
Abstract
Background
Goatpox virus (GTPV) and sheeppox virus (SPPV), which belong to the Capripoxvirus (CaPV), are economically important pathogens of small ruminants. Therefore, a sensitive, specific and rapid diagnostic assay for detection of GTPV and SPPV is necessary to accurately and promptly control these diseases.
Methods
Recombinase polymerase amplification (RPA) assays combined with a real-time fluorescent detection (real-time RPA assay) and lateral flow dipstick (RPA LFD assay) were developed targeting the CaPV G-protein-coupled chemokine receptor (GPCR) gene, respectively.
Results
The sensitivity of both CaPV real-time RPA assay and CaPV RPA LFD assay were 3 × 102 copies per reaction within 20 min at 38 °C. Both assays were highly specific for CaPV, with no cross-reactions with peste des petits ruminants virus, foot-and-mouth disease virus and Orf virus. The evaluation of the performance of these two assays with clinical sample (n = 107) showed that the CaPV real-time RPA assay and CaPV RPA LFD assay were able to specially detect SPPV or GTPV present in samples of ovine in liver, lung, kidney, spleen, skin and blood.
Conclusions
This study provided a highly time-efficient and simple alternative for rapid detection of GTPV and SPPV.
Similar content being viewed by others


Background
Sheeppox virus (SPPV) and goatpox virus (GTPV) belong to the genus Capripoxvirus (CaPV), subfamily Chordopoxvirinae, family Poxviridae, and cause serious pox diseases of domesticated small ruminants [1, 2]. Sheeppox and goatpox are endemic in most of Asia, the Middle East and North Africa and classified as notifiable diseases by World Organization for Animal Health (OIE, 2013, accessed 10.23.13) [3, 4]. For accurately and promptly controlling any outbreak, the foremost requirement is the sensitive, specific and rapid tool for detection of the causative agents [5, 6]. Although various methods such as virus isolation, serology tests and polymerase chain reaction (PCR) [7,8,9,10] are available for diagnosis of these diseases, these tests have certain limitations such as time-consuming, laborious and technical complexity [6, 11, 12]. PCR based-diagnostic assay relies on expensive instrumentation such as thermal cycler and usually takes over more than one hour to complete, which make it hard to be used in poorly equipped laboratories or as a pen-site test in the field.
Recombinase polymerase amplification (RPA) is a novel isothermal alternative to PCR, which can amplify detectable amount of DNA in 20 min or less with simple instrumentation. RPA employs recombinases to anneal oligonucleotide primers to template DNA for extension and amplification by a polymerase at an isothermal temperature [13]. Real-time detection of RPA amplicons could be performed through TwistAmp exo probes (TwistDx, Cambridge, UK). Fluorescence accumulation relies on the separation of fluorophore and quencher through exonuclease III. As an alternative to real-time detection, RPA amplicons could also be visualized on lateral flow dipstick with TwistAmp nfo probe. Currently, RPA assay has been successfully developed for rapid detection of different viruses of veterinary importance, including foot-and-mouth disease virus [13], avian influenza H5N1 [14], Orf virus [15, 16], bovine viral diarrhea virus [17] and canine parvovirus type 2 [18]. The purpose of this study was to develop a RPA assay based on either real-time fluorescent detection (real-time RPA assay) or lateral flow dipstick (RPA LFD assay) for rapid detection of GTPV and SPPV and evaluate its performance in clinical samples.
Methods
Virus strains, reference DNA, and samples preparing and extracting
Goatpox virus (GTPV) /AV40, GTPV/AV41, GTPV/GS-V1, sheeppox virus (SPPV) /Gulang2009, SPPV/Jingtai2011, SPPV/Hubei, Orf virus (ORFV) /Vaccine/CHA, ORFV/HB/CHA, peste des petits ruminants virus (PPRV)/Nigeria 75/1, foot-and-mouth disease virus (FMDV)/O/CHA and FMDV/A/CHA were preserved in our laboratory. To prepare reference DNA, 374 bp CaPV GPCR segments (ranging from 301 bp to 675 bp of KF661979.1) were synthesized by Genewiz (Suzhou, China) and cloned into a pUC57 vector, designated as pCaPV/RPA. The pCaPV/RPA DNA was extracted by Plasmid Mini kit I (Promega, USA) and then measured by Nanovue (GE life science). The DNA copy number was calculated using the following equation: DNA copy number = (ng × 6.02 × 1023 × 10−9)/ (Fragment length (bp) × 660). The DNA standard was then aliquoted and stored at −80 °C until used. To prepare CaPV-spiked tissue lysates, CaPV-free tissue samples of liver, lung, stomach, kidney, lymphatic node, spleen, nasal swab and skin (n = 24, three each sample type) were collected from three healthy sheep. 10% (w/vol) tissue suspensions (one milligram tissue samples in nine volumes PBS) were then prepared by homogenizing tissue samples in PBS using MP FastPrep-24. Following a brief centrifugation, the homogenized tissue samples were spiked with SPPV/Gulang 2009 at different concentrations from 107 copies to 104 copies per reaction and stored at −80 °C until used. During the period of October 2014 to August 2015, one hundred and seven clinical samples (liver, lung, kidney, spleen, skin and blood) were collected from fourteen suspected sheep and six suspected goats in Gansu province which were characterized by pyrexia, excessive salivation and generalized pock lesions in the skin. Tissue samples were collected by the animal disease investigating teams based on good animal practices of the Animal Ethics Procedures and Guidelines of the People’s Republic of China (AEPGPRC), and the samples were stored at −80 °C in our laboratory until used. Nucleic acids from virus strains, spiked samples and clinical samples were extracted using the viral DNA/RNA extraction kit (TaKaRa, Dalian, China), which could only be used in the laboratory. In order to employ an energy-free assay in the field, a single innuPREP MP basic kit A (Jena Analytik, Jena, Germany) with a magnetic bead separation rack was tested using spiked samples (n = 24), and the extraction efficiency were then evaluated by real-time RPA assay, CaPV RPA LFD assay and CaPV real-time qPCR assay, respectively.
Real-time qPCR assay
The real-time qPCR assay which could detect SPPV, GTPV and lumpy skin disease virus (LSDV) was carried out in an Agilent Technologies Stratagene Mx3005P thermocycler (Life technologies, USA) as previously reported [6]. Briefly, The PCR assay was carried out in a 25 μL reaction volume containing 2 × PCR buffer (a buffer containing 0.4 mM of each dNTP and 6 mM MgSO4, 12.5 μL), Taq DNA polymerase (5 U/μL, 0.5 μL), the probe (5`-CAATGGGTAAAAGATTTCTA-3`) (10 μM, 0.5 μL), the forward prime (5`-GGCGATGTCCATTCCCTG-3`) (10 μM, 1 μL), reverse primer (5`-AGCATTTCATTTCCGTGAGGA-3`) (10 μM, 1 μL), the DNA template (10 pg - 0.1 μg, 2 μL) and RNase-free water (9.5 μL). The cycling proceeded at 95 °C for 5 min, followed by 40 cycles of 95 °C for 50 s, 50 °C for 50 s and 72 °C for 1 min, and an additional extension for 5 min.
RPA oligonucleotides and conditions
After blasting GPCR gene of CaPV (number of access: KF661979.1 (SPPV), KF661976.1 (SPPV), JQ310666.1 (SPPV), FJ869364.1 (GTPV), FJ869361.1 (GTPV), KP663705.1 (GTPV), KP719918.1 (LSDV), FJ869376.1 (LSDV) and KR024780.1 (LSDV)), one probe and three different forward and reverse primers targeting the GPCR gene conserved region were designed for each assay (CaPV real-time RPA and CaPV RPA LFD assay) and synthesized by Sangon Biotech (Table 1). The RPA assay was carried out in a 50 μL freeze-dried reaction tube, using 29.5 μL rehydration buffers (TwistDx, Cambridge, UK), 2.1 μL of each primer (10 μM), 0.6 μL probe (10 μM), 11.2 μL ultrapure water, 2 μL template and 2.5 μL magnesium acetate (280 mM). The real-time CaPV RPA assay was carried out using the TwistAmp exo kit (TwistDx, Cambridge, UK), and the fluorescence signal in the FAM channel (Excitation 470 nm, Detection 520 nm) was detected in an Agilent Technologies Mx3005P thermocycler for 60 cycles at 38 °C for 20 s. The RPA reaction was completed in 20 min. A sample was deemed positive if all replicates were three and a half standard deviations (3.5SD) above the background during a defined time range (i.e. after 19 to 20 min of amplification). A threshold time range of 0 to 4 min and 30 s was used. The CaPV RPA LFD assay was performed using the TwistAmp nfo kit (TwistDx, Cambridge, UK) at 38 °C for 20 min in a water bath. The LFD strips (Milenia Biotec GmbH, Germany) were used to detect amplified products. One μL of the amplified product was diluted in 99 μL of the assay buffer (Tris-buffered saline). The LFD strip were directly inserted into the mixture and incubated at an upright position for 2 min. A test was considered positive when the detection line and the control line were visible. A test was considered negative when only the control line was visible.
Sensitivity and specificity of CaPV real-time RPA and CaPV RPA LFD assays
To determine sensitivity of CaPV real-time RPA assay and CaPV RPA LFD assay, the pCaPV/RPA DNA (ranging from 3 × 107 to 3 × 101 genome copies per reaction) was used in 8 replicates. RNase-free water was used as negative control reaction template in both CaPV real-time RPA assay and RPA LFD assay. The threshold time of CaPV real-time RPA assay was plotted against reference DNA molecules detected and the semi-log non-regression analysis was calculated with PRISM 5.0 software (GraphPad Software, USA). The probit analysis was performed using Statistica software (StatSoft, Hamburg, Germany). The specificity of both CaPV real-time RPA assay and RPA LFD assay was evaluated with nucleic acids extracted from different virus listed in Table 2. To determine the correlation of CaPV real-time RPA assay with CaPV real-time qPCR assay, both assays were tested with CaPV spiked samples (n = 24). The correction of CaPV real-time RPA assay threshold time (y axis) with CaPV real-time qPCR assay cycle threshold (CT) values (x axis) were generated by Excel software. To determine the optimum amplification temperature, CaPV RPA LFD assay was performed at a range of temperatures from 15 °C to 50 °C. The reaction time of CaPV RPA LFD assay was determined by terminating the RPA reaction at 0, 1, 5, 10, 15, 20, 25 and 30 min after the addition of magnesium acetate by immediate dilution and analysis on the lateral flow dipsticks.
Results
Sensitivity and specificity of CaPV real-time RPA assay
In initial optimization, nine combinations of candidate primers (3 forward and 3 reverse, Table 1) were generated and tested for the time to fluorescence threshold with the probe. Of these, five primer pairs produced signal with the probe, and three of the primer pairs (CaPV Fe3/CaPV Re1, CaPV Fe2/CaPV Re2 and CaPV Fe3/CaPV Re2) performed similarly and produced signal faster than other pairs (CaPV Fe1/CaPV Re1 and CaPV Fe1/CaPV Re3) (Fig. 1). Due to resource constraints, the primer pairs CaPV Fe2/CaPV Re2 were selected randomly for subsequent evaluation.

Optimal primers and probe combinations of CaPV real-time RPA assay. Three forward primers (CaPV Fe1 to CaPV Fe3), three reverse primers (CaPV Re1 to CaPV Re3) and one probe (CaPV Pe) were used to select the best combination. NC represents negative control
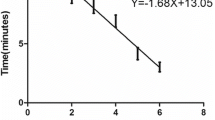
To test the sensitivity of CaPV real-time RPA assay, serial dilutions of the purified reference DNA were tested for 8 replicates. As shown in Fig. 2, the dynamic detection range of the assay spans 5 logs ranging from 7 to 2 log copies per reaction, with the corresponding threshold time ranging from 3 min at 3 × 107 copies per reaction to 7 min at 3 × 102 copies per reaction at 38 °C. This result indicates that CaPV real-time RPA assay has a wide dynamic range for quantifying target DNA (Fig. 2a, b). The detection limit of CaPV real-time RPA assay at 95% probability was 3 × 102 copies per reaction (probit analysis, p ≤ 0.05) (Fig. 2c). To further evaluate the sensitivity, the assay was tested with SPPV-spiked samples and compared with real-time qPCR assay. The results showed that both assays could detect viral DNA present in all the samples (Additional file 1: Table S1), and good correlation was found between threshold time (y axis) of CaPV real-time RPA assay and cycle threshold (CT) (x axis) of CaPV real-time qPCR assay (R squared 0.86, Fig. 3). In evaluation of the specificity of CaPV real-time RPA assay, consistent positive signal was only observed for GTPV strains (GTPV AV40, GTPV AV41, GTPV GS-V1) and SPPV strains (SPPV Gulang2009, SPPV Jingtai2011, SPPV Hubei), and no cross detection was observed with other viruses which can infect sheep and goats, including FMDV, ORFV and PPRV (Table 2). A simple DNA preparation method (single innuPREP MP basic kit with a magnetic bead separation rack) was also applied to spiked tissues samples. As shown in Table 3, CaPV real-time RPA assay performed well on these extracted DNA. There was no difference of the detection sensitivity between the two extraction methods when tested by CaPV real-time RPA assay (Table 3 and Additional file 1: Table S1).

Sensitivity of real-time RPA assay (a) Typical raw fluorescence data of CaPV real-time RPA assay using a dilution series of the pCaPV/RPA DNA. NC represents negative control; (b) Reproducibility of the CaPV real-time RPA assay; (c) The limit of detection in 95% probability based on eight replicates

Comparison between performances of CaPV real-time RPA assay with real-time qPCR assay. The correction of CaPV real-time RPA assay threshold time (y axis) with CaPV real-time qPCR assay cycle threshold (CT) values (x axis) on CaPV spiked samples (n = 24) were generated by Excel software
Sensitivity and specificity of CaPV RPA LFD assay
In exploring the optimal temperature for amplification in CaPV RPA LFD assay, it was found that the target viral DNA gene can be amplified well from 30 °C to 45 °C and could be detected in more than 10 min (Fig. 4a, b). The sensitivity of the CaPV RPA LFD assay was determined using serial dilutions of the purified reference DNA as described above. As shown in Fig. 5a, the sensitivity of CaPV RPA LFD assay was 3 × 102 copies per reaction (Fig. 5a). The limit of detection in 95% probability was 3 × 102 copies per reaction (probit analysis, p ≤ 0.05) (Fig. 5b). To further determine its sensitivity, it was evaluated using SPPV-spiked samples (n = 24). Positive band on the LFD was observed for all spiked samples (Additional file 1: Table S1). The detection limit of CaPV RPA LFD assay was also tested on SPPV/Gulang 2009 gDNA (genomic DNA), and the results showed that it could detect as low as 103 copies per reaction (Additional file 2: Figure S1). In investigation of the specificity of CaPV RPA LFD assay, the reactions were performed using a panel of genomes extracted from other important viruses of small ruminants as described above, which caused similar clinical signs. As shown in Table 2, the CaPV RPA LFD assay was specific for the detection of GTPV and SPPV. The CaPV RPA LFD assay also performed well on DNA extracted from spiked tissues samples using a simple DNA preparation method as described above (Table 3). And there was no difference of detection sensitivity between the two extraction methods when tested by CaPV RPA LFD assay (Table 3 and Additional file 1: Table S1).

Determination of reaction temperature and time (a) CaPV RPA LFD assay are performed at different temperatures as shown. b The test line is visible at 38 °C when the amplification time is longer than 10 min

Sensitivity of CaPV RPA LFD assay (a) The sensitivity of CaPV RPA LFD assay was performed using a dilution series of the pCaPV/RPA DNA, and NC represents negative control; (b) The limit of detection in 95% probability based on eight replicates of CaPV RPA LFD assay
Performance of CaPV real-time RPA and RPA LFD assay on clinical samples
All the clinical samples (n = 107) were detected simultaneously by CaPV real-time RPA assay, CaPV RPA LFD assay and CaPV real-time qPCR assay. Thirty-six samples were determined to be positive by CaPV real-time RPA assay (threshold time ranging from 4 to 6.6 min) and CaPV RPA LFD assay, while thirty-seven samples were positive by CaPV real-time qPCR assay (CT value ranging from 18 to 29). The clinical sensitivity and specificity of CaPV real-time RPA assay and CaPV RPA LFD assay for identification of GTPV and SPPV were 97% and 100%, respectively, when compared to CaPV real-time qPCR assay (Table 4).
Discussion
This study shows that the developed CaPV real-time RPA assay and CaPV RPA LFD assay are specific for detection of SPPV and GTPV with a detection limit of three hundred copies within 20 min. The sequence alignment shows these two methods would be also capable of detecting LSDV, which causes lumpy skin disease in cattle. However, their effectiveness for detecting LSDV in China remains to be determined as China is free of LSDV. RPA technology has multiple advantages over real-time qPCR assays. RPA does not require expensive equipment for amplification and has quicker time-to-answer under low temperature. These characteristics of the RPA assay make it much more applicable for field detection, in an infrastructure limited rural area, or for rapid diagnosis in less well-equipped laboratories. For the CaPV RPA LFD assay developed in this study, only a water bath is required for the RPA assay itself, and the amplicons analysis could be performed simply on the LFD strip which can be read by the naked eyes. A simple point-of-care scanner (ESEQuant tube scanner device, Germany) can also be used in the real-time RPA assay [13, 14]. The scanner is much cheaper and simpler than thermal cycler machine, and is also powered by battery which can be changed in the field.
For pen-site diagnostics, isothermal amplification technologies are of great interest due to their convenience or simplicity. Of these methods, a loop-mediated isothermal amplification (LAMP) has been studied for CaPV detection. The first CaPV LAMP assay targeted CaPV P32 gene was designed to detect both SPPV and GPPV as well as LDSV, and had a detection limit of 163 DNA copies per μL [19, 20], which is equivalent to the performance of the developed RPA assay in this study. In contrast to RPA assay, LAMP assay requires a longer time (45–60 min), a higher temperature (60–65 °C) and more complex primers (three pairs of primers) [19, 20]. Moreover, LAMP assay employs non-specific SYBR Green-based detection while RPA assay employs specific probes detection.
Conclusions
The CaPV real-time RPA assay and CaPV RPA LFD assay are successfully developed for the rapid and specific detection of SPPV and GTPV. Both assays could specially detect SPPV or GTPV present in liver, lung, kidney, spleen, skin and blood. The results are encouraging but the assay must be validated by analysis of a larger number of samples. With further optimization and validation, the RPA has potential to be a promising alternative to real-time qPCR or other isothermal methods for rapid detection of SPPV and GPPV or could be used in the field or in an infrastructure limited rural area.
Abbreviations
- CaPV:
-
Capripoxvirus
- GTPV:
-
Goatpox virus
- Real-time qPCR:
-
Real-time quantitative PCR
- Real-time RPA assay:
-
Recombinase polymerase amplification assay using real-time fluorescent detection
- RPA LFD assay:
-
Recombinase polymerase amplification assay combination with lateral flow dipstick
- RPA:
-
Recombinase polymerase amplification
- SPPV:
-
Sheeppox virus
References
Carn VM. Control of capripoxvirus infections. Vaccine. 1993;11:1275–9.
Lefkowitz EJ, Upton C, Changayil SS, Buck C, Traktman P, Buller RM. Poxvirus Bioinformatics Resource Center: a comprehensive Poxviridae informational and analytical resource. Nucleic Acids Res. 2005;33:D311–6.
Mangana O, Kottaridi C, Nomikou K. The epidemiology of sheep pox in Greece from 1987 to 2007. Rev Sci Tech. 2008;27:899–905.
Bowden TR, Babiuk SL, Parkyn GR, Copps JS, Boyle DB. Capripoxvirus tissue tropism and shedding: a quantitative study in experimentally infected sheep and goats. Virology. 2008;371:380–93.
Batra K, Kumar A, Kumar V, Nanda T, Maan NS, Maan S. Development and evaluation of loop-mediated isothermal amplification assay for rapid detection of Capripoxvirus. Vet World. 2015;8:1286–92.
Balinsky CA, Delhon G, Smoliga G, Prarat M, French RA, Geary SJ, Rock DL, Rodriguez LL. Rapid preclinical detection of sheeppox virus by a real-time PCR assay. J Clin Microbiol. 2008;46:438–42.
Zheng M, Liu Q, Jin N, Guo J, Huang X, Li H, Zhu W, Xiong Y. A duplex PCR assay for simultaneous detection and differentiation of Capripoxvirus and Orf virus. Mol Cell Probes. 2007;21:276–81.
Markoulatos P, Mangana-Vougiouka O, Koptopoulos G, Nomikou K, Papadopoulos O. Detection of sheep poxvirus in skin biopsy samples by a multiplex polymerase chain reaction. J Virol Methods. 2000;84:161–7.
Ireland DC, Binepal YS. Improved detection of capripoxvirus in biopsy samples by PCR. J Virol Methods. 1998;74:1–7.
Mangana-Vougiouka O, Markoulatos P, Koptopoulos G, Nomikou K, Bakandritsos N, Papadopoulos P. Sheep poxvirus identification from clinical specimens by PCR, cell culture, immunofluorescence and agar gel immunoprecipitation assay. Mol Cell Probes. 2000;14:305–10.
Stubbs S, Oura CA, Henstock M, Bowden TR, King DP, Tuppurainen ES. Validation of a high-throughput real-time polymerase chain reaction assay for the detection of capripoxviral DNA. J Virol Methods. 2012;179:419–22.
Lamien CE, Lelenta M, Goger W, Silber R, Tuppurainen E, Matijevic M, Luckins AG, Diallo A. Real time PCR method for simultaneous detection, quantitation and differentiation of capripoxviruses. J Virol Methods. 2011;171:134–40.
Abd El Wahed A, El-Deeb A, El-Tholoth M, Abd El Kader H, Ahmed A, Hassan S, Hoffmann B, Haas B, Shalaby MA, Hufert FT, Weidmann M. A portable reverse transcription recombinase polymerase amplification assay for rapid detection of foot-and-mouth disease virus. PLoS One. 2013;8:e71642.
Yehia N, Arafa AS, Abd El Wahed A, El-Sanousi AA, Weidmann M, Shalaby MA. Development of reverse transcription recombinase polymerase amplification assay for avian influenza H5N1 HA gene detection. J Virol Methods. 2015;223:45–9.
Yang Y, Qin X, Wang G, Zhang Y, Shang Y, Zhang Z. Development of a fluorescent probe-based recombinase polymerase amplification assay for rapid detection of Orf virus. Virol J. 2015;12:206.
Yang Y, Qin X, Wang G, Jin J, Shang Y, Zhang Z. Development of an isothermoal amplification-based assay for rapid visual detection of an Orf virus. Virol J. 2016;13:46.
Aebischer A, Wernike K, Hoffmann B, Beer M. Rapid genome detection of Schmallenberg virus and bovine viral diarrhea virus by use of isothermal amplification methods and high-speed real-time reverse transcriptase PCR. J Clin Microbiol. 2014;52:1883–92.
Wang J, Liu L, Li R, Fu Q, Yuan W. Rapid and sensitive detection of canine parvovirus type 2 by recombinase polymerase amplification. Arch Virol. 2016;161:1015–8.
Zhao Z, Fan B, Wu G, Yan X, Li Y, Zhou X, Yue H, Dai X, Zhu H, Tian B, et al. Development of loop-mediated isothermal amplification assay for specific and rapid detection of differential goat pox virus and sheep pox virus. BMC Microbiol. 2014;14:10.
Murray L, Edwards L, Tuppurainen ES, Bachanek-Bankowska K, Oura CA, Mioulet V, King DP. Detection of capripoxvirus DNA using a novel loop-mediated isothermal amplification assay. BMC Vet Res. 2013;9:90.
Acknowledgements
We thank Mr. Yuen Zhang from Medical School, UCL, UK for his help with editorial assistance.
Funding
Our work was supported by National Research and Development Program of China (2016YFD0500907), National Natural Science Foundation of China (NO. 31502096) and Innovation Fund of Chinese Academy of Agricultural Sciences (CAAS).
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and Additional file 1: Table S1.
Author information
Authors and Affiliations
Contributions
YY and XLZ performed the study and YY wrote the manuscript, XDQ, ZXZ, XLZ and GZC participated in preparation of samples. YML, XDQ and WZ participated in revision the manuscript, ZZ is the leader of the project. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This work was approved by the Animal Ethics Committee of the Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences (approval number LVRIAEC 2012–018). Tissue samples were collected from sheep and goat based on good animal practices of the Animal Ethics Procedures and Guidelines of the People’s Republic of China (AEPGPRC). Tissue samples were collected from the livestock with owner consent as routine disease surveillance. All study participants provided written and informed consent.
Consent for publication
Written informed consents for publication have been obtained from all the participants.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Table S1.
Comparison of CaPV real-time RPA assay and CaPV RPA LFD assay with real-time qPCR assay on spiked samples. (DOCX 18 kb)
Additional file 2: Figure S1.
The detection limit of CaPV RPA LFD assay. This assay was performed using a dilution series of the SPPV/Gulang 2009 genomic DNA, and NC represents negative control. (PPTX 393 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yang, Y., Qin, X., Zhang, X. et al. Development of real-time and lateral flow dipstick recombinase polymerase amplification assays for rapid detection of goatpox virus and sheeppox virus. Virol J 14, 131 (2017). https://doi.org/10.1186/s12985-017-0792-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-017-0792-7