
Abstract
The capsid core of HIV-1 is a large macromolecular assembly that surrounds the viral genome and is an essential component of the infectious virus. In addition to its multiple roles throughout the viral life cycle, the capsid interacts with multiple host factors. Owing to its indispensable nature, the HIV-1 capsid has been the target of numerous antiretrovirals, though most capsid-targeting molecules have not had clinical success until recently. Lenacapavir, a long-acting drug that targets the HIV-1 capsid, is currently undergoing phase 2/3 clinical trials, making it the most successful capsid inhibitor to-date. In this review, we detail the role of the HIV-1 capsid protein in the virus life cycle, categorize antiviral compounds based on their targeting of five sites within the HIV-1 capsid, and discuss their molecular interactions and mechanisms of action. The diverse range of inhibition mechanisms provides insight into possible new strategies for designing novel HIV-1 drugs and furthers our understanding of HIV-1 biology.
Graphical Abstract

Similar content being viewed by others


Introduction
As of 2020, it is estimated that 38 million individuals are living with human immunodeficiency virus type 1 (HIV-1) globally, and of these individuals, over 25 million are receiving antiretroviral therapy (ART) [1]. HIV-1 is a lentivirus that infects CD4+ human immune cells, such as T cells and macrophages, and is the causative agent of acquired immune deficiency syndrome (AIDS) [2,3,4]. The biological structure of the infectious HIV-1 particle, also known as the mature virion, has a viral envelope surrounding a “fullerene cone”-shaped capsid shell that encapsulates two copies of the positive-strand RNA genome along with associated cellular factors and viral proteins [4,5,6,7,8,9]. The capsid core is composed of capsid proteins (CA), also known as p24, that form pentameric and hexameric subunits, which assemble into the mature viral capsid. In this review we will refer to the capsid protein as “CA” and to the assembled viral conical core as “capsid” or “core”. Further, capsid serves critical roles in many aspects of the HIV-1 replication cycle such as reverse transcription, cytoplasmic transport, nuclear entry, and virion maturation in addition to interacting with over 20 host factors essential for infection [9,10,11,12,13,14]. These functions are fundamental to HIV-1 biology and therefore there is high interest in developing drugs that perturb capsid functions [15, 16]. Thus, this review is focused on the structure of CA and the compounds that specifically target it.
HIV-1 infection begins upon binding of the virion to a CD4 receptor and a CCR5 or CXCR4 coreceptor on the target cell surface. Receptor binding triggers a fusion event of the viral envelope with the host cell membrane, releasing the viral capsid and its contents into the cytoplasm [17, 18]. The precise timing, rate, and location at which the CA proteins are shed, a process at times referred to as “uncoating”, is still under investigation. Uncoating kinetics appear to be highly regulated, as mutations that either stabilize or destabilize CA–CA association have a negative impact on viral infectivity [19]. Regardless, it is known that CA serves multiple roles during and following reverse transcription, including trafficking to and through the nuclear pore complex (NPC), as well as integration of viral DNA into the host cell genome [12, 20,21,22,23,24]. Reverse transcription, the process of converting the HIV-1 RNA genome into double-stranded cDNA, is catalyzed by the enzyme reverse transcriptase (RT) located within the capsid, which prevents exposure of the viral nucleic acid to host proteins [12, 25]. For trafficking of viral particles to the NPC, CA interacts with the microtubule-associated proteins MAP1A and MAP1S, along with FEZ1, a kinesin-1 adaptor protein, and others [26,27,28,29,30]. At the NPC, CA interacts directly with nuclear pore proteins, including NUP358 and NUP153, both of which are essential for proper nuclear import [31,32,33,34]. Once the viral cDNA is inside the nucleus, integration can occur [35,36,37,38]. Integration is the incorporation of the viral cDNA, the product of reverse transcription, into the host cell’s genome forming the provirus, a process that is catalyzed by the HIV-1 enzyme integrase (IN) [39,40,41].
Provirus formation marks the end of the early phase of the HIV-1 life cycle [42]. The provirus is used as a template for transcription of nascent viral RNAs that may be translated into viral proteins or packaged as a viral genome. In some instances, the provirus enters a latent state where it remains transcriptionally inactive, enabling evasion of the immune response until reactivation of the host cell triggers expression of viral RNA [43, 44].
While CA does not have known catalytic activity, it can impact multiple viral enzymatic activities, including that of RT and IN, by mechanisms that are currently under investigation [11,12,13, 45,46,47,48]. Productive infection is further influenced by interactions of CA with host factors, including Cyclophilin A (CypA) and cleavage and polyadenylation specificity factor subunit 6 (CPSF6). Although these proteins bind CA at different sites, both CypA and CPSF6 have been shown to increase viral fitness and impact the location of integration events [34, 49,50,51,52,53,54,55,56]. This example is a limited view into the many complex roles of cellular factors for infection (recently reviewed in [7, 9, 57]).
The late phase of the HIV-1 life cycle begins with transcription of the provirus, followed by export of the synthesized RNA to the cytoplasm and translation of the Gag and GagPol polyproteins [42, 44, 58]. The Gag polyprotein precursor, also known as Pr55Gag, contains four major domains: Matrix (MA), CA, Nucleocapsid (NC), and p6, in addition to two small spacer peptides: SP1 and SP2 [59, 60]. In total, the molecular weight of Gag is ~ 55 kDa [61]. GagPol is a larger polyprotein precursor that is translated as a result of a programmed + 1 ribosomal frameshift in ~ 5% of cases compared to the relatively shorter Gag polyprotein [61, 62]. GagPol contains the aforementioned domains of Gag, except for a transframe domain that replaces the p6 domain, known as p6* or p6pol, following the frame shift located C-terminal region of NC [62]. Additionally, GagPol includes the protease (PR), RT, and IN domains at the C-terminal end, giving rise to a 160 kDa protein [60, 62].
Following translation, Gag proteins are trafficked to the inner leaflet of the plasma membrane [63, 64]. This localization is guided by the N-terminal MA domain that forms specific interactions with phosphatidyl-4,5-bisphosphate (PI(4,5)P2) and utilizes a myristic acid moiety as a membrane anchor [65,66,67,68,69,70]. The Gag protein interacts with the viral RNA at the plasma membrane; this is thought to nucleate Gag multimerization and particle assembly [61, 71]. Further, CA domain interactions serve as the driving force for this multimerization process, thus assembling the immature Gag lattice (reviewed in [61]). Due to the structural flexibility, the size, and the curved structure of the Gag-lattice, obtaining high-resolution structures of this protein assembly has posed a challenge. A low resolution structure of this lattice in virus-like particles was solved using cryogenic-electron tomography (cryo-ET) revealing that the lattice was formed from hexameric structures [72, 73]. An essential structural element of the Gag lattice is the CA-SP1 junction. The C-terminal residues of CA extended by SP1 form a dynamic 6-helix bundle that is necessary for the formation of the Gag lattice [74,75,76,77]. These interactions are further stabilized by the highly abundant metabolite inositol hexaphosphate (IP6) [78,79,80]. This site is also proposed to be a binding site for HIV-1 maturation inhibitors [61]. Several additional steps follow lattice assembly, including RNA packaging and envelope protein (Env) incorporation [81, 82], all leading to immature viral particles that bud from the plasma membrane, thus releasing the virion to the extracellular space [83].
The maturation process begins concomitantly with, or shortly following, budding [84,85,86]. Maturation is triggered by the PR cleavage of the Gag polyprotein. Gag processing is a multi-step process, the first of which involvescleavage between the SP1 and NC domains, followed by a series of cleavage events that further fragment Gag and Gag-Pol into individual proteins [61, 87]. Following polyprotein processing, HIV-1 maturation is characterized by large-scale structural rearrangements that lead to formation of the capsid, whereby CA hexamers and pentamers encapsidate two copies of the viral genomic RNA and other proteins, including RT, PR, and IN. The process of capsid maturation is incompletely understood, although there are several proposed mechanisms. The first and more widely supported mechanism hypothesizes that mature capsid reassembles from freed CA monomers present in the interior of the HIV-1 virion [88,89,90]. Another proposed mechanism posits that the mature capsid lattice forms without the freeing of CA monomers in the virus particle, called the displacive transition model [86, 91]. An alternative mechanism for capsid assembly combines elements from both proposed mechanisms [92].
After reassembly, the mature “fullerene cone”-shaped capsid shell comprises roughly 1500 CA proteins arranged in approximately 250 hexamers and precisely 12 pentamers [76, 93,94,95,96]. The pentamers are located on highly curved areas of the capsid core; specifically, seven pentamers are located at the wide end and five pentamers are located at the narrow end of the mature capsid. It is unknown whether it is the location of the pentamers that dictate the shape of the capsid or if it is the curvature of the capsid that determines the placement of the pentamers [61, 95, 97].
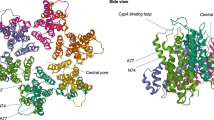
Individual CA monomers contain helical N-terminal domains (CANTD) and C-terminal domains (CACTD) connected through a flexible linker [93, 94, 98,99,100,101,102,103,104,105,106]. Specifically, CANTD contains seven α-helices (α1-7) and CACTD contains one short 310-helix and four α-helices (α8-11) (Fig. 1). Key structural elements of CA include an N-terminal β-hairpin that forms after proteolytic cleavage of the N-terminus, a proline-rich CypA-binding loop (CypA-BL) that interacts with human proline isomerase CypA, an interdomain linker, and a sequence known as the major homology region (MHR) which is conserved among orthoretroviruses [61, 97, 107, 108]. CA domains form interactions at the two-, three-, and sixfold symmetry regions of CA hexamers. These interactions are necessary for proper construction of the complete conical capsid. The central ring of the capsid hexamers is composed of six CANTD domains facing each other and held together by intrahexameric CANTD–CANTD and CANTD–CACTD interactions [98, 105]. It has also been reported that IP6 can interact with electropositive residues of this pore to stabilize the CA hexamers, thereby stabilizing the capsid core [80]. Along with stabilization, IP6 is thought to facilitate transport of deoxynucleotide triphosphates (dNTPs) into the capsid. IP6’s cellular functionality and interactions with capsid are topics of current research [80, 109,110,111,112]. In contrast to the inner hexamer ring, the outer ring of each hexamer is made up of six CACTDs. The exteriors of each CA hexamer can then make interactions with neighboring CA hexamers. These outer C-terminal domains participate in interhexamer interactions that involve CA residues at the two- and threefold symmetry axes [93, 98, 102,103,104, 113]. The capsid remains in this conformation until the next infection is established.

(Part C of this figure is based on Fig. 1 from Gres et al. [105]. Reprinted with permission from AAAS)
Structure of mature HIV-1 CA. A The HIV-1 CA monomer comprises CANTD (light gray) and CACTD (dark gray) domains (PDB ID: 4XFX). The view is rotated by 180°. B The same domains are shown in the context of the HIV-1 CA hexamer. Colors are as described in A, with neighboring CANTDʹ and CACTDʹ domains shown as light and dark cyan, respectively. The views are rotated by 90°. C Schematic outlining the sequence and secondary structural elements of HIV-1 CA (based on PDB ID: 4XFX)
Understanding the structural interactions at the interfaces of these capsid building blocks is crucial for learning about the dynamics that regulate capsid assembly and disassembly. Towards that end, structural biology techniques such as X-ray crystallography, cryogenic electron microscopy (cryo-EM), 3D nuclear magnetic resonance (NMR), and cryo-ET have been used. CA hexamers and pentamers, in cross-linked form, were first studied using X-ray crystallography. These early studies laid the foundation for the solution of many CA structures and revealed essential intrahexameric interactions [98, 102]. The X-ray structure of native CA in hexameric form revealed structural information of interhexameric interactions at the three- and twofold symmetry interfaces [105]. These CA–CA interactions are critical for the structural integrity and stability of the core [19, 91, 93, 94, 98, 102, 114,115,116,117,118,119,120,121,122,123]. Important low resolution information provided by cryo-EM in combination with NMR led to early useful molecular models of HIV-1 capsid [94]. Using new cryo-EM methodologies (ArbitrEM), a high-resolution structure of CA hexamers in tubular assemblies was solved [124]. Of note, cryo-EM may better recapitulate the curved nature of the core [97, 124]. Moreover, there are additional recent structures of CA tubes studied by cryo-ET [94, 97], cryo-EM or solid state NMR [94, 124,125,126]. Collectively, decades of structural work laid the foundation for understanding capsid structure and interactions that led to the discovery of numerous CA-targeting antivirals.
The interactions between CA proteins are the key determinants for the stability of the mature capsid, which is essential for the precise timing of the assembly and uncoating steps in the HIV-1 life cycle. Thus, creating small molecules that either stabilize or destabilize the capsid core is a promising strategy for the discovery of novel antiviral treatments.
Targeting HIV-1 by antivirals that bind to distinct sites of CA
Currently, there are 49 FDA-approved HIV medicines [127] that fall into seven categories: protease inhibitors (PIs), integrase strand transfer inhibitors (INSTIs), non-nucleoside reverse transcriptase inhibitors (NNRTIs), nucleoside (or nucleotide) reverse transcriptase inhibitors (NRTIs), fusion inhibitors, chemokine receptor antagonists, attachment inhibitors, and post-attachment inhibitors [128,129,130,131,132]. Although these drugs provide life-saving treatments for millions of individuals, there is no cure or vaccine presently available for HIV. Further, ART has several limitations including drug resistance, virologic failure, viral transmission, toxicity, and limited treatment options for treatment-experienced individuals [133, 134]. Therefore, it is essential to produce novel HIV drugs and to continue research on new therapeutic targets.
As such, the HIV-1 capsid has emerged as the next target of novel antiretrovirals due to its essential roles in both the early and late stages of the viral replication cycle [135]. Potential capsid effectors may interfere with CA–CA interactions within interhexameric or intrahexameric assemblies [136]. Such interference may modify the stability and morphology of the mature capsid, thus disrupting the processes of assembly and/or disassembly and resulting in the suppression of viral infectivity. Below, we review compounds that bind at five distinct sites of the CA protein (Fig. 2).

Binding sites of CA-targeting antivirals in mature HIV-1 CA monomers and hexamers. A The five CA-targeting antiviral binding sites are shown mapped onto the HIV-1 CA monomer (PDB ID: 4XFX). CANTDs are shown in light gray, CACTDs are shown in dark gray. Binding site 1 is shown in red, binding site 2 in orange, binding site 3 in yellow, binding site 4 in green, and binding site 5 in blue. The view is rotated by 180°. B The same domains and five CA-targeting antiviral binding sites are shown mapped onto the HIV-1 CA hexamer. Colors are as described in A. The views are rotated by 90°. C Schematic outlining the domains, binding sites, and secondary structure of HIV-1 CA. Compound binding sites are colored as described in A, and the Cyclophilin A binding loop (CypA-BL) is shown as pink circles. The CANTD and CACTD are shown as light and dark gray bars, respectively. The β-hairpin (βPin) and α-helices (α) shown as gray arrows and twists, respectively. Schematic generated by Protean 3D™ (V17.2.1) from DNASTAR, Inc
Site 1: FG binding site
The FG binding pocket is a primarily hydrophobic pocket that comprises structural elements of neighboring capsid monomers within a CA hexamer (Figs. 2, 3). Binding site 1 is formed by residues from the α3 and α4 α-helices of a CANTD subunit and by residues from the α8ʹ and α9ʹ α-helices of the CACTD of the adjacent subunit (Fig. 1) [105, 137,138,139]. This site has been the target of several small molecule antivirals. The compounds that bind CA at this pocket can compete with host factors that also bind at the same site, such as NUP153, CPSF6, and Sec24C, through interactions of phenylalanine–glycine (FG)-motifs present in these cellular proteins, hence the name of the pocket. Perturbation of NUP153, CPSF6, and Sec24C interactions with CA adversely affect nuclear import and viral infectivity [31, 32, 140, 141]. This pocket is also the binding site of small molecules that are discussed below: PF74 and analogs, BI-1 and BI-2, GS-CA1 and GS-6207, and others (Tables 1, 2).

(Parts of this figure are based on Fig. 4 from Gres et al. [105]. Reprinted with permission from AAAS)
Binding interactions of PF74 at binding site 1 of HIV-1 CA. A Chemical structure of PF74. B PF74 binding at the CANTD–CACTD interfaces of adjacent subunits within a CA hexamer (PDB ID: 4XFZ). The larger box shows a close-up view of the PF74 binding site in relation to the threefold (triangle) and twofold (oval) interfaces. CANTDs and the corresponding CACTDs are colored by the same colors (light and dark, respectively). C CANTD and a CACTD of a neighboring subunit (CACTDʹ) participate in PF74 binding (gray and dark cyan ribbons; site 1 residues are shown in red cartoon and pink sticks). PF74 is shown as yellow sticks. D PF74 binding geometries in CA-PF74 (PDB ID: 4XFZ; PF74 in yellow), CAXL-PF74 (PDB ID: 4U0E; PF74 in beige), and CANTD-PF74 (PDB ID: 2XDE; PF74 in brown). E Threefold interface of CA-PF74 superposed onto CA (aligned based on residues 1–219). Helices α10 of CA-PF74 and CA are in yellow and dark gray. Water molecules are shown as red spheres in CA; no water molecules were present in CA-PF74
PF74
PF-3450074 (PF74) (Fig. 3A) is a peptidomimetic capsid-targeting antiviral developed by Pfizer, USA (Tables 1, 2) [137]. This compound was shown to inhibit HIV-1 replication [52, 142, 143] with a submicromolar EC50 [137]. PF74 has a bimodal inhibition mechanism of action, whereby at lower concentrations (~ 2 µM or less), it interferes with CA-host factor interactions, whereas at concentrations greater than 10 µM, it triggers premature uncoating and blocks reverse transcription [144]. Structural studies of PF74 with CANTD [137], cross-linked CA hexamers [139], or native CA [105] have shown that PF74 binds at binding site 1 (Fig. 3B, C). Specifically, the R1 group of PF74 makes hydrophobic interactions with I73, A105, T107, T130 and a stacking interaction with N53. The benzyl group of R2 interacts with hydrophobic residues M66, L69, V59, I73, and L56. The R3 indole group interacts with the residues M66, Q67, K70, and Q63; the indole NH also forms a hydrogen bond with Q67 via a water molecule. Also, the amide bond of PF74 forms a hydrogen bond with N57 [137]. Intriguingly, the binding mode of PF74 to CANTD monomer is somehow different than its binding mode to the interface of adjacent CA monomers (Fig. 3D). Antiviral binding at this site is shared and competed for with the binding of host factors, including NUP153, Sec24C, and CPSF6 [32, 138, 140, 145]. PF74 has been known to affect both the stability of the capsid and the rate of CA assembly in vitro [138, 146]. Of note, the antiviral activity of PF74 is enhanced in the presence of CypA [144]. Though the mechanism has not been fully elucidated, there is antagonistic relationship between PF74 and cyclosporin, the molecules that bind CypA and sequester it from potential CA interactions, which may perturb other host factor interactions that PF74 mimics [34, 142, 143]. The structure of native CA in complex with PF74 has provided insight on the molecular interactions that may lead to stability changes of the capsid core; PF74 binding is accompanied with changes at the hydration layer and interactions between CA hexamers at the threefold interface of the CA lattice [105] (Fig. 3E).
The structure of PF74 comprises three aromatic moieties: R1 is based on a phenyl ring, R2 is a benzyl group, and R3 is a substituted indole ring (Fig. 3A). Comparison of the crystal structures of CA in complex with short peptides derived from CPSF6, NUP153, and Sec24C and of CA in complex with PF74 show that the peptides of the host factors interact with binding site 1 using their FG motifs, although the bound peptides had additional interactions (Fig. 4). The R2 benzyl group of PF74 serves as the surrogate for the phenylalanine group of the FG motif (Fig. 4) (reviewed in [7]).

Host factor FG motifs share a common binding conformation with the PF74 benzyl group. Crystal structures of HIV-1 CA hexamers in complex with CPSF6 (PDB ID: 6AY9; blue cartoon and sticks), NUP153 (PDB ID: 6AYA; orange cartoon and sticks), and Sec24C (PDB ID: 6PU1; magenta cartoon and sticks) peptides were aligned to a crystal structure of HIV-1 CA hexamer in complex with PF74 (PDB ID: 4XFZ; yellow sticks). The HIV-1 CA protein is omitted for clarity. The benzyl group of PF74 binds in a similar manner to the benzyl group of the phenylalanine in the FG motif of each host factor peptide
Several point mutations, such as S41A, Q67H, K70R, and T107N [143], have been identified in CA that decrease PF74 efficacy, in addition to decreasing the requirement for NUP153 interactions and generally decreasing HIV-1 infectivity [143, 147, 148]. Furthermore, PF74 has a very short half-life that predicts excessive first pass liver metabolism and poor oral bioavailability [149,150,151], making it a less-than ideal therapeutic compound. However, there have been recent advances in PF74-derived compounds. Chemical profiling has identified important characteristics of PF74 for developing further analogs, which include, for example, benzene rings with para-halogen substitutions and alkylation at the N-1 position of the indole ring [152]. Several reports have identified PF74-derived compounds that bind the same pocket, are more potent inhibitors of HIV-1, and have longer half-lives than PF74. The best performing compounds include 13m [149], II-10c [153], 11l [154], 6a-9 [155], H22 [156], CX06 [150], 10 [157], 12 [158], 15 [159], 20 [151], 32 [152], and 58 [152] (Tables 1, 2).
BI-1 and BI-2
4,5-Dihydro-1H-pyrrolo[3,4-c]pyrazol-6-one (pyrrolopyrazolone) antivirals were identified from a screen of compounds that impact the post-entry events of the HIV-1 life cycle [160]. From this screen, two lead compounds were reported: BI-2 (Fig. 5A) and BI-1 (Tables 1, 2). These compounds were found to stabilize capsid in vitro and impair nuclear entry. BI-1 and BI-2 were found to have EC50 values of 7.5–8.2 µM and 1.4–1.8 µM, respectively. Crystal structures and 1H–15N heteronuclear single quantum coherence (HSQC)-NMR revealed that BI-1 binds to the CANTD and interacts with the α4, α5, and α7 helices [160]. Although they bind at the same site as PF74, the BI compounds differ from PF74 as they only interact with the CANTD and not the CACTD (Fig. 5B). BI-1 and BI-2 are active only during the early phase of the HIV-1 life cycle [160], whereas PF74 is active in both early and late phases [137]. Similar to PF74, BI-2 was shown to decrease CPSF6 binding to CA [137, 161].

Different inter-subunit interactions of various CA-targeting compounds at CA binding site 1. A Chemical structures of BI-2 and GS-6207 (PF74 is shown in Fig. 3A). B BI-2 only interacts with the N-terminal domain of HIV-1 CA. The crystal structure of HIV-1 CA hexamer in complex with BI-2 (PDB ID: 4U0F; purple sticks) was superposed to a crystal structure of HIV-1 CA hexamer in complex with PF74 (PDB ID: 4XFZ; yellow sticks). The CANTD of one CA monomer is shown in light gray cartoon with binding site 1 shown in red, the CACTD of a neighboring monomer (CACTDʹ) is shown in dark green. Residues interacting with PF74 and/or BI-2 are shown as sticks. C GS-6207 forms extended interactions with the CANTD and CACTD of a neighboring CA monomer (CANTDʹ show in light cyan cartoon and CACTDʹ shown in dark green). The crystal structure of HIV-1 CA hexamer in complex with GS-6207 (PDB ID: 6V2F; green sticks) was superposed to a crystal structure of HIV-1 CA hexamer in complex with PF74 (PDB ID: 4XFZ; yellow sticks). The CANTD of one CA monomer is shown in light gray cartoon with binding site 1 shown in red. The novel interaction with CANTDʹ is shown as a black dotted line. The R1 and R2 groups of PF74 (see Fig. 3A) superpose well to GS-6207, while the R3 group adopts a different conformation
GS-CA1 and GS-6207
Developed by Gilead Sciences, both GS-CA1 and GS-6207 (lenacapavir) are complex molecules that contain a polyphenyl core and a linker region scaffold similar to PF74 (Fig. 5A) [162]. The three aromatic group regions of PF74 (R1, R2, and R3) are superposable with related structural elements of GS-CA1 or GS-6207 (Figs. 2A, 5A, C and Tables 1, 2). GS-CA1 and GS-6207 target the CANTD where FG-containing host factors interface with the CA hexamers and inhibits HIV-1 replication at post-entry steps, as well as production and maturation in virus-producer cells [141, 162]. GS-CA1, compared to PF74, is remarkably more potent (EC50 of 240 pM compared to ~ 600 nM), far more metabolically stable, less soluble, and has an outstanding EC50/CC50 selectivity index. Its attributes made it more appropriate for testing as a potential long-acting antiviral. Hence, when administered subcutaneously to a mouse model, one dose of GS-CA1 was able to maintain compound levels above its EC50 in the blood plasma for 56 days, outperforming the long-lasting NNRTI, rilpivirine, in potency and selectivity. During this study, all mice sustained viral suppression except for one who experienced virological failure at week 12 due to the mutations Q67H and K70R [162]. Mutations in CA residues L56I, N57S, M66I, Q67H, K70N, N74D, Al05T, and T107N have also been reported during in vitro resistance selection experiments with GS-CA1 exposure; the magnitudes of resistance vary from 2.3- to > 100-fold resistance [142, 144, 163]. Several of these CA mutations cause PF74 resistance and are located within the PF74 binding pocket [143, 148]. Some GS-CA1 resistance mutations significantly affect viral fitness, making their barrier to resistance higher. Overall, the studies with GS-CA1 showed excellent potential for its use as a long-acting antiviral therapeutic and led to the development of the more potent and metabolically stable GS-6207.
GS-6207 is the first capsid-targeting antiviral to make it into phase III clinical trials [135, 164]. Having a similar structural and mechanistic profile to GS-CA1, GS-6207 (Tables 1, 2, Fig. 5A) also has a remarkable potency, is very metabolically stable, and has an outstanding selectivity index. A crystal structure of GS-6207 bound to a cross-linked CA hexamer shows extensive hydrophobic and electrostatic interactions, seven hydrogen bonds, and two cation-π interactions between GS-6207 and residues of binding site 1 [135] (Fig. 5C). GS-6207 inhibits both early and late stages of the HIV-1 replication cycle, with exceptional EC50 values (~ 30 pM in target cells and ~ 440 pM in producer cells). The higher potency in early stages of the viral replication may be due to direct competition with host-cell nuclear import factors, such as NUP153 and CPSF6. In phase I clinical trials, a single ascending dose of GS-6207 was proven to be safe, well-tolerated, and demonstrated slow, sustained drug release, suggesting that a less frequent dosing regimen would be needed [135, 164, 165]. Phase 1b trials produced similar results in safety and effectivity. After 9 days of treatment, a single 450 mg dose of GS-6207 reduced the viral load in plasma by up to 2.2 log10. Notably, during this trial, treatment with GS-6207 led to the emergence of the Q67H resistance mutation [135]. Other mutations have appeared in in vitro resistance selection experiments against GS-6207, including L56I (239-fold resistance), M66I (> 3200 to > 85,000-fold resistance), Q67H (six- to tenfold resistance), K70N (24-fold resistance), N74D/S (22-fold resistance), Q67H/T107N (62-fold resistance), and Q67H/N74D (1099-fold resistance) double mutant. Both single and double resistance mutations have arisen from these studies, with M66I posing the greatest resistance to GS-6207 [125, 135], although the fitness of this virus is significantly suppressed. Recently, molecular dynamics simulations suggested that the mechanism of M66I resistance to GS-6207 is primarily a result of the high free energy cost required to reorganize the I66 side chain in order to minimize CA-GS-6207 clashes rather than reduced protein–ligand interactions [166]. Phase II clinical trials (NCT04143594) studied the safety and efficacy of orally and subcutaneously administered GS-6207 in combination with antiviral agents, tenofovir alafenamide (TAF) and emtricitabine. Currently, optimized 6-month oral administration regimens of GS-6207 are underway to evaluate the antiviral activity of this molecule on HIV-1 treatment-experienced patients with failing drug regimens due to multiple drug resistance (MDR) (NCT04150068) [167, 168]. The ongoing clinical trials with GS-6207 provide important validation that capsid is an effective antiviral drug target. Of note, a series of compounds structurally related to GS-6207 were also introduced through scaffold hopping, a computer-aided search for active compounds that contain different core structures [169], and were shown to also have picomolar potency [170,171,172]. Among them, compound ViiV-1 had an impressive EC50 of 25 pM (Tables 1, 2).
Other antivirals
In addition to the above compounds where binding information is provided by experimental structural approaches, two other compounds have been proposed to suppress HIV-1 replication by interacting at binding site 1 of CA. Specifically, 2-(benzothiazol-2-ylmethylthio)-4-methylpyrimidine (BMMP) (Tables 1, 2) was found to perturb Gag–Gag interactions through a yeast two-hybrid screen of membrane-associated Gag polyproteins [173]. Although the structure of CA-BMMP is not known, it was deduced that BMMP-mediated inhibition occurs through the CA domain of Gag and that the mechanism of action for BMMP is similar to PF74; both compounds impact events following cellular entry but prior to (or in conjunction with) nuclear entry [16, 137, 142, 173]. Additionally, coumermycin-A1 (C-A1) (Tables 1, 2) was shown by Fassati and colleagues to block HIV-1 by interfering with post-nuclear entry steps of the viral life cycle [174, 175]. In this work, it was shown that HIV-1 with A105S and N74D CA mutations conferred resistance to the integration block mediated by C-A1.
Site 2: CAP-1 binding site
Binding site 2 is located at the CANTD, proximal to binding site 1 (Fig. 2). This site has been targeted by the small molecule CAP-1 as well as various benzodiazepine and benzimidazole compounds. These antivirals bind at the apices of CA α-helices α1, α2, α3, α4, and α7 (Fig. 1). Inhibitor binding either displaces several residues within this site or it protrudes out of the pocket, thereby interfering with capsid assembly and preventing formation of the mature capsid [98, 176, 177].
CAP-1
CAP-1, N-(3-chloro-4-methylphenyl)-N0-(2-[((5-[(dimethylamino)-methyl]-2-furyl)-methyl)-sulfanyl]-ethyl) urea (Tables 1, 3 and Fig. 6), was the first discovered CA-targeting compound found in 2003 during a computational screen of small molecules against HIV-1 CA [178]. X-ray crystallography and NMR studies showed that CAP-1 binds to an induced hydrophobic pocket at the base of the CANTD helical bundle of the 5 α-helical apices of α1, α2, α3, α4, and α7 (Figs. 1, 6). The pocket is normally occupied by the aromatic side chain of F32 in structures of uninhibited CA hexamers, pentamers, and monomers. CAP-1 binding to this site induces conformational changes, displacing residues F32, H62, and Y145, and may disrupt the CANTD/CACTD interface, leading to inhibition of mature capsid formation [98, 176]. Structural data supports previous findings that CAP-1 interfered with the ability of CA to form tubular assemblies, reduced HIV-1 infectivity by 95% at 100 μM, caused pleiomorphic particle formation that did not contain capsid, and did not affect viral entry, reverse transcription, integration, and GagPol cleavage [178]. Collectively, these results suggested that CAP-1 inhibited mature capsid formation, a novel antiviral mechanism at the time. Because of its low binding affinity to CA, CAP-1 was not developed any further. Nonetheless, these studies established the proof of principle for the potential utility of this binding site [107, 178] that may eventually lead to the development of more potent compounds that act by an analogous mechanism of action.

HIV-1 antivirals targeting the CAP-1 binding site. A Two CA monomers from a CA hexamer (PDB ID: 4XFX) with CANTDs shown in light gray and light cyan, respective CACTDs in dark gray and dark cyan, and binding site 2 in orange. One subunit is outlined in red. B NMR structures of CANTD in complex with CAP-1 (PDB ID: 2JPR; CAP-1 in green sticks). Left: surface and cartoon views of CA site 2 interacting with pose 1 of CAP-1. Right: surface and cartoon views of CA site 2 interacting with pose 2 of CAP-1. C Crystal structure of CANTD in complex with BD-3 (PDB ID: 4E91; BD-3 in pink sticks). Left: surface and cartoon views of CA site 2 interacting with pose 1 of BD-3. Right: surface and cartoon views of CA site 2 interacting with pose 1 of BD-3
Benzodiazepines and benzimidazoles
Two other compound classes have been found to bind to the CAP-1 binding site located at the CANTD. Benzodiazepines (BDs) (Tables 1, 3), specifically with a 1,5-dihydrobenzo[b][1,4]diazepine-2,4-dione scaffold, were identified during a capsid assembly screen for compounds that bind to CANTD (Site 2, Table 1) [179]. Subsequently, using a novel high-throughput screen assay based on the in vitro association of CA-NC subunits on immobilized oligonucleotides, other BDs and benzimidazoles (BMs) were shown to bind to this location (Site 2) [177]. Optimization of these compounds produced a series of BDs and BMs that had high antiviral potency against wild-type HIV-1 as well as HIV-1 strains resistant to RT, PR, and IN inhibitors. The top two compounds, BD3 and BM4, had EC50 values of 0.4 µM and 46 µM, respectively (Tables 1, 3). NMR and X-ray crystallographic studies showed that these two classes of small molecules bind to the CAP-1 binding site (Fig. 6). Interestingly, BDs and BMs acted with different modes of action. Using EM and virus release studies, BD molecules were shown to prevent virus release, whereas BMs inhibited capsid assembly. BD compounds were shown to bind further into the helical bundle, leading to a displacement of the α1 helix. The α1 helix contributes to inter-subunit interactions within the CANTD of hexamers and pentamers (Fig. 1) [98, 102]. Its displacement upon BD binding can explain the effects on virus replication and capsid morphology. In contrast to BDs, BMs do not bind as deeply into the pocket and seem to protrude outside of the site. This extended binding leads to clashes with residues R162, D163, and D166 of the neighboring hexamer’s CTD. Within the pocket, the specific BM displaces residues F32 and H62, similar to CAP-1, and shifts the loop between α3 and α4 (Fig. 1) [177]. All of these interactions resulted in a loss of mature capsid assembly. These findings reveal that BDs and BMs bind at the same pocket, but in distinct modes, resulting in different mechanisms of action.
Passage of HIV-1 with these compounds led to resistance mutations in the highly conserved binding pocket or at the CACTD [177]. This study demonstrated distinct patterns of selected resistance mutations with potent vs. weaker inhibitors and with BD vs. BM antivirals. Whereas the V36T and G61E mutants were selected using BD compounds, K30R and S33G were obtained only upon selection with BM antivirals. V27A/I changes were observed for the BD series only. Common resistance mutations outside of the pocket included T58I and G208I. Although these mutations either stabilized the mature capsid or decreased BD and BM binding affinity, many mutations also resulted in a less fit virus with impaired replication abilities or low resistance to the compounds [177]. Optimization of targeted antivirals for this pocket is challenging due to the pocket’s hydrophobicity and flexibility. Moreover, these compounds are not suitable antiviral agents due to their low solubility and limited bioavailability. Regardless, these studies identified two compound classes, BDs and BMs, that target the CANTD and reaffirmed that the HIV-1 capsid can be a target for viral inhibition.
Site 3: The “twofold” binding site
CA is a critical component of viral maturation by driving Gag multimerization through dimeric interactions between CACTDs from neighboring hexamers at the twofold symmetry interface [115] (Fig. 2B). The dimer interface involves the residues 179–192 and those of the 310 helix (150–152) (Fig. 1) [99, 180, 181]. Additionally, the CACTD also contains the highly conserved MHR (residues 153–172), which is conserved among orthoretroviruses and participates in virus assembly. The MHR is not a part of the dimerization interface but maps to the binding region of some compounds that bind at this site [61, 97, 107, 108]. Mutagenesis studies have shown that the dimerization interface is critical for viral particle formation [123], and thus the high level of conservation of this site and the reliance on the CACTD for dimerization makes this site a potential site for drug development.
Several compounds that target this site are synthetic peptides. Though small-molecules dominate the HIV-1 therapeutic armamentarium, protein-based therapeutics have been characterized for many clinical purposes [182, 183]. As such, these peptides inhibit virion assembly by perturbing CACTD dimerization and subsequently HIV-1 maturation.
CAI
One example of a peptide-based inhibitor is Capsid assembly inhibitor (CAI) that was discovered through a phage-display screen that identified 12-mer peptides with affinity for CA (Tables 1, 4) [184]. CAI (primary sequence: ITFEDLLDYYGP) was found to interact with the hydrophobic groove of the CACTD at the twofold interhexamer symmetry axis through an induced fit mechanism, resulting in an α-helical structure (Fig. 7) [100, 184]. Subsequent analysis revealed that CAI inhibits assembly of both immature and mature HIV-1 in vitro, however, it did not inhibit viral assembly in vivo [184]. Superposition of the CACTD-CAI complex onto the CANTD/CACTD interface of assembled CA suggested that CAI binding would likely sterically hinder the CANTD/CACTD interaction. It was also suggested that CAI may also allosterically alter the CACTD dimer geometry, thus affecting propagation of the mature CA lattice [177].

HIV-1 antivirals targeting the twofold binding site. A Two CA monomers from a CA hexamer (PDB ID: 4XFX) with CANTDs shown in light gray and light cyan, with the respective CACTDs in dark gray and dark cyan, and binding site 3 in yellow. B Crystal structure of CACTD in complex with CAI (PDB ID: 2BUO; CAI in cyan cartoon and sticks). Surface (left) and cartoon (right) views of CA site 3 interacting with CAI. C NMR structure of CACTD in complex with NYAD-13 (PDB ID: 2L6E; NYAD-13 in pink cartoon and sticks). Left: Surface (left) and cartoon (right) views of CA site 3 interacting with NYAD-13, which is stabilized through hydrocarbon stapling with (S)-2-(2ʹ-pentenyl) alanine (magenta cartoon and sticks)
The early success of this compound led to the design of several improved derivatives. One such example is NYAD-1 (ITFxDLLxYYGP; x = (S)-2-(2ʹ-pentenyl)Ala), which strategically modified the sequence of CAI (Tables 1, 4) [185]. NYAD-1 replaces CAI residues D4 and G8 with a non-canonical amino acid, (S)-2-(2ʹ-pentenyl) alanine, to form a hydrocarbon staple. Hydrocarbon stapling is an established technique used to stabilize the secondary structure of helical peptides by crosslinking the i and i + 4 positions with an olefinic chain via alanine analogues [186]. NYAD-1 formed a stable helix in solution and was cell-permeable, overcoming a large issue of CAI [184, 185]. Further, NYAD-1 binds the CACTD from residues 169–191, the same interface as CAI, and specifically disrupted the formation of mature HIV-1 particles in a dose-dependent manner with an EC50 ~ 4–15 μM and a CC50 > 135–300 μM [185]. To further study the stabilized helical structures, NYAD-13 was derived from NYAD-1 by replacing the C-terminal proline with three lysine residues (ITFxDLLxYYGKKK; x = (S)-2-(2ʹ-pentenyl)Ala) (Tables 1, 4) [185, 187]. The positive charges increased the solubility of the peptide and facilitated structural studies. The high-resolution NMR structure of the CACTD/NYAD-13 complex showed that the intermolecular interactions were mediated by the packing of hydrophobic side chains at the buried interface and remained unperturbed due to the hydrocarbon stapling chain. NYAD-13 has antiviral activity (IC50 ~ 10.5 μM, CC50 = 40.5 μM) [185, 186]. These structural analyses provided further insight for the design of higher affinity selective inhibitors of HIV-1 particle assembly.
CAC1
The antiviral peptide CAC1 (primary sequence: EQASQEVKNWMTETLLVQNA) was designed based on the premise that helical mimics of CA would inhibit capsid assembly (Tables 1, 4) [188]. Its sequence mimics helix α9 of the twofold symmetry interface. CAC1 was shown to bind to CACTD using NMR, size exclusion chromatography, thermal denaturation measurements, and circular dichroism (Kd of 50 μM). CAC1 binding is thought to disrupt the dimer interface, resulting in the loss of key interactions that are crucial for capsid assembly. Bocanegra et al. subsequently designed other helical mimics, CAC1C, CAC1M, and H8, aiming to increase solubility and binding affinity. Using 2D 1H–15N HSQC-NMR, CAC1C (ESASSSVKAWMTETLLVQNA) and CAC1M (SESAASSVKAWMTETLLVANTSS) were shown to bind the CACTD at the same site as CAC1 [189]. The binding affinities of CAC1C and CAC1M were higher than that of CAC1, with Kds of 19 μM and 8 μM, respectively. H8 (KEPFRDYVDRFYKTLRAEQ) was designed to mimic helix α8 of the CACTD and was able to inhibit viral assembly in vitro, albeit to a lesser extent than CAC and CACM. Inhibition of HIV-1 by CAC1/CAC1M, H8, and CAI (mentioned above), alone and in combination, was then tested in vitro and in vivo and these compounds worked best in combination with some synergistic relationships identified [188]. While the twofold binding site is potentially important for viral replication, the available peptides do not have potent antiviral activity.
To further stabilize the helical mimics, NYAD-201 (AQEVKxWMTxTLLVA; x = (S)-2-(2ʹ-pentenyl)Ala) was designed in a similar fashion to CAC1 by modifying the CACTD dimerization domain (SQEVKNWMTETLLVQ) (Tables 1, 4) [190]. N6 and G11 from the interhexameric dimer interface sequence were replaced to allow hydrocarbon stapling to generate a more stable, α-helical, cell permeable antiviral with an EC50 of 2.8–5.2 μM and a CC50 > 115 μM [190]. The observation that relatively weak CA binders, such as NYAD-201, are sufficient to dissociate and deform the virion cores offers encouragement for the exploration of a broader class of peptide antivirals for targeting CA.
Other antivirals
MKN-1A
A highly conserved Tryptophan (W)–Methionine (M) dipeptide (W184/M185) motif in H9 of CACTD is engaged in hydrophobic interactions with the corresponding W184/M185 residues of a CA in a neighboring hexamer, at the twofold symmetry axis. Not surprisingly, mutations of these residues impair viral assembly [123, 191]. A small molecule was selected by in silico screening as a peptidomimetic compound of W184/M185 in the interaction site. It was synthesized and its anti-HIV-1 activity was demonstrated. The lead compound from this study was MKN-1A (EC50 = 8 µM; CC50 = 38 µM) (Tables 1, 4). Of note, the chirality of this molecule was important, as an MKN-1A diastereomer had decreased potency. Additionally, the trypsin-based indoyl and the methionine-based sulfidyl groups were essential for antiviral activity [191]. Hence, these efforts further confirmed that this site is a potential therapeutic target for subsequent drug design.
Compound 16
Machara et al. were the first to identify a set of small molecules that bind the CAI pocket [192]. Their high-throughput screening method was based on the amplified luminescence proximity assay system (Alphascreen). A high signal was produced when CAI attached to a donor bead came into close proximity of CACTD on an acceptor bead. This signal could then be competed by molecules that were able to interact with CACTD. Out of > 7000 compounds screened, they identified a family of 2-arylquinazolines able to compete with CAI and presumably bind at the same binding site. Compound 16 was shown to be the most potent 2-arylquinazoline (IC50 = 5 µM) (Tables 1, 4) and was also shown to inhibit HIV-1 in tissue culture at low micromolar concentrations.
Ebselen
Ebselen was identified from a high-throughput screen for inhibitors of CACTD–CACTD dimerization (Tables 1, 4) [193]. Liquid chromatography–electrospray ionization–mass spectrometry (LC–ESI–MS) confirmed that ebselen binds covalently to CA by forming a selenylsulfide (-Se-S-) bond with cysteines C198 and C218 at the CACTD, thus promoting a conformation of an ebselen-linked monomer that is susceptible to aggregation. While ebselen may not directly interact with residues at the twofold interface, NMR studies confirmed that its binding changes the chemical shift of residues in helix α9, which is involved in CA dimerization. Ebselen binds with an IC50 of 47 nM and has antiviral activity with an EC50 of 4.3 μM [193].
I-XW-053
Kortagere et al. [194] used a hybrid structure-based screening method and identified 900 compounds of interest. Further docking and scoring analysis and a single-round infection study identified CK026, which was modified to yield DMJ-I-073, I-XW-091, and I-XW-053 (Tables 1, 4). The most potent of the series, I-XW-053, appear to have modest potency (48.4 to 93.6 µM), (Tables 1, 4). Mutational analysis identified residues R173 and I37 to be essential for I-XW-053 binding [194] suggesting proximity to α9 helix, which is at the twofold interface.
Site 4: Apical binding site
Benzimidazoles (BM)
As mentioned above, some BM compounds can interact with CA at the binding site 2 (Fig. 8, Tables 1, 5). However, other BMs can bind to a unique capsid pocket located at the apical binding site of CANTD, near the CypA-BL, in this review referred to as binding site 4 [195]. The CA residues that comprise this relatively shallow binding pocket and interact with these compounds were revealed using 1H–15N HSQC-NMR and confirmed using X-ray crystallography. They are at the top of helix α4, the base of the CypA-BL, within helices α5 and α6, the loops joining helices α5, α6, and α7, and at the top of helix α7 (Figs. 1, 8). While the BM compounds bind near the CypA-BL, it was reported that CypA does not compete with binding of BM inhibitors to the CANTD. The EC50 and CC50 values of these small molecules ranged from 0.95–2.8 μM and 57–80 μM, respectively. Interestingly, molecules of this type were also able to bind CA in the presence of CAP-1. However, a significant proportion of the apical binding site is made from residues that are polymorphic among various strains, H120 for example in Fig. 8C. These polymorphisms were shown to affect the potency of these inhibitors, eliminating them as candidates for possible HIV-1 therapies. Nonetheless, these studies established the apical binding site as a novel HIV-1 CA binding site and further support that CA can be simultaneously targeted by more than one CA-targeted antiviral (BM and CAP-1) [195].

HIV-1 antivirals targeting the apical binding site. A Chemical structure of benzimidazole compound 4 (from [195]). B Crystal structure of CANTD in complex with 4 (PDB ID: 4E91; compound 4 in orange sticks) in surface view. CANTD is shown in light gray, with site 4 colored in green. C Close-up view of compound 4 at site 4, colors as in (B). Residues having strong hydrophobic interactions with compound 4 are shown as light green sticks. Hydrogen bonds with the side chain of R132 are shown as black dotted lines
Site 5: Central pore binding site
At the center of the sixfold symmetry axis in CA hexamers, there is a positively charged central pore lined by six arginine residues (R18) from each of the CANTDs in the CA hexamer, having the appearance of an iris [196] (Fig. 9). At the top of the pore, the conformation of the flexible N-terminal β-hairpin changes in a pH-dependent manner, with the pore appearing open under acidic conditions and blocked by the hairpin under basic conditions. The pore has been proposed to serve as a gate through which dNTPs enter inside the core [112, 196]. Abolishing positive charges impaired viral infectivity and reverse transcription in a dose-dependent manner.

HIV-1 CA central pore binding site. A CA hexamer (PDB ID: 4XFX) is shown with CANTDs in light gray and light cyan, CACTDs in dark gray and dark cyan, and R18 side chains at the sixfold symmetry axis in alternating blue and light blue colors. B IP6 (brown sticks) is shown bound to CA hexamer (PDB ID: 6BHT). Right, IP6 above and below R18 ring. C Hexacarboxybenzene (HCB; magenta sticks) bound to CA hexamer (PDB ID: 5HGP). Right, HCB interacting with R18
IP6, the anionic metabolite essential for immature Gag assembly, also interacts with the ring of the six R18 residues in the mature capsid (Table 1) [80]. Additional interactions were recently reported for a more internal, basic ring made by six K25 residues, which based on its observed binding of inositol pentaphosphate (IP5) was speculated to also bind IP6 [109]. Crystallographic studies suggest that IP6 assumes conformations that are almost parallel or at an angle, with respect to the ring of six R18 or K25 residues. Of note, based on molecular dynamics (MD) simulations it was proposed that IP6 assumes a binding mode perpendicular to the R18 ring, thus allowing unhindered dNTP entry through the pore (Fig. 9B) [80, 112]. While the central pore binding site has been proposed to be the binding site in the mature capsid, simulation studies report that IP6 may additionally bind to other pockets, including site 1 [197].
Carboxybenzenes
It was reported that the symmetrical, polyanionic carboxybenzene compounds bind to the R18 ring within the six-fold axis [196] (Fig. 9). The potency of these compounds was low in in vitro studies, as the compounds decreased, but did not abolish, reverse transcription. The most effective of these molecules is hexacarboxybenzene (HCB), also known as benzenehexacarboxylic acid or mellitic acid (Tables 1, 6). MD simulations found that HCB has a preference to lay parallel to the R18 ring, in contrast to IP6 that seems to generally bind at an angle (Fig. 9C) [112]. The parallel positioning of HCB was proposed to block the pore and prevent dNTPs from entering the interior of capsid where reverse transcription takes place [112, 196]. Although these compounds are not useful as potential therapeutics, they provide support to the hypothesis that the central pore binding site may be useful for future drug discovery efforts.
ACAi-028
Recently a small molecule, ACAi-028, was shown to inhibit HIV-1 by binding to a hydrophobic pocket located at the CANTD, interacting with the β-hairpin end, the flexible linker, and the front edge of helix α1 [198] (Tables 1, 6, Fig. 1). Amano et al. found that the addition of a small amino acid sequence in the CANTD, specifically at R18 and T19, resulted in CA degradation [199]. Based on this information, Chia et al. hypothesized that small molecules binding at this area would affect the properties of the core [198]. Using in silico docking, they identified 40 compounds that are predicted to bind this hydrophobic pocket and inhibit HIV-1 replication with relatively low cytotoxicity. ACAi-028 emerged as the top candidate (EC50 = 0.55 µM). This small molecule was proposed to form hydrogen bonds with Q13, T19, and the backbone of S16. These results suggest that ACAi-028 may bind to monomeric HIV-1 CA. ACAi-028 was shown to inhibit the early stages of HIV-1 replication, likely the capsid uncoating process. ACAi-028 is unable to inhibit HIV-2 likely because of differences in amino acid sequence (residues 2–15) at the proposed binding sites and a shallower hydrophobic pocket. This study highlights the HIV-1 CANTD hydrophobic pocket and β-hairpin as potential antiviral targets and presents small molecules that may target this site [198].
Conclusion
Despite the great success of ART, complications can emerge during this life-long treatment. Antiretrovirals put a selective pressure on HIV-1 to develop mutations that cause drug resistance. To combat the emergence of drug resistance, there is continued interest in developing novel therapeutic targets. Of the possible HIV-1 proteins, CA is an excellent candidate for antiviral design because of its influential, and often indispensable roles in HIV-1 cytoplasmic trafficking, immune evasion, reverse transcription, nuclear entry, integration, assembly, and maturation.
As such, in 2003 CAP-1 was the first compound reported to target CA and bind in what we described here as Site 2. Although CAP-1 did not have clinical success due to its low binding affinity, this molecule experimentally demonstrated that CA is a valid target for drug design. Almost 20 years later, the field of HIV-1 capsid inhibitors has expanded greatly, as we have discussed over 40 small molecules and peptides that exert antiviral activity by targeting five functional binding sites on HIV-1 capsid.
Of the five CA antiviral binding sites, site 1 has been targeted with marked success by compounds such as PF74 and GS-6207. In particular, the development of PF74 by Pfizer was a major landmark in the development of CA-targeting compounds. While it never reached the clinic, PF74 demonstrated sub-micromolar potency and serves as the backbone for some of the most promising current compounds, including PF74 analogues with increased potency, particularly against CA mutants that display resistance to PF74. The core peptidic backbone of PF74 is also at the heart of the most promising CA targeting compound to have been developed thus far, GS-6207, developed by Gilead Sciences. GS-6207 is extremely potent and is currently in phase 2/3 clinical trials. Although this compound is the most successful capsid inhibitor to-date, drug resistant mutants have been selected in passaging experiments and it remains to be seen whether resistance will emerge during therapeutic use. Further research into compound binding at Site 1, and the impact of resistance associated mutations, will promote the development of antivirals with improved resistance profiles via structure-based design. Overall, HIV-1 capsid inhibitors are a diverse set of compounds with excellent therapeutic potential. Each of the five targeted sites described here contributes to at least one of the many functions of CA in HIV-1 replication. Mutations at these sites often impair viral fitness, demonstrating their usefulness as antiviral targets. Continued work on capsid inhibitors and their targeted sites will further our understanding of HIV-1 biology and aid in the development of next generation antiretroviral drugs.
Future perspectives
The new developments in capsid-targeting antivirals have generated excitement and have validated HIV-1 CA monomers and the HIV-1 capsid core as a therapeutic target. Future work may focus on the development of later generation antivirals to address challenges of solubility, metabolic stability, and barrier to resistance, while maintaining potency. Importantly, in a recent development, Gilead Sciences and Merck & Co. announced investigational treatment combinations of lenacapavir (GS-6207) and islatravir (EFdA), enabling powerful combinations of long-acting therapeutics that target HIV-1 CA and reverse transcriptase [200,201,202,203].
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Abbreviations
- AIDS:
-
Acquired immune deficiency syndrome
- ART:
-
Antiretroviral therapy
- BD:
-
Benzodiazepines
- BM:
-
Benzimidazoles
- CA:
-
Capsid protein or domain
- CACTD :
-
Capsid protein C-terminal domains
- CAI:
-
Capsid assembly inhibitor
- CANTD :
-
Capsid protein N-terminal domains
- CPSF6:
-
Cleavage and polyadenylation specificity factor subunit 6
- Cryo-EM:
-
Cryogenic-electron microscopy
- Cryo-ET:
-
Cryogenic-electron tomography
- CypA:
-
Cyclophilin A
- CypA-BL:
-
Cyclophilin A-binding loop
- dNTPs:
-
Deoxynucleotide triphosphates
- EFdA:
-
Islatravir
- Env:
-
Envelope protein
- FG:
-
Phenylalanine-glycine dipeptide
- GS-6207:
-
Lenacapavir
- HCB:
-
Hexacarboxybenzene
- HIV:
-
Human immunodeficiency virus
- HSQC:
-
Heteronuclear single quantum coherence
- IN:
-
Integrase
- INSTI:
-
Integrase strand transfer inhibitors
- IP5:
-
Inositol pentaphosphate
- IP6:
-
Inositol hexaphosphate
- LC–ESI–MS:
-
Liquid chromatography–electrospray ionization–mass spectrometry
- MA:
-
Matrix protein or domain
- MD:
-
Molecular dynamics
- MDR:
-
Multiple drug resistance
- MHR:
-
Major homology region
- NC:
-
Nucleocapsid protein or domain
- NMR:
-
Nuclear magnetic resonance
- NNRTI:
-
Non-nucleoside reverse transcriptase inhibitors
- NPC:
-
Nuclear pore complex
- NRTI:
-
Nucleoside reverse transcriptase inhibitors
- PF74:
-
PF-3450074
- PI:
-
Protease inhibitors
- PI(4,5)P2 :
-
Phosphatidyl-4,5-bisphosphate
- PR:
-
Protease protein or domain
- RNP:
-
Ribonucleoprotein
- RT:
-
Reverse transcriptase
- SP:
-
Spacer peptide
- TAF:
-
Tenofovir alafenamide
- α:
-
α-Helix
- βPin:
-
β-Hairpin
References
Global HIV & AIDS statistics—2020 fact sheet. https://www.unaids.org/en/resources/fact-sheet.
Moir S, Chun T-W, Fauci AS. Pathogenic mechanisms of HIV disease. Annu Rev Pathol. 2011;6:223–48.
Weiss RA. How does HIV cause AIDS? Science. 1993;260:1273–9.
Fanales-Belasio E, Raimondo M, Suligoi B, Buttò S. HIV virology and pathogenetic mechanisms of infection: a brief overview. Annali dell’Istituto superiore di sanita. 2010;46:5–14.
Pornillos O, Ganser-Pornillos BK. HIV-1 virion structure. In: Hope TJ, Stevenson M, Richman D, editors. Encyclopedia of AIDS. New York: Springer; 2014. p. 1–6.
Ganser BK, Li S, Klishko VY, Finch JT, Sundquist WI. Assembly and analysis of conical models for the HIV-1 core. Science. 1999;283:80–3.
Zhuang S, Torbett BE. Interactions of HIV-1 capsid with host factors and their implications for developing novel therapeutics. Viruses. 2021;13:417.
Lingappa JR, Lingappa VR, Reed JC. Addressing antiretroviral drug resistance with host-targeting drugs—first steps towards developing a host-targeting HIV-1 assembly inhibitor. Viruses. 2021;13:451.
Wilbourne M, Zhang P. Visualizing HIV-1 capsid and its interactions with antivirals and host factors. Viruses. 2021;13:246.
Yamashita M, Engelman AN. Capsid-dependent host factors in HIV-1 infection. Trends Microbiol. 2017;25:741–55.
Christensen DE, Ganser-Pornillos BK, Johnson JS, Pornillos O, Sundquist WI. Reconstitution and visualization of HIV-1 capsid-dependent replication and integration in vitro. Science. 2020;370:eabc8420.
Novikova M, Zhang Y, Freed EO, Peng K. Multiple roles of HIV-1 capsid during the virus replication cycle. Virol Sin. 2019;34:119–34.
Hulme AE, Perez O, Hope TJ. Complementary assays reveal a relationship between HIV-1 uncoating and reverse transcription. Proc Natl Acad Sci USA. 2011;108:9975–80.
AlBurtamani N, Paul A, Fassati A. The role of capsid in the early steps of HIV-1 infection: new insights into the core of the matter. Viruses. 2021;13:1161.
Carnes SK, Sheehan JH, Aiken C. Inhibitors of the HIV-1 capsid, a target of opportunity. Curr Opin HIV AIDS. 2018;13:359–65.
Tedbury PR, Freed EO. HIV-1 gag: an emerging target for antiretroviral therapy. Curr Top Microbiol Immunol. 2015;389:171–201.
Berger EA, Murphy PM, Farber JM. CHEMOKINE RECEPTORS AS HIV-1 CORECEPTORS: roles in viral entry, tropism, and disease. Annu Rev Immunol. 1999;17:657–700.
Chen B. Molecular mechanism of HIV-1 entry. Trends Microbiol. 2019;27:878–91.
Forshey BM, von Schwedler U, Sundquist WI, Aiken C. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J Virol. 2002;76:5667.
Campbell EM, Hope TJ. HIV-1 capsid: the multifaceted key player in HIV-1 infection. Nat Rev Microbiol. 2015;13:471–83.
Francis AC, Melikyan GB. Single HIV-1 imaging reveals progression of infection through CA-dependent steps of docking at the nuclear pore, uncoating, and nuclear transport. Cell Host Microbe. 2018;23:536-548.e536.
Peng K, Muranyi W, Glass B, Laketa V, Yant SR, Tsai L, Cihlar T, Müller B, Kräusslich H-G. Quantitative microscopy of functional HIV post-entry complexes reveals association of replication with the viral capsid. Elife. 2014;3:e04114.
Francis AC, Marin M, Prellberg MJ, Palermino-Rowland K, Melikyan GB. HIV-1 uncoating and nuclear import precede the completion of reverse transcription in cell lines and in primary macrophages. Viruses. 2020;12:1234.
Yamashita M, Emerman M. Capsid is a dominant determinant of retrovirus infectivity in nondividing cells. J Virol. 2004;78:5670–8.
Rasaiyaah J, Tan CP, Fletcher AJ, Price AJ, Blondeau C, Hilditch L, Jacques DA, Selwood DL, James LC, Noursadeghi M, Towers GJ. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature. 2013;503:402–5.
Fernandez J, Portilho DM, Danckaert A, Munier S, Becker A, Roux P, Zambo A, Shorte S, Jacob Y, Vidalain P-O, et al. Microtubule-associated proteins 1 (MAP1) promote human immunodeficiency virus type I (HIV-1) intracytoplasmic routing to the nucleus. J Biol Chem. 2015;290:4631–46.
Malikov V, da Silva ES, Jovasevic V, Bennett G, de Souza Aranha Vieira DA, Schulte B, Diaz-Griffero F, Walsh D, Naghavi MH. HIV-1 capsids bind and exploit the kinesin-1 adaptor FEZ1 for inward movement to the nucleus. Nat Commun. 2015;6:6660.
Malikov V, Naghavi MH. Localized phosphorylation of a kinesin-1 adaptor by a capsid-associated kinase regulates HIV-1 motility and uncoating. Cell Rep. 2017;20:2792–9.
Dharan A, Campbell EM. Role of microtubules and microtubule-associated proteins in HIV-1 infection. J Virol. 2018;92:e00085-00018.
Naghavi MH. HIV-1 capsid exploitation of the host microtubule cytoskeleton during early infection. Retrovirology. 2021;18:19.
Matreyek KA, Engelman A. The requirement for nucleoporin NUP153 during human immunodeficiency virus type 1 infection is determined by the viral capsid. J Virol. 2011;85:7818.
Matreyek KA, Yücel SS, Li X, Engelman A. Nucleoporin NUP153 phenylalanine-glycine motifs engage a common binding pocket within the HIV-1 capsid protein to mediate lentiviral infectivity. PLOS Pathog. 2013;9:e1003693.
Meehan AM, Saenz DT, Guevera R, Morrison JH, Peretz M, Fadel HJ, Hamada M, van Deursen J, Poeschla EM. A cyclophilin homology domain-independent role for Nup358 in HIV-1 infection. PLOS Pathog. 2014;10:e1003969.
Schaller T, Ocwieja KE, Rasaiyaah J, Price AJ, Brady TL, Roth SL, Hué S, Fletcher AJ, Lee K, KewalRamani VN, et al. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 2011;7:e1002439.
Burdick RC, Li C, Munshi M, Rawson JMO, Nagashima K, Hu WS, Pathak VK. HIV-1 uncoats in the nucleus near sites of integration. Proc Natl Acad Sci USA. 2020;117:5486–93.
Li C, Burdick RC, Nagashima K, Hu WS, Pathak VK. HIV-1 cores retain their integrity until minutes before uncoating in the nucleus. Proc Natl Acad Sci USA. 2021;118:e2019467118.
Zila V, Margiotta E, Turoňová B, Müller TG, Zimmerli CE, Mattei S, Allegretti M, Börner K, Rada J, Müller B, et al. Cone-shaped HIV-1 capsids are transported through intact nuclear pores. Cell. 2021;184:1032-1046.e1018.
Müller TG, Zila V, Peters K, Schifferdecker S, Stanic M, Lucic B, Laketa V, Lusic M, Müller B, Kräusslich HG. HIV-1 uncoating by release of viral cDNA from capsid-like structures in the nucleus of infected cells. Elife. 2021;10:e64776.
Craigie R, Bushman FD. HIV DNA integration. Cold Spring Harb Perspect Med. 2012;2:a006890–a006890.
Cook NJ, Li W, Berta D, Badaoui M, Ballandras-Colas A, Nans A, Kotecha A, Rosta E, Engelman AN, Cherepanov P. Structural basis of second-generation HIV integrase inhibitor action and viral resistance. Science. 2020;367:806–10.
Andrake MD, Skalka AM. Retroviral integrase: then and now. Annu Rev Virol. 2015;2:241–64.
Kirchhoff F. HIV life cycle: overview. In: Hope TJ, Stevenson M, Richman D, editors. Encyclopedia of AIDS. New York: Springer; 2013. p. 1–9.
Siliciano RF, Greene WC. HIV latency. Cold Spring Harbor Perspect Med. 2011;1:a007096.
Van Lint C, Bouchat S, Marcello A. HIV-1 transcription and latency: an update. Retrovirology. 2013;10:67.
Koh Y, Wu X, Ferris AL, Matreyek KA, Smith SJ, Lee K, KewalRamani VN, Hughes SH, Engelman A. Differential effects of human immunodeficiency virus type 1 capsid and cellular factors nucleoporin 153 and LEDGF/p75 on the efficiency and specificity of viral DNA integration. J Virol. 2013;87:648.
Balasubramaniam M, Zhou J, Addai A, Martinez P, Pandhare J, Aiken C, Dash C. PF74 inhibits HIV-1 integration by altering the composition of the preintegration complex. J Virol. 2019;93:e01741-01718.
Hulme AE, Hope TJ. The cyclosporin A washout assay to detect HIV-1 uncoating in infected cells. Methods Mol Biol. 2014;1087:37–46.
Miller MD, Farnet CM, Bushman FD. Human immunodeficiency virus type 1 preintegration complexes: studies of organization and composition. J Virol. 1997;71:5382–90.
Braaten D, Luban J. Cyclophilin A regulates HIV-1 infectivity, as demonstrated by gene targeting in human T cells. EMBO J. 2001;20:1300–9.
Lee K, Ambrose Z, Martin TD, Oztop I, Mulky A, Julias JG, Vandegraaff N, Baumann JG, Wang R, Yuen W, et al. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe. 2010;7:221–33.
Luban J, Bossolt KL, Franke EK, Kalpana GV, Goff SP. Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell. 1993;73:1067–78.
Saito A, Henning MS, Serrao E, Dubose BN, Teng S, Huang J, Li X, Saito N, Roy SP, Siddiqui MA, et al. Capsid-CPSF6 interaction is dispensable for HIV-1 replication in primary cells but is selected during virus passage in vivo. J Virol. 2016;90:6918.
Selyutina A, Persaud M, Simons LM, Bulnes-Ramos A, Buffone C, Martinez-Lopez A, Scoca V, Di Nunzio F, Hiatt J, Marson A, et al. Cyclophilin A prevents HIV-1 restriction in lymphocytes by blocking human TRIM5α binding to the viral core. Cell Rep. 2020;30:3766-3777.e3766.
Achuthan V, Perreira JM, Sowd GA, Puray-Chavez M, McDougall WM, Paulucci-Holthauzen A, Wu X, Fadel HJ, Poeschla EM, Multani AS, et al. Capsid-CPSF6 interaction licenses nuclear HIV-1 trafficking to sites of viral DNA integration. Cell Host Microbe. 2018;24:392-404.e398.
Francis AC, Marin M, Singh PK, Achuthan V, Prellberg MJ, Palermino-Rowland K, Lan S, Tedbury PR, Sarafianos SG, Engelman AN, Melikyan GB. HIV-1 replication complexes accumulate in nuclear speckles and integrate into speckle-associated genomic domains. Nat Commun. 2020;11:3505.
Zhong Z, Ning J, Boggs EA, Jang S, Wallace C, Telmer C, Bruchez MP, Ahn J, Engelman AN, Zhang P, et al. Cytoplasmic CPSF6 regulates HIV-1 capsid trafficking and infection in a Cyclophilin A-dependent manner. MBio. 2021;12:e03142-03120.
Engelman AN. HIV capsid and integration targeting. Viruses. 2021;13:125.
Guerrero S, Batisse J, Libre C, Bernacchi S, Marquet R, Paillart J-C. HIV-1 replication and the cellular eukaryotic translation apparatus. Viruses. 2015;7:199–218.
Freed EO. HIV-1 gag proteins: diverse functions in the virus life cycle. Virology. 1998;251:1–15.
Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW Jr, Swanstrom R, Burch CL, Weeks KM. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature. 2009;460:711–6.
Freed EO. HIV-1 assembly, release and maturation. Nat Rev Microbiol. 2015;13:484–96.
Jacks T, Power MD, Masiarz FR, Luciw PA, Barr PJ, Varmus HE. Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature. 1988;331:280–3.
Jouvenet N, Neil SJD, Bess C, Johnson MC, Virgen CA, Simon SM, Bieniasz PD. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 2006;4:e435.
Ono A. HIV-1 assembly at the plasma membrane: gag trafficking and localization. Futur Virol. 2009;4:241–57.
Bryant M, Ratner L. Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. Proc Natl Acad Sci. 1990;87:523.
Göttlinger HG, Sodroski JG, Haseltine WA. Role of capsid precursor processing and myristoylation in morphogenesis and infectivity of human immunodeficiency virus type 1. Proc Natl Acad Sci. 1989;86:5781.
Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc Natl Acad Sci USA. 2004;101:14889.
Zhou W, Parent LJ, Wills JW, Resh MD. Identification of a membrane-binding domain within the amino-terminal region of human immunodeficiency virus type 1 Gag protein which interacts with acidic phospholipids. J Virol. 1994;68:2556.
Tang C, Loeliger E, Luncsford P, Kinde I, Beckett D, Summers MF. Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc Natl Acad Sci USA. 2004;101:517–22.
Saad JS, Miller J, Tai J, Kim A, Ghanam RH, Summers MF. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc Natl Acad Sci USA. 2006;103:11364–9.
Deng Y, Hammond JA, Pauszek R, Ozog S, Chai I, Rabuck-Gibbons J, Lamichhane R, Henderson SC, Millar DP, Torbett BE, Williamson JR. Discrimination between functional and non-functional cellular gag complexes involved in HIV-1 assembly. J Mol Biol. 2021;433:166842.
Briggs JA, Riches JD, Glass B, Bartonova V, Zanetti G, Kräusslich H-G. Structure and assembly of immature HIV. Proc Natl Acad Sci. 2009;106:11090–5.
Wright ER, Schooler JB, Ding HJ, Kieffer C, Fillmore C, Sundquist WI, Jensen GJ. Electron cryotomography of immature HIV-1 virions reveals the structure of the CA and SP1 Gag shells. EMBO J. 2007;26:2218–26.
Bayro MJ, Ganser-Pornillos BK, Zadrozny KK, Yeager M, Tycko R. Helical conformation in the CA-SP1 junction of the immature HIV-1 lattice determined from solid-state NMR of virus-like particles. J Am Chem Soc. 2016;138:12029–32.
Accola MA, Höglund S, Göttlinger HG. A putative α-helical structure which overlaps the capsid-p2 boundary in the human immunodeficiency virus type 1 gag precursor is crucial for viral particle assembly. J Virol. 1998;72:2072–8.
Schur FK, Obr M, Hagen WJ, Wan W, Jakobi AJ, Kirkpatrick JM, Sachse C, Kräusslich H-G, Briggs JA. An atomic model of HIV-1 capsid-SP1 reveals structures regulating assembly and maturation. Science. 2016;353:506–8.
Wagner JM, Zadrozny KK, Chrustowicz J, Purdy MD, Yeager M, Ganser-Pornillos BK, Pornillos O. Crystal structure of an HIV assembly and maturation switch. Elife. 2016;5:e17063.
Kräusslich HG, Fäcke M, Heuser AM, Konvalinka J, Zentgraf H. The spacer peptide between human immunodeficiency virus capsid and nucleocapsid proteins is essential for ordered assembly and viral infectivity. J Virol. 1995;69:3407.
Mallery DL, Faysal KMR, Kleinpeter A, Wilson MSC, Vaysburd M, Fletcher AJ, Novikova M, Böcking T, Freed EO, Saiardi A, James LC. Cellular IP6 levels limit HIV production while viruses that cannot efficiently package IP6 are attenuated for infection and replication. Cell Rep. 2019;29:3983-3996.e3984.
Dick RA, Zadrozny KK, Xu C, Schur FKM, Lyddon TD, Ricana CL, Wagner JM, Perilla JR, Ganser-Pornillos BK, Johnson MC, et al. Inositol phosphates are assembly co-factors for HIV-1. Nature. 2018;560:509–12.
Checkley MA, Luttge BG, Freed EO. HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J Mol Biol. 2011;410:582–608.
Rein A. RNA Packaging in HIV. Trends Microbiol. 2019;27:715–23.
Sundquist WI, Kräusslich H-G. HIV-1 assembly, budding, and maturation. Cold Spring Harb Perspect Med. 2012;2:a006924.
Kaplan AH, Manchester M, Swanstrom R. The activity of the protease of human immunodeficiency virus type 1 is initiated at the membrane of infected cells before the release of viral proteins and is required for release to occur with maximum efficiency. J Virol. 1994;68:6782.
Carlson L-A, de Marco A, Oberwinkler H, Habermann A, Briggs JAG, Kräusslich H-G, Grünewald K. Cryo electron tomography of native HIV-1 budding sites. PLoS Pathog. 2010;6:e1001173.
Frank GA, Narayan K, Bess JW, Del Prete GQ, Wu X, Moran A, Hartnell LM, Earl LA, Lifson JD, Subramaniam S. Maturation of the HIV-1 core by a non-diffusional phase transition. Nat Commun. 2015;6:5854.
Pettit SC, Moody MD, Wehbie RS, Kaplan AH, Nantermet PV, Klein CA, Swanstrom R. The p2 domain of human immunodeficiency virus type 1 Gag regulates sequential proteolytic processing and is required to produce fully infectious virions. J Virol. 1994;68:8017.
Briggs JA, Grünewald K, Glass B, Förster F, Kräusslich H-G, Fuller SD. The mechanism of HIV-1 core assembly: insights from three-dimensional reconstructions of authentic virions. Structure. 2006;14:15–20.
Keller PW, Huang RK, England MR, Waki K, Cheng N, Heymann JB, Craven RC, Freed EO, Steven AC. A two-pronged structural analysis of retroviral maturation indicates that core formation proceeds by a disassembly-reassembly pathway rather than a displacive transition. J Virol. 2013;87:13655–64.
Woodward CL, Cheng SN, Jensen GJ. Electron cryotomography studies of maturing HIV-1 particles reveal the assembly pathway of the viral core. J Virol. 2015;89:1267–77.
Meng X, Zhao G, Yufenyuy E, Ke D, Ning J, DeLucia M, Ahn J, Gronenborn AM, Aiken C, Zhang P. Protease cleavage leads to formation of mature trimer interface in HIV-1 capsid. PLoS Pathog. 2012;8:e1002886.
Ning J, Erdemci-Tandogan G, Yufenyuy EL, Wagner J, Himes BA, Zhao G, Aiken C, Zandi R, Zhang P. In vitro protease cleavage and computer simulations reveal the HIV-1 capsid maturation pathway. Nat Commun. 2016;7:1–12.
Ganser-Pornillos BK, Yeager M, Pornillos O. Assembly and architecture of HIV. Adv Exp Med Biol. 2012;726:441–65.
Zhao G, Perilla JR, Yufenyuy EL, Meng X, Chen B, Ning J, Ahn J, Gronenborn AM, Schulten K, Aiken C, Zhang P. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature. 2013;497:643–6.
Li S, Hill CP, Sundquist WI, Finch JT. Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature. 2000;407:409–13.
Briggs JA, Simon MN, Gross I, Kräusslich HG, Fuller SD, Vogt VM, Johnson MC. The stoichiometry of Gag protein in HIV-1. Nat Struct Mol Biol. 2004;11:672–5.
Mattei S, Glass B, Hagen WJ, Kräusslich H-G, Briggs JA. The structure and flexibility of conical HIV-1 capsids determined within intact virions. Science. 2016;354:1434–7.
Pornillos O, Ganser-Pornillos BK, Kelly BN, Hua Y, Whitby FG, Stout CD, Sundquist WI, Hill CP, Yeager M. X-ray structures of the hexameric building block of the HIV capsid. Cell. 2009;137:1282–92.
Worthylake DK, Wang H, Yoo S, Sundquist WI, Hill CP. Structures of the HIV-1 capsid protein dimerization domain at 2.6 Å resolution. Acta Crystallogr Sect D Biol Crystallogr. 1999;55:85–92.
Ternois F, Sticht J, Duquerroy S, Kräusslich H-G, Rey FA. The HIV-1 capsid protein C-terminal domain in complex with a virus assembly inhibitor. Nat Struct Mol Biol. 2005;12:678–82.
Bartonova V, Igonet S, Sticht J, Glass B, Habermann A, Vaney M-C, Sehr P, Lewis J, Rey FA, Kraüsslich H-G. Residues in the HIV-1 capsid assembly inhibitor binding site are essential for maintaining the assembly-competent quaternary structure of the capsid protein. J Biol Chem. 2008;283:32024–33.
Pornillos O, Ganser-Pornillos BK, Yeager M. Atomic-level modelling of the HIV capsid. Nature. 2011;469:424–7.
Du S, Betts L, Yang R, Shi H, Concel J, Ahn J, Aiken C, Zhang P, Yeh JI. Structure of the HIV-1 full-length capsid protein in a conformationally trapped unassembled state induced by small-molecule binding. J Mol Biol. 2011;406:371–86.
Deshmukh L, Schwieters CD, Grishaev A, Ghirlando R, Baber JL, Clore GM. Structure and dynamics of full-length HIV-1 capsid protein in solution. J Am Chem Soc. 2013;135:16133–47.
Gres AT, Kirby KA, KewalRamani VN, Tanner JJ, Pornillos O, Sarafianos SG. X-ray crystal structures of native HIV-1 capsid protein reveal conformational variability. Science. 2015;349:99–103.
Shin R, Tzou Y-M, Krishna NR. Structure of a monomeric mutant of the HIV-1 capsid protein. Biochemistry. 2011;50:9457–67.
Kleinpeter AB, Freed EO. HIV-1 maturation: lessons learned from inhibitors. Viruses. 2020;12:940.
Chang Y-F, Wang S-M, Huang K-J, Wang C-T. Mutations in capsid major homology region affect assembly and membrane affinity of HIV-1 Gag. J Mol Biol. 2007;370:585–97.
Renner N, Mallery DL, Faysal KMR, Peng W, Jacques DA, Böcking T, James LC. A lysine ring in HIV capsid pores coordinates IP6 to drive mature capsid assembly. PLoS Pathog. 2021;17:e1009164.
Dick RA, Xu C, Morado DR, Kravchuk V, Ricana CL, Lyddon TD, Broad AM, Feathers JR, Johnson MC, Vogt VM, et al. Structures of immature EIAV Gag lattices reveal a conserved role for IP6 in lentivirus assembly. PLoS Pathog. 2020;16:e1008277.
Dick RA, Mallery DL, Vogt VM, James LC. IP6 regulation of HIV capsid assembly, stability, and uncoating. Viruses. 2018;10:640.
Xu C, Fischer DK, Rankovic S, Li W, Dick RA, Runge B, Zadorozhnyi R, Ahn J, Aiken C, Polenova T, et al. Permeability of the HIV-1 capsid to metabolites modulates viral DNA synthesis. PLoS Biol. 2020;18:e3001015.
Grime JMA, Dama JF, Ganser-Pornillos BK, Woodward CL, Jensen GJ, Yeager M, Voth GA. Coarse-grained simulation reveals key features of HIV-1 capsid self-assembly. Nat Commun. 2016;7:11568.
Byeon I-JL, Meng X, Jung J, Zhao G, Yang R, Ahn J, Shi J, Concel J, Aiken C, Zhang P, Gronenborn AM. Structural convergence between Cryo-EM and NMR reveals intersubunit interactions critical for HIV-1 capsid function. Cell. 2009;139:780–90.
Ganser-Pornillos BK, von Schwedler UK, Stray KM, Aiken C, Sundquist WI. Assembly properties of the human immunodeficiency virus type 1 CA protein. J Virol. 2004;78:2545–52.
Jiang J, Ablan SD, Derebail S, Hercík K, Soheilian F, Thomas JA, Tang S, Hewlett I, Nagashima K, Gorelick RJ, Freed EO. The interdomain linker region of HIV-1 capsid protein is a critical determinant of proper core assembly and stability. Virology. 2011;421:253–65.
Manocheewa S, Swain JV, Lanxon-Cookson E, Rolland M, Mullins JI. Fitness costs of mutations at the HIV-1 capsid hexamerization interface. PLoS ONE. 2013;8:e66065.
Rihn SJ, Wilson SJ, Loman NJ, Alim M, Bakker SE, Bhella D, Gifford RJ, Rixon FJ, Bieniasz PD. Extreme genetic fragility of the HIV-1 capsid. PLoS Pathog. 2013;9:e1003461.
Bocanegra R, Rodríguez-Huete A, Fuertes MÁ, Del Álamo M, Mateu MG. Molecular recognition in the human immunodeficiency virus capsid and antiviral design. Virus Res. 2012;169:388–410.
Neira JL, del Álamo M, Mateu MG. Thermodynamic dissection of a low affinity protein-protein interface involved in human immunodeficiency virus assembly. J Biol Chem. 2003;278:27923–9.
Wacharapornin P, Lauhakirti D, Auewarakul P. The effect of capsid mutations on HIV-1 uncoating. Virology. 2007;358:48–54.
Gamble TR, Yoo S, Vajdos FF, Von Schwedler UK, Worthylake DK, Wang H, McCutcheon JP, Sundquist WI, Hill CP. Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science. 1997;278:849–53.
von Schwedler UK, Stray KM, Garrus JE, Sundquist WI. Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J Virol. 2003;77:5439–50.
Ni T, Gerard S, Zhao G, Dent K, Ning J, Zhou J, Shi J, Anderson-Daniels J, Li W, Jang S, et al. Intrinsic curvature of the HIV-1 CA hexamer underlies capsid topology and interaction with cyclophilin A. Nat Struct Mol Biol. 2020;27:855–62.
Bester SM, Wei G, Zhao H, Adu-Ampratwum D, Iqbal N, Courouble VV, Francis AC, Annamalai AS, Singh PK, Shkriabai N, van Blerkom P. Structural and mechanistic bases for a potent HIV-1 capsid inhibitor. Science. 2020;370:360–4.
Lu M, Russell RW, Bryer AJ, Quinn CM, Hou G, Zhang H, Schwieters CD, Perilla JR, Gronenborn AM, Polenova T. Atomic-resolution structure of HIV-1 capsid tubes by magic-angle spinning NMR. Nat Struct Mol Biol. 2020;27:863–9.
FDA-approved HIV medicines. https://hivinfo.nih.gov/understanding-hiv/fact-sheets/fda-approved-hiv-medicines.
Arts EJ, Hazuda DJ. HIV-1 antiretroviral drug therapy. Cold Spring Harbor Perspect Med. 2012;2:a007161.
Iacob SA, Iacob DG. Ibalizumab targeting CD4 receptors, an emerging molecule in HIV therapy. Front Microbiol. 2017;8:2323.
Meanwell NA, Krystal MR, Nowicka-Sans B, Langley DR, Conlon DA, Eastgate MD, Grasela DM, Timmins P, Wang T, Kadow JF. Inhibitors of HIV-1 attachment: the discovery and development of temsavir and its prodrug fostemsavir. J Med Chem. 2018;61:62–80.
Jacobson JM, Kuritzkes DR, Godofsky E, DeJesus E, Larson JA, Weinheimer SP, Lewis ST. Safety, pharmacokinetics, and antiretroviral activity of multiple doses of ibalizumab (formerly TNX-355), an anti-CD4 monoclonal antibody, in human immunodeficiency virus type 1-infected adults. Antimicrob Agents Chemother. 2009;53:450–7.
Henrich TJ, Kuritzkes DR. HIV-1 entry inhibitors: recent development and clinical use. Curr Opin Virol. 2013;3:51–7.
Garbelli A, Riva V, Crespan E, Maga G. How to win the HIV-1 drug resistance hurdle race: running faster or jumping higher? Biochem J. 2017;474:1559–77.
Phanuphak N, Gulick RM. HIV treatment and prevention 2019: current standards of care. Curr Opin HIV AIDS. 2020;15:4–12.
Link JO, Rhee MS, Tse WC, Zheng J, Somoza JR, Rowe W, Begley R, Chiu A, Mulato A, Hansen D, et al. Clinical targeting of HIV capsid protein with a long-acting small molecule. Nature. 2020;584:614–8.
Li G, Verheyen J, Rhee S-Y, Voet A, Vandamme A-M, Theys K. Functional conservation of HIV-1 Gag: implications for rational drug design. Retrovirology. 2013;10:1–11.
Blair WS, Pickford C, Irving SL, Brown DG, Anderson M, Bazin R, Cao J, Ciaramella G, Isaacson J, Jackson L, et al. HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog. 2010;6:e1001220.
Bhattacharya A, Alam SL, Fricke T, Zadrozny K, Sedzicki J, Taylor AB, Demeler B, Pornillos O, Ganser-Pornillos BK, Diaz-Griffero F, Ivanov DN. Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proc Natl Acad Sci. 2014;111:18625–30.
Price AJ, Jacques DA, McEwan WA, Fletcher AJ, Essig S, Chin JW, Halambage UD, Aiken C, James LC. Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLoS Pathog. 2014;10:e1004459.
Rebensburg SV, Wei G, Larue RC, Lindenberger J, Francis AC, Annamalai AS, Morrison J, Shkriabai N, Huang SW, KewalRamani V, et al. Sec24C is an HIV-1 host dependency factor crucial for virus replication. Nat Microbiol. 2021;6:435–44.
Price AJ, Fletcher AJ, Schaller T, Elliott T, Lee K, KewalRamani VN, Chin JW, Towers GJ, James LC. CPSF6 defines a conserved capsid interface that modulates HIV-1 replication. PLoS Pathog. 2012;8:e1002896.
Shi J, Zhou J, Shah VB, Aiken C, Whitby K. Small-molecule inhibition of human immunodeficiency virus type 1 infection by virus capsid destabilization. J Virol. 2011;85:542–9.
Zhou J, Price AJ, Halambage UD, James LC, Aiken C. HIV-1 resistance to the capsid-targeting inhibitor PF74 results in altered dependence on host factors required for virus nuclear entry. J Virol. 2015;89:9068–79.
Saito A, Ferhadian D, Sowd GA, Serrao E, Shi J, Halambage UD, Teng S, Soto J, Siddiqui MA, Engelman AN, et al. Roles of capsid-interacting host factors in multimodal inhibition of HIV-1 by PF74. J Virol. 2016;90:5808–23.
McArthur C, Gallazzi F, Quinn TP, Singh K. HIV capsid inhibitors beyond PF74. Diseases. 2019;7:56.
Rankovic S, Ramalho R, Aiken C, Rousso I. PF74 reinforces the HIV-1 capsid to impair reverse transcription-induced uncoating. J Virol. 2018;92:e00845-00818.
Siddiqui MA, Saito A, Halambage UD, Ferhadian D, Fischer DK, Francis AC, Melikyan GB, Ambrose Z, Aiken C, Yamashita M. A novel phenotype links HIV-1 capsid stability to cGAS-mediated DNA sensing. J Virol. 2019;93:e00706-00719.
Shi J, Zhou J, Halambage UD, Shah VB, Burse MJ, Wu H, Blair WS, Butler SL, Aiken C. Compensatory substitutions in the HIV-1 capsid reduce the fitness cost associated with resistance to a capsid-targeting small-molecule inhibitor. J Virol. 2015;89:208.
Wu G, Zalloum WA, Meuser ME, Jing L, Kang D, Chen C-H, Tian Y, Zhang F, Cocklin S, Lee K-H, Liu X. Discovery of phenylalanine derivatives as potent HIV-1 capsid inhibitors from click chemistry-based compound library. Eur J Med Chem. 2018;158:478–92.
Xu JP, Francis AC, Meuser ME, Mankowski M, Ptak RG, Rashad AA, Melikyan GB, Cocklin S. Exploring modifications of an HIV-1 capsid inhibitor: design, synthesis, and mechanism of action. J Drug Des Res. 2018;5:1070.
Wang L, Casey MC, Vernekar SKV, Sahani RL, Kirby KA, Du H, Zhang H, Tedbury PR, Xie J, Sarafianos SG, Wang Z. Novel PF74-like small molecules targeting the HIV-1 capsid protein: balance of potency and metabolic stability. Acta Pharm Sin B. 2021;11:810–22.
Wang L, Casey MC, Vernekar SKV, Do HT, Sahani RL, Kirby KA, Du H, Hachiya A, Zhang H, Tedbury PR, et al. Chemical profiling of HIV-1 capsid-targeting antiviral PF74. Eur J Med Chem. 2020;200:112427.
Jiang X, Wu G, Zalloum WA, Meuser ME, Dick A, Sun L, Chen CH, Kang D, Jing L, Jia R, et al. Discovery of novel 1,4-disubstituted 1,2,3-triazole phenylalanine derivatives as HIV-1 capsid inhibitors. RSC Adv. 2019;9:28961–86.
Sun L, Dick A, Meuser ME, Huang T, Zalloum WA, Chen CH, Cherukupalli S, Xu S, Ding X, Gao P, et al. Design, synthesis, and mechanism study of benzenesulfonamide-containing phenylalanine derivatives as novel HIV-1 capsid inhibitors with improved antiviral activities. J Med Chem. 2020;63:4790–810.
Sun L, Huang T, Dick A, Meuser ME, Zalloum WA, Chen C-H, Ding X, Gao P, Cocklin S, Lee K-H, Zhan P. Design, synthesis and structure-activity relationships of 4-phenyl-1H-1, 2, 3-triazole phenylalanine derivatives as novel HIV-1 capsid inhibitors with promising antiviral activities. Eur J Med Chem. 2020;190:112085.
Xu JP, Branson JD, Lawrence R, Cocklin S. Identification of a small molecule HIV-1 inhibitor that targets the capsid hexamer. Bioorg Med Chem Lett. 2016;26:824–8.
Vernekar SKV, Sahani RL, Casey MC, Kankanala J, Wang L, Kirby KA, Du H, Zhang H, Tedbury PR, Xie J, et al. Toward structurally novel and metabolically stable HIV-1 capsid-targeting small molecules. Viruses. 2020;12:452.
Wang L, Casey MC, Vernekar SKV, Sahani RL, Kankanala J, Kirby KA, Du H, Hachiya A, Zhang H, Tedbury PR, et al. Novel HIV-1 capsid-targeting small molecules of the PF74 binding site. Eur J Med Chem. 2020;204:112626.
Sahani RL, Diana-Rivero R, Vernekar SKV, Wang L, Du H, Zhang H, Castaner AE, Casey MC, Kirby KA, Tedbury PR, et al. Design, synthesis and characterization of HIV-1 CA-targeting small molecules: conformational restriction of PF74. Viruses. 2021;13:479.
Lamorte L, Titolo S, Lemke CT, Goudreau N, Mercier J-F, Wardrop E, Shah VB, von Schwedler UK, Langelier C, Banik SS, Aiken C. Discovery of novel small-molecule HIV-1 replication inhibitors that stabilize capsid complexes. Antimicrob Agents Chemother. 2013;57:4622–31.
Fricke T, Buffone C, Opp S, Valle-Casuso J, Diaz-Griffero F. BI-2 destabilizes HIV-1 cores during infection and prevents binding of CPSF6 to the HIV-1 capsid. Retrovirology. 2014;11:120.
Yant SR, Mulato A, Hansen D, Winston CT, Niedziela-Majka A, Zhang JR, Stepan GJ, Jin D, Wong MH, Perreira JM, Singer E. A highly potent long-acting small-molecule HIV-1 capsid inhibitor with efficacy in a humanized mouse model. Nat Med. 2019;25:1377–84.
Perrier M, Bertine M, Le Hingrat Q, Joly V, Visseaux B, Collin G, Landman R, Yazdanpanah Y, Descamps D, Charpentier C. Prevalence of gag mutations associated with in vitro resistance to capsid inhibitor GS-CA1 in HIV-1 antiretroviral-naive patients. J Antimicrob Chemother. 2017;72:2954–5.
Sager J, Begley R, Rhee M, West S, Ling J, Escobar J, Mathias A. Safety and PK of subcutaneous GS-6207, a novel HIV-1 capsid inhibitor. In: HIV Medicine: 17th European AIDS Conference; Novermber 6–9; Basel: Wiley; 2019: 39.
Begley R, Rhee M, West S, Worth A, Ling J, German P. PK, food effect, and safety of oral GS-6207, a novel HIV-1 capsid inhibitor. In: Proceedings of the conference on retroviruses and opportunistic infections 2020, Abstract #470.
Sun Q, Levy RM, Kirby KA, Wang Z, Sarafianos SG, Deng N. Molecular dynamics free energy simulations reveal the mechanism for the antiviral resistance of the M66I HIV-1 capsid mutation. Viruses. 2021;13:920.
Coffin JM. Structure, replication, and recombination of retrovirus genomes: some unifying hypotheses. J Gen Virol. 1979;42:1–26.
Pham HT, Yoo S, Mesplède T. Combination therapies currently under investigation in phase I and phase II clinical trials for HIV-1. Expert Opin Investig Drugs. 2020;29:273–83.
Hu Y, Stumpfe D, Bajorath J. Recent advances in scaffold hopping. J Med Chem. 2017;60:1238–46.
Babu S, Belema M, Bender JA, Iwuagwu C, Kadow JF, Kumaravel S, Nagalakshmi P, Naidu BN, Patel M, Peese KM, et al. Preparation of heterocyclyl substituted acetamides as inhibitors of human immunodeficiency virus replication. Brentford: ViiV Healthcare UK No.5 Limited; 2018. p. 536.
Gillis EP, Parcella KE, Patel M, Peese KM. Indazolylquinazolinones as inhibitors of human immunodeficiency virus replication and their preparation. Brentford: ViiV Healthcare UK No.5 Limited; 2020. p. 277.
Belema M, Bender JA, Frennesson DB, Gillis EP, Iwuagwu C, Kadow JF, Naidu BN, Parcella KE, Peese KM, Rajamani R, et al. Preparation of 4-oxo-3,4-dihydroquinazoline compounds as inhibitors of human immunodeficiency virus replication. Brentford: ViiV Healthcare UK No.5 Limited; 2019. p. 313.
Urano E, Kuramochi N, Ichikawa R, Murayama SY, Miyauchi K, Tomoda H, Takebe Y, Nermut M, Komano J, Morikawa Y. Novel postentry inhibitor of human immunodeficiency virus type 1 replication screened by yeast membrane-associated two-hybrid system. Antimicrob Agents Chemother. 2011;55:4251.
Chen N-Y, Zhou L, Gane PJ, Opp S, Ball NJ, Nicastro G, Zufferey M, Buffone C, Luban J, Selwood D, et al. HIV-1 capsid is involved in post-nuclear entry steps. Retrovirology. 2016;13:28.
Vozzolo L, Loh B, Gane PJ, Tribak M, Zhou L, Anderson I, Nyakatura E, Jenner RG, Selwood D, Fassati A. Gyrase B inhibitor impairs HIV-1 replication by targeting Hsp90 and the capsid protein. J Biol Chem. 2010;285:39314–28.
Kelly BN, Kyere S, Kinde I, Tang C, Howard BR, Robinson H, Sundquist WI, Summers MF, Hill CP. Structure of the antiviral assembly inhibitor CAP-1 complex with the HIV-1 CA protein. J Mol Biol. 2007;373:355–66.
Lemke CT, Titolo S, von Schwedler U, Goudreau N, Mercier J-F, Wardrop E, Faucher A-M, Coulombe R, Banik SS, Fader L, Gagnon A. Distinct effects of two HIV-1 capsid assembly inhibitor families that bind the same site within the N-terminal domain of the viral CA protein. J Virol. 2012;86:6643–55.
Tang C, Loeliger E, Kinde I, Kyere S, Mayo K, Barklis E, Sun Y, Huang M, Summers MF. Antiviral inhibition of the HIV-1 capsid protein. J Mol Biol. 2003;327:1013–20.
Fader LD, Bethell R, Bonneau P, Bös M, Bousquet Y, Cordingley MG, Coulombe R, Deroy P, Faucher A-M, Gagnon A, Goudreau N. Discovery of a 1, 5-dihydrobenzo[b][1,4]diazepine-2,4-dione series of inhibitors of HIV-1 capsid assembly. Bioorg Med Chem Lett. 2011;21:398–404.
Wong HC, Shin R, Krishna NR. Solution structure of a double mutant of the carboxy-terminal dimerization domain of the HIV-1 capsid protein. Biochemistry. 2008;47:2289–97.
Mateu MG. The capsid protein of human immunodeficiency virus: intersubunit interactions during virus assembly. FEBS J. 2009;276:6098–109.
Dimitrov DS. Therapeutic proteins. Methods Mol Biol. 2012;899:1–26.
Leader B, Baca QJ, Golan DE. Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discov. 2008;7:21–39.
Sticht J, Humbert M, Findlow S, Bodem J, Müller B, Dietrich U, Werner J, Kräusslich H-G. A peptide inhibitor of HIV-1 assembly in vitro. Nat Struct Mol Biol. 2005;12:671–7.
Zhang H, Zhao Q, Bhattacharya S, Waheed AA, Tong X, Hong A, Heck S, Curreli F, Goger M, Cowburn D, et al. A cell-penetrating helical peptide as a potential HIV-1 inhibitor. J Mol Biol. 2008;378:565–80.
Schafmeister CE, Po J, Verdine GL. An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides. J Am Chem Soc. 2000;122:5891–2.
Bhattacharya S, Zhang H, Debnath AK, Cowburn D. Solution structure of a hydrocarbon stapled peptide inhibitor in complex with monomeric C-terminal domain of HIV-1 capsid. J Biol Chem. 2008;283:16274–8.
Garzón MT, Lidón-Moya MC, Barrera FN, Prieto A, Gómez J, Mateu MG, Neira JL. The dimerization domain of the HIV-1 capsid protein binds a capsid protein-derived peptide: a biophysical characterization. Protein Sci. 2004;13:1512–23.
Bocanegra R, Nevot M, Doménech R, López I, Abián O, Rodríguez-Huete A, Cavasotto CN, Velázquez-Campoy A, Gómez J, Martínez MÁ, Neira JL. Rationally designed interfacial peptides are efficient in vitro inhibitors of HIV-1 capsid assembly with antiviral activity. PLoS ONE. 2011;6:e23877.
Zhang H, Curreli F, Zhang X, Bhattacharya S, Waheed AA, Cooper A, Cowburn D, Freed EO, Debnath AK. Antiviral activity of α-helical stapled peptides designed from the HIV-1 capsid dimerization domain. Retrovirology. 2011;8:28.
Kobayakawa T, Yokoyama M, Tsuji K, Fujino M, Kurakami M, Boku S, Nakayama M, Kaneko M, Ohashi N, Kotani O, et al. Small-molecule anti-HIV-1 agents based on HIV-1 capsid proteins. Biomolecules. 2021;11:208.
Machara A, Lux V, Kožíšek M, Grantz Šašková KR, Štěpánek OE, Kotora M, Parkan K, Pávová M, Glass BR, Sehr P, Lewis J. Specific inhibitors of HIV capsid assembly binding to the C-terminal domain of the capsid protein: evaluation of 2-arylquinazolines as potential antiviral compounds. J Med Chem. 2016;59:545–58.
Thenin-Houssier S, de Vera IMS, Pedro-Rosa L, Brady A, Richard A, Konnick B, Opp S, Buffone C, Fuhrmann J, Kota S, et al. Ebselen, a small-molecule capsid inhibitor of HIV-1 replication. Antimicrob Agents Chemother. 2016;60:2195.
Kortagere S, Madani N, Mankowski MK, Schön A, Zentner I, Swaminathan G, Princiotto A, Anthony K, Oza A, Sierra L-J, Passic SR. Inhibiting early-stage events in HIV-1 replication by small-molecule targeting of the HIV-1 capsid. J Virol. 2012;86:8472–81.
Goudreau N, Lemke CT, Faucher A-M, Grand-Maitre C, Goulet S, Lacoste J-E, Rancourt J, Malenfant E, Mercier J-F, Titolo S, Mason SW. Novel inhibitor binding site discovery on HIV-1 capsid N-terminal domain by NMR and X-ray crystallography. ACS Chem Biol. 2013;8:1074–82.
Jacques DA, McEwan WA, Hilditch L, Price AJ, Towers GJ, James LC. HIV-1 uses dynamic capsid pores to import nucleotides and fuel encapsidated DNA synthesis. Nature. 2016;536:349–53.
Yu A, Lee EM, Jin J, Voth GA. Atomic-scale characterization of mature HIV-1 capsid stabilization by inositol hexakisphosphate (IP6). Sci Adv. 2020;6:eabc6465.
Chia T, Nakamura T, Amano M, Takamune N, Matsuoka M, Nakata H. A small molecule, ACAi-028, with anti-HIV-1 activity targets a novel hydrophobic pocket on HIV-1 capsid. Antimicrob Agents Chemother. 2021;65:e01039-21.
Amano M, Bulut H, Tamiya S, Nakamura T, Koh Y, Mitsuya H. Amino-acid inserts of HIV-1 capsid (CA) induce CA degradation and abrogate viral infectivity: insights for the dynamics and mechanisms of HIV-1 CA decomposition. Sci Rep. 2019;9:1–17.
Roy M, Reuters Staff: Gilead, Merck collaborate to develop long-acting HIV treatment. Reuters; 2021.
Markowitz M, Sarafianos SG. 4′-Ethynyl-2-fluoro-2′-deoxyadenosine, MK-8591: a novel HIV-1 reverse transcriptase translocation inhibitor. Curr Opin HIV AIDS. 2018;13:294–9.
Salie ZL, Kirby KA, Michailidis E, Marchand B, Singh K, Rohan LC, Kodama EN, Mitsuya H, Parniak MA, Sarafianos SG. Structural basis of HIV inhibition by translocation-defective RT inhibitor 4′-ethynyl-2-fluoro-2′-deoxyadenosine (EFdA). Proc Natl Acad Sci. 2016;113:9274–9.
Michailidis E, Huber AD, Ryan EM, Ong YT, Leslie MD, Matzek KB, Singh K, Marchand B, Hagedorn AN, Kirby KA, et al. 4′-Ethynyl-2-fluoro-2′-deoxyadenosine (EFdA) inhibits HIV-1 reverse transcriptase with multiple mechanisms. J Biol Chem. 2014;289:24533–48.
Acknowledgements
We would like to thank Philip Patenall for creating the graphical abstract. We would like to thank Graham Johnson and David Goodsell for assistance with the graphical abstract.
Funding
This work was supported in part by National Institutes of Health (NIH) Grants R01 AI120860 (S.G.S. and Z.W.), U54 AI150472 (S.G.S.), R35 GM133719 (M.R.) and National Science Foundation (NSF) Grant CHE-1752654 (M.R.). W.M.M., A.A.S. acknowledge support from training Grant T32 GM135060. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the National Science Foundation. S.G.S. also acknowledges funding from the Nahmias-Schinazi Distinguished Chair in Research.
Author information
Authors and Affiliations
Contributions
AAS and WMM prepared the manuscript with guidance from SGS, KAK, and PRT. WMM, AAS, KAK, PRT, MR, ZW, and SGS edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
McFadden, W.M., Snyder, A.A., Kirby, K.A. et al. Rotten to the core: antivirals targeting the HIV-1 capsid core. Retrovirology 18, 41 (2021). https://doi.org/10.1186/s12977-021-00583-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12977-021-00583-z