
Abstract
Background
The high-altitude-adapted frog Rana kukunoris, occurring on the Tibetan plateau, is an excellent model to study life history evolution and adaptation to harsh high-altitude environments. However, genomic resources for this species are still underdeveloped constraining attempts to investigate the underpinnings of adaptation.
Results
The R. kukunoris genome was assembled to a size of 4.83 Gb and the contig N50 was 1.80 Mb. The 6555 contigs were clustered and ordered into 12 pseudo-chromosomes covering ~ 93.07% of the assembled genome. In total, 32,304 genes were functionally annotated. Synteny analysis between the genomes of R. kukunoris and a low latitude species Rana temporaria showed a high degree of chromosome level synteny with one fusion event between chr11 and chr13 forming pseudo-chromosome 11 in R. kukunoris. Characterization of features of the R. kukunoris genome identified that 61.5% consisted of transposable elements and expansions of gene families related to cell nucleus structure and taste sense were identified. Ninety-five single-copy orthologous genes were identified as being under positive selection and had functions associated with the positive regulation of proteins in the catabolic process and negative regulation of developmental growth. These gene family expansions and positively selected genes indicate regions for further interrogation to understand adaptation to high altitude.
Conclusions
Here, we reported a high-quality chromosome-level genome assembly of a high-altitude amphibian species using a combination of Illumina, PacBio and Hi-C sequencing technologies. This genome assembly provides a valuable resource for subsequent research on R. kukunoris genomics and amphibian genome evolution in general.
Similar content being viewed by others

Background
Recent advances in sequencing techniques have led to the rapid increase in high-quality vertebrate genomes available to the evolutionary biology community [1,2,3]. However, complete genome assemblies of anurans remain relatively rare due to their large size (> 4 GB, range: 4 GB to more than 8 Gb GB) and complexity due to repetitive elements which makes them expensive to sequence and challenging to assemble [4]. Studies focused on the evolution and ecology of anurans are important due to their sensitivity to environmental changes, but the lack of high-quality genomic resources and large-scale re-sequencing projects is limiting progress towards understanding the genetic underpinnings of their adaptation to new and changing environments [4, 5]. To date, out of ca. 8100 amphibian species described [6], about10 anuran genomes have been published including Xenopus tropicalis [7], Nanorana parkeri [5], Xenopus laevis [8], Rana catesbeiana [9], Rhinella marina [10], Oophaga pumilio [11], Leptobrachium leishanense [2], Vibrissaphora ailaonica [12], Bufo gargarizans [3] and Rana temporaria [13]. The genomes that are already available provide a great resource for amphibian comparative genomic research, but to advance our understanding of amphibian genome evolution and amphibian adaptation to their current and future habitats, additional genomes are required. In particular, additional genomes from species closely related to already sequenced species but adapted to unique environmental conditions would be of interest to decipher the genomic basis of adaptations.
Rana kukunoris, which is known as the Plateau brown frog, is a true frog from the family Ranidae and is an endemic species living in the Qinghai-Tibetan Plateau of China and distributed in altitudes ranging from 2200 to 4400 m [14]. Living at such high altitudes exposes R. kukunoris to extreme environmental conditions including hypoxia, high levels of UV-B radiation and dramatic temperature fluctuations [5]. Consequently, R. kukunoris provides an excellent biological model to study amphibian adaptations to extreme high-altitude conditions. However, current research has mainly been limited to a few phylogenetic studies which have tried to resolve its status as a valid species [15], larger scale biogeographic studies of Chinese brown frogs [16] and sex-chromosome evolution and turnover in true frogs [17].
The genome of R. kukunoris is thought to consist of 2n = 24 chromosomes [18], with Rana species in general having 26 diploid chromosomes (n = 13) but chromosome number has been observed vary from 2n = 22 to 2n = 24 [19]. Its mitochondrial genome has been assembled which revealed that R. kukunoris is closely related to R. temporaria and R. chensinensis [20]. Recent genomic studies of this species have focused on generating transcriptomic data to identify differentially expressed genes associated with high altitude [20]. However, having a well assembled, high-quality, chromosome-level genome would facilitate future research to study the genomic underpinnings of adaptation to differing environments and anuran genome evolution in general [21,22,23].
In this study, we report the assembly and annotation of the complex and large de novo genome of a male R. kukunoris using Illumina short-read, PacBio long-range and Hi-C sequencing and comparison of this assembly to that of the ten available anuran genomes. To this end, we assessed whole-genome synteny, transposable elements and their distribution, amphibian-specific highly conserved elements (HCEs), and changes in functionally important gene families among species. This genome will facilitate future in-depth phylogenomic studies, whole-genome resequencing studies, investigations into how species adapt to extreme high-altitude environments and how genomes of amphibians evolve.
Results
Genome survey
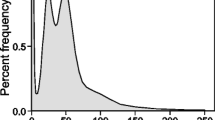
The Illumina sequencing produced 322.83 Gb of high-quality sequences at a total sequencing depth of 71.91×. The haploid genome size of R. kukunoris with 21 k-mer coverage evaluation was estimated to be ~ 4.49 Gb (4,489,353,974 bp, peak = 29, SI Appendix, Additional file 1: Table S2 and Fig. 1) and the estimated heterozygosity rate was ~ 0.3%. Repetitive DNA content was estimated at 59.81% and GC content at 43.62%, suggesting that R. kukunoris has a large and complex genome.

21-mer analysis of Rana kukunoris genome characteristics
Genome assembly
PacBio Sequel II sequencing results generated a total of ~ 437.68 Gb (437,678,042,147 bp) sequencing data. The average read length and N50 length of PacBio reads were 20.53 Kb (20,527 bp) and 30.65 Kb (30,654 bp), respectively (Additional file 1: Tables S3, S4), and total sequencing depth was 90.61×. After polishing the assembly using a combination of PacBio and Illumina sequencing data to improve its accuracy, we obtained a genome of ~ 4.83 Gb (4,830,373,361 bp) with contig N50 of ~ 1.80 Mb (1,798,518 bp) and a GC content of 44.41% (Table 1; Additional file 1: Table S5). The contig N50, total genome size and coverage in this assembly were relatively high compared to the amphibian genomes reported to date (Additional file 1: Table S6). The constructed Hi-C libraries produced 1,170,517,199 validated interaction pairs to achieve a chromosome-level R. kukunoris genome, being clustered in 6555 scaffolds with the final genome length of ~ 4.81 Gb (4,814,345,922 bp) and grouped into 12 pseudochromosomes (Fig. 2; Additional file 1: Table S7).

The multidimensional landscape of Rana kukunoris genome. Rings, from outside to inside, correspond to: (a) Chromosome ideograms; (b) TE density; (c) gene density and (d) GC content. Lines inside the circle display collinear synteny
Up to 99.50% of the Illumina raw reads (NCBI Accession No.: SRR17818264-17818270) could be mapped back to the assembled R. kukunoris genome (Additional file 1: Table S8), suggesting that most reads could be successfully assembled into the genome. According to the CEGMA database (version 2.5), 93.45% of 458 Core Eukaryotic Conserved Genes (CEGs) were present in the assembly and 72.18% of 248 highly conserved CEGs were present. In addition, ~ 92.31% of the complete BUSCO genes were recovered including 87.81% complete and single-copy BUSCOs(S), 4.50% complete and duplicated BUSCOs (D), 1.88% fragmented BUSCOs(F) and 5.81% missing BUSCOs (M) (Additional file 1: Table S9). In general, the R. kukunoris genome showed high genomic integrity.
Synteny analysis
To explore the genome evolution, synteny analysis was performed between the R. kukunoris (high-altitude species) and R. temporaria (low-altitude species) genomes. A high degree of collinearity was observed, each chromosome of R. kukunoris having corresponding chromosome in R. temporaria. For instance, most of the synteny blocks of R. kukunoris in the pseudo-chromosomes p-chr1, p-chr2, p-chr3 p-chr4, p-chr5, p-chr6, p-chr7, p-chr8, p-chr9, p-chr10, and p-chr12 were matched to R. temporaria chr1, chr2 and chr3, chr4, chr5, chr6, chr7, chr8, chr9, chr10 and chr12, respectively. However, the R. kukunoris p-chr11 appeared to be fusion of R. temporaria chr11 and chr13, suggesting chromosome fusion/fission events between the two species (Fig. 3).

Chromosome synteny between R. kukunoris and R. temporaria genomes
Genome annotation
In total, 32,304 protein-coding genes were predicted according to ab initio prediction (Stanke et al., 2006), homology-based prediction and RNA-seq data (NCBI Accession No.: SRR17284458; Additional file 1: Table S9). In total, 31,957 of the 32,304 genes, up to 98.93% of the R. kukunoris genes, were annotated across eight databases including KEGG, GO, KOG, SwissProt, Pfam, TrEMBL, eggNOG and NR (Additional file 1: Tables S10, S11). Also, 8554 tRNA, 2510 rRNA, 210 miRNA, 2144 snoRNA and 184 snRNA were predicted. Approximately 61.46% (2,968,809,293/4,830,373,361 bp) of genome appears to consist of TEs, of which retroelements and DNA transposons accounted for 38.60% and 22.85%, respectively (Additional file 1: Table S12). Approximately 9.47% (457,368,139 bp) of the R. kukunoris genome was indicated to be tandem repeats (Additional file 1: Table S6). Based on the comparison with 10 other anuran genomes used in this study, there was no correlation neither between genome size and number of protein-coding genes, nor between genome size and the proportion of repetitive elements in the genome across the species (p > 0.5 in both comparisons, Additional file 1: Table S5).
Phylogenetic relationships, evolution and expansion of gene families
Comparison of the R. kukunoris genome with 10 other anuran genomes revealed that 30,050 of the 32,304 protein coding genes belonged to 17,575 orthologous groups (Fig. 4), of which 176 were single-copy orthologous genes. Reconstruction of phylogenetic relationships using these 176 single-copy genes revealed that R. kukunoris clustered with R. temporaria and R. catesbeiana forming their sister lineage (Fig. 5). The estimated divergence time between R. kukunoris and R. temporaria was 17.7 Ma (13.8–23.5 Ma; Fig. 5). We also identified 279 expanded and 16 contracted gene families in R. kukunoris (Fig. 5). Functional enrichment analysis of expanded gene families revealed DNA recombination GO (Gene Ontology) terms that were significantly enriched. The expanded gene families were mainly related to the innate immune system, DNA repair, and taste sense (Additional file 1: Table S13). The KEGG annotation of the expanded genes suggested that the pathway of the NOD-like receptor signaling pathway had the highest ratio, followed by nucleotide excision repair (Fig. 6).

A Venn diagram showing the clustering of gene families among B. gargarizans, N. parkeri, R. catesbeianus, R. temporaria and R. kukunoris

The phylogenetic relationships and estimated divergence times among 11 amphibian species based on 176 single-copy orthologus genes, Danio rerio was used an outgroup species. Pie diagrams indicate the expansion and contraction of gene family in different species. Purple = expanded gene families, Green = contracted gene families

KEGG pathway and functional enrichment analysis of expanded gene families. The size of the dots are indicative of gene counts enriched in the pathway, and the color of the dot shows the significance of the enrichment. a Kegg pathway; b Cellular component; c Biological process; d Molecular function
Functional enrichment and KEGG annotation of the positively selected genes
We detected 97 single-copy orthologous genes having been under positive selection in the R. kukunoris genome (Additional file 1: Table S14). The positively selected genes (PSGs) were classified by GO annotation into three different function categories. Cellular component annotations were primarily related to RNA polymerase complex and catalytic complex. Molecular functions were mainly protein domain specific binding, SH3 domain binding and peptidase activity. Biological process annotations were mainly negative regulation of developmental growth, interaction with symbionts and regulation of gluconeogenesis. The results regarding KEGG annotation showed that p53 signaling pathway and FoxO signaling pathway had the highest ratios (Additional file 1: Fig. S1).
Discussion
Our results showed that R. kukunoris has a complex and large genome with a size intermediate (4.83 Gb) to other sequenced Rana genomes. Its genome is the third largest of all anuran genomes sequenced so far and like other assembled anuran genomes (Additional file 1: Table S5), over half of the R. kukunoris genome consisted of repetitive elements. In size and structure, the R. kukunoris genome appears to be similar to that of its close relative, R. temporaria [13]. In addition, we also detected several proteins in KEGG pathway of the positively selected genes, which may be associated to the adaptation of high-altitude conditions.
Amphibian genomes contain a lot of repetitive sequences [5]. Similar to the results of previous studies [3, 12], we found that the genome assembly of R. kukunoris consisted of 70.91% repetitive DNA with TEs making up 61.46% of its genome. Hence, the large genome of R. kukunoris seems to be mainly due to the accumulation of interspersed repeats. To this end, our results are consistent with those of previous studies suggesting that LTR expansions underlie genome gigantism in amphibians [3, 5, 9].
Both ultimate and proximate cause of genome size variation among species remain challenging to understand [24,25,26,27,28]. Our results align with the common finding that species with large genomes do not necessarily have more genes, especially protein-coding genes, than species with smaller genomes [29,30,31]. Based on the comparison of 11 anuran genomes used in this study, we did not find any correlation neither between genome size and number protein-coding genes, nor between genome size and the proportion of repetitive elements in the genome across the species. One possible reason for latter finding is that the old repeat elements may have accumulated enough mutations so that they are not recognized anymore as repetitive elements.
Hypoxia, high levels of UV-B radiation and dramatic temperature fluctuations are typical characteristics of high-altitude environments [20, 32] and they are believed to be the driving forces of genome evolution of organisms in these habitats [5, 33]. Fluctuating temperatures will impact on energy metabolism and can activate certain amino acid and carbohydrate metabolism related genes such as hpd, ALPL, Aldh6a1, g6pd, PCK2, NADSYN1, Aldh6a1, and G6PD[5, 33,34,35,36]. We predicted that these genes would also be important for adaptation of the Tibetan tree frog to its high-altitude habitat, however no direct evidence for positive selection on these genes were found. In addition, melanogenesis (the process of producing melanin pigments) may be important for adaptation to higher UV radiation intensity and is a complex process [34]. However, we did find evidence for positive selection of the creb3l2 protein in the kegg pathway of melanogenesis. nucleotide We did find evidence for positive selection for excision repair (DDB1 protein) which may reflect adaptation to high UV radiation intensity. Furthermore, although we would have predicted EPAS1 to be positively selected in R. kukunoris because it has been demonstrated to be under selection for adaptation to hypoxia at high altitudes in Tibetan species [33, 35, 36], we did not detect EPAS1 in KEGG pathway of the positively selected genes.. Likewise, we did not found the FEN1 genes coding for proteins involved in DNA damage repair among the positively selected genes. Taken all this together, the results indicate that different genetic mechanisms may underly different plateau living organisms’ adaptation to their environment [5, 33]. Finally, we note that the abovementioned genes and/or proteins should be the focus of future studies and further functional validation.
Conclusions
In conclusion, we have described a high-quality genome assembly of R. kukunoris which is the 34th amphibian genome assembled (17th chromosome-level genome assembly) so far and the first chromosome-level genome assembly from a high-altitude species. This chromosome-level genome assembly should provide a valuable comparable genomic resource for studies of amphibian genome evolution particularly because it provides opportunity to focus on amphibian genome evolution at much shorter timescales than has been previously possible (cf. Fig. 5). The assembly also provides a resource for population genomic studies of this species, aiding attempts to uncover genomic underpinnings of their adaptation to high-altitude environments.
Methods
Sample collection
An adult male of R. kukunoris was collected from Yushu, western China (32° 32′ 8.35″ N, 96° 25′ 0.63″ E, 4000 m a.s.l). To obtain sufficient high-quality DNA for the PacBio Sequel II platform (Pacific Biosciences, USA) and Illumina Novaseq 6000, the frog was euthanized with MS-222 and dissected (Ethical proof No. IACUC(AHU)-2022-007). Fresh tissues were retrieved and were snap-frozen in liquid nitrogen for 10 min, and then delivered to the sequencing company on dry ice (the Biomarker Technologies Company, Qingdao, China). DNA was extracted from the muscle and liver tissues. To aid the protein-coding gene annotation of R. kukunoris, RNA was extracted from the brain, heart, testis, stomach, intestines, fat, lungs, and skin tissues for transcriptome sequencing.
Illumina and PacBio sequencing
Extracted DNA was sequenced using the Illumina Novaseq 6000 and PacBio Sequel II platforms and extracted RNA was sequenced using the Illumina Novaseq 6000. The PacBio long-reads were used for genome assembly with Smartdenovo software (version 1.06) (https://github.com/ruanjue/smartdenovo), whereas Illumina short reads were used to estimate genome size and to correct errors in the assembled genome. Library preparations of Illumina and PacBio sequencing were conducted following the manufacturer’s protocols. In brief, for the Illumina sequencing, DNA was sonicated to a fragment size of 350 bp, then three libraries were built by terminal repairing, poly-A and adaptor adding, target fragment selection and PCR amplification. To generate PacBio libraries, DNA was sheared using a Covaris g-Tube and a SMRT-bell library was prepared using the SMRTbellR Express Template Preparation Kit (PacBio DNA template prep kit 2.0; 20–30 Kb). Hi-C libraries were constructed using the NEBNext® Ultra™ RNA Library Prep Kit (NEB, UK) according to the manufacturer’s instructions and sequenced using the Illumina NovaSeq 6000 to anchor scaffolds and facilitate genome annotation. Illumina RNA-seq libraries were also prepared following the manufacturer’s instructions and extracted RNA was pool-sequenced using the Illumina NovaSeq 6000 with a read length of PE150. All sequencing was performed by the Biomarker Technologies Company (Qingdao, China).
Genomic data processing and genome survey
Adapters were removed from the sequence reads and low-quality reads were filtered out using fastp (parameters: -q 10 -u 50 -y -g -Y 10 -e 20 -l 100 -b 150 -B 150) [37]. PCR duplicates were removed with samtools (version 1.9) [38]. The filtered reads were used to estimate genome size and other characteristics including heterozygosity, repetitive DNA content and GC content. The k-mer distribution was estimated using the jellyfish software (version 2.1.4) with parameter -h 1,000,000,000 [39]. The heterozygosity ratio of the genome and genome size were estimated using GenomeScope (version 2.0) with the following parameters -k 21 -p 2 -m 100,000 [40]. We followed the rule of best k-mer selection: 4 K-mer ≥ genome size [41]. As this is 421 ≈ 4398 Gb, the appropriate selected k-mer size chosen was 21-mers. The genome size was estimated using the following modified formula: G = Nk mer/Daverage k-mer, where G is genome size, Nk-mer the total number of k-mers, Daverage k-mer the average depth of k-mers [42].
Genome assembly
The raw data from PacBio SequelII platform was corrected with Canu (version 1.5) [43] and the error-corrected PacBio long reads were de novo assembled with Smartdenovo version 1.0 (https://github.com/ruanjue/smartdenovo). To further improve the accuracy of the assembly, the final assembled genome sequence was polished by pilon (version 1.18) based on Illumina raw data with default parameters [44]. To evaluate the accuracy of the assembled genomic sequence, the Illumina sequencing data (NCBI Accession No.: SRR17818264-17818270) was mapped back to the assembled genome using bwa (version 0.7.12) with default parameters [45]. To evaluate the completeness of the genome assembly, Benchmarking Universal Single-Copy Orthologs (BUSCO) (parameters: –evalue 1e−03 (E-value cutoff for BLAST searches), -sp human) was used to search for annotated genes in the assembled genome based on the vertebrata_odb9 database [46]. In addition, the assembly was also investigated with the Core Eukaryotic Genes Mapping Approach (CEGMA version: 2.5) with default parameters [47].
To anchor scaffolds into pseudo-chromosomes, adapter sequences and low-quality raw reads were filtered out using fastp (parameters: -q 10 -u 50 -y -g -Y 10 -e 20 -l 100 -b 150 -B 150). After this, the clean paired-end reads were mapped back to the draft assembly with bwa aligner (version: 0.7.10-r789) and default parameter settings to obtain the unique mapped paired-end reads [48]. Only uniquely mapped reads with a mapping quality > 20 were retained for further analysis. Invalid read pairs, including dangling-end and self-cycle, re-ligation and dumped products, were filtered by HiC-Pro (version 2.10.0) [49]. The uniquely mapped read pairs were used for correction of scaffolds by clustering, ordering and orientating scaffolds into chromosomes by the LACHESIS de novo assembly pipeline (version 1.0) [50] using the parameters: CLUSTER_MIN_RE_SITES = 94; CLUSTER_MAX_LINK_DENSITY = 2; ORDER_MIN_N_RES_IN_TRUNK = 51; ORDER_MIN_N_RES_IN_SHREDS = 51.
Synteny analysis
To determine genetic differences between low-altitude (R. temporaria) and high-altitude species (R. kukunoris), the assembled chromosomes of R. kukunoris were aligned to the well-assembled chromosomes of R. temporaria (accession no. GCF_007399415.2) using Diamond (version 0.9.29.130) with the following parameters e < 1e−5 and C score > 0.5 [51] and the collinearity blocks between species were investigated using MCScanX [52]. After filtering the aligned blocks shorter than 2 Mb in length, we plotted the results using circos [53] and JCVI (version 0.9.13) [54].
Gene prediction and annotation
Three approaches (de novo prediction, homology search, and transcript-based assembly, respectively) were integrated to predict protein-coding genes in the R. kukunoris genome. The de novo gene models were predicted using two ab initio gene-prediction software tools, including Augustus (version 2.4) with the default parameters [55] and SNAP (version 2006-07-28)[56]. For the homolog-based approach, GeMoMa (version 1.7) [57] was performed using the reference gene model from seven species including B. gargarizans, N. parkeri, R. catesbeianus, R. marina, R. temporaria, X. laevis, and X. tropicalis (Additional file 1: Table S1). For the transcript-based prediction, Trinity software (version 2.11) was used to assemble genes [58] and the PASA software (version 2.0.2) [59] was used to predict genes. Gene models from these different approaches were combined using the EVM software (version 1.1.1) [60] and updated by PASA (version 2.0.2) [59]. The tRNAscan-SE (version 2.0.9) algorithm was used to predict tRNA genes [61] and the miRNAs were identified by searching miRBase [62]. Within the Rfamdatabase (version 12.0) [63], identification of the rRNA genes was conducted, and the snoRNA and snRNA genes were also predicted using INFERNAL (version 1.1.1) [64] against the Rfam database (version 14.5) [63]. Pseudogenes were predicted using GenBlastA (version 1.0.4) [65] and GeneWise (version 2.4.1) [66]. GenBlastA was used to scan the whole genomes after masking predicted functional genes [65], and the putative candidates were then analyzed by searching for premature stop codons and frame-shift mutations using GeneWise [66].
The predicted genes were annotated by searching the GenBank Non-Redundant (NR, 20200921, ftp://ftp.ncbi.nlm.nih.gov/blast/db), EggNOG (5.0, http://eggnog5.embl.de/download/eggnog_5.0/) [67], TrEMBL (202005, https://www.uniprot.org), SWISS-PROT (202005, http://ftp.ebi.ac.uk/pub/databases/swissprot) [68], eukaryotic orthologous groups (KOG, 20110125), gene ontology (GO, 20200615, http://geneontology.org) and Kyoto Encyclopedia of Genes and Genomes (KEGG, 20191220, http://www.genome.jp/kegg; 25), and Pfam (version33.1) (http://pfam.xfam.org) [69] databases.
Transposable element and tandem repeat annotation
Transposable elements (TEs) were identified by a combination of homology-based and de novo approaches. We first customized a de novo repeat library of the genome using RepeatModeler (version 2.0.1) [70], RECON (version 1.08) [71] and RepeatScout (version 1.05) [72]. Then full-length long terminal repeat retrotransposons (fl-LTR-RTs) were identified using both LTRharvest (version 1.5.9) [73] with the parameters:-minlenltr 100 -maxlenltr 40,000 -mintsd 4 -maxtsd 6 -motif TGCA -motifmis 1 -similar 85 -vic 10 -seed 20 -seqids yes and LTR_finder (version 1.0) [74] with the parameters -D 40,000 -d 100 -L 9000 -l 50 -p 20 -C -M 0.9. The high-quality intact fl-LTR-RTs and non-redundant LTR libraries were then produced by LTR_retriever (version 2.8) [75]. A non-redundant species-specific TE library was constructed by combining the de novo TE sequence library above with the Repbase (version 19.06) [76], REXdb (version 3.0) [77], and Dfam (version 3.2) [78] databases. Final TE sequences in the given genomes were identified and classified by a homology search against the library using RepeatMasker (version 4.10). Tandem repeats were annotated by MIcroSAtellite identification tool (MISA) (version 2.1) [79], and Tandem Repeats Finder (version 4.09) [80] with the parameters: 1 1 2 80 5 200 2000 –d -h. We also investigated the correlation between genome size and protein-coding gene numbers as well as the correlation between genome size and the proportion of repetitive elements in the genome based on data on 11 anuran genomes (10 previously published genomes and our newly assembled genome).
Gene family identification
To identify putative paralogous and orthologous gene clusters, proteins from the longest transcripts of genes from R. kukunoris, Rhinatrema bivittatum, B. gargarizans, N. parkeri, R. catesbeianus, R. marina, R. temporaria, X. laevis, X. tropicalis, V. ailaonica, Microcaecilia Unicolor and D. rerio were compared using Orthofinder software (version 2.4.0) [81] and Diamond was used to align these protein sequences with an e-value of 0.001. The PANTHER database (version 14.0) was used to annotate the obtained gene families [82].
To resolve phylogenetic relationships among R. kukunoris and other amphibian species including R. bivittatum, B. gargarizans, N. parkeri, R. catesbeianus, R. marina, R. temporaria, X. laevis, X. tropicalis, V. ailaonica, M. unicolor, protein sequences from 176 R. kukunoris single-copy ortholog genes were used for phylogenetic tree reconstruction, with D. rerio used as the outgroup. The protein sequences of the single-copy ortholog genes were aligned with the program mafft (version 5) with the parameters –localpair –maxiterate 1000 [83] and protein alignment was transformed with codon alignment with PAL2NAL (version 14) [84]. The sequence regions with poor alignments were removed using Gblocks (version 0.91b) with the parameters: -b5 = h [85]. After this, all the corresponding coding DNA Sequences (CDS) were concatenated. The best model (GTR + F + I + G4) was determined with ModelFinder [86], and IQtree (version 1.6.11) was used to construct the Maximum Likelihood phylogenetic tree [87] with bootstrap sets of 1000 and GTR + F + I + G4 model.
Divergence times were estimated using MCMCTREE in PAML (version 4.9i) with the model JC69 and the correlated molecular clock [88]. The two parameters (gradient and hessian) were estimated using MCMCTREE. The consistency of the two repeated calculations was 1, and iteration parameters of a Markov chain were -burnin 5,000,000 -sampfreq 30 -nsample 10,000,000. The divergence times were calibrated with estimates from timetree (http://www.timetree.org/) including data for R. kukunoris and R. bivittatum (311–330 Ma), X. laevis and R. temporaria (193–223 Ma), and R. kukunoris and R. marina (145–160 Ma). MCMCTreeR (version 1.1) was used for graphical presentation [89].
To provide more insight into the evolutionary dynamics of the genes, based on the identified gene families and the constructed phylogenetic tree with the predicted divergence times among species, we used CAFE (version 4.2) [90] to analyze gene family expansion and contraction. In CAFE, a random birth and death process was adopted to study gene gain or loss across a specified phylogenetic tree. The gene families with both family-wide and viterbi p-values less than 0.05 were considered to have an accelerated rate for gene gain or loss. Identified gene family expansions and contractions in R. kukunoris were mapped to KEGG pathways for functional enrichment analysis, which was conducted using the enrichment methods implementing hypergeometric test algorithms and the Q-value (FDR, False Discovery Rate) was calculated to adjust the p-values using R-package qvalue [91].
Several genes (e.g., 4-hydroxyphenylpyruvate dioxygenase(hpd), Alkaline Phosphatase(ALPL), Aldehyde dehydrogenase 6 family member A1(Aldh6a1), Glucose-6-phosphate dehydrogenase(G6pd), Phosphoenolpyruvate Carboxykinase 2(PCK2), NAD synthetase 1(NADSYN1), aldehyde dehydrogenase 6 family member A1 (Aldh6a1), endothelial PAS domain protein 1(EPAS1), Flap structure-specific endonuclease 1 (FEN1) and Glucose-6-phosphate dehydrogenase(G6PD) have been shown to be positively selected in high-altitude environments[5, 33, 35, 36]. In order to test whether any of these genes are important in adaptation to high-altitudes in R. kukunoris, we identified genes showing signals of positive selection in the R. kukunoris genome. Single-copy genes of N. parkeri, R. catesbeiana, R. kukunoris, R. temporaria, B. gargarizans, and R. marina were obtained using Orthofinder (version 2.4). then MAFFT (parameters:–localpair –maxiterate 1000) was used for protein alignment of each gene family, and the protein alignment was transformed with codon alignment in PAL2NAL (version 14) after which the CodeML program in PAML (version 4.9i; F3 × 4 model of codon frequencies) was used to detect positively selected genes in the clade containing N. parkeri, R. catesbeiana, R. kukunoris, R. temporaria, B. gargarizans, and R. marina. Among them, the branch-site model was used, and likelihood ratio tests (LRTs) were calculated (p < 0.05) between Model A (foreground clade ω > 1) and null Model (any sites forbidden ω > 1). Posterior probability was calculated using empirical Bayes method (BEB) and posterior probability > 0.95 was considered as positively selected gene. Finally, the R-package clusterProfile was used for the GO and KEGG enrichment analysis of positively selected genes [92].
Availability of data and materials
All sequence data and assembled scaffolds are deposited in NCBI with BioProject accession number PRJNA787055. All data using this study are deposited in the Additional file 1.
References
Beauclair L, Ramé C, Arensburger P, Piégu B, Guillou F, Dupont J, et al. Sequence properties of certain GC rich avian genes, their origins and absence from genome assemblies: case studies. BMC Genomics. 2019;20:734.
Li Y, Ren Y, Zhang D, Jiang H, Wang Z, Li X, et al. Chromosome-level assembly of the mustache toad genome using third-generation DNA sequencing and Hi-C analysis. GigaScience. 2019;8:giz114.
Lu B, Jiang J, Wu H, Chen X, Song X, Liao W, et al. A large genome with chromosome-scale assembly sheds light on the evolutionary success of a true toad (Bufo gargarizans). Mol Ecol Resour. 2021;21:1256–73.
Funk WC, Zamudio KR, Crawford AJ. Advancing understanding of amphibian evolution, ecology, behavior, and conservation with massively parallel sequencing. In Population genomics: wildlife (pp. 211–254). Springer, Cham; 2018.
Sun YB, Xiong ZJ, Xiang XY, Liu SP, Zhou WW, Tu XL, et al. Whole-genome sequence of the Tibetan frog Nanorana parkeri and the comparative evolution of tetrapod genomes. PNAS. 2012;112:1257–62.
Duellman WE, Zug GR. "amphibian". Encyclopedia Britannica, 4 Feb. 2020, https://www.britannica.com/animal/amphibian. Accessed 22 March 2022.
Hellsten U, Harland RM, Gilchrist MJ, Hendrix D, Jurka J, Kapitonov V, et al. The genome of the Western clawed frog Xenopus tropicalis. Science. 2010;328:633–6.
Session AM, Uno Y, Kwon T, Chapman JA, Toyoda A, Takahashi S, et al. Genome evolution in the allotetraploid frog Xenopus laevis. Nature. 2016;538:336–43.
Hammond SA, Warren RL, Vandervalk BP, Kucuk E, Khan H, Gibb EA, et al. The North American bullfrog draft genome provides insight into hormonal regulation of long noncoding RNA. Nat Commun. 2017;8:1433.
Edwards RJ, Tuipulotu DE, Amos TG, O’Meally D, Richardson MF, Russell TL, et al. Draft genome assembly of the invasive cane toad, Rhinella marina. GigaScience. 2018;7:giy095.
Rogers RL, Zhou L, Chu C, Márquez R, Corl A, Linderoth T, et al. Genomic takeover by transposable elements in the strawberry poison frog. Mol Biol Evol. 2018;35:2913–27.
Li J, Yu H, Wang W, Fu C, Zhang W, Han F, et al. Genomic and transcriptomic insights into molecular basis of sexually dimorphic nuptial spines in Leptobrachium leishanense. Nat Commun. 2019;10:1–13.
Streicher JW, Wellcome Sanger Institute Tree of Life programme, Wellcome Sanger Institute Scientific Operations: DNA Pipelines collective et al. The genome sequence of the common frog, Rana temporaria Linnaeus 1758 [version 1; peer review: 2 approved]. Wellcome Open Res. 2021; 6: 286.
Chen W, Wang X, Fan X. Do anurans living in higher altitudes have higher prehibernation energy storage? Investigations from a high-altitude frog. Herpetol J. 2013;23:45–9.
Wang J, Li Z, Gao H, Liu Z, Teng L. The complete mitochondrial genome of the Rana kukunoris (Anura: Ranidae) from Inner Mongolia, China. Mitochondrial DNA B. 2020;5:586–7.
Zhou Y, Wang SR, Zhu HD, Li PP, Yang BT, Ma JZ. Phylogeny and biogeography of South Chinese brown frogs (Ranidae, Anura). PLoS ONE. 2017;12:e0175113.
Jeffries DL, Lavanchy G, Sermier R, Sredl MJ, Miura I, Borzée A, et al. A rapid rate of sex-chromosome turnover and non-random transitions in true frogs. Nat Commun. 2018;9:4088.
Jiang JP, Xie F, Zheng ZH. Phylogenetic relationships of Chinese brown frogs with discussion on the Karyotype Evolution. J Sichuan Univ (Natl Sci Edn). 2002;39:85–9.
Spasić-Bošković O, Tanić N, Blagojević J, Vujošević M. Comparative cytogenetic analysis of European brown frogs: Rana temporaria, R dalmatina and R graeca. Caryologia. 1997;50:139–49.
Yang W, Qi Y, Bi K, Fu JZ. Toward understanding the genetic basis of adaptation to high-elevation life in poikilothermic species: a comparative transcriptomic analysis of two ranid frogs, Rana chensinensis and R. kukunoris. BMC Genomics. 2012;13:588.
Savolainen O, Lascoux M, Merilä J. Ecological genomics of local adaptation. Nat Rev Genet. 2013;14:807–20.
Hoban S, Kelley JL, Lotterhos KE, Antolin MF, Bradburd G, Lowry DB, et al. Finding the genomic basis of local adaptation: Pitfalls, practical solutions, and future directions. Am Nat. 2016;188:379–97.
Rees JS, Castellano S, Andrés AM. The genomics of human local adaptation. Trends Genet. 2020;36:415–28.
Petrov DA. Evolution of genome size: new approaches to an old problem. Trends Genet. 2001;17:23–8.
Shapiro JA, Sternberg RV. Why repetitive DNA is essential to genome function. Biol Rev. 2005;80:227–50.
Gregory TR. Coincidence, coevolution, or causation? DNA content, cell size, and the C-value enigma. Biol Rev. 2001;76:65–101.
Gregory TR. Genome size and developmental parameters in the homeothermic vertebrates. Genome. 2002;45:833–8.
Gregory TR. Variation across amphibian species in the size of the nuclear genome supports apluralistic, hierarchical approach to the C-value enigma. Biol J Linn Soc. 2003;79:329–39.
Ohno S. “So much junk” DNA in our genome. In: Smith HH, editor. Evolution of genetic systems. , New York: Gordon and Breach; 1972. p. 366–70.
Pagel M, Johnstone RA. Variation across species in the size of the nuclear genome supports the junk-DNA explanation for the C-value paradox. P Roy Soc B-Biol Sci. 1992;249:119–24.
Gregory TR. Macroevolution, hierarchy theory, and the C-value enigma. Paleobiology. 2004;30:179–202.
Zhou WW, Yan F, Fu JZ, Wu SF, Murphy RW, Che J, et al. River islands, refuge, and genetic structuring in the endemic brown frog Rana kukunoris (Anura, Ranidae) of the Qinghai-Tibetan Plateau. Mol Ecol. 2013;22:130–42.
Li JT, Gao YD, Xie L, Deng C, Shi P, Guan ML, et al. Comparative genomic investigation of high-elevation adaptation in ectothermic snakes. PNAS. 2018;115:8406–11.
D’Mello SAN, Finlay GJ, Baguley BC, Askarian-Amiri ME. Signaling pathways in melanogenesis. Int J Mol Sci. 2016;17:1144.
Hanaoka M, Droma Y, Basnyat B, Ito M, Kobayashi N, Katsuyama Y, et al. Genetic variants in EPAS1 contribute to adaptation to high-altitude hypoxia in Sherpas. PLoS ONE. 2012;7: e50566.
Miao B, Wang Z, Li Y. Genomic analysis reveals hypoxia adaptation in the Tibetan mastiff by introgression of the gray wolf from the Tibetan plateau. Mol Biol Evol. 2017;34:734–43.
Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:884–90.
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10:gia008.
Marçais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–70.
Vurture GW, Sedlazeck FJ, Nattestad M, Underwood CJ, Fang H, Gurtowski J, et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics. 2017;33:2202–4.
Liu JJ, Liu SQ, Zheng K, Tang M, Gu LP, Young J, et al. Chromosome-level genome assembly of the Chinese three-keeled pond turtle (Mauremys reevesii) provides insights into freshwater adaptation. Mol Ecol Resour. 2021;22:1596–605.
Cui F, Taier G, Li M, Dai X, Hang N, Zhang X, et al. The genome of the warm-season turfgrass African bermudagrass (Cynodon transvaalensis). Hortic Res. 2021;8:93.
Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–36.
Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE. 2014;9: e112963.
Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25:1754–60.
Simão FA, Waterhouse RM, Panagiotis I, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–2.
Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23:1061–7.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
Servant N, Varoquaux N, Lajoie BR, Viara E, Chen CJ, Vert JP, et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16:259.
Burton JN, Adey A, Patwardhan RP, Qiu R, Kitzman JO, Shendure J. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol. 2013;31:1119–25.
Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12:59–60.
Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40:70–82.
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–45.
Tang HB, Bowers J, Wang XY, Ming R, Alam M, Paterson A. Synteny and collinearity in plant genomes. Science. 2008;320:486–8.
Stanke M, Diekhans M, Baertsch R, Haussler D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 2008;24:637–44.
Korf I. Gene finding in novel genomes. BMC Bioinform. 2004;5:59.
Keilwagen J, Wenk M, Erickson JL, Schattat MH, Jan G, Frank H. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 2016;44: e89.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Amit I. Trinity: reconstructing a full-length transcriptome without a genome from Rna-seq data. Nat Biotechnol. 2013;29:644–52.
Haas BJ, Delcher AL, Mount SM, Wortman JR, Smith RK, Hannick LI, et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003;31:5654–66.
Haas BJ, Salzberg SL, Zhu W, Pertea M, Allen JE, Orvis J, et al. Automated eukaryotic gene structure annotation using evidencemodeler and the program to assemble spliced alignments. Genome Biol. 2008;9:R7.
Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–64.
Griffiths-Jones S, Grocock RJ, Van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:140–4.
Griffiths-Jones S, Moxon S, Marshall M, Khanna A, Eddy SR, Bateman A. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005;33:121–4.
Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold Faster RNA Homology Searches. Bioinformatics. 2013;29:2933–5.
She R, Chu JS, Wang K, Pei J, Chen N. GenBlastA: Enabling BLAST to identify homologous gene sequences. Genome Res. 2009;19:143–9.
Birney E, Clamp M, Durbin R. GeneWise and genomewise. Genome Res. 2004;14:988–95.
Huerta-Cepas J, Szklarczyk D, Heller D, Hernández-Plaza A, Forslund SK, Cook H, et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019;47:309–14.
Boeckmann B, Bairoch A, Apweiler R, Blatter MC, Estreicher A, Gasteiger E, et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365–70.
Finn RD, Mistry J, Schuster-Böckler B, Griffiths-Jones S, Hollich V, Lassmann T, et al. Pfam: clans, web tools and services. Nucleic Acids Res. 2006;34:247–51.
Flynn JM, Hubley R, Goubert C, Rosen J, Clark AG, Feschotte C, Smit AF. RepeatModeler2 for automated genomic discovery of transposable element families. PNAS. 2020;117:9451–7.
Bao Z, Eddy SR. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002;12:1269–76.
Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21:351–8.
Ellinghaus D, Kurtz S, Willhoeft U. LTRharvest, an efficient and flexible software for De Novo detection of LTR retrotransposons. BMC Bioinform. 2008;9:18.
Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2008;35:265–8.
Ou S, Jiang N. LTR_retriever: A highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 2018;176:1410–22.
Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–7.
Neumann P, Novák P, Hoštáková N, Macas J. Systematic survey of plant LTR-retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mob DNA. 2019;10:1.
Wheeler TJ, Clements J, Eddy SR, Hubley R, Jones TA, Jurka J, et al. Dfam: a database of repetitive DNA based on profile hidden Markov models. Nucleic Acids Res. 2013;41:70–82.
Beier S, Thiel T, Münch T, Scholz U, Mascher M. MISA-web: a web server for microsatellite prediction. Bioinformatics. 2017;33:2583–5.
Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–80.
Emms DM, Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 2019;20:238.
Mi H, Muruganujan A, Huang X, Ebert D, Mills C, Guo X, Thomas PD. Protocol update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat protocols. 2019;14:703–21.
Katoh K, Kuma KI, Toh H, Miyata T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005;33:511–8.
Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34:609–12.
Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56:564–77.
Kalyaanamoorthy S, Minh BQ, Wong TKF, Haeseler AV, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587–9.
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74.
Yang Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–91.
Puttick MN. MCMCtreeR: functions to prepare MCMCtree analyses and visualize posterior ages on trees. Bioinformatics. 2019;35:5321–2.
Han MV, Thomas GW, Lugo-Martinez J, Hahn MW. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol Biol Evol. 2013;30:1987–97.
Storey JD, Bass AJ, Dabney A, Robinson D. qvalue: Q-value estimation for false discovery rate control. R package version 2.26.0; 2021.
Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation. 2021;2:100141.
Acknowledgements
We thank the Biomarker Technologies Company for sequencing the whole genomes and providing support with data analysis.
Funding
This study was funded by National Natural Science Foundation of China (No. 32270457) and Key Foundation of Sichuan Provincial Department of Education (18ZA0255).
Author information
Authors and Affiliations
Contributions
WC and LCJ designed research; HZC, HFQ, JHL, ZKZ, LXZ, MT, XYL, YFW and BWZ performed research; WC, KR and JM wrote the paper, and JM, KR, BHF and XYF corrected the draft. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures used for this study were approved by the Animal Ethics Committee of the Anhui University(Ethical proof No. IACUC(AHU)-2022-007).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supplemental Information.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, W., Chen, H., Liao, J. et al. Chromosome-level genome assembly of a high-altitude-adapted frog (Rana kukunoris) from the Tibetan plateau provides insight into amphibian genome evolution and adaptation. Front Zool 20, 1 (2023). https://doi.org/10.1186/s12983-022-00482-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12983-022-00482-9