then diluted it to 106 vRNA templates per reaction. Serial dilutions of quantified vRNA were converted to viral cDNA by reverse-transcription. The multiplex PCR was performed on the viral cDNA. For Method 2, we prepared total cDNA from 107 vRNA templates of each stock. We then quantified the viral cDNA with a qPCR reaction specific for gag. The quantified viral cDNA was diluted to 106 cDNA gag copies per reaction and the multiplex PCR was then performed. After multiplex PCR for either Method 1 or 2, 75 ng of each pool of PCR products were combined into a single tube to generate a 150 ng DNA pool containing all the generated PCR amplicons. This amplicon library was then tagged using an Illumina TruSeq kit, and sequenced on an Illumina MiSeq.
Detection of false positives in clonal SIVmac239
We first sequenced clonal SIVmac239 to determine the frequency of false positives when using either Method 1 or 2. We used serially diluted 100% SIVmac239 vRNA or viral cDNA for this part of the project. For each replicate using Method 1, new cDNA was prepared and then multiplex PCR and sequencing were performed. These experiments were performed in triplicate. For each replicate using Method 2, the same prepared cDNA was used for all of the multiplex PCR reactions. These experiments were performed in duplicate.
FASTQ sequences were examined using a modified version of a custom pipeline previously used to analyze multiplex PCR ZIKV sequences [22], and uploaded to a Docker container in order to ensure reproducibility. Using this tool, we randomly subsampled up to 2000 reads per amplicon across each data set and mapped them to SIVmac239 (Accession: M33262), as described in the Materials and Methods. Amplification of each PCR product does not occur equally, so by subsampling up to 2000 reads, we could attempt to informatically normalize the depth of coverage, while not oversampling any one single amplicon. VarScan (https://sourceforge.net/projects/varscan/) was then used to identify nucleotides present in the virus population that were different from the reference at a frequency of 1% or greater and had a depth of coverage of at least 1800 nucleotides, or 90% of our maximum subsampled depth. SNPeff [23]was used to annotate variants and their effect on each coding sequence. Any single nucleotide variant (SNV) present at a frequency of 1% or greater and with a depth of coverage of at least 1800 nucleotides was categorized as a false positive for our analysis. These thresholds are more conservative than the 3% cutoff and 400 × coverage required by Grubaugh et al. [24].
We began by assessing false positives present in sequences generated by Method 1. The average rate of false positives in a single replicate was related to the number of input templates, with samples containing 103 input copies having a higher average rate of false positives at 1.13 × 10–2 false positives per nucleotide, and samples containing 106 input copies having a lower average rate of 2.6 × 10–3 false positives per nucleotide Fig. 2a, left panel, closed circles, p < 0.0001, Tukey’s multiple comparisons test). We then determined whether the rate of false positives declined when considering two or more replicates. We found there was not a significant copy-dependent decrease in false positives when we used two replicates compared to a single replicate Fig. 2a, left panel, open circles, p = 0.83, Tukey’s multiple comparisons test).

a Number of false positives detected per nucleotide with coverage of at least 1800 and a variant frequency of at least 1% in at least one replicate per input copy (closed circle) or at least two replicates per input copy (open circle) for our Method 1 (vRNA) (left) and Method 2 (cDNA) (right) data sets. Lines represent median ± 95% confidence interval. No significant differences were identified between data sets by Kruskal–Wallis tests. b False positive variant frequency of variants identified in all input templates for Method 1 data sets. c False positive variant frequency of variants identified in all input templates for Method 2 data sets. cDNA input template numbers denoted by colors. Lines represent mean and standard deviation for each variant’s replicate. All variants shown are present at a frequency of 1% or greater, have a nucleotide depth of at least 1800, and are detected in at least two samples
We investigated the individual nucleotide positions where we detected false positives in multiple replicates when using Method 1 Fig. 2b. We found 11 positions with false positives at all input copy numbers, with 4 being insertions at nucleotide positions 1254, 1480, 5428, and 7396, and 9 being substitutions at nucleotide positions 6181, 6186, 6188, 6190, 6192, 6201, 6205, 6207, and 6713. We found the median false positive frequency did not depend on the number of input copies (p = 0.92, Kruskal–Wallis) Fig. 2b. Each of the insertions occurred in a poly-A region containing 6 consecutive adenines. Although the chemistry of Illumina sequencing does not lead to the same errors in homopolymers that are notorious in other sequencing platforms [25], there can still be PCR-based errors in homopolymeric regions [26, 27]. All substitutions, aside from position 6713, were present within a stretch of 27 nucleotides that are adjacent to a primer binding site. These SNVs are contained within an overlap region between Amplicons 20 and 21. Notably, these variants were present in Amplicon 21, but not Amplicon 20, suggesting that Amplicon 21 may be more prone to the incorporation of PCR-based substitutions than Amplicon 20. While unfortunate, inaccuracies in variant reporting is not an uncommon phenomenon at the ends of amplicons and has been reported previously [28, 29]. We also observed that when using a different analysis pipeline that does not normalize coverage across the genome through subsampling, these variants were not reported in the vcf file, highlighting the importance of validating the analysis methods prior to calling variants as true variants. However, we felt that the benefit of standardizing variant calling with normalized coverage across the genome outweighed the complexity associated with variabilities related to relative oversampling of individual amplicons.
We then used the same metrics to identify false positives using Method 2 Fig. 2c. Similar to Method 1, the average number of false positives per nucleotide in at least one replicate was related to the number of input templates, with 103 input cDNA templates having an average of 1.05 × 10–2 false positives per nucleotide and 106 input cDNA templates having an average of 1.52 × 10–3 false positives per nucleotide Fig. 2a, right panel, closed circles, p < 0.001, Tukey’s multiple comparisons test). When only including false positives detected in at least two replicates, there was no difference in the rate of false positives between 103 and 106 cDNA templates Fig. 2a, right panel).
We identified 9 nucleotide positions with false positives in at least one replicate of all input template levels using Method 2 Fig. 2c. All of the false positives detected by Method 2 were also detected by Method 1. Since these false positives are present in nearly every sample and this is a clonal virus stock Fig. 2b, c, it is likely an artifact of the method rather than true variants, highlighting the importance of validating novel methods with virus stocks of known composition. Additionally, it is important to understand the effects of nucleotide sequence and primer binding sites on false positive detection, as primer slippage may be a confounding factor.
To help determine if the rate of false positives was related to coverage depth, we calculated the frequency of nucleotide sites that had sufficient coverage (a nucleotide depth of at least 1800) for our cDNA and vRNA data sets. There was no significant difference between the percentage of bases with at least 1800 × coverage using Method 1 or 2 (Method 1 mean = 76.27% nucleotides over 1800, Method 2 mean = 75.03% nucleotides over 1800; p = 0.95, Mann–Whitney, data not shown), indicating that the differences in false positive frequency are more likely a result of starting template than coverage alone.
Detection of genome-wide variants using multiplex SIV sequencing
We then examined the sensitivity and reproducibility of detecting individual SNVs in SIV by Methods 1 and 2. We used two stock viruses, SIVmac239 and SIVmac239-24x, that differed at 24 nucleotides throughout the entire viral coding sequence Fig. 1b, Table 2. Viral RNA was isolated from these two stocks and quantified with a gag qPCR assay. We proceeded with Method 1 by mixing the two stocks of vRNA to a total number of 106 copies at the following SIVmac239 to SIVmac239-24 × ratios: 100:0, 95:5, 90:10, 75:25, 50:50, and 0:100 Fig. 3a. Each mixture of vRNA was serially diluted to 105, 104, and 103 templates per 11 ul. We also tested Method 2 by first preparing viral cDNA from 107 vRNA templates of each of the two stocks, quantifying viral cDNA, and then mixing the cDNA templates to a total of 106 templates in the same ratios as the vRNA templates were mixed Fig. 3b. The same quantified vRNA or viral cDNA mixtures were used for the entire experiment.

Schematic of experimental design. a Method 1: viral RNA was isolated from original stock and quantified via qRT-PCR. SIVmac239 and SIVmac239-24 × were diluted to 106 copies/reaction and mixed at the following SIVmac239:SIVmac239-24 × ratios: 100:0, 95:5, 90:10, 75:25, 40:60, and 0:100. Serial dilutions were preformed to 105, 104, and 103 copies per reaction. Viral cDNA was generated from viral RNA mixes, with one cDNA reaction per vRNA mix. b Method 2: viral RNA was isolated and approximately 107 viral RNA copies were added to each cDNA synthesis reaction. Viral cDNA copies were quantified using qRT-PCR and each was diluted to 106 copies per reaction. SIVmac239:SIVmac239-24 × mixes were generated at the following ratios: 100:0, 95:5, 90:10, 75:25, 50:50, and 0:100. cDNA mixes were then serially diluted to 105, 104, and 103 copies per reaction. c cDNA was used for multiplex PCR. PCR products were then combined at equimolar ratios and library prepped according to TruSeq Library Preparation documentation (Illumina). Libraries were quantified, pooled, and sequenced using a 2 × 250 v2 MiSeq cartridge
The remaining multiplex PCR procedures were performed for the different numbers of input templates and for each of the individual ratios. PCR products were tagged, and sequencing was performed on the Illumina MiSeq. FASTQ reads were mapped to SIVmac239 and the frequencies of each individual SNV relative to SIVmac239 were determined as described for the clonal SIVmac239 data.
We compared the observed to the expected variant frequencies for all 24 positions in the genome for both Methods 1 and 2. We generated a linear regression for each number of input templates Fig. 4ato determine if the relationship between the expected and observed SNV frequency was the same. We did not find a significant difference when we compared the slopes for all four linear regression lines with either Method 1 (p = 0.069, Fig. 4aor Method 2 (p = 0.185, Fig. 4b. Notably, all of these data sets had an SNV present at position 9110 Fig. 4a, b, open circles) that was consistently detected inaccurately. While there did appear to be a slight increase in observed variant frequency when compared to expected variant frequency, site 9110 was a clear outlier in the data sets Fig. 4c.

a Observed versus expected variant frequencies identified in the Method 1 (vRNA) mixed data sets. Observed variant frequency indicates percent SIVmac239-24× identified. Error bars indicate standard deviation for each replicate. Linear regressions colored by vRNA templates. Open circles indicate SNV 9110. b Observed versus expected variant frequencies identified in Method 2 (cDNA) mixed data sets. Observed variant frequency indicates percent SIVmac239-24× identified. Error bars indicate standard deviation for each replicate. Linear regressions colored by cDNA templates. Open circles indicate SNV 9110. No significant difference is observed between input templates and slope. c Observed versus expected variant frequency for SNV at 9110 for vRNA data sets. Lines represent medians and error bars indicate 95% confidence interval. Asterisk indicate p-value less than 0.05 as determined by Kruskal–Wallis
To further understand how the number of templates and the type of quantified starting material affects the reproducibility of the detected SNV frequencies, we compared the observed frequencies of each of the 24 individual SNVs across all the data sets. We found that when using Method 2, there was less variability in variant frequencies across the number of input templates when compared to using Method 1 Fig. 5a–f. This observation is consistent with data indicating that the process of reverse transcription is inefficient and variable [30], such that when 103 vRNA input templates are used in the assay, it is unlikely that there are actually 103 viral cDNA templates available for subsequent PCR. For both input types, it was not surprising that as the number of templates increased, the SNV frequencies tended to be more consistent and reproducible across the genome.

a–f Observed variant frequency of SIVmac239-24 × SNPs in varying SIVmac239:SIVmac239-24 × ratios by vRNA (left) or cDNA (right) input templates. Dotted line indicates expected variant frequency. Significance determined by Kruskal–Wallis and designated by asterisks. p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), p < 0.0001 (****)
Detection of variants within biological samples
To ensure the that multiplex PCR can be used with biological samples, we sequenced vRNA isolated from plasma and a lymph node (LN) of an SIV+ cynomolgus macaque at the same time point. We diluted quantified vRNA to a starting number of 103, 104, and 105 input templates. Each sample was sequenced in triplicate. Again, we required a variant to be present at a frequency of 1% or greater and with a sample depth of 1800 to be considered a true variant. While we hoped to readily detect the same variants in sequences generated from all three numbers of input templates, we detected a substantial amount of amplicon dropout for the samples starting with 103 vRNA templates. Still, we confidently obtained sufficient coverage over at least 80% of the genome from the 104 and 105 vRNA template data sets, and used these sets for further analysis.
We found 24 SNVs were present in at least 2 out of 3 replicates in each data set Fig. 6. However, as a result of insufficient coverage in some of the replicates, none of the variants shown in Fig. 6 were present in all three replicates of each data set. Notably, the frequencies of these 24 SNVs were similar across samples. Similar to what was shown in Fig. 2b, there was considerable variation in the nucleotide region between 6181 and 6205. Ultimately, we show that variants are detectable consistently at a minimum of 104 vRNA input templates, and that variants are similar between the LN and plasma in a given animal, which is consistent with prior reports [31, 32].

a Variant frequency of SNVs identified in at least two of three replicates for that input copy and sample type data set. vRNA input template numbers denoted by colors, with darker shades representing LN and lighter colors representing the plasma. Bars indicate 95% CI. All variants shown are present at a frequency of 1% or greater and have a nucleotide depth of at least 1800
Discussion
The goal of this study was to adapt a multiplex PCR and sequencing approach [8]to sequence SIV from low quality starting material. This would include occasions where SIV is present at low titer or as partially degraded vRNA. Recognizing that different sequencing methods have their limitations, we set out to validate this approach in a series of assays described in this study.
We first sequenced clonal SIVmac239 to determine the false positive rate using both Methods 1 and 2. We found 4 nucleotide sites (1254, 2480, 5428, and 7396) in homopolymeric regions with consecutive adenines where false positive indels were detected. We predict that these insertions were introduced during the PCR step. We also found 8 individual false positives in the stock that were attributed to substitutions consistently present in the same 27-nucleotide region of Amplicon 21, but not in the adjacent Amplicon 20. We hypothesize that these substitutions are specific to the generation of Amplicon 21 and the analysis pipeline, rather than actually being real substitutions.
We also found that sequencing replicates reduced the detection of false positives, particularly when there are low numbers of input templates, consistent with previous results [24]. While we realize that there are not always enough resources available to sequence a sample in duplicate, our data highlights that caution should be taken when interpreting data from a single assay of a sample with low virus titer. Importantly, the process of validating a method with a known clonal virus stock is key to distinguishing between false positives, sequencing error, and true variants. Without doing the validation assays in this study, it would be impossible to know the benefits and technical limitations of using the multiplex PCR approach to sequence virus isolated from animals infected with SIVmac239. Detecting these method-dependent systematic errors by characterizing false positives in a clonal stock is important so that investigators using this method can perform the assay with knowledge of which variants are real and which are technical artifacts.
By mixing SIVmac239 and SIVmac239-24x, we detected variants at a frequency of 5% with as few as 1000 input copies. We opted for this conservative threshold because we already knew that there were some false positives detected when a threshold of 1% was used Fig. 2, and the most relevant variants accumulate over time to a higher frequency. Thus, detection of variants at a frequency of < 5% was less critical for broad analyses of SIV population diversity. Future studies that require more sensitive variant detection could address whether variants present between 1 and 5% can be accurately detected.
For these mixing studies, we chose two viruses with variants scattered throughout the genome, with at least one variant present in each gene. This let us determine whether we could effectively detect variants throughout the genome and across a large number of PCR amplicons generated by either Method 1 or 2. We were surprised to find it difficult to interpret the SNV frequency at position 9110. This site lies in a region dense with adenines and guanines which may contribute to some inconsistencies as a result of PCR slippage or misincorporation of nucleotides during PCR amplification [33]. In addition, the forward primer for Amplicon 33 is one nucleotide different from its complementary sequence in SIVmac239-24x due to the modified nucleotide 9110 present in the SIVmac239-24 × sequence. While we did trim primers computationally, this would not prevent PCR error from occurring. As a result, some SIVmac239-24 × templates may not be amplified as efficiently because of a single nucleotide difference, which may also lead to amplicon dropout and skewed results.
Throughout our study, we compared the results obtained using Methods 1 and 2, which used quantified vRNA and quantified viral cDNA, respectively. Reverse transcription is inefficient [30], so we wanted to determine if there were fewer false positives and more consistent detection of SNVs when quantified cDNA was used as the starting material rather than vRNA. We found the observed SNV frequencies were more similar to expected frequencies when quantified cDNA was used as a starting template Fig. 4b and, not surprisingly, when increased numbers of vRNA or cDNA templates were used. Even though our quantification of viral cDNA was based only on the copies of gag, we found that using quantified viral cDNA as the input improved the reproducibility of variant detection when we mixed two clonal virus inocula at predefined ratios, even when using only 103 quantified templates. This observation further raises concerns that using quantified vRNA as starting material gives an overestimation of the number of vRNA templates that are actually converted to cDNA and amplified to yield the reported sequence data.
By testing this method with biological samples, we provide evidence for the use of SIV multiplex PCR in animal studies as a way to characterize viral populations in vivo. Unfortunately, we were unable to fully amplify the biological samples with the lowest input titer. This may be a consequence of host RNA or DNA that may have reduced the efficiency of primer binding. Nonetheless, we could still amplify from samples containing 104 input vRNA templates isolated from plasma or host cells. We detected variants with similar frequencies that passed our threshold in two of three replicates in each data set Fig. 6. The variants that were not present in all three replicates typically did not have enough coverage in all three replicates to pass our threshold, which is stringent compared to other similar analyses [24]. Importantly, we found the variant frequencies were similar between tissues and plasma, consistent with previous reports [31, 32], and that a biologically relevant amount of virus (104 total vRNA input templates) is sufficient to provide reproducible variant detection across nearly all of the SIV genome.
Overall, we found that the multiplex PCR approach could be successfully used to generate genome wide sequences of SIV, but our results strongly imply that any new sequencing and analysis methods be validated before using them widely to characterize variant frequency in a virus population. While it was possible to generate sequence data from 103 vRNA templates, the use of quantified cDNA was more consistent. Further, although this method could be used to successfully detect SNVs across the genome, we found there were key features in the viral genome that affected the accuracy of the multiplex PCR approach. Thus, while the multiplex PCR method has many advantages for deep sequencing virus populations, validation experiments and visualization of the output alignments are essential for correct data reporting, as expected for any sequencing approach.
Conclusions
Our initial goal of this study was to generate a sequencing approach that was able to characterize viral population diversity with low input templates. Multiplex PCR has been used to accurately sequence other viruses, including Zika [8], Dengue, and Chikungunya [34], at titers between 103 and 106 vRNA copies per mL, and most recently with SARS-CoV-2 [9, 10] and we were hoping this would extend to SIV. However, many publications fail to state the viral input titer when describing their sequencing methods. We learned that increasing numbers of input SIV templates and utilization of quantified cDNA as a starting material improved reproducibility of variant calling. Further, our data suggests that the multiplex PCR and sequencing approach may not be as sensitive at low numbers of input templates for SIV, when compared to other using low numbers of templates for other viruses. We additionally were able to demonstrate that this model may be used with biological samples and at biologically relevant levels of circulating virus. Most importantly, our study demonstrates the need to validate new sequencing approaches because the same method may not be viable for sequencing all viruses with the same sensitivity and reproducibility. We now understand the limitations of the assay so that experiments can be designed to maximize the likelihood of success and minimize the overinterpretation of data.
Methods
Primer design
Primers were designed using Primal Scheme, as previously described by [8]. FASTA files of SIVmac239 and three consensus sequences of virus populations isolated from animals infected with SIVmac239 were used as the foundation for the Primal Scheme tool. 37 primer pairs Table 1 were generated to span the entire SIV genome. The lengths of the resulting amplicons ranged from 285 to 397 bp, with an average length of 351 bp. The number of overlapping nucleotides for each amplicon ranged from 40 to 149 bp, with an average length of 100 bp. Primer pairs were split into two pools to ensure that the amplicons generated within each pool would not overlap. Primer sequences, pools, and concentrations can be found in Table 1. Final concentration of Pool 1 was 35 µM and Pool 2 was 24 µM.
Isolation of vRNA for sequencing
SIVmac239 and SIVmac239-24 × vRNA were isolated from clonal virus stocks. Briefly, 1 ml of each virus stock was centrifuged at 13,000 rpm for 30 s to pellet any cells that were present. Plasma vRNA was obtained from an animal described in Ellis et al. [35] #20499, and lymph node (LN) vRNA was isolated from a necropsy tissue homogenate by adding TriZol and conducting a standard phenol–chloroform RNA extraction. The supernatant of each sample was transferred to a 1.5 mL Eppendorf tube and spun at 13,000 rpm for 1 h at 4C to concentrate virus particles. After spinning the sample, we removed all the supernatant, except 200 µL of liquid, so as not to disturb the viral pellet. The vRNA was then extracted using the Qiagen MinElute vRNA extraction kit, according to manufacturer’s protocols (Qiagen). Prior to elution, 25 µL of Buffer AVE was added directly to the MinElute Column membrane and incubated for 5 min.
Preparation of viral cDNAs
The vRNA isolated from the SIVmac239 and SIVmac239-24 × virus stocks were each diluted to 106 copies/11ul in nuclease-free water. They were mixed at SIVmac239: SIVmac239-24 × ratios of 100:0, 95:5, 90:10, 75:25, 50:50, and 0:100. These mixtures were diluted 1:10 in nuclease-free water to generate vRNA template concentration dilution series of 106, 105, 104, and 103 templates per 11uL. From each mixture, we used 11ul of vRNA and performed cDNA synthesis using SuperScript IV Reverse Transcriptase (Invitrogen), according to manufacturer’s protocol. For experiments where quantified viral cDNA was used as starting material, approximately 107 viral templates were used for cDNA synthesis using SuperScript IV Reverse Transcriptase (Invitrogen), according to manufacturer’s protocol. Viral cDNA and vRNA was then quantified using a gag qPCR assay as previously described [36].
Multiplex PCR reactions
Each tube of viral cDNA generated from the virus stocks or biological samples was split equally, such that 10uL of viral cDNA was PCR amplified with the two separate primer pools. Amplification was performed with the Q5 polymerase and the following reaction conditions: 98 °C for 30 s, 35 cycles of 95 °C for 15 s and 65 °C for 5 min, and then cooled to 4 °C. Products were verified using a 1% agarose gel and were quantified using the Qiagen High Sensitivity DNA kit (Thermo Fisher).
Library preparation and sequencing
After the two amplicon pools were generated, 75 ng of each pool was mixed to generate a total of 150 ng DNA. This pool of PCR products was tagged with the Illumina TruSeq Nano HT kit, according to the manufacturer’s protocol (Illumina). Following tagging and purifying, the libraries were quantified using the Qiagen High Sensitivity DNA kit. The quality of each library was characterized with a High Sensitivity DNA kit (Agilent) on an Agilent Bioanalzyer. If unligated adapter dimers were detected at 140 bp, an additional bead clean up step was performed. The average tagged library size was approximately 503 bp (range 491–512). Tagged libraries were pooled at equimolar concentrations and diluted so that the final concentration of DNA molecules per run was 10 pM. This diluted pool and 10 pM PhiX were denatured with 0.2 N sodium hydroxide for 5 min at room temperature. Denatured PhiX was then added to the pool at a final frequency of 10 percent. Each pool was loaded at 10 pM concentration onto a 500-cycle v2 MiSeq cartridge and sequenced.
Data analysis
FASTQ reads were demultiplexed and then processed using a modified pipeline from David O’Connor’s lab, called the Zequencer, with the initial scripts available at https://bitbucket.org/dholab/ and referenced in Dudley et al. (2017. All scripts used, and their documentation, can be found on our github repository, https://github.com/SLO-Lab/SIV_MultiplexPCR. Briefly, reads were trimmed, merged, and normalized using bbtools (https://jgi.doe.gov/data-and-tools/bbtools/) and Seqtk (https://github.com/lh3/seqtk). A FASTA file was generated that contained the nucleotide reference sequences for all 37 amplicons, as they would exist in SIVmac239 (Accession: M33262). Up to 2000 merged reads that mapped at low sensitivity to each of the 37 reference amplicons were extracted from the data set. These reads were then aligned to SIVmac239 using NovoAlign (http://www.novocraft.com/products/novoalign/). A pileup file was generated from the BAM alignment. Variants with a frequency of 1% or higher were called by VarScan (https://sourceforge.net/projects/varscan/) and annotated by SNPeff y[23]. VCF files were processed and analyzed in R(v3.6.1). Variants with a sample depth less than 1800 were discarded to reduce bias as a result of poor sample depth. Position 9609 codes for a stop codon in the nef protein in the M33262 Genbank reference for SIVmac239, but our stock virus is SIVmac239-nef-open, which has a T to G transversion at this position, converting the stop codon (TAA) to a glutamate (GAA) amino acid.
Availability of data and materials
Code used to generate data can be found on the lab’s GitHub page (see Data analysis section).
Abbreviations
- HIV:
-
Human immunodeficiency virus
- SIV:
-
Simian immunodeficiency virus
- ZIKV:
-
Zika virus
- vRNA:
-
Viral RNA
- SNV:
-
Single nucleotide variant
References
Poirier EZ, Vignuzzi M. Virus population dynamics during infection. Curr Opin Virol. 2017;23:82–7.
Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, Andino R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature. 2006;439(7074):344–8.
Dudley DM, Bailey AL, Mehta SH, Hughes AL, Kirk GD, Westergaard RP, et al. Cross-clade simultaneous HIV drug resistance genotyping for reverse transcriptase, protease, and integrase inhibitor mutations by Illumina MiSeq. Retrovirology. 2014;11(1):122.
Henn MR, Boutwell CL, Charlebois P, Lennon NJ, Power KA, Macalalad AR, et al. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog. 2012;8(3):e1002529.
Lessler J, Chaisson LH, Kucirka LM, Bi Q, Grantz K, Salje H, et al. Assessing the global threat from Zika virus. Science. 2016;353(6300):aaf8160.
Hadfield J, Megill C, Bell SM, Huddleston J, Potter B, Callender C, et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. 2018;34(23):4121–3.
Carroll MW, Matthews DA, Hiscox JA, Elmore MJ, Pollakis G, Rambaut A, et al. Temporal and spatial analysis of the 2014–2015 Ebola virus outbreak in West Africa. Nature. 2015;524(7563):97–101.
Quick J, Grubaugh ND, Pullan ST, Claro IM, Smith AD, Gangavarapu K, et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc. 2017;12(6):1261–76.
DC COVID-19 Response Team, Jorden MA, Rudman SL, Villarino E, Hoferka S, Patel MT, et al. Evidence for limited early spread of COVID-19 within the United States, January–February 2020. MMWR Morb Mortal Wkly Rep. 2020;69(22):680–4.
Fauver JR, Petrone ME, Hodcroft EB, Shioda K, Ehrlich HY, Watts AG, et al. Coast-to-coast spread of SARS-CoV-2 during the early epidemic in the United States. Cell. 2020;181:990–6.
Sanjuán R, Domingo-Calap P. Mechanisms of viral mutation. Cell Mol Life Sci. 2016;73(23):4433–48.
Zanini F, Puller V, Brodin J, Albert J, Neher RA. In vivo mutation rates and the landscape of fitness costs of HIV-1. Virus Evol. 2017;3(1):vex003.
Bangsberg DR, Kroetz DL, Deeks SG. Adherence-resistance relationships to combination HIV antiretroviral therapy. Curr HIV/AIDS Rep. 2007;4(2):65.
Oikonomopoulos S, Wang YC, Djambazian H, Badescu D, Ragoussis J. Benchmarking of the Oxford Nanopore MinION sequencing for quantitative and qualitative assessment of cDNA populations. Sci Rep. 2016;6:31602.
Tyler AD, Mataseje L, Urfano CJ, Schmidt L, Antonation KS, Mulvey MR, et al. Evaluation of Oxford Nanopore’s MinION sequencing device for microbial whole genome sequencing applications. Sci Rep. 2018;8(1):10931.
Adey A, Morrison HG, Asan A, Xun X, Kitzman JO, Turner EH, et al. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol. 2010;11(12):R119.
Arias A, Watson SJ, Asogun D, Tobin EA, Lu J, Phan MVT, et al. Rapid outbreak sequencing of Ebola virus in Sierra Leone identifies transmission chains linked to sporadic cases. Virus Evol. 2016;2(1):vew016.
Gambhira R, Keele BF, Schell JB, Hunter MJ, Dufour JP, Montefiori DC, et al. Transmitted/founder simian immunodeficiency virus envelope sequences in vesicular stomatitis and Semliki forest virus vector immunized rhesus macaques. PLoS ONE. 2014;9(10):e109678.
Harris M, Burns CM, Becker EA, Braasch AT, Gostick E, Johnson RC, et al. Acute-phase CD8 T cell responses that select for escape variants are needed to control live attenuated simian immunodeficiency virus. J Virol. 2013;87(16):9353–64.
Hassounah SA, Mesplède T, Quashie PK, Oliveira M, Sandstrom PA, Wainberg MA. Effect of HIV-1 integrase resistance mutations when introduced into SIVmac239 on susceptibility to integrase strand transfer inhibitors. J Virol. 2014;88(17):9683–92.
Kumar N, Chahroudi A, Silvestri G. Animal models to achieve an HIV cure. Curr Opin HIV AIDS. 2016;11(4):432–41.
Dudley DM, Newman CM, Lalli J, Stewart LM, Koenig MR, Weiler AM, et al. Infection via mosquito bite alters Zika virus tissue tropism and replication kinetics in rhesus macaques. Nat Commun. 2017;8(1):2096.
Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6(2):80–92.
Grubaugh ND, Gangavarapu K, Quick J, Matteson NL, De Jesus JG, Main BJ, et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019;20(1):8.
Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, Boutell JM, Bryant J, Carter RJ, Keira Cheetham R, Cox AJ, Ellis DJ, Flatbush MR, Gormley NA, Humphray SJ, Irving LJ, Karbelashvili MS, Kirk SM, Li H, Liu X, Maisinger KS, Murray LJ, Obradovic B, Ost T, Parkinson ML, Pratt MR, Rasolonjatovo IM, Reed MT, Rigatti R, Rodighiero C, Ross MT, Sabot A, Sankar SV, Scally A, Schroth GP, Smith ME, Smith VP, Spiridou A, Torrance PE, Tzonev SS, Vermaas EH, Walter K, Wu X, Zhang L, Alam MD, Anastasi C, Aniebo IC, Bailey DM, Bancarz IR, Banerjee S, Barbour SG, Baybayan PA, Benoit VA, Benson KF, Bevis C, Black PJ, Boodhun A, Brennan JS, Bridgham JA, Brown RC, Brown AA, Buermann DH, Bundu AA, Burrows JC, Carter NP, Castillo N, Chiara E Catenazzi M, Chang S, Neil Cooley R, Crake NR, Dada OO, Diakoumakos KD, Dominguez-Fernandez B, Earnshaw DJ, Egbujor UC, Elmore DW, Etchin SS, Ewan MR, Fedurco M, Fraser LJ, Fuentes Fajardo KV, Scott Furey W, George D, Gietzen KJ, Goddard CP, Golda GS, Granieri PA, Green DE, Gustafson DL, Hansen NF, Harnish K, Haudenschild CD, Heyer NI, Hims MM, Ho JT, Horgan AM, Hoschler K, Hurwitz S, Ivanov DV, Johnson MQ, James T, Huw Jones TA, Kang GD, Kerelska TH, Kersey AD, Khrebtukova I, Kindwall AP, Kingsbury Z, Kokko-Gonzales PI, Kumar A, Laurent MA, Lawley CT, Lee SE, Lee X, Liao AK, Loch JA, Lok M, Luo S, Mammen RM, Martin JW, McCauley PG, McNitt P, Mehta P, Moon KW, Mullens JW, Newington T, Ning Z, Ling Ng B, Novo SM, O'Neill MJ, Osborne MA, Osnowski A, Ostadan O, Paraschos LL, Pickering L, Pike AC, Pike AC, Chris Pinkard D, Pliskin DP, Podhasky J, Quijano VJ, Raczy C, Rae VH, Rawlings SR, Chiva Rodriguez A, Roe PM, Rogers J, Rogert Bacigalupo MC, Romanov N, Romieu A, Roth RK, Rourke NJ, Ruediger ST, Rusman E, Sanches-Kuiper RM, Schenker MR, Seoane JM, Shaw RJ, Shiver MK, Short SW, Sizto NL, Sluis JP, Smith MA, Ernest Sohna Sohna J, Spence EJ, Stevens K, Sutton N, Szajkowski L, Tregidgo CL, Turcatti G, Vandevondele S, Verhovsky Y, Virk SM, Wakelin S, Walcott GC, Wang J, Worsley GJ, Yan J, Yau L, Zuerlein M, Rogers J, Mullikin JC, Hurles ME, McCooke NJ, West JS, Oaks FL, Lundberg PL, Klenerman D, Durbin R, Smith AJ. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456(7218):53–9. https://doi.org/10.1038/nature07517.
McInerney P, Adams P, Hadi MZ. Error rate comparison during polymerase chain reaction by DNA polymerase. Mol Biol Int. 2014;2014:287430.
Potapov V, Ong JL. Examining sources of error in PCR by single-molecule sequencing. PLoS ONE. 2017;12(1):e0169774.
Schirmer M, D’Amore R, Ijaz UZ, et al. Illumina error profiles: resolving fine-scale variation in metagenomic sequencing data. BMC Bioinform. 2016;17:125. https://doi.org/10.1186/s12859-016-0976-y.
McCoy RC, Taylor RW, Blauwkamp TA, Kelley JL, Kertesz M, et al. Illumina TruSeq synthetic long-reads empower de novo assembly and resolve complex, highly-repetitive transposable elements. PLoS ONE. 2014;9(9):e106689. https://doi.org/10.1371/journal.pone.0106689.
Bustin S, Dhillon HS, Kirvell S, Greenwood C, Parker M, Shipley GL, et al. Variability of the reverse transcription step: practical implications. Clin Chem. 2015;61(1):202–12.
Immonen TT, Camus C, Reid C, Fennessey CM, Del Prete GQ, Davenport MP, Lifson JD, Keele BF. Genetically barcoded SIV reveals the emergence of escape mutations in multiple viral lineages during immune escape. Proc Natl Acad Sci USA. 2020. https://doi.org/10.1073/pnas.1914967117.PMID:31843933;PMCID:PMC6955354.
Vanderford TH, Bleckwehl C, Engram JC, Dunham RM, Klatt NR, Feinberg MB, Garber DA, Betts MR, Silvestri G. Viral CTL escape mutants are generated in lymph nodes and subsequently become fixed in plasma and rectal mucosa during acute SIV infection of macaques. PLoS Pathog. 2011. https://doi.org/10.1371/journal.ppat.1002048.PMID:21625590;PMCID:PMC3098234.
Pfeiffer F, Gröber C, Blank M, Händler K, Beyer M, Schultze JL, et al. Systematic evaluation of error rates and causes in short samples in next-generation sequencing. Sci Rep. 2018;8(1):10950.
Kafetzopoulou LE, Efthymiadis K, Lewandowski K, Crook A, Carter D, Osborne J, et al. Assessment of metagenomic Nanopore and Illumina sequencing for recovering whole genome sequences of chikungunya and dengue viruses directly from clinical samples. Euro Surveill. 2018;23(50):1800228.
Ellis AL, Balgeman AJ, et al. MAIT cells are functionally impaired in a mauritian cynomolgus macaque model of Siv and Mtb co-infection. PLoS Pathog. 2020;16(5):e1008585.
Cline AN, Bess JW, Piatak M, Lifson JD. Highly sensitive SIV plasma viral load assay: practical considerations, realistic performance expectations, and application to reverse engineering of vaccines for AIDS. J Med Primatol. 2005;34(5–6):303–12.
Acknowledgements
We would like to thank the Virology Services unit at the Wisconsin National Primate Research Center for quantifying SIV vRNA and cDNA, which allowed us to dilute our samples to predetermined levels. We would also like to thank Josh Quick and Nick Loman for helping us with primer design.
Funding
Research reported in this publication was supported by the Office Of The Director, National Institutes of Health under Award Number P51OD011106 to the Wisconsin National Primate Research Center, University of Wisconsin-Madison. This work was also supported by an Australian National Health and Medical Research Council (NHMRC) grants 1080001 and 1052979 (to MPD) and NHMRC Career Development Fellowship 1067590 (to VV).
Author information
Authors and Affiliations
Contributions
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. RVM and SLO wrote the manuscript. Primer design and validation by NF and MSS. Data analysis was aided by MPD, TS, and VV, and conducted by RVM and SLO. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent to publication
Not applicable.
Competing interests
All authors read and approved the manuscript and declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Moriarty, R.V., Fesser, N., Sutton, M.S. et al. Validation of multiplex PCR sequencing assay of SIV. Virol J 18, 21 (2021). https://doi.org/10.1186/s12985-020-01473-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-020-01473-0