
Abstract
Background
Myasthenia gravis (MG) is an autoimmune disease characterized by pathogenic antibodies that target structures of the neuromuscular junction. The evidence suggests that the regulation of long noncoding RNAs (lncRNAs) that is mediated by transcription factors (TFs) plays a key role in the pathophysiology of MG. Nevertheless, the detailed molecular mechanisms of lncRNAs in MG remain largely undetermined.
Methods
Using microarray analysis, we analyzed the lncRNA levels in MG. By bioinformatics analysis, LINC01566 was found to potentially play an important role in MG. First, qRT‒PCR was performed to verify the LINC1566 expressions in MG patients. Then, fluorescence in situ hybridization was conducted to determine the localization of LINC01566 in CD4 + T cells. Finally, the impact of LINC01566 knockdown or overexpression on CD4 + T-cell function was also analyzed using flow cytometry and CCK-8 assay. A dual-luciferase reporter assay was used to validate the binding of the TF FOSL1 to the LINC01566 promoter.
Results
Based on the lncRNA microarray and differential expression analyses, we identified 563 differentially expressed (DE) lncRNAs, 450 DE mRNAs and 19 DE TFs in MG. We then constructed a lncRNA-TF-mRNA network. Through network analysis, we found that LINC01566 may play a crucial role in MG by regulating T-cell-related pathways. Further experiments indicated that LINC01566 is expressed at low levels in MG patients. Functionally, LINC01566 is primarily distributed in the nucleus and can facilitate CD4 + T-cell apoptosis and inhibit cell proliferation. Mechanistically, we hypothesized that LINC01566 may negatively regulate the expressions of DUSP3, CCR2, FADD, SIRPB1, LGALS3 and SIRPB1, which are involved in the T-cell activation pathway, to further influence the cellular proliferation and apoptosis in MG. Moreover, we found that the effect of LINC01566 on CD4 + T cells in MG was mediated by the TF FOSL1, and in vitro experiments indicated that FOSL1 can bind to the promoter region of LINC01566.
Conclusions
In summary, our research revealed the protective roles of LINC01566 in clinical samples and cellular experiments, illustrating the potential roles and mechanism by which FOSL1/LINC01566 negatively regulates CD4 + T-cell activation in MG.
Similar content being viewed by others


Introduction
Myasthenia gravis (MG) is an autoimmune disease in which antibodies bind to functional molecules in the postsynaptic membrane at neuromuscular junctions [1]. Autoantibodies against the acetylcholine receptor (AChR), muscle-specific kinase (MuSK), and lipoprotein-related protein 4 (LRP4) have been identified as pathogenic factors in MG [2]. Antibodies induce skeletal muscle weakness, which typically increases with exercise and repetitive muscle use (fatigue) and varies over the course of a day and from day to day [1]. However, the origin of the autoimmune dysfunction that leads to the onset of MG remains unclear, and the molecular mechanisms regulating its progression are highly complex.
It has been widely accepted that the MG autoantibody response is T-cell-dependent. Specifically, CD4 + T cells are overactivated in MG patients [3, 4] and stimulate B cells to produce autoantibodies via a complex network involving interactions among cells and cytokines. CD4 + T cells can be subdivided into Th1, Th2, Th17, and Treg cells [5]. Abnormal CD4 + T-cell selection and activation have been intensively investigated in MG [6, 7]. Analysis of the T-cell subset composition in the blood revealed an increased proportion of mature CD4 + T cells in MG patients compared to healthy controls [8, 9]. MG patients have more Th1 and Th17 cells and their associated cytokines, namely, IL-1, IL-6, IL-17, interferon-gamma (IFN-γ), and tumour necrosis factor alpha (TNF-α) [10,11,12]. Furthermore, studies have confirmed that there are defects in or dysfunction of Foxp3(+) CD25(+) CD4(+) Treg cells in MG patients compared to those in healthy subjects [13, 14]. Therefore, abnormal CD4 + T-cell activation and cytokine production are important in the pathogenesis of MG. In recent years, long noncoding RNAs (lncRNAs) have attracted increasing attention in the field of autoimmune disease [15,16,17] and thus have provided novel insight into the pathogenesis, diagnosis, and treatment of MG.
LncRNAs are defined as transcripts of more than 200 nucleotides that are not translated into proteins [18] and have also been found to play important roles in different biological functions, including the regulation of immune responses [19]. LncRNAs have been proposed to carry out diverse functions, including transcriptional regulation when cis- or trans-acting, the organization of nuclear domains, and the regulation of proteins or RNA molecules [20]. There have been many studies on the molecular mechanism underlying the effects of lncRNAs and T cells on disease status [21,22,23]. It has also been reported that the lncRNA GAS5 can regulate the Th17/Treg balance by targeting miR-23a expression in MG [24]. The influence of the lncRNA IFNG-AS1 on the expression level of CD40L and CD4+ T-cell activation in MG patients partly depends on its effect on HLA-DRB1 expression [25]. However, few lncRNAs have been identified in MG, and the correlations between these lncRNAs and CD4 + T-cell activation have not been actively investigated. Therefore, it is necessary to systematically explore the regulatory mechanisms of CD4 + T cells involving lncRNAs in MG.
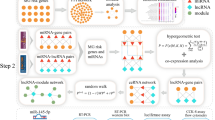
In our research, we designed an integrative analysis based on lncRNA and mRNA microarray sequencing in MG (Fig. 1). We first showed that LINC01566 is expressed at low levels in peripheral blood mononuclear cells (PBMCs) obtained from MG patients and that its expression is mediated by the transcription factor (TF) FOSL1. Intriguingly, the expression level of LINC01566 was positively correlated with the neutrophil count and neutrophil-lymphocyte ratio (NLR) but negatively correlated with the lymphocyte and monocyte counts in MG patients. Functionally, LINC01566 is primarily distributed in the nucleus and can facilitate CD4 + T-cell apoptosis and inhibit CD4 + T-cell proliferation. Mechanistically, we hypothesized that LINC01566 may negatively regulate the expression of the T-cell activation pathway molecules DUSP3, CCR2, FADD, SIRPB1, LGALS3 and P2RX7 to further influence cellular proliferation and apoptosis in MG. Our study revealed the potential roles of LINC01566 and provided new insight into the regulatory mechanism of lncRNAs in MG.

Flowchart of this study
Materials and methods
Microarray analysis
Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood using lymphocyte isolation fluid (TBD, Tianjin, China) according to the manufacturer’s instructions. Total RNA was isolated from PBMCs with TRIzol® Reagent (Invitrogen, Carlsbad, CA, USA) and quantified using a NanoDrop ND-1000 (Thermo Fisher Scientific) system. RNA integrity was assessed by standard denaturing agarose gel electrophoresis. Each RNA sample was amplified and transcribed into fluorescent cDNA along the entire length of the transcripts without 3’ bias utilizing a random priming method. The labeled cDNAs were hybridized onto the Human LncRNA Array, version 5.0 (8 × 60 K, Arraystar). After hybridization and washing, the arrays were scanned using an Agilent G2505C scanner and Agilent Feature Extraction software, version 11.1, to analyze the acquired array images. Quantile normalization and subsequent data processing were performed using Agilent GeneSpring GX software, version 12.1. The lncRNA array data have been deposited in the Gene Expression Omnibus database under accession GSE263220.
Differential expression analysis
We identified the significantly DE lncRNAs and DE mRNAs through volcano plot filtering and calculated a P value using a student’s t test. The thresholds for the up- and downregulated genes were a fold change of 2.0 or greater and a P value of 0.05 or less. Heatmaps representing the differentially regulated genes were generated using the “pheatmap” package in R.
Functional enrichment analysis
To screen the potential functions of the differentially expressed genes (DEGs), gene ontology (GO) functional enrichment analysis was performed with the Database for Annotation Visualization and Integrated Discovery (DAVID) [26], an online bioinformatics tool, to interpret the gene ontology biological processes (GO_BP) of those DEGs. The R package “ggplot” was used for visualization of the functional clustering of gene sets or gene clusters. A p value < 0.05 and FDR < 0.05 were set as the cutoff criteria.
Clinical samples
Seventy MG patients and fifty healthy controls (HCs) were recruited at the Neurology Department of the Second Affiliated Hospital of Harbin Medical University between September 2021 and February 2023. The first cohort, which included MG patients (n = 8) and age- and sex-matched HCs (n = 8), was used for the microarray analysis. The second cohort, which included MG patients (n = 62) and HCs (n = 42), was used for qRT‒PCR analysis. The P-value for sex and age between the two group showed no statistical significance (p > 0.05) in second cohort. MG was diagnosed based on a combination of fluctuating muscle weakness with a positive neostigmine test, abnormal single-fiber EMG test, or positivity for serum antibodies [27]. The autoantibody results included AChR and MUSK antibodies. All eight MG patients in the first cohort were positive for AChR antibodies. The clinical samples in the second cohort consisted of 59 that were AChR antibody-positive, one that was MuSK antibody-positive, and two seronegative MG patients. 9 MG patients were diagnosed with or were suspected to have thymoma among seventy MG patients. None of the patients had undergone hormone or immunosuppressant treatment during the previous 6 months. Fifty healthy subjects with no autoimmune disease were enrolled as controls. All patients and HCs signed informed consent forms. This study was approved by the Ethics Committee of the Second Affiliated Hospital of Harbin Medical University.
Coexpression analysis
To explore the regulatory mode between lncRNAs and their host mRNAs, we calculated their Pearson correlation coefficients (PCCs) and classified their relationships into three groups: positively correlated (PCC > 0.9 and P value ≤ 0.01), negatively correlated (PCC < − 0.9 and P value ≤ 0.01) and noncorrelated based on the PCC.
Identification of lncRNA-gene and TF-target interactions
First, the interactions of lncRNA-mRNA pairs were acquired from the above coexpression analysis results. The promoter sequences (-2000 bp of the transcription start site) of the DE lncRNAs and coexpressed mRNAs were collected from the UCSC Table Browser [28]. Furthermore, using the JASPAR database tools [29], we computed the transcription factor binding site (TFBS) enrichments in the collected promoter sequences to obtain the target genes of the DE TFs in MG. The significant sites with relative profile scores > 85% within the promoter of genes were retained.
Construction of the TF-lncRNA-gene network
A regulatory network was built by integrating all the pairs obtained from the previous analysis for MG. Network visualization was performed with Cytoscape software version 3.9.1. In the network, nodes denote genes, TFs, and lncRNAs, and edges denote their interactions. After constructing the network, network analysis was performed in Cytoscape.
RNA isolation and qRT‒PCR analysis
A quantitative reverse transcription polymerase chain reaction (qRT‒PCR) was performed to evaluate the validity of the key lncRNAs. Total RNA was extracted using TRIzol reagent (Sigma Life Science, Darmstadt, Germany) according to the manufacturer’s instructions. The RNA purity and concentration of each sample were evaluated and quantified using spectrometry. The purified RNA was reverse-transcribed to generate cDNA using a Transcriptor First Strand cDNA Synthesis Kit (Roche, Basel, Switzerland). qRT‒PCR was performed using FastStar Universal SYBR Green Master Mix (Roche, Basel, Switzerland). The 2−ΔΔCT method was used to determine the relative gene expressions, which were normalized against the level of GAPDH.
Fluorescence in situ hybridization (FISH)
FISH assays were carried out using a Fluorescent in Situ Hybridization Kit (RiboBio Biotechnology, Guangzhou, China). The suspended cells that were fixed on slides were washed with phosphate-buffered saline (PBS) and fixed with 4% formaldehyde at room temperature for 10 min. Then, the cells were permeabilized with PBS containing 0.5% Triton X-100 on ice for 5 min. Hybridization was performed by incubating the cells with a Cy3-labeled probe at 37 ℃ overnight. After the probes were removed, the cells were stained with 4’,6-diamidino-2-phenylindole (DAPI) at room temperature for 5 min. Images were acquired with a confocal microscope (LSM980, Zeiss) to analyze the localization of LINC01566.
Cell culture and transfection
Jurkat cells (a CD4 + T-cell line) and 293T cells were purchased from the BeNa Culture Collection (Henan, China). The Jurkat cells were cultured in RPMI 1640 (Gibco, Grand Island, NY, USA) medium and were supplemented with 10% fetal bovine serum (Excell Bio, Suzhou, China) and 1% penicillin/streptomycin (Beyotime Biotechnology, Nanjing, China). The 293T cells were cultured in DMEM (Gibco, NY, Grand Island, USA) and were supplemented with 10% fetal bovine serum (Excell Bio, Suzhou, China) and 1% penicillin/streptomycin (Beyotime Biotechnology, Nanjing, China). All cell lines were cultured in a 5% CO2 humidified incubator at 37 °C.
PLenti-CMV-LINC00566 was constructed by Gene Pharma (Shanghai, China). LINC00566 antisense oligonucleotide (ASO_LINC01566) was purchased from RiboBio (Guangdong, China). Jurkat cells in the exponential growth phase were centrifuged (1000 r, 5 min), inoculated in a 6-well plate at a density of 1 × 105 cells/well, and then incubated for subsequent transfections. The pLenti-CMV-LINC00566 plasmid, ASO_LINC00566, or corresponding negative controls were used for cell transfection in accordance with the different purposes of the respective experiments. All transfection processes were conducted using LipofectamineTM 3000 transfection reagent (Invitrogen, Carlsbad, CA, USA) according to the relevant protocols. The transfected cells were incubated for 48 h for subsequent analysis. In addition, the transfection efficiency was evaluated by extracting total RNA for qRT‒PCR analysis.
Dual-luciferase reporter assay
FOSL1 binding site prediction to the LINC01566 promoter was performed using the JASPAR database. The 2.0 kb LINC01566 promoter fragment (WT) and mutant (MUT) sections containing binding sites for LINC01566 were amplified, and the resulting products were subsequently cloned and inserted into the corresponding vectors. Functional assays between FOSL1 and the LINC01566 promoter were performed via transient transfections of 293T and Jurkat cells. The transfections included the pGL4.10-LINC01566 WT/vec-FOSL1/pRL-TK group, pGL4.10-LINC01566 WT/vec/pRL-TK group, pGL4.10-LINC01566 MUT/vec-FOSL1/pRL-TK group, pGL4.10-LINC01566 WT/vec/pRL-TK group, pGL4.10/vec-FOSL1/pRL-TK group, and pGL4.10/vec/pRL-TK group. After 48 h of incubation, the luciferase activities of firefly and Renilla were detected in harvested 293T and Jurkat cells using a dual-luciferase reporter assay system (Promega, Madison, WI, USA) according to the manufacturer’s instructions. All experiments were repeated three times independently.
Flow cytometry
Cell apoptosis was determined with an FITC Annexin V Apoptosis Detection Kit I (Becton-Dickinson, FL, NJ, USA) according to the manufacturer’s instructions. The cells were washed twice with cold 1× PBS and resuspended in binding buffer at a concentration of 1 × 106 cells/ml. Five microliters of annexin V-FITC and 5 µL of propidium iodide (PI) were added to a total of 100 µL of cell suspension. The mixtures were incubated for 15 min at RT in the dark and then subjected to flow cytometry analysis (Becton Dickinson FACSCalibur, Franklin, NJ, USA).
Cell proliferation
Cell proliferation was assessed using a Cell Counting Kit-8 (CCK-8) assay (Seven, Beijing, China). After transfection, Jurkat cells at 24 h were seeded into 96-well plates at a density of 3000 cells/well in 100 µl of complete medium and cultured in a humidified atmosphere containing 5% CO2 at 37 ℃. Then, 10 µl of CCK8 reagent was added to each well at 24, 48, 72, and 96 h after transfection, and the cells were further cultured for 2 h at 37 °C. Subsequently, the optical density (OD) was measured using a microplate reader (BioTek, Vermont, New England, USA).
Western blotting analysis
The cells were collected and lysed in radioimmunoprecipitation assay lysis buffer (P0013B, Beyotime) supplemented with 1% protease and phosphatase inhibitors (Roche, Basel, Switzerland). The total protein concentrations were measured using a Bicinchoninic Acid Protein Assay Kit (Beyotime Biotechnology, Nanjing, China). Sample proteins were separated by SDS‒polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes (Roche, Basel, Switzerland). Then, the membranes were blocked in protein-free rapid blocking buffer (PS108, Epizyme Biotech) for 0.5 h at room temperature before being incubated with various primary antibodies overnight at 4 °C. The following primary antibodies were used: rabbit anti-Bcl-2 (A0208, 1:1000, ABclonal); anti-BAX (5032S, 1:1000 CST); anti-DUSP3 (ab125077, 1:100000, Abcam); anti-P2RX7 (ab307718, 1:100000, Abcam); anti-FADD (A5819, 1:1000, ABclonal); anti-LGALS3 (A22442, 1:500, ABclonal); anti-CCR2 (A2385, 1:1000, ABclonal); anti-SIRPB1 (DF4507, 1:800, Affinity); anti-beta-actin (AF7018, 1:10000, Affinity); and anti-beta-tubulin (T0023, 1:20000, Affinity). The membranes were then incubated with the corresponding secondary antibodies (Immunoway, 1:10000, China) for one hour on the following day. After the membranes were washed with TBST, the protein expressions were visualized with enhanced chemiluminescence reagent, and ImageJ software was used for density measurements and quantification.
Statistical analysis
Statistical analysis and plotting of the statistical graphs were performed with GraphPad Prism 9.0 software. For qRT-PCR and western blot results, statistical analysis was performed on the 2−ΔΔCT values and protein studied/β-actin data comparing the negative control group and experimental group. Student’s t test was used for comparisons of the differences between two groups. Correlation analysis was performed using Pearson correlation. All the data are presented as the mean ± standard deviation (SD). When the P values were < 0.05, the differences were considered to be statistically significant.
Results
LncRNA and mRNA expression profiles in MG
To determine the expression patterns of the lncRNAs associated with MG, we performed lncRNA and mRNA microarray analysis on PBMCs that were isolated from MG patients and healthy controls (HCs). First, the samples were separated into two groups through hierarchical clustering based on similar expression patterns, and this difference could distinguish MG patients from healthy individuals (Fig. 2A, B). The results also showed that the expression patterns of the lncRNAs were mostly downregulated in MGs compared to HCs. We observed that the expressions of 83 lncRNAs were significantly upregulated and that the expressions of 444 lncRNAs were downregulated in the MG group compared to the HCs group (Fig. 2C). Moreover, 469 DE mRNAs were identified, of which 189 mRNAs were highly expressed and 280 mRNAs were expressed at low levels (Fig. 2D).
The functional GO term enrichment analysis revealed that the upregulated DEGs were significantly enriched in the regulation of the Fc receptor signaling pathway, T-cell proliferation, positive regulation of T-cell activation and regulation of the extrinsic apoptotic signaling pathway (Fig. 2E). The downregulated DEGs were significantly enriched in the insulin-like growth factor pathway, the L-lysine process to acetyl-CoA, neuronal cell to specific neuron, skeletal system morphogenesis and binding of sperm to the zona pellucida (Fig. 2F). These findings indicate that the upregulated DEGs were enriched in the immune system process, inflammatory response, and innate immune response in MG patients. These findings were consistent with the pathogenesis of MG. Therefore, we used the upregulated genes for follow-up analysis.

Differential expression and functional analysis of lncRNAs and mRNAs in MG microarray data. A, C. Heatmap plot and volcano plot of the differentially expressed lncRNAs between MG patients and controls (fold change > 2, P value < 0.05). B, D. Heatmap plot and volcano plot of the differentially expressed mRNAs between MG patients and controls. E. The top 20 most enriched GO_BP terms for the upregulated mRNAs. F. The top 20 most enriched GO_BP terms for downregulated mRNAs
Construction of the lncRNA-TF-mRNA regulatory network associated with MG and identification of the key lncRNAs
TFs were screened from the JASPAR database, and 19 differentially expressed TFs were identified among the 469 DE mRNAs (Fig. 3A). A heatmap of the expressions of 19 DE TFs, including 19 upregulated TFs and 8 downregulated TFs, is shown in Fig. 3B. The TFs were CDX2, DRGX, E2F8, ESR1, FEZF2, FOSL1, HES5, HOXA9, HOXB8, HOXC8, LEF1, LHX1, NEUROD2, NR1I3, NR3C1, PLAGL2, SOX12, SOX9 and TFEC. First, we obtained lncRNA‒mRNA pairs via coexpression analysis. That the functional analysis of upregulated mRNAs revealed that these genes are significantly enriched in T-cell-related pathways. Therefore, lncRNA‒mRNA coexpression analysis was performed to explore the relationships among DE lncRNAs and upregulated mRNAs in MG. The results revealed 20,616 lncRNA‒mRNA pairs consisting of 388 DE lncRNAs and 164 DE mRNAs. Functional enrichment analysis revealed that the 164 DE mRNAs coexpressed with DE lncRNAs were enriched in T-cell activation, regulation of T-cell activation and T-cell proliferation (Fig. 3C).
Next, by combining the above coexpression analysis results, we predicted the possible targets of 19 DE TFs. We obtained 2942 TF-lncRNA pairs and 2310 TF-mRNA pairs and constructed a lncRNA-TF-mRNA network to illustrate the potential functions of the lncRNAs in MG. This network consisted of 568 nodes and 25,569 edges (Fig. 3D). To characterize the topological features of the network, we calculated the degree of the lncRNA-TF-mRNA network. We primarily analyzed the degree distribution of the lncRNAs and ranked the top 10 lncRNAs according to degree (Fig. 3E). LINC01566 had the highest degree. In terms of LINC01566, the target genes were significantly enriched in several terms that are closely related to MG-related biological processes (Fig. 4C). Furthermore, we found that the expression level of LINC01566 was downregulated in the MG microarray data.

Construction of the lncRNA-TF-mRNA network in MG. (A) Venn diagram showing DE TFs. (B) Heatmap of the expressions of 17 DE TFs. (C) The top 10 most enriched GO terms for the upregulated mRNAs that were coexpressed with DE lncRNAs. (D) The lncRNA-TF-mRNA network. The red ellipses represent TFs, the orange triangles represent mRNAs, and the blue diamond represent lncRNAs. Lines represent their regulatory interactions. (E) The top 10 lncRNAs according to the degree ranking in the network
LINC01566 was expressed at low levels in MG patients and was associated with clinical indices in MG patients
In this study, we focused on the lncRNA LINC01566, which is encoded by a gene at chromosome position 16p11.1 in the genome. qRT‒PCR confirmed the significant decrease in LINC01566 expressions in the PBMCs of MG patients compared with those in the PBMCs of HCs (p < 0.0001, Fig. 4A). The decreased expression of LINC01566 in PBMCs was found in these subgroups including AChR Ab + MG patients (85.8%)、seronegative MG patients (100%) and thymoma-associated MG patients (89.8%). However, the expression level of LINC01566 was higher in 100% (1/1) of the MuSK Ab + MG patient than in normal healthy controls. We performed receiver operating characteristic (ROC) curve analysis to evaluate the diagnostic performance of the LINC01566 signature in the validation set, the results demonstrated that the lncRNA precisely distinguished patients with MG from healthy people (AUC = 0.873, Fig. 4B). To further explore the biological function of LINC01566, we obtained lncRNA coexpressed genes from the previous lncRNA-TF-gene network to perform GO functional analysis, and the results revealed significant enrichment of genes related to neutrophil activation involved in the immune response, leukocyte cell‒cell adhesion, T-cell activation, monocyte chemotaxis and regulation of the leukocyte apoptotic process (Fig. 4C).
In addition, we also investigated the association between the LINC01566 expressions and routine clinical blood data from patients. There was a correlation between higher expression levels of LINC01566 and higher NLR (R = 0.427, p < 0.01; Fig. 4E), and the neutrophil counts were also positively correlated with the expression levels of LINC01566 (R = 0.472, p < 0.01; Fig. 4F). Moreover, the LINC01566 expressions were negatively correlated with the lymphocyte and monocyte counts (R = -0.415, p < 0.01; R = -0.511, p < 0.001; Fig. 4G, H). These data demonstrate that LINC01566 may be an important biological signature in MG.

Validation experiment and analysis of LINC01566 in clinical samples (A) The expressions of LINC01566 were detected in MG patients and control subjects by qRT‒PCR. (B) Receiver operating characteristic (ROC) curve analysis showed that the LINC01566 expression levels have diagnostic value in MG patients. (C) The top 20 most enriched GO_BP terms for coexpressed mRNAs of LINC01566. (D) Molecules related to T-cell proliferation and activation terms among the coexpressed genes of LINC01566. E, F. Relationships between the expression levels of LINC01566 and the neutrophil-to-lymphocyte ratios and neutrophil counts in MG patients. G, H. Relationships between the expression levels of LINC01566 and the lymphocyte and monocyte counts in MG patients
Localization of LINC01566 in CD4 + T cells and LINC01566 inhibition of CD4 + T-cell proliferation
Our results showed that LINC01566 was down-regulated in MG. However, the detailed molecular mechanism that contributes to these roles has not yet been investigated. That functional analysis of LINC01566 coexpressed genes revealed that LINC01566 was significantly enriched in T-cell-related terms. To clarify the molecular mechanism of LINC01566 in T lymphocytes, we first evaluated the subcellular localization of LINC01566 in Jurkat cells, which are a common CD4 + T-cell line. FISH was performed to determine the localization of LINC01566 in Jurkat cells, and the results indicated that LINC01566 predominantly localized to the nucleus (Fig. 5A).
To further confirm the functional connection between LINC01566 and T-cell activation in MG, we next analyzed the effect of LINC01566 knockdown (KD) or overexpression (OE) on CD4 + T-cell function using flow cytometry and western blotting analysis. Jurkat cells were transfected with ASO negative control (ASO_NC) or ASO_LINC01566, and the qRT‒PCR results revealed that LINC01566 KD decreased LINC01566 expressions. In addition, Jurkat cells were transfected with pLenti-CMV-control or pLenti-CMV-LINC01566. The qRT‒PCR results indicated that LINC01566 OE significantly increased LINC01566 expressions (Fig. 5B). We found that the apoptosis rate of the LINC01566 OE group increased, while that of the LINC01566 KD group decreased compared to that of the negative control group (Fig. 5C、D). Similarly, compared to those in the negative control group, the levels of the proapoptotic molecules BAX in the LINC01566 OE group were significantly increased, and the level of the antiapoptotic protein Bcl-2 was significantly decreased. The expressions of apoptotic proteins in the LINC01566 KD group showed the opposite trend, which further confirmed the above results (Fig. 5E、F). Moreover, the CCK-8 assay showed that the proliferation of Jurkat cells was notably repressed in the LINC01566 OE group compared with that in the negative control group; in contrast, cell proliferation was promoted in the LINC01566 KD group (Fig. 5G). The results showed that LINC01566 could inhibit CD4 + T-cell proliferation and promote apoptosis.

LINC01566 inhibited CD4 + T-cell proliferation and promoted CD4 + T-cell apoptosis (A) Subcellular localization of LINC01566 in Jurkat cells. (B) The efficiency of LINC01566 knockdown and overexpression was measured by qRT‒PCR. C, D. Flow cytometric analysis was performed using cells stained with Annexin-V-FITC/PI after transfecting negative controls, pLenti-CMV-LINC01566, or ASO_LINC01566 into Jurkat cells. E, F. Western blot analysis and relative protein levels of Blc-2 and Bax in the pLenti-CMV-LINC01566 group, pLenti-CMV-vector group, ASO_LINC01566 group and ASO_NC group. The data are presented as the mean ± SD (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). G. The proliferation of Jurkat cells was analyzed using CCK-8 assays by transfecting negative controls, pLenti-CMV-LINC01566, or ASO_LINC01566 into Jurkat cells
LINC00566 influences CD4+ T-cell activation and proliferation by regulating six molecules
Next, we investigated the molecular mechanism by which LINC01566 regulates CD4 + T-cell function. According to previous functional analysis results of the coexpressed LINC01566 genes, we identified T-cell activation-related molecules, such as DUSP3, FADD, CCR2, LGALS3, P2RX7, and SIRPB1 (Fig. 4D). The microarray analysis results showed that all six molecules were upregulated. Given that LINC01566 is located in the nucleus, we speculated that LINC01566 may regulate T-cell activation-related proteins through the nuclear pathway. To verify whether LINC01566 regulates these six molecules, Jurkat cells were transfected with pLenti-CMV-LINC01566, pLenti-CMV-vector, ASO_LINC01566 or ASO_NC. The results showed that the mRNA levels of DUSP3, CCR2, FADD, LGALS3, P2RX7 and SIRPB1 in the pLenti-CMV-LINC01566 group were lower than those in the pLenti-CMV-vector control group, while the mRNA levels of DUSP3, CCR2, FADD, LGALS3, P2RX7 and SIRPB1 in the ASO_LINC01566 group were greater than those in the ASO_NC group (Fig. 6A). Western blot analysis further confirmed that LINC01566 downregulated the protein levels of DUSP3, CCR2, FADD, LGALS3, P2RX7 and SIRPB1 (Fig. 5B, C). Our data showed that LINC01566 regulated the expressions of six key molecules related to T-cell activation and proliferation, which further explored the molecular mechanism of LINC01566 in MG.

LINC01566 regulated six key molecules related to T-cell activation. A. The mRNA expression levels of CCR2, FADD, DUSP3, P2RX7, LGALS3 and SIRPB1 were measured by qRT‒PCR after transfecting negative controls, pLenti-CMV-LINC01566, or ASO_LINC01566, into Jurkat cells. The data are presented as the mean ± SD (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). B, C. Western blot analysis and relative protein levels of CCR2, FADD, DUSP3, P2RX7, LGALS3 and SIRPB1 in the pLenti-CMV-LINC01566, pLenti-CMV-vector, ASO_LINC01566 and ASO_NC groups. The data are presented as the mean ± SD (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001)
The transcription factor FOSL1 regulates LINC01566 expression
Our results showed that LINC01566 plays a crucial role in MG. Consequently, we aimed to identify the regulators of LINC01566. TFs are fundamental players in regulating gene expressions. To further determine whether the TF regulates LINC01566 expressions, we extracted potential TFs that might interact with LINC01566 promoters from the previous lncRNA-TF-mRNA network and identified 16 candidate TFs (Fig. 7A). Among those TFs, FOSL1 was significantly downregulated in MG (Fig. 7B). The qRT‒PCR results also demonstrated that FOSL1 overexpression significantly contributed to LINC01566 upregulation (Fig. 7C). Localization analysis of FOSL1 was performed in CD4 + T-cell lines, and FOSL1 was found to be located in the nucleus, consistent with its regulatory role as a transcription factor in MG (Fig. 7D).
To confirm that LINC01566 is a transcriptional target of FOSL1, we cloned a 2.0-kb region of the LINC01566 promoter into the pGL4.10 vector and measured the luciferase activity after transfection into 293T cells and Jurkat cells. A significantly increased luciferase activity was obtained using a plasmid containing a 2.0-kb section of the LINC01566 promoter in 293T cells that were cotransfected with FOSL1 overexpression plasmids (Fig. 7E). Furthermore, to identify specific binding sites of FOSL1 in the promoter of LINC01566, we predicted potential binding sites using the JASPAR database tools and selected the sites with the highest scores for subsequent experimental verification. Through dual-luciferase reporter assays using mutated LINC01566 reporter constructs, we identified the promoter region at -300~-288 nt as the likely specific binding site of FOSL1 (Fig. 7E, G). Similarly, we demonstrated these results in Jurkat cells (Fig. 7F). Our results revealed that LINC01566 downregulation is indeed mediated by FOSL1 in MG.

The transcription factor FOSL1 regulates LINC01566 expression. (A) The potential TFs targeting LINC01566. (B) The expression level of FOSL1 in MG clinical samples was measured by qRT‒PCR. (C) The expression levels of LINC01566 and FOSL1 were detected after transfection with pLenti-CMV-FOSL1 or pLenti-CMV-vector. The data are presented as the mean ± SD (*p < 0.05). (D) FISH of LINC01566 (red stain) and IF of FOSL1 (green stain) in Jurkat cells. F. HEK-293T cells and Jurkat cells were cotransfected with luciferase reporters containing sections containing the WT or MUT LINC01566 promoter and vec-FOSL1 to test the effect of FOSL1 overexpression on LINC01566 promoter activity. G. Target binding sequences of FOSL1 in the LINC01566 promoter region
Discussion
MG is a T-cell-dependent antibody-mediated disease. Abnormal lncRNA expression can influence the emergence, development, and prognosis of various immune diseases by controlling CD4 + T-cell differentiation [30]. In this study, we aimed to understand lncRNA transcriptional changes and elucidate the underlying mechanisms of the effects of key lncRNAs on CD4 + T-cell activation in MG. We revealed that a subset of aberrant lncRNAs can be used to distinguish MG patients from healthy individuals. Subsequently, we constructed lncRNA–TF–gene networks and determined that LINC01566 might play a critical role in MG. The potential regulatory mechanism of LINC01566 in MG was also further investigated.
Jin et al. reported that LINC01566 may be involved in the pathogenesis of periodontitis [31]. To date, the role and mechanism of LINC01566 in MG have not been reported. We discovered that LINC01566 levels are considerably lower in the peripheral blood of MG patients than in that of healthy controls, which suggested that LINC01566 might play an important protective role in MG. Precisely annotating the functions of lncRNAs remains complex. Here, the biological function of LINC01566 was significantly enriched in neutrophil activation involved in the immune response, leukocyte cell‒cell adhesion, T-cell activation, monocyte chemotaxis and regulation of the leukocyte apoptotic process. In our study, we found that LINC01566 was positively correlated with neutrophil counts and the NLR. The NLR is a reliable indicator of systemic inflammatory status and disease activity [32]. Previous research has shown that the NLR is significantly increased in patients with severe MG [32, 33]. Accumulating evidence has indicated that inflammation could be a key factor in MG [34]. There is genetic evidence from genome-wide association studies that the neutrophil respiratory burst and ROS production are important risk factors for autoimmune disease [35]. Studies have also shown that lncRNAs can promote neutrophil inflammation or neutrophil infiltration [36, 37]. Therefore, we suspected that LINC01566 may influence the number or function of neutrophils to mediate the inflammatory effects of MG. In addition, LINC01566 expression was correlated with lymphocyte and monocyte counts, which illustrated that LINC01566 may be related to the presence and activity of MG.
MG is considered a largely T-cell-dependent antibody-mediated disease, and the activation and proliferation of T cells play significant roles in the pathogenesis of MG. Functional analysis revealed that there was enrichment in the genes coexpressed with LINC01566 in T-cell-related pathways. Hence, an experiment was designed to validate whether LINC01566 affects the apoptosis and proliferation of CD4 + T cells. Jurkat cells were used for functional verification. Functionally, we discovered that LINC01566 facilitates T-cell proliferation and represses apoptosis. The results showed that LINC01566 is involved in MG pathogenesis by inhibiting CD4 + T-cell proliferation and promoting apoptosis. LncRNAs are themselves functional units, making their subcellular localization critical to their function. Different subcellular localizations may lead to lncRNAs exerting their functions via various mechanisms. FISH experiments showed that LINC01566 was predominantly localized to the nuclei of Jurkat cells. This finding suggested that LINC01566 could be an essential regulator in the nuclei of CD4 + T cells.
However, the underlying molecular mechanism by which LINC01566 regulates T-cell function in MG remains unclear. Furthermore, we hypothesized that LINC01566 might regulate T-cell activation-related proteins to affect T-cell function. We identified T-cell activation-related molecules, namely, DUSP3, FADD, CCR2, LGALS3, P2RX7, and SIRPB1, from LINC01566 coexpression analysis. Kim et al. reported increased anti-AChR antibody titres in patients with the CCR2 G/G genotype, which suggests that the CCR2 gene plays a role in the pathophysiology of MG [38]. FADD constitutes a mechanism to maintain TCR-induced programmed necrotic signaling during the early phases of T-cell clonal expansion [39]. Research by Aruna et al. strongly indicated that the upregulation of apoptosis via the Fas-FasL pathway is one of the mechanisms by which dual APL reverses EAMG manifestations in C57BL/6 mice [40], while DUSP3, P2RX7, LGALS3 and SIRPB1 have not been reported to be involved in MG pathogenesis. However, the specific correlations between LINC01566 and these T-cell activation-related molecules have yet to be identified. In our present study, qRT‒PCR and western blot analysis demonstrated that LINC01566 regulates six T-cell activation-related proteins. DUSP3 is known to negatively regulate the ERK and JNK pathways [41, 42], and previous studies have confirmed that the proteins p38 and ERK1/2 are overexpressed in MG thymic epithelial cells (TECs) [43]. Therefore, it is reasonable to suspect that altering the expression of DUSP3 may regulate CD4 + T-cell activation via the p38/ERK pathway in MG. In addition, we hypothesized that LINC01566 influences CD4 + T-cell apoptosis by regulating the expression of FADD, which is a crucial molecule in the Fas-FasL pathway [44]. These findings indicated that low LINC01566 expression is key in the pathogenesis of MG. In addition, we investigated the mechanism underlying the low expression of LINC01566 and discovered that FOSL1 regulates LINC01566 expression in MG. Our results provide a new understanding of the role of LINC01566 in CD4 + T-cell activation and a new direction for future research.
Our study has several limitations. Although we found that the expression level of LINC01566 decreased in the PBMCs of MG patients, the expression of LINC01566 in a specific type of immune cell in MG patients was not further explored. In addition, we studied the ability of LINC01566 to inhibit CD4 + T-cell proliferation, but we did not explore the role of LINC01566-mediated inflammation in MG. Further research will be needed to explore the specific roles of LINC01566 in immune cells and inflammation and its potential mechanisms in MG.
Conclusion
In summary, we showed that nuclear LINC01566 is downregulated in MG and is correlated with neutrophil counts, lymphocyte counts, monocyte counts and NLR in MG patients. We described its regulatory function in the proliferation and apoptosis of CD4 + T cells through the regulation of six T-cell activation-related molecules. Our study suggested that low expressions of LINC01566 in MG might contribute to the T lymphocyte proliferation and activation that characterize the immune abnormalities in MG. Furthermore, we found that the transcription factor FOSL1 regulates LINC01566 expression in MG. Our findings describe a potential mechanism underlying the protective effect of LINC01566 on MG and reveal that LINC01566 may be a valuable biomarker and therapeutic target in MG.
Data availability
Datasets generated using microarray analysis are available at NCBI Gene Expression Omnibus Accession GSE263220.
Abbreviations
- MG:
-
Myasthenia gravis
- lncRNAs:
-
Long noncoding RNAs
- TFs:
-
Transcription factors
- qRT-PCR:
-
Quantitative reverse transcription polymerase chain reaction
- FISH:
-
Fluorescence in situ hybridization
- DEGs:
-
Differentially expressed genes
- NLR:
-
Neutrophil-to-lymphocyte ratio
- GO_ BP:
-
Gene ontology biological process
- AChR:
-
Acetylcholine receptor
- MuSK:
-
Muscle-specific kinase
- LRP4:
-
Lipoprotein-related protein 4
- PBMCs:
-
Peripheral blood mononuclear cells
- HCs:
-
Healthy controls
- ASO:
-
Antisense oligonucleotide
- CCK-8:
-
Cell Counting Kit-8
References
Gilhus NE, Longo DL. Myasthenia Gravis. N Engl J Med. 2016;375:2570–81.
Gilhus NE, Verschuuren JJ. Myasthenia gravis: subgroup classification and therapeutic strategies. Lancet Neurol. 2015;14:1023–36.
Conti-Fine BM, Milani M, Kaminski HJ. Myasthenia gravis: past, present, and future. J Clin Invest. 2006;116:2843–54.
Gilhus NE, Tzartos S, Evoli A, Palace J, Burns TM, Verschuuren JJGM. Myasthenia gravis. Nat Reviews Disease Primers 2019, 5.
Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood. 2008;112:1557–69.
Marx A, Pfister F, Schalke B, Saruhan-Direskeneli G, Melms A, Ströbel P. The different roles of the thymus in the pathogenesis of the various myasthenia gravis subtypes. Autoimmun rev. 2013;12:875–84.
Marx A, Porubsky S, Belharazem D, Saruhan-Direskeneli G, Schalke B, Ströbel P, Weis C-A. Thymoma related myasthenia gravis in humans and potential animal models. Exp Neurol. 2015;270:55–65.
Camilla Buckley M, Daniel Douek J, Newsom-Davis FRS, Angela Vincent. FRCPath, and Nicholas Willcox: mature, long-lived CD41 and CD81 T cells are generated by the Thymoma in Myasthenia Gravis. Ann Neurol. 2001;50:64–72.
Viola Hoffacker AS, James J, Tiesinga R, Gold B, Schalke W, Nix R, Kiefer. Hans Konrad Müller-Hermelink and Alexander Marx: Thymomas alter the T-cell subset composition in the blood: a potential mechanism for thymoma-associated autoimmune disease. blood 2000, 96:3872–3879.
Berrih-Aknin S. Myasthenia gravis: Paradox versus paradigm in autoimmunity. J Autoimmun. 2014;52:1–28.
Raphael I, Nalawade S, Eagar TN, Forsthuber TG. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine. 2015;74:5–17.
Yi JS, Guidon A, Sparks S, Osborne R, Juel VC, Massey JM, Sanders DB, Weinhold KJ, Guptill JT. Characterization of CD4 and CD8 T cell responses in MuSK myasthenia gravis. J Autoimmun. 2014;52:130–8.
Balandina A, Lécart S, Dartevelle P, Saoudi A, Berrih-Aknin S. Functional defect of regulatory CD4 + CD25 + T cells in the thymus of patients with autoimmune myasthenia gravis. Blood. 2005;105:735–41.
Thiruppathi M, Rowin J, Ganesh B, Sheng JR, Prabhakar BS, Meriggioli MN. Impaired regulatory function in circulating CD4 + CD25highCD127low/– T cells in patients with myasthenia gravis. Clin Immunol. 2012;145:209–23.
Elhai M, Micheroli R, Houtman M, Mirrahimi M, Moser L, Pauli C, Bürki K, Laimbacher A, Kania G, Klein K et al. The long non-coding RNA HOTAIR contributes to joint-specific gene expression in rheumatoid arthritis. Nat Commun 2023, 14.
Hao H, Nakayamada S, Ohkubo N, Yamagata K, Zhang M, Shan Y, Iwata S, Zhang T, Tanaka Y. Involvement of lncRNA IL21-AS1 in interleukin-2 and T follicular regulatory cell activation in systemic lupus erythematosus. Arthritis Res Therapy 2021, 23.
Haridy SFA, Shahin NN, Shabayek MI, Selim MM, Abdelhafez MA, Motawi TK. Diagnostic and prognostic value of the RUNXOR/RUNX1 axis in multiple sclerosis. Neurobiol Dis 2023, 178.
Fernandes J, Acuña S, Aoki J, Floeter-Winter L, Muxel S. Long non-coding RNAs in the regulation of Gene expression: physiology and disease. Non-Coding RNA 2019, 5.
Chen YG, Satpathy AT, Chang HY. Gene regulation in the immune system by long noncoding RNAs. Nat Immunol. 2017;18:962–72.
Kopp F, Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. Cell. 2018;172:393–407.
Huang D, Chen J, Yang L, Ouyang Q, Li J, Lao L, Zhao J, Liu J, Lu Y, Xing Y, et al. NKILA lncRNA promotes tumor immune evasion by sensitizing T cells to activation-induced cell death. Nat Immunol. 2018;19:1112–25.
Wheeler BD, Gagnon JD, Zhu WS, Muñoz-Sandoval P, Wong SK, Simeonov DS, Li Z, DeBarge R, Spitzer MH, Marson A, Ansel KM. The lncRNA Malat1 inhibits miR-15/16 to enhance cytotoxic T cell activation and memory cell formation. eLife 2023, 12.
Wu M, Sun J, Wang L, Wang P, Xiao T, Wang S, Liu Q. The lncRNA HOTAIR via Mir-17-5p is involved in arsenite-induced hepatic fibrosis through regulation of Th17 cell differentiation. J Hazard Mater 2023, 443.
Xu Y, Ouyang Y. Long non-coding RNA growth arrest specific 5 regulates the T helper 17/regulatory T balance by targeting miR-23a in myasthenia gravis. J Int Med Res 2022, 50.
Luo M, Liu X, Meng H, Xu L, Li Y, Li Z, Liu C, Luo Y-B, Hu B, Xue Y, et al. IFNA-AS1 regulates CD4 + T cell activation in myasthenia gravis though HLA-DRB1. Clin Immunol. 2017;183:121–31.
Sherman BT, Hao M, Qiu J, Jiao X, Baseler MW, Lane HC, Imamichi T, Chang W. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022;50:W216–21.
Mantegazza R, Cavalcante P. Diagnosis and treatment of myasthenia gravis. Curr Opin Rheumatol. 2019;31:623–33.
Nassar LR, Barber GP, Benet-Pagès A, Casper J, Clawson H, Diekhans M, Fischer C, Gonzalez JN, Hinrichs Angie S, Lee Brian T, et al. The UCSC Genome Browser database: 2023 update. Nucleic Acids Res. 2023;51:D1188–95.
Castro-Mondragon JA, Riudavets-Puig R, Rauluseviciute I, Berhanu Lemma R, Turchi L, Blanc-Mathieu R, Lucas J, Boddie P, Khan A, Manosalva Pérez N, et al. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2022;50:D165–73.
Liu C, Zhang Y, Ma Z, Yi H. Long noncoding RNAs as orchestrators of CD4 + T-Cell fate. Front Cell Dev Biology 2022, 10.
Jin SH, Zhou RH, Guan XY, Zhou JG, Liu JG. Identification of novel key lncRNAs involved in periodontitis by weighted gene co-expression network analysis. J Periodontal Res. 2019;55:96–106.
Duan Z, Jia A, Cui W, Feng J. Correlation between neutrophil-to-lymphocyte ratio and severity of myasthenia gravis in adults: a retrospective study. J Clin Neurosci. 2022;106:117–21.
De-Hao Yang M-ZQ, Wei M-M, Li J4, Yu M-M, Lu X-M, Yang H, Lin H, Li X. Jun-Yan Zhu and Xu Zhang: The correlation of neutrophil-to-lymphocyte ratio with the presence and activity of myasthenia gravis. Oncotarget 2017, 8.
Huda R. Inflammation and autoimmune myasthenia gravis. Front Immunol 2023, 14.
Wigerblad G, Kaplan MJ. Neutrophil extracellular traps in systemic autoimmune and autoinflammatory diseases. Nat Rev Immunol. 2022;23:274–88.
Liu D, Sun W, Zhang D, Yu Z, Qin W, Liu Y, Zhang K, Yin J. Long noncoding RNA GSEC promotes neutrophil inflammatory activation by supporting PFKFB3-involved glycolytic metabolism in sepsis. Cell Death Dis 2021, 12.
Premadasa LS, Lee E, McDew-White M, Alvarez X, Jayakumar S, Ling B, Okeoma CM, Byrareddy SN, Kulkarni S, Mohan M. Cannabinoid enhancement of lncRNA MMP25-AS1/MMP25 interaction reduces neutrophil infiltration and intestinal epithelial injury in HIV/SIV infection. JCI Insight 2023, 8.
Hyun Sook Kim D-SK, Lee EY, Sunwoo I-N, Choi Y-C. CCR2-64I and CCR5∆32 polymorphisms in Korean patients with Myasthenia Gravis. J Clin Neurol. 2007;3:133–8.
Osborn SL, Diehl G, Han S-J, Xue L, Kurd N, Hsieh K, Cado D, Robey EA, Winoto A. Fas-associated death domain (FADD) is a negative regulator of T-cell receptor–mediated necroptosis. Proceedings of the National Academy of Sciences 2010, 107:13034–13039.
Aruna BV, Ben-David H, Sela M, Mozes E. A dual altered peptide ligand down‐regulates myasthenogenic T cell responses and reverses experimental autoimmune myasthenia gravis via up‐regulation of Fas–FasL‐mediated apoptosis. Immunology. 2006;118:413–24.
Qiu W, Guo X, Li B, Wang J, Qi Y, Chen Z, Zhao R, Deng L, Qian M, Wang S, et al. Exosomal miR-1246 from glioma patient body fluids drives the differentiation and activation of myeloid-derived suppressor cells. Mol Ther. 2021;29:3449–64.
Tambe MB, Narvi E, Kallio M. Reduced levels of Dusp3/Vhr phosphatase impair normal spindle bipolarity in an Erk1/2 activity-dependent manner. FEBS Lett. 2016;590:2757–67.
Michaela Colombara VA, Riviera AP, Mainiero F, Strippoli R, Merola M, Fracasso G, Poffe O, Brutti N, Tridente G. Marco Colombatti, and Dunia Ramarli3: constitutive activation of p38 and ERK1/2 MAPKs in epithelial cells of myasthenic Thymus leads to IL-6 and RANTES overexpression: effects on Survival and Migration of Peripheral T and B cells. J Immunol. 2005;175:7021–8.
Sun H, Yang Y, Gu M, Li Y, Jiao Z, Lu C, Li B, Jiang Y, Jiang L, Chu F, et al. The role of Fas-FasL-FADD signaling pathway in arsenic-mediated neuronal apoptosis in vivo and in vitro. Toxicol Lett. 2022;356:143–50.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China [grant numbers 82071407, 82171396]; Heilongjiang Provincial Natural Science Foundation [grant number YQ2021H012].
Author information
Authors and Affiliations
Contributions
JW and LW conceived and supervised the project. LL and DL performed the bioinformatics analysis. XW, NH, LT and BC collected the clinical samples. LL, JJ, FX and YR conducted the experiments. LL, ZC, LZ, XL and LQ performed the statistical analyses. LL wrote the paper. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The studies involving human participants were approved by the Ethics Committee of the Second Affiliated Hospital of Harbin Medical University (Ky2020-104) and performed in accordance with the latest version of the Helsinki Declaration. The patients/participants provided their written informed consent to participate in this study.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, L., Li, D., Jin, J. et al. FOSL1-mediated LINC01566 negatively regulates CD4+ T-cell activation in myasthenia gravis. J Neuroinflammation 21, 197 (2024). https://doi.org/10.1186/s12974-024-03194-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-024-03194-5