
Abstract
Two HIV virus types exist: HIV-1 is pandemic and aggressive, whereas HIV-2 is confined mainly to West Africa and less pathogenic. Despite the fact that it has been almost 40 years since the discovery of AIDS, there is still no cure or vaccine against HIV. Consequently, the concepts of functional vaccines and cures that aim to limit HIV disease progression and spread by persistent control of viral replication without life-long treatment have been suggested as more feasible options to control the HIV pandemic. To identify virus-host mechanisms that could be targeted for functional cure development, researchers have focused on a small fraction of HIV-1 infected individuals that control their infection spontaneously, so-called elite controllers. However, these efforts have not been able to unravel the key mechanisms of the infection control. This is partly due to lack in statistical power since only 0.15% of HIV-1 infected individuals are natural elite controllers. The proportion of long-term viral control is larger in HIV-2 infection compared with HIV-1 infection. We therefore present the idea of using HIV-2 as a model for finding a functional cure against HIV. Understanding the key differences between HIV-1 and HIV-2 infections, and the cross-reactive effects in HIV-1/HIV-2 dual-infection could provide novel insights in developing functional HIV cures and vaccines.
Similar content being viewed by others

HIV-1 and HIV-2 epidemiology and pathogenesis
HIV-2 was first discovered in 1986 in West Africa [1]. Although HIV-2 has been found in other parts of Africa, Europe, India and the United States; West Africa has consistently had the largest HIV-2 prevalence [2,3,4,5]. In 1994, the first studies of HIV-2 reported a reduced rate of disease progression compared with HIV-1 among female sex workers in Senegal [6]. In 1997, it was reported that HIV-2 infected individuals had twice as high mortality compared with HIV negative individuals in Guinea-Bissau [7]. This was confirmed in later studies that showed mortality rates two to fivefold higher in HIV-2 infected individuals compared with HIV-negative individuals in Guinea-Bissau [8, 9]. Other studies, from The Gambia and France, compared HIV-1 and HIV-2 infection and reported a generally slower CD4+ T-cell decline in HIV-2 infected individuals [10, 11]. HIV-2 infected individuals therefore exhibit longer asymptomatic stages than HIV-1 infected individuals [12]. However, in individuals reaching AIDS, HIV-1 and HIV-2 share a similar clinical spectrum, with the exception of a lower incidence of Kaposi’s sarcoma in HIV-2 infected individuals [13, 14]. Interestingly, studies have also reported that similar baseline viral load and CD4+ T-cell levels predict similar prognosis for HIV-1 and HIV-2 infected individuals [15, 16]. This could indicate that disease prognosis is determined during the early stages of both types of HIV infections.
The viral set-point has been suggested to be 10–28 fold lower in HIV-2, with lower levels of viraemia persisting into clinical stages of disease [17, 18]. As a consequence, HIV-2 infection is characterized by lower transmission rates at both the horizontal and vertical levels [19, 20]. Moreover, AIDS seem to occur at a relatively lower viral load level in HIV-2 compared with HIV-1 infection, although the CD4 count is often higher in HIV-2-infected subjects when AIDS-defining illnesses develop [12, 14, 21]. The lower transmission rates of HIV-2 compared with HIV-1 is further highlighted by the parallel HIV-1 increase and HIV-2 decline seen in West Africa between 1990 and 2010 [2, 4, 5, 22].
Several reports have indicated that only approximately 15–25% of HIV-2 infected individuals will progress to AIDS if following a natural course of disease [3, 23, 24]. However, these assumptions were based on data from HIV-2 infected individuals without information on infection date. On the one hand, lack of infection date will inevitably select for individuals with a slower disease progression rate than the average. On the other hand, such population will also enter the study at a more advanced stage. These biases will create a contradiction that will be difficult to adjust for when estimating true disease progression rates. In 2018, data from individuals with an estimated date of infection showed that the disease trajectory was almost identical between HIV-1 and HIV-2 infections, albeit at approximately half the rate among HIV-2 infected individuals [12, 25, 26]. Importantly, this showed that AIDS will develop among the majority of HIV-2 infected individuals without antiretroviral treatment (ART). Nevertheless, although no such indication was seen in the study, the existence of a subset of HIV-2-infected subjects who maintain long-term viral control and have a normal life expectancy cannot be entirely excluded since this would require a complete follow-up to the end-stage (AIDS or death) of all study participants [27]. However, in such subgroup, the time to AIDS would be longer than the predicted human lifespan, meaning that the age at HIV-2 infection would be a determining factor for the size of the group. In fact, the median age at infection was 38 years in the HIV-2 infected group [27]. This, together with the lack of information on infection date, could explain previous results of the high proportion of HIV-2 infected individuals not developing HIV-related disease.
HIV-1 and HIV-2 virology and immunology
The HIV-1 and HIV-2 epidemics constitute multiple different introductions of simian immunodeficiency viruses (SIV) into the human population [28]. HIV-1 has its origin from SIV of the chimpanzee, whereas HIV-2 originated from the SIV of the sooty mangabey [29, 30]. Due to the parallel evolution of SIV and HIV in simian and human populations there is a distinct genetic diversity between HIV-1 and HIV-2. To date, a large number of groups, subtypes, subsubtypes and circulating recombinant forms have been described for HIV-1, and at least nine groups of HIV-2 have been described (termed A–I) [28]. Group A and B are most common in HIV-2 infection, although intergroup recombinants between group A and B has been described [30]. However, and despite their different origins, HIV-1 and HIV-2 are related retroviruses and show approximately 55% similarity in Gag and Pol, and 35% similarity in Env on the protein level (the overall similarity level is approximately 55% on the nucleotide level) [31]. Although the virus types share transmission routes and target cells, contrasting results in terms of replicative fitness and cytopathicity have been reported [32, 33].
It is well established that blood plasma viral load is lower in HIV-2 compared with HIV-1 infection [34]. It would therefore make sense that viral replication could largely explain the difference in pathogenicity between the two viruses. Studies of natural disease progression caused by HIV-1 have indicated large variations in viral loads between individuals and the difference in plasma viral load may not fully explain the difference in rate of disease progression between the two virus infections. Interestingly, a recent study showed that CD4+ T-cell levels during the asymptomatic stage of infection was stronger associated with HIV-2 disease progression rate than with CD4+ T-cell decline [35]. Further studies are therefore needed to determine the causative effects and predictive values of viral load and CD4+ T-cell levels in natural disease progression of both HIV-1 and HIV-2 infection [36, 37].
It has been suggested that untreated HIV-1 and HIV-2 infected individuals with similar CD4+ T-cell levels have similar levels of gag mRNA transcripts, indicating that substantial viral transcription occurs in HIV-2 infected individuals despite the generally lower viral loads [38]. Interestingly, the tat/gag ratio between HIV-1 and HIV-2 infections has been shown to differ [39,40,41]. Altogether, these studies suggest that tat mRNA levels are reduced compared with gag mRNA levels in cells from untreated HIV-2 infected individuals, whereas tat mRNA levels are more abundant than gag mRNA levels in cells from HIV-1 infected individuals. Since tat mRNA represents early transcripts, these results could reflect a reduced rate of recent cell infections in HIV-2 infection. It is also possible that post-transcriptional control of viral production could be involved in differences in HIV-1 and HIV-2 pathogenesis [42].
Lower virus production in HIV-2 compared with HIV-1 infection may also reflect a lower activation state in infected cells, or that HIV-2 is less responsive to activation. The long terminal repeat (LTR) of both HIV-1 and HIV-2 regulates the expression of the virus in response to cellular transcription signals. The HIV-2 LTR differs from HIV-1 in numbers and type of transcription binding elements and enhancers, leading to reduced responsiveness to transcription factors present in activated T-cells [43]. It has been shown that the HIV-2 LTR does not respond as well as the HIV-1 LTR to tumour necrosis factor alpha [44]. Similar results were obtained in experiments measuring viral replication [45]. There could also be differences in activation of HIV-1 and HIV-2 infected cells. In contrast to HIV-1, the HIV-2 envelope glycoprotein was found to stimulate production of higher levels of gamma interferon and interleukin 16 (both inhibit viral replication), and lower levels of interleukin 4 (stimulates viral replication) [46]. Further studies may result in novel molecular targets for functional HIV cure strategies.
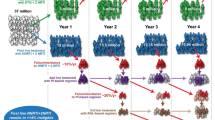
The latent HIV-1 reservoir has been studied extensively, and the HIV-1 reservoir establishment is associated with the seeding of virus during the acute HIV-1 infection before the adaptive arm of the immune system starts to partially control the virus replication [47, 48]. It is also well established that HIV-1 remain quiescent in long-lived CD4+ memory T-cells. Moreover, viral rebound is normally seen only a few weeks after secession of ART even in patients with previous long-term virus suppression. Hence, virus latency in these cells remain one of the main challenges for finding a functional cure against HIV. The size of the virus reservoir has been measured using different protocols, including qPCR of cell-bound virus DNA and mRNA, quantification of ex vivo reactivation of virus mRNA and proteins, as well as in ex vivo virus outgrowth assays. In HIV-1 long-term non-progressors (LTNP) and elite controllers (EC), that have been suggested as models for functional cure, the reservoir of latently infected cells is reduced compared with HIV-1 viraemic and treated individuals [49, 50]. Interestingly, conflicting results about proviral DNA levels in HIV-2 compared with HIV-1 infection have been reported. Two studies indicated similar proviral levels after adjusting for disease stage [51, 52]; whereas a study by Gueudin et al. [40] reported the opposite. Thus, further studies are needed to establish proviral DNA loads at different disease stages of HIV-2 infection, and how they differ from HIV-1 infection. Moreover, even though quantification of virus DNA by qPCR correlate with the size of the latent HIV-1 reservoir, these assays often overestimate the size of the replication competent latent HIV-1 reservoir. Although few studies have characterized the HIV-2 reservoir, it was recently reported that HIV-2 DNA could be quantified in transitional-memory cells from four of 14 ART naive HIV-2-infected individuals, and in central-memory cells from one of 14 ART naive HIV-2-infected individuals [41]. Approximately 100 HIV-2 DNA copies/106 cells were detected in each of the specific memory cell subsets, respectively. However, HIV-2 in vitro reactivation was only observed in cells from three of the 14 individuals, suggesting presence of defective proviruses. In line with this, the predominance of defective proviral DNA in HIV-2 infected individuals on successful ART has recently been reported from studies of three virally suppressed individuals [53]. In this study, most of the HIV-2 genomes had large deletions, whereas hypermutations were noted in a smaller fraction of the sequences. However, complete understanding of the HIV-2 reservoir will require larger studies and the use of different protocols. Moreover, both in vitro and ex vivo studies of latency reversal agents used in HIV-1 clinical settings are needed for HIV-2 (reviewed in [54]). Still, from available proviral load data and studies of HIV-1 in LTNP and EC (that in many ways resemble HIV-2) it is plausible that the remission of HIV-2 would be relapse-free or delayed, and less frequent compared with the general HIV-1 case (Fig. 1).

Schematic of potential differences between HIV progressor groups in frequency of HIV reactivation from latency. Clearance or control of the latent HIV reservoir remains one of the main obstacles to achieve a functional HIV cure. Although the viral reservoir in HIV-1 infection has been extensively studied, much less is known about the reservoir size or reactivation frequency from this reservoir in HIV-2, and HIV-1 and HIV-2 dual-infection. This figure outlines possible differences in HIV reservoir size and reactivation frequency between the main HIV infection types and progressor groups discussed in this review. The importance of the order of infection in HIV-1 and HIV-2 dual-infection has been highlighted in the figure, and it is likely that the HIV reservoir size and reactivation frequency will differ between depending of the order of HIV infections types
Explanations for lower virus loads and reduced pathogenicity in HIV-2 compared with HIV-1 infections have also been attributed to different types of virus-controlling immune responses. Robust, broadly cross-reactive and polyfunctional virus-specific responses of both CD4+ and CD8+ T-cells have been shown to distinguish HIV-2 from HIV-1 infections [55,56,57,58,59,60,61,62,63]. In particular, HIV-2 Gag-specific T-cell responses have been shown to correlate with virus control [59, 61, 62]. Similarly, CD8+ T-cells co-expressing CD28 have been associated with controlled HIV-2 infection [18, 64]. Strikingly, when subjects from the “Caió cohort” with high and undetectable viral loads were compared, the main distinguishing feature was CD8+ T-cell responses to Gag, which were absent in 52% of viraemic participants (the “Caió cohort” was a large community-based cohort from the small rural village Caió, Guinea-Bissau) [63]. Gag-specific responses in HIV-2-infected subjects often show unusually high functional avidity, with the capacity to respond to picomolar quantities of antigen, and are at an earlier stage of differentiation than HIV-1-specific CD8+ T-cells, presumably retaining their ability to proliferate [58, 63]. In line with these observations, HIV-2-specific T-cells from LTNPs in the French cohort showed potent suppression of viral replication, in many cases exceeding the suppressive abilities of HIV-1-specific T-cells from elite controllers [64]. Given that high potency HIV-specific cytotoxic T-lymphocytes (CTL) without features of exhaustion and broad cross-reactivity seem to be required for killing of the latent reservoir, there are good arguments to suggest that CTL from HIV-2 infected viral controllers would meet these requirements [47].
The cellular involvement in modulation of HIV-2 infections within West African populations also comes from HLA associations with viral control (HLA-B*58:01, HLA-DPB1*10:01 and HLA-DRB1*11:01) or disease progression (HLA-B*15:03 and HLA-B*35) [65,66,67]. Furthermore, the function of natural killer (NK) cells appears more well-preserved during asymptomatic HIV-2 compared with HIV-1 infection, whereas the functionality of these cells seem to drop to levels found during HIV-1 infection with declining CD4+ T-cells [68]. Similarly to HIV-1 infection, chronic immune activation is linked to immunopathogenesis and progressive disease in HIV-2 infection [18, 69,70,71,72,73,74,75]. Elevated frequencies of T-cells expressing markers of activation and exhaustion appear to distinguish HIV-2 infected individuals that progress despite no or low level viraemia, which could be the consequence of long infection duration and/or virus replication not mirrored by plasma RNA levels [18, 70,71,72]. Although these findings could suggest that aviraemic HIV-2 infected individuals should be offered ART, there remains a group of aviraemic individuals with HIV-2 infection without evidence of immune activation who may not necessarily benefit from therapy [70].
Both potent and broadly neutralizing antibodies have been detected at different disease stages in HIV-2 infection [51, 76,77,78,79]. Moreover, although susceptibility of HIV-2 to neutralizing antibodies seem to vary according to the infecting virus strain, it is in general significantly elevated compared to HIV-1 [77,78,79,80]. Furthermore, mutants escaping from neutralizing antibodies seem to emerge less frequently in HIV-2 infection and do not reach fixation [79, 81, 82]. Fc-mediated effector functions, such as the effect of complement on antibody antiviral activity, have also been reported to be potent in HIV-2 infection [76]. In addition, antibodies with a broad ability to mediate antibody dependent cellular cytotoxicity (ADCC), and even cross-react with HIV-1, are found in HIV-2 infected individuals [83, 84]. Thus, despite low-level viraemia, strong antibody responses in HIV-2 infection are sustained during both asymptomatic and progressive HIV-2 infections and do not distinguish between patient groups in different disease stages [79]. The impact of antibodies on disease progression during HIV-2 chronic infection is, therefore, not entirely clear.
Finally, animal models for HIV analysis of virus transmission and AIDS development are rare, and chimpanzees are the only non-human primates that are susceptible to HIV-1 infection. However, due to ethics, risk of extinction, and an infection that rarely results in progressive disease, these animals are not used as an infection model for HIV [85]. Experimental HIV-2 infection models have instead been established using rhesus and pigtail macaques. Still, these infections mainly result in low or non-pathogenic infections [86, 87]. In contrast, serial in vivo passages of HIV-2 in baboons have resulted in the development of an AIDS-like disease model [88, 89]. It has also been reported that HIV-2 infection of humanized mice results in persistent viraemia and CD4+ T-cell loss [90]. These could therefore represent alternative models for testing efficacy of antiretroviral and latency reversal strategies. As for in vitro models, competition assays between HIV-1 and HIV-2 have demonstrated that the replication fitness of most HIV-2 isolates was lower and outcompeted by HIV-1 isolates [32], and others have shown that HIV-2 isolates from aviraemic individuals have reduced in vitro replication capacity compared to HIV-2 isolated during viraemia [91].
HIV-1 and HIV-2 dual-infection
Dual-infection with HIV-1 and HIV-2 has been reported with a prevalence of up to 3.2% in West Africa [4, 92]. However, cross-reactivity in antibody testing and limited molecular testing to distinguish dual-infections have hampered the accuracy of prevalence estimates. Intriguingly, a possible protective effect of HIV-2 on subsequent incident HIV-1 infection was reported in 1995, and several studies reported that HIV-2 could alter HIV-1 infectivity and replication in vitro [93,94,95]. Moreover, HIV-2 infection has been shown to inhibit immunosuppression and simian AIDS after subsequent challenge with pathogenic SIV or SHIV in the Macaque model [96, 97]. In 2012, it was shown that HIV-2 could inhibit HIV-1 disease progression also in humans, resulting in almost twice as long time to AIDS and mortality among HIV-1 and HIV-2 dual-infected individuals compared with HIV-1 single-infected individuals [98, 99]. Importantly, the results showed that the slower disease progression was determined during the establishment of infection, and that the inhibitory effect was strongest among study participants where the HIV-2 infection preceded the HIV-1 infection (indicating the importance of the order of infections, Fig. 1). Moreover, in-depth analyses of CD4+ T-cell counts and HIV-1 diversity evolution indicated that the main difference between single and dual-infected individuals was determined during early HIV-1 infection. In support, results from the Bissau HIV cohort showed that the median CD4+ T-cell count was higher and the mortality lower in dual-infected individuals compared with HIV-1 single-infected individuals [100]. In contrast, a meta-analysis by Prince et al. [101] did not show any difference between HIV-1 single, and HIV-1 and HIV-2 dual-infected individuals. However, the data used was extracted from studies that were not designed for comparing survival between single and dual-infected individuals, and lacked information on estimated infection date and infection order among the dual-infected individuals. Most studies suffered from short periods of patient follow-up or observation time, and some were based on hospitalized patients with severe disease already at enrollment. Altogether, if identified, the determinants of the inhibition and slower disease progression in HIV-1 and HIV-2 dual-infection compared with HIV-1 single-infection could represent novel targets suitable for HIV cure strategies or vaccines. Moreover, it is possible that the viral reservoir in HIV-1 and HIV-2 dual-infected individuals is reduced compared to that in the majority of HIV-1 single-infected individuals. Cure strategies in dual-infected individuals may therefore be more successful compared with HIV-1 single-infected individuals (Fig. 1).
Current functional cure strategies and the possibility of such studies in HIV-2 endemic areas
As the research field of HIV cure has matured over recent years, it has been necessary to define different concepts of the term ‘cure’ [48]. Within HIV-1 cure research, the aspect of ‘functional cure’ has emerged, or lately ‘relapse-free remission’ to define sustained suppression of virus without the need for ART. This means that a ‘functional cure’ does not have to result in complete absence of HIV in the body. Several different cure strategies have been suggested, e.g. stimulation of the latently infected cells to reduce the reservoir size; gene therapy to reduce the number of target cells; and immunotherapy to ameliorate the HIV-specific immune response [102,103,104,105]. An example of naturally occurring ‘functional cure’ are so-called elite controllers, which has been described in a small minority of HIV-1 patients [106,107,108]. Interestingly, this phenomenon seems to be much more frequent among HIV-2 infected individuals, and it is largely unknown why this is the case [27]. Therefore, there are important lessons to be learned from HIV-2 pathogenesis, and HIV-2 may represent a model to study relapse-free remission and open up new avenues towards how to induce relapse-free remission in HIV-1 infection (Fig. 1).
Antiretroviral treatment effectively suppresses, but does not cure HIV infection. Multiple therapeutic strategies have been pursued in HIV cure research, but there has been a particular focus on using latency-reversing-agents (LRAs) such as histone deacetylase inhibitors (HDACi), disulfiram, Protein C kinase agonists and Toll-like Receptor agonists, to activate HIV-expression in latently infected cells, and thereby exposing their infected status to the immune system and potentially facilitating immune or virus-mediated cell lysis [109]. This is usually termed “shock-and-kill” [103]. Yet, although clinical trials of many of these compounds have demonstrated that HIV latency can be disrupted in individuals on suppressive ART, this does not lead to a reduction in the frequency of latently infected cells or delayed viral rebound during analytical interruption of ART [110]. More recently, attempts to reverse latency with compounds that both activate virus and modulate immunity to enhance clearance of infected cells (so-called immunomodulatory LRAs), have been made [89]. The inability of latency-reversal interventions to impact the latent HIV reservoir in clinical trials has increased the scientific focus on immune-enhancement strategies towards a concept of relapse-free remission after cessation of therapy. This is also supported by the demonstration that even in individuals with no or extremely low levels of HIV in cells or plasma, there is rebound viraemia when ART is stopped [111, 112]. This emphasizes that an effective strategy to achieve long-term ART-free remission should include both a component that reduces the amount of HIV that persists on ART, and a component that improves anti-HIV immune surveillance of residual viruses. This could involve immune-based therapies with immune checkpoint inhibitors, TLR agonists, or HIV-specific broadly neutralising antibodies, which are currently under intense investigation for application in HIV prevention, treatment and cure [105, 113,114,115]. Additionally, starting ART shortly after infection has been a focus area, as this is associated with both a lower frequency of latently infected CD4+ T-cells in blood and tissue, and a better preserved T-cell function [116, 117]. Furthermore, early ART increases the likelihood of post-treatment control, i.e. the ability to achieve durable remission after interruption of ART, which was started in primary infection—a phenomenon initially described in the French VISCONTI cohort [118].
Despite recent disappointments on the possibility of LRAs leading to longstanding remission in HIV-1, there could be a case for trying LRAs and “shock-and-kill” therapies in HIV-2, since it may be a less fit and more sensitive virus. HIV-2 cytotoxic CD8+ T-cell responses and possibly antibody responses, either broadly neutralizing or mediating ADCC, may partly explain the delayed progression of HIV-1 in patients firstly infected with HIV-2 and later superinfected with HIV-1 [56, 59, 77,78,79, 84, 98, 99, 119,120,121,122,123]. Hence, if HIV-2 immune responses play a role in controlling the rate of HIV-1 disease progression in individuals with dual-infection, it is plausible that boosting immunity may be able to induce relapse-free remission in HIV-2. There is a need to elicit studies on remission-strategies among HIV-2 infected individuals as these studies hold important promises for achieving an increased understanding of how to achieve remission in HIV-1.
An important question is if there is sufficient capacity to undertake a functional cure study based on HIV-2 in the field sites where there is a sufficient number of relevant HIV-2 cases. There are undoubtedly numerous factors to take into consideration before embarking on such highly complex clinical trials, and a number of relevant concerns need to be taken into account (summarised in Table 1). Yet, these are all manageable challenges that have been overcome in previous studies (Table 2). For example, both the Bissau HIV cohort and the Guinea-Bissau police cohort have built a high standard trial capacity during the last decades. Moreover, a therapeutic HIV-1 vaccine trial has already been completed in the Bissau HIV cohort [124, 125]. Despite limited settings, large HIV treatment trials such as the PIONA trial have been possible through an experienced clinical trial unit that is still in place in Bissau, Guinea-Bissau [126]. This trial unit has the capacity to handle 10–20 annual project visits that such trials may entail, as well as provide complex treatments requiring long infusions. The set-up for advanced analyses of immune-mediated processes is in place, both locally and among external partners [65, 71, 73,74,75,76,77, 80, 84, 119, 127,128,129]. Finally, more collaborations are needed in order to fully take on the many possibilities within this emerging field of HIV-2 cure research, and the Bissau cohorts are open to any collaborative efforts in this area and possible HIV cure applications.
Conclusion
HIV-2 is a less pathogenic virus than HIV-1, disease progression is slower and the proportions of controllers and slow progressors are higher. Both cellular and humoral immune responses, particularly HIV-2-specific CD8+ T-cell responses, are likely to play a role in controlling the rate of disease progression in individuals with dual-infection. A main immunological correlate for the substantial proportion of the aviraemic slow progressors seen in HIV-2 infection is the presence of highly avid, early-differentiated polyfunctional Gag-specific CTL (potentially more effective at targeting reactivated latent virus than HIV-1-specific T-cells). Therefore, there are important lessons to be learned from HIV-2 pathogenesis, and HIV-2 may represent a model to study relapse-free remission in HIV-1 infection (Fig. 1). Understanding the key differences between HIV-1 and HIV-2 infections, and the cross-reactive effects in HIV-1/HIV-2 dual-infection could provide novel insights in developing functional HIV cures and vaccines. There is a clear need to conduct studies on remission-strategies among HIV-2 infected individuals as these studies would provide valuable insights for achieving HIV cure.
Availability of data and materials
Not applicable.
References
Clavel F, Guetard D, Brun-Vezinet F, Chamaret S, Rey M, Santos-Ferreira M, et al. Isolation of a new human retrovirus from West African patients with AIDS. Science. 1986;233:343–6.
da Silva ZJ, Oliveira I, Andersen A, Dias F, Rodrigues A, Holmgren B, et al. Changes in prevalence and incidence of HIV-1, HIV-2 and dual infections in urban areas of Bissau, Guinea-Bissau: is HIV-2 disappearing? Aids. 2008;22(10):1195–202.
de Silva TI, Cotten M, Rowland-Jones SL. HIV-2: the forgotten AIDS virus. Trends Microbiol. 2008;16(12):588–95.
Mansson F, Biague A, da Silva ZJ, Dias F, Nilsson LA, Andersson S, et al. Prevalence and incidence of HIV-1 and HIV-2 before, during and after a civil war in an occupational cohort in Guinea-Bissau, West Africa. Aids. 2009;23(12):1575–82.
van der Loeff MF, Awasana AA, Sarge-Njie R, van der Sande M, Jaye A, Sabally S, et al. Sixteen years of HIV surveillance in a West African research clinic reveals divergent epidemic trends of HIV-1 and HIV-2. Int J Epidemiol. 2006;35(5):1322–8.
Richard M, Phyllis K, Ibou T, Karin T, Geoffrey E, Tidiane S, et al. Reduced rate of disease development after HIV-2 infection as compared to HIV-1. Science. 1994;265(5178):1587.
Poulsen A-G, Aaby P. 9-year HIV-2-associated mortality in an urban community in Bissau, west Africa. Lancet. 1997;349(9056):911.
Holmgren B, da Silva Z, Vastrup P, Larsen O, Andersson S, Ravn H, et al. Mortality associated with HIV-1, HIV-2, and HTLV-I single and dual infections in a middle-aged and older population in Guinea-Bissau. Retrovirology. 2007;4:85.
Norrgren H, Da Silva ZJ, Andersson S, Biague JA, Dias F, Biberfeld G, et al. Clinical features, immunological changes and mortality in a cohort of HIV-2-infected individuals in Bissau, Guinea-Bissau. Scand J Infect Dis. 1998;30(4):323–9.
Jaffar S, Wilkins A, Ngom PT, Sabally S, Corrah T, Bangali JE, et al. Rate of decline of percentage CD4+ cells is faster in HIV-1 than in HIV-2 infection. J Acquir Immune Defic Syndr Hum Retrovirol. 1997;16(5):327–32.
Drylewicz J, Matheron S, Lazaro E, Damond F, Bonnet F, Simon F, et al. Comparison of viro-immunological marker changes between HIV-1 and HIV-2-infected patients in France. Aids. 2008;22(4):457–68.
Esbjornsson J, Mansson F, Kvist A, da Silva ZJ, Andersson S, Fenyo EM, et al. Long-term follow-up of HIV-2-related AIDS and mortality in Guinea-Bissau: a prospective open cohort study. Lancet HIV. 2018;6:e25–31.
Ariyoshi K, Schim van der Loeff M, Cook P, Whitby D, Corrah T, Jaffar S, et al. Kaposi’s sarcoma in the Gambia, West Africa is less frequent in human immunodeficiency virus type 2 than in human immunodeficiency virus type 1 infection despite a high prevalence of human herpesvirus 8. J Hum Virol. 1998;1(3):193–9.
Martinez-Steele E, Awasana AA, Corrah T, Sabally S, van der Sande M, Jaye A, et al. Is HIV-2- induced AIDS different from HIV-1-associated AIDS? Data from a West African clinic. Aids. 2007;21(3):317–24.
Gottlieb GS, Sow PS, Hawes SE, Ndoye I, Redman M, Coll-Seck AM, et al. Equal plasma viral loads predict a similar rate of CD4+ T cell decline in human immunodeficiency virus (HIV) type 1- and HIV-2-infected individuals from Senegal, West Africa. J Infect Dis. 2002;185(7):905–14.
Hansmann A, Loeff MF, Kaye S, Awasana AA, Sarge-Njie R, O’Donovan D, et al. Baseline plasma viral load and CD4 cell percentage predict survival in HIV-1- and HIV-2-infected women in a community-based cohort in The Gambia. J Acquir Immune Defic Syndr. 2005;38(3):335–41.
Andersson S, Norrgren H, da Silva Z, Biague A, Bamba S, Kwok S, et al. Plasma viral load in HIV-1 and HIV-2 singly and dually infected individuals in Guinea-Bissau, West Africa: significantly lower plasma virus set point in HIV-2 infection than in HIV-1 infection. Arch Intern Med. 2000;160(21):3286–93.
Honge BL, Petersen MS, Jespersen S, Medina C, Te David DS, Kjerulff B, et al. T and B-cell perturbations identify distinct differences in HIV-2 compared with HIV-1 induced immunodeficiency. Aids. 2019;33:1131–41.
Kanki PJ, Travers KU. Slower heterosexual spread of HIV-2 than HIV-1. Lancet. 1994;343(8903):93.
O’Donovan D, Ariyoshi K, Milligan P, Ota M, Yamuah L, Sarge-Njie R, et al. Maternal plasma viral RNA levels determine marked differences in mother-to-child transmission rates of HIV-1 and HIV-2 in The Gambia. AIDS. 2000;14(4):441–8.
Nyamweya S, Hegedus A, Jaye A, Rowland-Jones S, Flanagan KL, Macallan DC. Comparing HIV-1 and HIV-2 infection: lessons for viral immunopathogenesis. Rev Med Virol. 2013;23(4):221–40.
Olesen JS, Jespersen S, da Silva ZJ, Rodrigues A, Erikstrup C, Aaby P, et al. HIV-2 continues to decrease, whereas HIV-1 is stabilizing in Guinea-Bissau. Aids. 2018;32(9):1193–8.
Saleh S, Vranckx L, Gijsbers R, Christ F, Debyser Z. Insight into HIV-2 latency may disclose strategies for a cure for HIV-1 infection. J Virus Erad. 2017;3(1):7–14.
Tchounga B, Ekouevi DK, Balestre E, Dabis F. Mortality and survival patterns of people living with HIV-2. Curr Opin HIV AIDS. 2016;11(5):537–44.
Esbjornsson J, Mansson F, Lindman J, Rowland-Jones SL, Jansson M, Medstrand P, et al. New insights are game-changers in HIV-2 disease management—Authors’ reply. Lancet HIV. 2019;6(4):e214–5.
Wejse C, Honge BL. Is it time to revise the notion that HIV-2 is benign? Lancet HIV. 2018;6:e3–4.
van der Loeff MF, Larke N, Kaye S, Berry N, Ariyoshi K, Alabi A, et al. Undetectable plasma viral load predicts normal survival in HIV-2-infected people in a West African village. Retrovirology. 2010;7:46.
Sauter D, Kirchhoff F. Key viral adaptations preceding the AIDS pandemic. Cell Host Microbe. 2019;25(1):27–38.
Gao F, Bailes E, Robertson DL, Chen Y, Rodenburg CM, Michael SF, et al. Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes. Nature. 1999;397(6718):436–41.
Santiago ML, Range F, Keele BF, Li Y, Bailes E, Bibollet-Ruche F, et al. Simian immunodeficiency virus infection in free-ranging sooty mangabeys (Cercocebus atys atys) from the Tai Forest, Cote d’Ivoire: implications for the origin of epidemic human immunodeficiency virus type 2. J Virol. 2005;79(19):12515–27.
Motomura K, Chen J, Hu WS. Genetic recombination between human immunodeficiency virus type 1 (HIV-1) and HIV-2, two distinct human lentiviruses. J Virol. 2008;82(4):1923–33.
Arien KK, Abraha A, Quinones-Mateu ME, Kestens L, Vanham G, Arts EJ. The replicative fitness of primary human immunodeficiency virus type 1 (HIV-1) group M, HIV-1 group O, and HIV-2 isolates. J Virol. 2005;79(14):8979–90.
Rowland-Jones S. Protective immunity against HIV infection: lessons from HIV-2 infection. Future Microbiol. 2006;1(4):427–33.
Popper SJ, Sarr AD, Travers KU, Gueye-Ndiaye A, Mboup S, Essex ME, et al. Lower human immunodeficiency virus (HIV) type 2 viral load reflects the difference in pathogenicity of HIV-1 and HIV-2. J Infect Dis. 1999;180(4):1116–21.
Palm AA, Lemey P, Jansson M, Mansson F, Kvist A, Szojka Z, et al. Low postseroconversion CD4(+) T-cell level is associated with faster disease progression and higher viral evolutionary rate in HIV-2 infection. MBio. 2019;10(1):e01245-18.
Mellors JW, Rinaldo CR Jr, Gupta P, White RM, Todd JA, Kingsley LA. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science. 1996;272(5265):1167–70.
Rodriguez B, Sethi AK, Cheruvu VK, Mackay W, Bosch RJ, Kitahata M, et al. Predictive value of plasma HIV RNA level on rate of CD4 T-cell decline in untreated HIV infection. JAMA. 2006;296(12):1498–506.
Soares RS, Tendeiro R, Foxall RB, Baptista AP, Cavaleiro R, Gomes P, et al. Cell-associated viral burden provides evidence of ongoing viral replication in aviremic HIV-2-infected patients. J Virol. 2011;85(5):2429–38.
Sonza S, Mutimer HP, O’Brien K, Ellery P, Howard JL, Axelrod JH, et al. Selectively reduced tat mRNA heralds the decline in productive human immunodeficiency virus type 1 infection in monocyte-derived macrophages. J Virol. 2002;76(24):12611–21.
Gueudin M, Damond F, Braun J, Taieb A, Lemee V, Plantier JC, et al. Differences in proviral DNA load between HIV-1- and HIV-2-infected patients. Aids. 2008;22(2):211–5.
Samri A, Charpentier C, Diallo MS, Bertine M, Even S, Morin V, et al. Limited HIV-2 reservoirs in central-memory CD4 T-cells associated to CXCR41 co-receptor expression in attenuated HIV-2 infection. PLoS Pathog. 2019;15(5):e1007758.
Nunes-Cabaco H, Matoso P, Foxall RB, Tendeiro R, Pires AR, Carvalho T, et al. Thymic HIV-2 infection uncovers posttranscriptional control of viral replication in human thymocytes. J Virol. 2015;89(4):2201–8.
Clark NM, Hannibal MC, Markovitz DM. The peri-kappa B site mediates human immunodeficiency virus type 2 enhancer activation in monocytes but not in T cells. J Virol. 1995;69(8):4854–62.
Hannibal MC, Markovitz DM, Clark N, Nabel GJ. Differential activation of human immunodeficiency virus type 1 and 2 transcription by specific T-cell activation signals. J Virol. 1993;67(8):5035–40.
Tong-Starksen SE, Welsh TM, Peterlin BM. Differences in transcriptional enhancers of HIV-1 and HIV-2. Response to T cell activation signals. J Immunol. 1990;145(12):4348–54.
Sekigawa I, Kaneko H, Neoh LP, Takeda-Hirokawa N, Akimoto H, Hishikawa T, et al. Differences of HIV envelope protein between HIV-1 and HIV-2: possible relation to the lower virulence of HIV-2. Viral Immunol. 1998;11(1):1–8.
Sengupta S, Siliciano RF. Targeting the latent reservoir for HIV-1. Immunity. 2018;48(5):872–95.
Davenport MP, Khoury DS, Cromer D, Lewin SR, Kelleher AD, Kent SJ. Functional cure of HIV: the scale of the challenge. Nat Rev Immunol. 2019;19(1):45–54.
Garcia M, Gorgolas M, Cabello A, Estrada V, Ligos JM, Fernandez-Guerrero M, et al. Peripheral T follicular helper cells make a difference in HIV reservoir size between elite controllers and patients on successful cART. Sci Rep. 2017;7(1):16799.
Tarancon-Diez L, Dominguez-Molina B, Viciana P, Lopez-Cortes L, Ruiz-Mateos E. Long-term persistent elite HIV-controllers: the Right model of functional cure. EBioMedicine. 2018;28:15–6.
Rodriguez SK, Sarr AD, MacNeil A, Thakore-Meloni S, Gueye-Ndiaye A, Traore I, et al. Comparison of heterologous neutralizing antibody responses of human immunodeficiency virus type 1 (HIV-1)- and HIV-2-infected Senegalese patients: distinct patterns of breadth and magnitude distinguish HIV-1 and HIV-2 infections. J Virol. 2007;81(10):5331–8.
Popper SJ, Sarr AD, Gueye-Ndiaye A, Mboup S, Essex ME, Kanki PJ. Low plasma human immunodeficiency virus type 2 viral load is independent of proviral load: low virus production in vivo. J Virol. 2000;74(3):1554–7.
Bender AM, Simonetti FR, Kumar MR, Fray EJ, Bruner KM, Timmons AE, et al. The Landscape of persistent viral genomes in ART-treated SIV, SHIV, and HIV-2 infections. Cell Host Microbe. 2019;26(1):73–85.e4.
Rasmussen TA, Sogaard OS. Clinical interventions in HIV cure research. Adv Exp Med Biol. 2018;1075:285–318.
Duvall MG, Jaye A, Dong T, Brenchley JM, Alabi AS, Jeffries DJ, et al. Maintenance of HIV-specific CD4+ T cell help distinguishes HIV-2 from HIV-1 infection. J Immunol. 2006;176(11):6973–81.
Duvall MG, Precopio ML, Ambrozak DA, Jaye A, McMichael AJ, Whittle HC, et al. Polyfunctional T cell responses are a hallmark of HIV-2 infection. Eur J Immunol. 2008;38(2):350–63.
Gillespie GM, Pinheiro S, Sayeid-Al-Jamee M, Alabi A, Kaye S, Sabally S, et al. CD8+ T cell responses to human immunodeficiency viruses type 2 (HIV-2) and type 1 (HIV-1) gag proteins are distinguishable by magnitude and breadth but not cellular phenotype. Eur J Immunol. 2005;35(5):1445–53.
Leligdowicz A, Onyango C, Yindom LM, Peng Y, Cotten M, Jaye A, et al. Highly avid, oligoclonal, early-differentiated antigen-specific CD8+ T cells in chronic HIV-2 infection. Eur J Immunol. 2010;40(7):1963–72.
Leligdowicz A, Yindom LM, Onyango C, Sarge-Njie R, Alabi A, Cotten M, et al. Robust Gag-specific T cell responses characterize viremia control in HIV-2 infection. J Clin Invest. 2007;117(10):3067–74.
Andersson S, Larsen O, Da Silva Z, Linder H, Norrgren H, Dias F, et al. Human immunodeficiency virus (HIV)-2-specific T lymphocyte proliferative responses in HIV-2-infected and in HIV-2-exposed but uninfected individuals in Guinea-Bissau. Clin Exp Immunol. 2005;139(3):483–9.
Zheng NN, Kiviat NB, Sow PS, Hawes SE, Wilson A, Diallo-Agne H, et al. Comparison of human immunodeficiency virus (HIV)-specific T-cell responses in HIV-1- and HIV-2-infected individuals in Senegal. J Virol. 2004;78(24):13934–42.
Foxall RB, Cortesao CS, Albuquerque AS, Soares RS, Victorino RM, Sousa AE. Gag-specific CD4+ T-cell frequency is inversely correlated with proviral load and directly correlated with immune activation in infection with human immunodeficiency virus type 2 (HIV-2) but not HIV-1. J Virol. 2008;82(19):9795–9.
de Silva TI, Peng Y, Leligdowicz A, Zaidi I, Li L, Griffin H, et al. Correlates of T-cell-mediated viral control and phenotype of CD8(+) T cells in HIV-2, a naturally contained human retroviral infection. Blood. 2013;121(21):4330–9.
Angin M, Wong G, Papagno L, Versmisse P, David A, Bayard C, et al. Preservation of lymphopoietic potential and virus suppressive capacity by CD8+ T cells in HIV-2-infected controllers. J Immunol. 2016;197(7):2787–95.
Thomsen D, Erikstrup C, Jespersen S, Medina C, Te DDS, Correira FG, et al. The influence of human leukocyte antigen-types on disease progression among HIV-2 infected patients in Guinea-Bissau. Aids. 2018;32(6):721–8.
Yindom LM, Leligdowicz A, Martin MP, Gao X, Qi Y, Zaman SM, et al. Influence of HLA class I and HLA-KIR compound genotypes on HIV-2 infection and markers of disease progression in a Manjako community in West Africa. J Virol. 2010;84(16):8202–8.
Diouf K, Sarr AD, Eisen G, Popper S, Mboup S, Kanki P. Associations between MHC class I and susceptibility to HIV-2 disease progression. J Hum Virol. 2002;5(1):1–7.
Nuvor SV, van der Sande M, Rowland-Jones S, Whittle H, Jaye A. Natural killer cell function is well preserved in asymptomatic human immunodeficiency virus type 2 (HIV-2) infection but similar to that of HIV-1 infection when CD4 T-cell counts fall. J Virol. 2006;80(5):2529–38.
Jaffar S, Van der Loeff MS, Eugen-Olsen J, Vincent T, Sarje-Njie R, Ngom P, et al. Immunological predictors of survival in HIV type 2-infected rural villagers in Guinea-Bissau. AIDS Res Hum Retrovir. 2005;21(6):560–4.
Leligdowicz A, Feldmann J, Jaye A, Cotten M, Dong T, McMichael A, et al. Direct relationship between virus load and systemic immune activation in HIV-2 infection. J Infect Dis. 2010;201(1):114–22.
Buggert M, Frederiksen J, Lund O, Betts MR, Biague A, Nielsen M, et al. CD4+ T cells with an activated and exhausted phenotype distinguish immunodeficiency during aviremic HIV-2 infection. Aids. 2016;30(16):2415–26.
Sousa AE, Carneiro J, Meier-Schellersheim M, Grossman Z, Victorino RM. CD4 T cell depletion is linked directly to immune activation in the pathogenesis of HIV-1 and HIV-2 but only indirectly to the viral load. J Immunol. 2002;169(6):3400–6.
Bachle SM, Malone DF, Buggert M, Karlsson AC, Isberg PE, Biague AJ, et al. Elevated levels of invariant natural killer T-cell and natural killer cell activation correlate with disease progression in HIV-1 and HIV-2 infections. Aids. 2016;30(11):1713–22.
Nowroozalizadeh S, Mansson F, da Silva Z, Repits J, Dabo B, Pereira C, et al. Microbial translocation correlates with the severity of both HIV-1 and HIV-2 infections. J Infect Dis. 2010;201(8):1150–4.
Nowroozalizadeh S, Mansson F, da Silva Z, Repits J, Dabo B, Pereira C, et al. Studies on toll-like receptor stimuli responsiveness in HIV-1 and HIV-2 infections. Cytokine. 2009;46(3):325–31.
Ozkaya Sahin G, Holmgren B, Sheik-Khalil E, da Silva Z, Nielsen J, Nowroozalizadeh S, et al. Effect of complement on HIV-2 plasma antiviral activity is intratype specific and potent. J Virol. 2013;87(1):273–81.
Ozkaya Sahin G, Holmgren B, da Silva Z, Nielsen J, Nowroozalizadeh S, Esbjornsson J, et al. Potent intratype neutralizing activity distinguishes human immunodeficiency virus type 2 (HIV-2) from HIV-1. J Virol. 2012;86(2):961–71.
Kong R, Li H, Bibollet-Ruche F, Decker JM, Zheng NN, Gottlieb GS, et al. Broad and potent neutralizing antibody responses elicited in natural HIV-2 infection. J Virol. 2012;86(2):947–60.
de Silva TI, Aasa-Chapman M, Cotten M, Hue S, Robinson J, Bibollet-Ruche F, et al. Potent autologous and heterologous neutralizing antibody responses occur in HIV-2 infection across a broad range of infection outcomes. J Virol. 2012;86(2):930–46.
Ozkaya Sahin G, Mansson F, Palm AA, Vincic E, da Silva Z, Medstrand P, et al. Frequent intratype neutralization by plasma immunoglobulin a identified in HIV type 2 infection. AIDS Res Hum Retrovir. 2013;29(3):470–8.
Bjorling E, Scarlatti G, von Gegerfelt A, Albert J, Biberfeld G, Chiodi F, et al. Autologous neutralizing antibodies prevail in HIV-2 but not in HIV-1 infection. Virology. 1993;193(1):528–30.
Shi Y, Brandin E, Vincic E, Jansson M, Blaxhult A, Gyllensten K, et al. Evolution of human immunodeficiency virus type 2 coreceptor usage, autologous neutralization, envelope sequence and glycosylation. J Gen Virol. 2005;86(Pt 12):3385–96.
Ljunggren K, Biberfeld G, Jondal M, Fenyo EM. Antibody-dependent cellular cytotoxicity detects type- and strain-specific antigens among human immunodeficiency virus types 1 and 2 and simian immunodeficiency virus SIVmac isolates. J Virol. 1989;63(8):3376–81.
Karlsson I, Tingstedt JL, Sahin GO, Hansen M, Szojka Z, Buggert M, et al. Cross-reactive antibodies with the capacity to mediate HIV-1 envelope glycoprotein-targeted antibody-dependent cellular cytotoxicity identified in HIV-2-infected individuals. J Infect Dis. 2019;219:1749–54.
Van Rompay KK. The use of nonhuman primate models of HIV infection for the evaluation of antiviral strategies. AIDS Res Hum Retrovir. 2012;28(1):16–35.
Putkonen P, Bottiger B, Warstedt K, Thorstensson R, Albert J, Biberfeld G. Experimental infection of cynomolgus monkeys (Macaca fascicularis) with HIV-2. J Acquir Immune Defic Syndr. 1989;2(4):366–73.
Franchini G, Markham P, Gard E, Fargnoli K, Keubaruwa S, Jagodzinski L, et al. Persistent infection of rhesus macaques with a molecular clone of human immunodeficiency virus type 2: evidence of minimal genetic drift and low pathogenetic effects. J Virol. 1990;64(9):4462–7.
Locher CP, Witt SA, Herndier BG, Abbey NW, Tenner-Racz K, Racz P, et al. Increased virus replication and virulence after serial passage of human immunodeficiency virus type 2 in baboons. J Virol. 2003;77(1):77–83.
Locher CP, Witt SA, Herndier BG, Tenner-Racz K, Racz P, Levy JA. Baboons as an animal model for human immunodeficiency virus pathogenesis and vaccine development. Immunol Rev. 2001;183:127–40.
Hu S, Neff CP, Kumar DM, Habu Y, Akkina SR, Seki T, et al. A humanized mouse model for HIV-2 infection and efficacy testing of a single-pill triple-drug combination anti-retroviral therapy. Virology. 2017;501:115–8.
Blaak H, van der Ende ME, Boers PH, Schuitemaker H, Osterhaus AD. In vitro replication capacity of HIV-2 variants from long-term aviremic individuals. Virology. 2006;353(1):144–54.
Hamel DJ, Sankale JL, Eisen G, Meloni ST, Mullins C, Gueye-Ndiaye A, et al. Twenty years of prospective molecular epidemiology in Senegal: changes in HIV diversity. AIDS Res Hum Retrovir. 2007;23(10):1189–96.
Al-Harthi L, Owais M, Arya SK. Molecular inhibition of HIV type 1 by HIV type 2: effectiveness in peripheral blood mononuclear cells. AIDS Res Hum Retrovir. 1998;14(1):59–64.
Arya SK, Gallo RC. Human immunodeficiency virus (HIV) type 2-mediated inhibition of HIV type 1: a new approach to gene therapy of HIV-infection. Proc Natl Acad Sci USA. 1996;93(9):4486–91.
Kokkotou EG, Sankale JL, Mani I, Gueye-Ndiaye A, Schwartz D, Essex ME, et al. In vitro correlates of HIV-2-mediated HIV-1 protection. Proc Natl Acad Sci USA. 2000;97(12):6797–802.
Putkonen P, Walther L, Zhang YJ, Li SL, Nilsson C, Albert J, et al. Long-term protection against SIV-induced disease in macaques vaccinated with a live attenuated HIV-2 vaccine. Nat Med. 1995;1(9):914–8.
Otten RA, Adams DR, Kim CN, Pullium JK, Sawyer T, Jackson E, et al. Chronic HIV-2 infection protects against total CD4+ cell depletion and rapid disease progression induced by SHIV89.6p challenge. Aids. 2004;18(8):1127–35.
Esbjornsson J, Mansson F, Kvist A, Isberg PE, Nowroozalizadeh S, Biague AJ, et al. Inhibition of HIV-1 disease progression by contemporaneous HIV-2 infection. N Engl J Med. 2012;367(3):224–32.
Esbjornsson J, Mansson F, Kvist A, Isberg PE, Biague AJ, da Silva ZJ, et al. Increased survival among HIV-1 and HIV-2 dual-infected individuals compared to HIV-1 single-infected individuals. Aids. 2014;28(7):949–57.
Oliveira I, Andersen A, Furtado A, Medina C, da Silva D, da Silva ZJ, et al. Assessment of simple risk markers for early mortality among HIV-infected patients in Guinea-Bissau: a cohort study. BMJ Open. 2012;2:e001587.
Prince PD, Matser A, van Tienen C, Whittle HC, Schim van der Loeff MF. Mortality rates in people dually infected with HIV-1/2 and those infected with either HIV-1 or HIV-2: a systematic review and meta-analysis. Aids. 2014;28(4):549–58.
Dash PK, Kaminski R, Bella R, Su H, Mathews S, Ahooyi TM, et al. Sequential LASER ART and CRISPR treatments eliminate HIV-1 in a subset of infected humanized mice. Nat Commun. 2019;10(1):2753.
Deeks SG. HIV: shock and kill. Nature. 2012;487(7408):439–40.
Borducchi EN, Cabral C, Stephenson KE, Liu J, Abbink P, Ng’ang’a D, et al. Ad26/MVA therapeutic vaccination with TLR7 stimulation in SIV-infected rhesus monkeys. Nature. 2016;540(7632):284–7.
Borducchi EN, Liu J, Nkolola JP, Cadena AM, Yu WH, Fischinger S, et al. Antibody and TLR7 agonist delay viral rebound in SHIV-infected monkeys. Nature. 2018;563(7731):360–4.
Migueles SA, Connors M. Long-term nonprogressive disease among untreated HIV-infected individuals: clinical implications of understanding immune control of HIV. JAMA. 2010;304(2):194–201.
Autran B, Descours B, Avettand-Fenoel V, Rouzioux C. Elite controllers as a model of functional cure. Curr Opin HIV AIDS. 2011;6(3):181–7.
Walker BD. Elite control of HIV Infection: implications for vaccines and treatment. Top HIV Med. 2007;15(4):134–6.
Rasmussen TA, Tolstrup M, Sogaard OS. Reversal of latency as part of a cure for HIV-1. Trends Microbiol. 2016;24(2):90–7.
Pitman MC, Lau JSY, McMahon JH, Lewin SR. Barriers and strategies to achieve a cure for HIV. Lancet HIV. 2018;5(6):e317–28.
Henrich TJ, Hanhauser E, Marty FM, Sirignano MN, Keating S, Lee TH, et al. Antiretroviral-free HIV-1 remission and viral rebound after allogeneic stem cell transplantation: report of 2 cases. Ann Intern Med. 2014;161(5):319–27.
Luzuriaga K, Gay H, Ziemniak C, Sanborn KB, Somasundaran M, Rainwater-Lovett K, et al. Viremic relapse after HIV-1 remission in a perinatally infected child. N Engl J Med. 2015;372(8):786–8.
Mendoza P, Gruell H, Nogueira L, Pai JA, Butler AL, Millard K, et al. Combination therapy with anti-HIV-1 antibodies maintains viral suppression. Nature. 2018;561(7724):479–84.
Wykes MN, Lewin SR. Immune checkpoint blockade in infectious diseases. Nat Rev Immunol. 2018;18(2):91–104.
Xu L, Pegu A, Rao E, Doria-Rose N, Beninga J, McKee K, et al. Trispecific broadly neutralizing HIV antibodies mediate potent SHIV protection in macaques. Science. 2017;358(6359):85–90.
Ananworanich J, Chomont N, Eller LA, Kroon E, Tovanabutra S, Bose M, et al. HIV DNA set point is rapidly established in acute HIV Infection and dramatically reduced by early ART. EBioMedicine. 2016;11:68–72.
Crowell TA, Fletcher JL, Sereti I, Pinyakorn S, Dewar R, Krebs SJ, et al. Initiation of antiretroviral therapy before detection of colonic infiltration by HIV reduces viral reservoirs, inflammation and immune activation. J Int AIDS Soc. 2016;19(1):21163.
Frange P, Faye A, Avettand-Fenoel V, Bellaton E, Descamps D, Angin M, et al. HIV-1 virological remission lasting more than 12 years after interruption of early antiretroviral therapy in a perinatally infected teenager enrolled in the French ANRS EPF-CO10 paediatric cohort: a case report. Lancet HIV. 2016;3(1):e49–54.
Honge BL, Petersen MS, Jespersen S, Medina C, Te DDS, Kjerulff B, et al. T-cell and B-cell perturbations are similar in ART-naive HIV-1 and HIV-1/2 dually infected patients. Aids. 2019;33(7):1143–53.
Vidyavijayan KK, Cheedarala N, Babu H, Precilla LK, Sathyamurthi P, Chandrasekaran P, et al. Cross type neutralizing antibodies detected in a unique HIV-2 infected individual from India. Front Immunol. 2018;9:2841.
Esbjornsson J, Mansson F, Kvist A, Isberg PE, Nowroozalizadeh S, Biague AJ, et al. Effect of HIV-2 infection on HIV-1 disease progression and mortality. Aids. 2014;28(4):614–5.
Bertoletti A, Cham F, McAdam S, Rostron T, Rowland-Jones S, Sabally S, et al. Cytotoxic T cells from human immunodeficiency virus type 2-infected patients frequently cross-react with different human immunodeficiency virus type 1 clades. J Virol. 1998;72(3):2439–48.
Rowland-Jones S, Sutton J, Ariyoshi K, Dong T, Gotch F, McAdam S, et al. HIV-specific cytotoxic T-cells in HIV-exposed but uninfected Gambian women. Nat Med. 1995;1(1):59–64.
Borggren M, Jensen SS, Heyndrickx L, Palm AA, Gerstoft J, Kronborg G, et al. Neutralizing antibody response and antibody-dependent cellular cytotoxicity in HIV-1-infected individuals from Guinea-Bissau and Denmark. AIDS Res Hum Retrovir. 2016;32(5):434–42.
Roman VR, Jensen KJ, Jensen SS, Leo-Hansen C, Jespersen S, da Silva Te D, et al. Therapeutic vaccination using cationic liposome-adjuvanted HIV type 1 peptides representing HLA-supertype-restricted subdominant T cell epitopes: safety, immunogenicity, and feasibility in Guinea-Bissau. AIDS Res Hum Retrovir. 2013;29(11):1504–12.
Jespersen S, Honge BL, Krarup H, Medstrand P, Sorensen A, Medina C, et al. Protease inhibitors or NNRTIs as first-line HIV-1 treatment in West Africa (PIONA): a randomized controlled trial. J Acquir Immune Defic Syndr. 2018;79(3):386–93.
Andersen MN, Honge BL, Jespersen S, Medina C, da Silva Te D, Laursen A, et al. Soluble macrophage mannose receptor (sCD206/sMR) as a biomarker in human immunodeficiency virus infection. J Infect Dis. 2018;218(8):1291–5.
Honge BL, Andersen MN, Jespersen S, Medina C, Correira FG, Jakobsen MR, et al. Brief report: macrophage activation in HIV-2-infected patients is less affected by antiretroviral treatment-sCD163 in HIV-1, HIV-2, and HIV-1/2 dually infected patients. J Acquir Immune Defic Syndr. 2016;72(3):254–8.
Wejse C, Furtado A, Camara C, Luneborg-Nielsen M, Sodemann M, Gerstoft J, et al. Impact of tuberculosis treatment on CD4 cell count, HIV RNA, and p24 antigen in patients with HIV and tuberculosis. Int J Infect Dis. 2013;17(10):e907–12.
Jespersen S, Honge BL, Oliveira I, Medina C, da Silva Te D, Correia FG, et al. Challenges facing HIV treatment in Guinea-Bissau: the benefits of international research collaborations. Bull World Health Organ. 2014;92(12):909–14.
Jespersen S, Honge BL, Oliveira I, Medina C, da Silva Te D, Correira FG, et al. Cohort profile: the Bissau HIV cohort—a cohort of HIV-1, HIV-2 and co-infected patients. Int J Epidemiol. 2015;44(3):756–63.
Esbjornsson J, Mild M, Mansson F, Norrgren H, Medstrand P. HIV-1 molecular epidemiology in Guinea-Bissau, West Africa: origin, demography and migrations. PLoS ONE. 2011;6(2):e17025.
Jespersen S, Honge BL, Medina C, da Silva Te D, Correira FG, Laursen AL, et al. Lack of awareness of treatment failure among HIV-1-infected patients in Guinea-Bissau—a retrospective cohort study. J Int AIDS Soc. 2015;18:20243.
Honge BL, Jespersen S, Aunsborg J, Mendes DV, Medina C, da Silva Te D, et al. High prevalence and excess mortality of late presenters among HIV-1, HIV-2 and HIV-1/2 dually infected patients in Guinea-Bissau—a cohort study from West Africa. Pan Afr Med J. 2016;25:40.
Jespersen S, Honge BL, Esbjornsson J, Medina C, da Silva Te D, Correira FG, et al. Differential effects of sex in a West African cohort of HIV-1, HIV-2 and HIV-1/2 dually infected patients: men are worse off. Trop Med Int Health. 2016;21(2):253–62.
Thomsen D, Hviid CJ, Honge BL, Medina C, Te DDS, Correira FG, et al. Increased mortality among HIV infected patients with cryptococcal antigenemia in Guinea-Bissau. Pan Afr Med J. 2018;29:18.
Denton PW, Sogaard OS, Tolstrup M. Using animal models to overcome temporal, spatial and combinatorial challenges in HIV persistence research. J Transl Med. 2016;14:44.
Esbjornsson J, Mansson F, Martinez-Arias W, Vincic E, Biague AJ, da Silva ZJ, et al. Frequent CXCR138 tropism of HIV-1 subtype A and CRF02_AG during late-stage disease-indication of an evolving epidemic in West Africa. Retrovirology. 2010;7:23.
James KL, de Silva TI, Brown K, Whittle H, Taylor S, McVean G, et al. Low-bias RNA sequencing of the HIV-2 genome from blood plasma. J Virol. 2018;93(1):e00677-18.
Palm AA, Esbjornsson J, Mansson F, Biague A, da Silva ZJ, Norrgren H, et al. Cocirculation of several similar but unique HIV-1 recombinant forms in Guinea-Bissau revealed by near full-length genomic sequencing. AIDS Res Hum Retrovir. 2015;31(9):938–45.
Palm AA, Esbjornsson J, Mansson F, Kvist A, Isberg PE, Biague A, et al. Faster progression to AIDS and AIDS-related death among seroincident individuals infected with recombinant HIV-1 A3/CRF02_AG compared with sub-subtype A3. J Infect Dis. 2014;209(5):721–8.
Lindman JL, Mansson F, Biague A, Da Silva ZJ, Andersson S, Norrgren H. Declining prevalence rates of syphilis among police officers in Guinea-Bissau, West Africa, 1990–2010. Sex Transm Dis. 2013;40(10):794–6.
Olsen B, Mansson F, Camara C, Monteiro M, Biai A, Alves A, et al. Phenotypic and genetic characterisation of bacterial sexually transmitted infections in Bissau, Guinea-Bissau, West Africa: a prospective cohort study. BMJ Open. 2012;2(2):e000636.
Linderholm L, Biague A, Mansson F, Norrgren H, Bergman A, Jakobsson K. Human exposure to persistent organic pollutants in West Africa—a temporal trend study from Guinea-Bissau. Environ Int. 2010;36(7):675–82.
Norrgren H, Bamba S, Da Silva ZJ, Koivula T, Andersson S. Higher mortality in HIV-2/HTLV-1 co-infected patients with pulmonary tuberculosis in Guinea-Bissau, West Africa, compared to HIV-2-positive HTLV-1-negative patients. Int J Infect Dis. 2010;14(Suppl 3):e142–7.
Gronborg HL, Jespersen S, Egedal JH, Correia FG, Medina C, Krarup H, et al. Prevalence and clinical characteristics of CMV coinfection among HIV infected individuals in Guinea-Bissau: a cross-sectional study. Trop Med Int Health. 2018;23(8):896–904.
Honge BL, Jespersen S, Medina C, da Silva Te D, da Silva ZJ, Lewin SR, et al. Hepatitis C prevalence among HIV-infected patients in Guinea-Bissau: a descriptive cross-sectional study. Int J Infect Dis. 2014;28:35–40.
Honge BL, Jespersen S, Medina C, Te Dda S, da Silva ZJ, Lewin S, et al. Hepatitis B and Delta virus are prevalent but often subclinical co-infections among HIV infected patients in Guinea-Bissau, West Africa: a cross-sectional study. PLoS ONE. 2014;9(6):e99971.
Jensen MM, Olesen JS, Kjerulff B, Byberg S, da Silva ZJ, Rodrigues A, et al. HTLV prevalence is no longer following the decreasing HIV prevalence—20 years of retroviral surveillance in Guinea-Bissau, West Africa. Acta Trop. 2019;192:144–50.
Sorensen A, Jespersen S, Katzenstein TL, Medina C, Te Dda S, Correira FG, et al. Clinical presentation and opportunistic infections in HIV-1, HIV-2 and HIV-1/2 dual seropositive patients in Guinea-Bissau. Infect Dis. 2016;48(8):604–11.
Steiniche D, Jespersen S, Erikstrup C, Krarup H, Handberg A, Ostergaard L, et al. Diabetes mellitus and impaired fasting glucose in ART-naive patients with HIV-1, HIV-2 and HIV-1/2 dual infection in Guinea-Bissau: a cross-sectional study. Trans R Soc Trop Med Hyg. 2016;110(4):219–27.
Wilhelmson S, Mansson F, Lopatko Lindman J, Biai A, Esbjornsson J, Norrgren H, et al. Prevalence of HIV-1 pretreatment drug resistance among treatment naive pregnant women in Bissau, Guinea Bissau. PLoS ONE. 2018;13(10):e0206406.
Jespersen S, Tolstrup M, Honge BL, Medina C, Te Dda S, Ellermann-Eriksen S, et al. High level of HIV-1 drug resistance among patients with HIV-1 and HIV-1/2 dual infections in Guinea-Bissau. Virol J. 2015;12:41.
Lindman J, Honge BL, Kjerulff B, Medina C, da Silva ZJ, Erikstrup C, et al. Performance of Bio-Rad HIV-1/2 Confirmatory Assay in HIV-1, HIV-2 and HIV-1/2 dually reactive patients—comparison with INNO-LIA and immunocomb discriminatory assays. J Virol Methods. 2019;268:42–7.
Honge BL, Bjarnason Obinah MP, Jespersen S, Medina C, Te Dda S, da Silva ZJ, et al. Performance of 3 rapid tests for discrimination between HIV-1 and HIV-2 in Guinea-Bissau, West Africa. J Acquir Immune Defic Syndr. 2014;65(1):87–90.
Honge BL, Jespersen S, Medina C, Te David DS, da Silva ZJ, Christiansen M, et al. Discriminatory rapid tests cause HIV-type misclassification-evaluation of three rapid tests using clinical samples from Guinea-Bissau. Trans R Soc Trop Med Hyg. 2019. https://doi.org/10.1093/trstmh/trz041.
Honge BL, Jespersen S, Medina C, Te DS, da Silva ZJ, Christiansen M, et al. The challenge of discriminating between HIV-1, HIV-2 and HIV-1/2 dual infections. HIV Med. 2018;19(6):403–10.
Honge BL, Jespersen S, Mendes DV, Wejse C, Erikstrup C. Comment on Gautheret-Dejean et al.: Performance of rapid tests for discrimination between HIV-1 and/or HIV-2 infections. J Med Virol. 2016;88(3):367–8.
Andersson S, da Silva Z, Norrgren H, Dias F, Biberfeld G. Field evaluation of alternative testing strategies for diagnosis and differentiation of HIV-1 and HIV-2 infections in an HIV-1 and HIV-2-prevalent area. Aids. 1997;11(15):1815–22.
Walther-Jallow L, Andersson S, da Silva Z, Biberfeld G. High concordance between polymerase chain reaction and antibody testing of specimens from individuals dually infected with HIV types 1 and 2 in Guinea-Bissau, West Africa. AIDS Res Hum Retrovir. 1999;15(11):957–62.
Dyrehave C, Rasmussen DN, Honge BL, Jespersen S, Correia FG, Medina C, et al. Nonadherence is associated with lack of HIV-Related knowledge: a cross-sectional study among HIV-infected individuals in Guinea-Bissau. J Int Assoc Provid AIDS Care. 2016;15(4):350–8.
Honge BL, Jespersen S, Nordentoft PB, Medina C, da Silva D, da Silva ZJ, et al. Loss to follow-up occurs at all stages in the diagnostic and follow-up period among HIV-infected patients in Guinea-Bissau: a 7-year retrospective cohort study. BMJ Open. 2013;3(10):e003499.
Nordentoft PB, Engell-Sorensen T, Jespersen S, Correia FG, Medina C, da Silva Te D, et al. Assessing factors for loss to follow-up of HIV infected patients in Guinea-Bissau. Infection. 2017;45(2):187–97.
Rasmussen DN, da Silva Te D, Rodkjaer L, Oliveira I, Medina C, Barfod T, et al. Barriers and facilitators to antiretroviral therapy adherence among patients with HIV in Bissau, Guinea-Bissau: a qualitative study. Afr J AIDS Res. 2013;12(1):1–8.
Acknowledgements
The authors thank Dr. Thomas Asgaard for valuable input on the manuscript before submission.
Funding
J.E. was supported by the Swedish Research Council (No. 2016-01417), the Swedish Society of Medical Research (SA-2016), and the Medical Faculty at Lund University.
Author information
Authors and Affiliations
Contributions
All authors contributed in writing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Esbjörnsson, J., Jansson, M., Jespersen, S. et al. HIV-2 as a model to identify a functional HIV cure. AIDS Res Ther 16, 24 (2019). https://doi.org/10.1186/s12981-019-0239-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12981-019-0239-x