
Abstract
A key pathological factor of Alzheimer’s disease (AD), the most prevalent form of age-related dementia in the world, is excessive β-amyloid protein (Aβ) in extracellular aggregation in the brain. And in the peripheral blood, a large amount of Aβ is derived from platelets. So far, the causality between the levels of peripheral blood Aβ and its aggregation in the brain, particularly the role of the peripheral blood Aβ in the pathology of AD, is still unclear. And the relation between the peripheral blood Aβ and tau tangles of brain, another crucial pathologic factor contributing to the pathogenesis of AD, is also ambiguous. More recently, the anti-Aβ monoclonal antibodies are approved for treatment of AD patients through declining the peripheral blood Aβ mechanism of action to enhance plasma and central nervous system (CNS) Aβ clearance, leading to a decrease Aβ burden in brain and improving cognitive function, which clearly indicates that the levels of the peripheral blood Aβ impacted on the Aβ burden in brain and involved in the pathogenesis of AD. In addition, the role of peripheral innate immune cells in AD remains mostly unknown and the results obtained were controversial. In the present review, we summarize recent studies on the roles of peripheral blood Aβ and the peripheral innate immune cells in the pathogenesis of AD. Finally, based on the published data and our own work, we believe that peripheral blood Aβ plays an important role in the development and progression of AD by impacting on the peripheral innate immune cells.
Similar content being viewed by others

Introduction
Alzheimer’s disease (AD) is the most common age-associated dementia with progressive loss of memory and cognitive functions [1]. It is predicted that by 2050, approximately 115 million people will be affected by AD [2]. Without question, AD has become a major public health dilemma and the largest health and social crisis in the world [3]. The neuropathological features of AD include amyloid-β (Aβ) plaques and neurofibrillary tangles formed by intracellular accumulation of hyperphosphorylated tau protein (p-Tau), neuroinflammation, as well as neuron and synapse loss, etc. [4, 5]. Aβ42 peptide is the main component of senile plaques in the brain of AD patients. All long major pathogenic hypotheses of AD focus on the Aβ cascade and p-Tau accumulation. For a long time in the past, all efforts in the treatment of AD targeting the pathogenic Aβ or tau have failed to demonstrate clinical efficiency unfortunately [6, 7], suggesting that the pathogenesis of AD should be quite complex and multifactorial [8].
Although the hypotheses of Aβ cascade and p-Tau have been challenged, there is growing evidence that the hypotheses of Aβ cascade play a key role in the pathogenesis of AD [9]. Neuroimaging studies showed that Aβ plaques begin to deposit in brain ten years or more before the onset of cognitive decline [10], which is consistent with the fact that AD pathologic change is a chronic long-term neurodegenerative process. Based on the Aβ cascade hypothesis, the clearance of brain Aβ plaques through different pathway should be able to treat AD and cease its progression, which promotes the development of innovative anti-Aβ drugs in order to lower Aβ production and prevent Aβ aggregation in brain.
Excitingly, in July 2023, the U.S. Food and Drug Administration (FDA) granted traditional approval to lecanemab, an anti-Aβ monoclonal antibody (mAb), according to a confirmatory clinical trial demonstrating clinical benefits for AD patients. In general, the anti-Aβ monoclonal antibodies (mAbs), such as lecanemab can enhance plasma and central nervous system (CNS) Aβ clearance, leading to a decline Aβ burden through the peripheral sink mechanism of action [11, 12], by which mAbs binding to Aβ in peripheral blood may alter the balance between circulation and brain Aβ levels, thereby enhancing outflow of soluble Aβ from the brain via low density lipoprotein receptor related protein 1 (LRP1) expressed on the blood brain barrier (BBB) [13, 14]. At the same time, binding of mabs to Aβ in peripheral blood also might reduce Aβ levels in the brain by lowering Aβ inflow into the brain through binding to the receptor for advanced glycation end products [14,15,16]. Lecanemab can selectively bind to large, soluble Aβ protofibrils that are the most neurotoxic and contribute to the pathogenesis of AD, and reduces Aβ plaques formed in brain, and slows down the rate of cognitive decline in early AD patients by 27% [17,18,19]. However, the mechanisms and pathways that the peripheral blood Aβ affects the progression of AD are not fully understood, which is an increasingly interesting topic that deserves in-depth research.
AD has been considered as a systemic disease that involves the peripheral and central immune responses. Dysregulation of immune response is also one of the pathological features of AD [20, 21]. In the past decade, emerging evidence has shown that the peripheral innate immunity has pathological effects on AD via the alterations of the function and quantity of peripheral immune cells in the blood and promotion of these cells infiltrating into the brain, exacerbating neuropathy in AD [21,22,23]. Notwithstanding, the precise effects of the peripheral immune system on AD are still unclear and controversial, and the studies to explore the role of peripheral immune cells in AD brain are still at a relatively nascent stage. So far there is lack of sufficient study on the correlations between peripheral immunity and AD, especially, how the peripheral blood Aβ impacts on brain of AD.
In the present article, we primarily review the role of the peripheral blood Aβ in AD patients and its animal models, and the role of the peripheral innate immune cells in AD pathogenesis. Finally, we focus on exploring the mechanism behind whether the peripheral blood Aβ contributes to AD through impacting on the peripheral innate immune cell functions.
Peripheral blood Aβ source
In human peripheral blood, platelets are the primary source of Aβ peptides and a large amount (more than 90%) of Aβ is derived from circulating platelets. Thus, platelets are direct contributors to the amount of peripheral blood Aβ. Among them Aβ40 peptide is considered predominant form in some reported studies [24,25,26,27]. The level of platelet APP isoforms is comparable to it in the brain [28]. It has been revealed that Aβ metabolism is associated with platelet [29]. More than 90% of blood Aβ40 and over 97% of blood Aβ42 are related to plasma lipoproteins in human [30]. In mice, the amount of Aβ in platelets is increased with age [31].
In addition, the liver is another origin of brain Aβ deposits and is involved in peripheral clearance of circulating Aβ in the blood [32], therefore, the liver can affect indirectly the peripheral blood Aβ levels. Recent study reported that the liver was the earliest affected organ in AβPP/PS1 mice, the animal model of AD. During Aβ pathology progression [33], the liver was the major organ responsible for plasma clearance of Aβ40/42 [34, 35]. In our recent study, we found that the liver weights of APP/PS-1 transgenic (Tg) mice, the animal model for AD, decreased significantly, and liver cell necrosis as well as lymphocyte infiltration increased obviously compared to wild type mice [36], which indicates that the liver plays important roles in the pathogenesis of AD through affecting on Aβ metabolism [32, 37, 38]. Other peripheral sources of Aβ are endothelial vascular cells [39] and skeletal muscle [40, 41], but they are not the main source of the peripheral blood Aβ.
Aβ deposition in the brain may potentially correlate to the level in the peripheral circulatory system [42]. The discrepancy of Aβ isoforms levels in blood may mirror alterations in brain efflux of Aβ from cerebrospinal fluid (CSF) to blood [43]. However, some studies indicated that blood Aβ mainly presents changes in peripheral synthesis, secretion and metabolism of Aβ as an apoprotein of lipoproteins [44,45,46]. Deane et al. demonstrated that Aβ peptides are able to proactively transit across the BBB [47, 48]. When the BBB was damaged, the exchange of Aβ peptides between brain and peripheral blood/tissues was increased clearly. Reduction of the peripheral blood Aβ is enough to decline Aβ levels in brain, and reconfirming peripheral blood Aβ does not come from the brain [49].
Role of peripheral Aβ in the pathogenesis of AD
Circulating activated platelets can produce Aβ and proinflammatory mediators. Both of them amplify peripheral inflammation and endothelial senescence, leading to change the permeability of the BBB and contributing to Aβ across BBB into brain, accelerating Aβ deposition in the brain and enhancing Aβ level in peripheral blood [50, 51], which leads to a decline in learning and memory. All human platelets can produce a large amount of Aβ in the peripheral blood, but not everyone will develop AD, which should involve other factors and is worth further exploring.
A retrospective cohort study reported that aspirin, a traditional antipyretic analgesic, applied to prevent and treat ischemic heart disease and cerebral thrombosis, can inhibit platelet aggregation in vitro and activation in vivo. Thus aspirin may lower the risk of AD [52, 53], since inhibition of platelet aggregation can decrease Aβ release [54]. The underlying mechanisms may be aspirin inhibiting cyclooxygenase (COX) activity, improving synaptic dysfunction by impacting COX-dependent action [55, 56]. Human platelets can regulate Aβ release by the cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA) pathway, by which enhanced cAMP can block the processing and secretion of Aβ [57].
Additionally, the mice received the platelets from aged APP/PS1 mice (AD mouse model) through the tail vein injection caused significantly learning and memory deficits and raised Aβ deposition when compared with the mice injected with plasma [31], suggesting that in peripheral blood, Aβ derived from platelets is able to enter into the brain through the BBB and deposits in the hippocampus, playing an crucial role in the pathogenesis of AD [58]. Unfortunately, so far the effect of aspirin on AD has been controversial and no clinical evidence was confirmed that aspirin was efficacious in reducing cognitive decline in patients [59]. However, aspirin can indeed diminish the levels of Aβ40, Aβ42 and tau in both platelets and plasma of mice, and lower Aβ40 level obviously in hippocampi of AD mice accompanied by a tendency to decline Aβ42 deposition [31].
Clearing brain-derived Aβ is through transporting to the periphery, while the liver is the largest organ responsible for the clearance of metabolites, including Aβ in the periphery probably. A systemic failure of cell-mediated Aβ clearance is a pivotal event in the pathogenesis of AD and contributes to AD occurrence and progression. More recently, Cheng et al. reported that approximately 13.9% of Aβ42 and 8.9% of Aβ40 were cleared from the blood when flowing through the liver. When Aβ receptor LRP-1 expression was down-regulated in hepatocytes of the aged animals, this clearing ability was declined. Aβ levels in both blood and brain interstitial fluid were increased when hepatic blood flow reduced significantly [34], which showed that physiologically, the liver can clear peripheral blood Aβ and regulate brain Aβ levels, suggesting that during aging, the decrease in Aβ clearance in the liver might be associated with the pathogenesis of AD [34]. In the livers of AD patients, Aβ degradation capacity decreased significantly due to the reducing expression of Aβ-degrading enzymes, including cathepsin D and insulin-degrading enzyme compared with normal people [45].
An increasing number of studies indicated that the early diagnosis of AD might be conducted via assessment of peripheral plasma contains biomarkers that predict the levels of Aβ in the brain [60, 61]. There are now well-validated blood biomarkers for Aβ and tau pathology, as well as neurodegeneration and astrocytic activation in AD, including Aβ40, Aβ42, glial fibrillary acidic protein (GFAP), neurofilament light chain (NfL) and P-tau181 as well as the ratio Aβ42/Aβ40 [60, 62, 63]. Young people’s blood or plasma transfused to elderly patients can effectively lighten AD symptoms [64]. Overall, the above studies clearly evidenced the association between the peripheral blood Aβ and Aβ level/deposit in brain of AD, and the peripheral blood Aβ is a crucial causative factor in AD involving in the occurrence and progression of AD definitely. Furthermore, Aβ deposition in the brain may potentially correlate to the level in the peripheral circulatory system [65]. Based on this fact, the BBB could be one possible way of communication between the brain and periphery [66]. It has been reported that the BBB permeability of AD patients is higher than that of non-AD patients, so the peripheral blood Aβ is more likely to impact the stable environment of the CNS, and lead to the pathology of AD, such as Aβ deposit, inflammation and neurotoxicity [67].
Impacting of the peripheral Aβ on peripheral innate immune cells
To date, dissecting the role of immune cells in AD pathogenesis has been challenging, particularly for the peripheral innate immune cells due to the fact that the exact role of the immune system in AD is still unclear and controversial. Previously immune dysfunction in the CNS was considered as a cause of the pathogenesis and progression of AD, while accumulating results showed key contributions of the peripheral immune system in AD as well. A consensus reached was that aberrant immune response was a cardinal feature of AD; meanwhile, a large amount of evidence suggested that pathological changes occurred in the central and peripheral immune responses throughout the entire AD process [23]. However, immune process raises a notable question how communication between the peripheral and central compartments occurs? Here, we emphasize to clarify the effects of the peripheral Aβ on peripheral innate immune cells, including neutrophils, monocytes, macrophages and natural killer (NK) cells. Understanding the causative roles (protective or harmful) of peripheral innate immune cells impacted by peripheral Aβ in AD will hopefully instruct therapeutic avenues to target the immune system at different stages of AD.
Effect on neutrophils
Neutrophils are formed by bone marrow precursors in the bone marrow and are the most abundant white blood cells in humans and in mice [68]. Neutrophils are crucial in curbing invasive pathogens, antibacterial and antiviral responses, tissue repair, and mediating inflammation have become evident by phagocytosis, the release of antimicrobial molecules through degranulation, and containment and killing of pathogens via release of nuclear DNA [69]. Compared with young people, the activity of blood neutrophils in the healthy elderly is declined [70]. Particularly, lowered neutrophil function in AD was even greater in the later stages of the disease. Nevertheless, more neutrophils were observed in the early stages of AD than in age-matched healthy controls [70].
More than a decade ago, Baik, et al. have observed the peripheral neutrophils infiltrating into the brains of both AD patients and its Tg models (5xFAD and 3xTg-AD mice), and existing in the areas with Aβ deposits [71, 72]. At that time, it was obscure what factors touched off the recruitment of blood neutrophils toward Aβ plaques in brain? Because Aβ was not possible to directly recruit neutrophils, therefore, it was obscure what factors touched off the recruitment of blood neutrophils toward Aβ plaques in brain at that time. Recently, it has been found that Aβ42 triggered the lymphocyte function-associated antigen-1 (LFA-1) integrin high-affinity state and rapid neutrophil adhesion to integrin ligands. LFA-1 integrin can regulate neutrophil extravasation into the brain in vivo [72]. Particularly, Aβ is a formylpeptide receptor 2 agonist and a potent chemoattractant for attracting the peripheral blood leukocytes, monocytes as well as other immune cells entry into the brain and activated these cells there [73]. The brain is vulnerable to the effects of reactive oxygen species (ROS), while neutrophils produce a large amount of ROS in AD brain. Therefore, neutrophils appear to be the driving force behind AD.
In periphery, inflammatory mediators released by neutrophils can further cause changes in peripheral neutrophils count. Also BBB allows neutrophils to enter the brain and promotes the accumulations of neutrophils and Aβ there [74, 75]. For example, in acute colitis, neutrophils accelerated the accumulation of Aβ in the brain of the mouse model of AD and Kaneko et al. considered that neutrophil targeted therapy in AD may be a new strategy [75]. Recent studies have again demonstrated that neutrophils contribute to inflammation and disease progression in AD [72, 76,77,78,79], indicating that these effects of neutrophils via Aβ chemical attraction were harmful in AD. However, recently Sas et al. found a subset of neutrophils contributing to neuronal survival in the CNS [80]. Therefore, neutrophils may play double roles in the pathogenesis of AD. Anyway, pathogenic infiltration of peripheral immune cells, including neutrophils into the brain exacerbates AD pathology definitely [23, 81, 82], in which process, Aβ plays a pioneering role in pathogenicity due to attracting peripheral immune cells. Specially, depletion of infiltrating neutrophils by anti-Ly 6G or anti–Gr-1 antibody or suppressing neutrophil trafficking via LFA-1 alleviated pathological changes in two mouse models of AD (5XFAD and 3xTg) mice, which improved memory in mice those have cognitive impaired already and diminished the amyloid burden [72]. Moreover, infiltrating neutrophils in brain can induce neurotoxicity by releasing IL-17 that is a cytotoxic cytokine of neurons and mediates the destruction BBB, neutrophil extracellular traps and myeloperoxidase [72].
Aβ as a potent chemoattractant attracts the peripheral blood immune cells entry into the brain through three possible routes: (1) BBB that has been evidenced breakdown and dysfunction in AD [83]; (2) meninges that are enable immune cells to bypass BBB entering the brain through special skull bone marrow channels [84]; (3) choroid plexus that acts as a portal for immune cells from bone marrow to enter the brain [85]. Therefore, peripheral immune cells can play a role in both healthy and diseased brains [86, 87]. Several studies showed that the ratio of neutrophil and lymphocyte in the blood was correlated with cognitive decline in AD [65, 88] and neutrophils are involved in the pathogenesis of AD at the early stages via mediating BBB damage, infiltration and intravascular adhesion of the CNS [72]. Hou et al. reported that the increase in neutrophil count and neutrophil lymphocyte ratio were related to a decrease in cognitive, memory and executive functions. At the same time, they found that raised neutrophil–lymphocyte ratio was associated with a lower level of Aβ and higher level of total tau (T-tau) of CSF, as well as the atrophy of the hippocampus [22]. The latest research further emphasizes that the high proportion of peripheral neutrophils to lymphocytes may reflect an imbalance between innate and adaptive immunity, and is related to greater Aβ deposition and longitudinal cognitive decline [89]. When compared with mild cognitive impairment (MCI) patients, there were higher proportion of harmful and highly reactive aging and immunosuppressive neutrophils in AD [23]. The high proportion of peripheral neutrophils to lymphocytes may reflect an imbalance between innate and adaptive immunity, and a larger proportion of Aβ deposition is related to a decrease in longitudinal cognitive ability [89].
In addition, there are significant changes in neutrophil homeostasis when compared the patients with slower decline cognitive function with the patients with faster decline cognitive function, indicating that the neutrophil immune phenotype reflects the stage of the disease, but also showed a decrease in cognitive ability [90]. The specific impact of peripheral Aβ on neutrophils is still unclear, but the pathway of the role of the peripheral Aβ in neutrophils revealed a complex network involving Aβ clearance [91].
Effect on monocytes
Monocytes, an innate immune cell population, usually account for 3–8% of the total number of white blood cells in the blood. They are derived from bone marrow hematopoietic stem cells and eventually enter tissues and transform into macrophages. Monocytes are less often in the CNS. In the blood, they will float in the blood until they reach the site that requires an immune response, then they will transform into macrophages, clearing away harmful substances such as bacteria and viruses. The peripheral monocytes are heterogeneous cells that are divided into multiple subpopulations with diverse surface markers, heterogeneous transcriptional profiles, and diverse functions. Up to now, the study on the role of monocytes in AD and impacting of the peripheral Aβ on monocytes has been limited. More recently, Liu et al. reported that the ability of blood monocyte phagocytosis Aβ in AD declined and monocyte in AD mice showed decreased in energy metabolism accompanied by cellular senescence, aging related secretory phenotype and dysfunction of phagocytic function [92]. However, increasing blood monocyte Aβ phagocytosis by improving energy metabolism in vivo can reduce brain Aβ deposition and neuroinflammation, ultimately improving cognitive function [92]. Additionally, removal of Ly6C monocyte in APP/PS1 mice can induce an obvious increase of Aβ deposit in the cortex and hippocampus, uncovering the ability of Ly6C(lo) monocytes eliminating Aβ [93]. Town et al. found presence of crawling monocytes carrying Aβ in veins and their ability to circulate back into the bloodstream in AD mice observed by live intravital two-photon microscopy [93]. Although circulating monocyte can penetrate into the brain and clear Aβ in AD patients, when compared with monocyte in the healthy subjects, the effect of these monocytes on alleviating AD pathology was significantly poor, accompanied by limited phagocytosis and phenotype, and have been adjusted to an inflammatory state, which was in line with the study of AD mice [94]. In 2021, Better et al. uncovered that Aβ42-induced monocytes lowed IL-1β secretion in healthy elderly adults and MCI, but not effected on AD, moreover, Aβ42 stimulated monocytes of healthy older to produce IL-10 only, suggesting the impact of monocytes on AD pathology was very limited [23]. Transforming growth factor-β (TGF-β) signaling expressing on peripheral macrophages was blocked to lead to substantial infiltration and clearance of cerebral Aβ in the Tg2576 mice, the animal model for AD [93]. Monocytes infiltrating the brain of AD Tg mice (APP/PS1 and 5XFAD) reduced Aβ burden, and improved cognitive performance [95, 96], indicating a beneficial role of monocytes in AD pathology.
Furthermore, the lack of methyltransferase like 3 (METTL3) in monocytes derived macrophages improved Aβ caused cognitive function impairment in AD mice, which is due to METTL3 ablation attenuating the m6A modification in DNA methyltransferase 3A (Dnmt3a) mRNAs and consequently impairing YTH N6-methyladenosine RNA binding protein 1 (YTHDF1)-mediated translation of DNMT3A [97]. More recently, the emerging evidence found that angiotensin-converting enzyme (ACE) can degrade Aβ42 neurotoxic 42-residue long alloform, while monocytes overexpressing ACE has neuroprotective properties in AD [98]. In addition, the peripheral myeloid cells infiltrating into brain tissue alleviated Aβ deposition and improved cognitive function in AD mouse models [93, 99, 100]. But, using brain-resident myeloid cells did not alter Aβ deposition in two mouse models of AD (APP23 and APP/PS1 mice) [101, 102], indicating that peripheral monocytes and microglia play their respective roles in clearing Aβ.
The latest findings indicate the interaction between cytokine tissue inhibitor of metalloproteinases-1 (TIMP-1) that triggered glucose uptake and proinflammatory cytokine expression in human monocytes, and members of the amyloid precursor protein (APP) family, namely APP and amyloid precursor protein-2 (APLP2) proved by confocal microscopy. TIMP-1 expression positively correlated with monocyte activation and proinflammatory cytokine production in cancer patients [103]. However, the impact of TIMP-1 on AD monocytes activation and its potential molecular mechanisms are largely unclear, at least one point can be understood: in AD, TIMP-1 activation of human monocytes is related to inflammation caused by Aβ. Infiltration of AD monocytes into the CNS relied on C–C motif chemokine receptor 2 (CCR2) and blockade of CCR2 in AD mice (Appswe/PS1 and Tg2576) enhanced Aβ pathology and deteriorated memory impairment [99, 100]. In the process of Aβ42 recruiting peripheral monocytes, the translocator protein-18 kDa (TSPO) that is a transmembrane protein and overexpressed in response to neuroinflammation, involved in modulating this process [104]. It has been found in 1999 that benzodiazepine-induced chemotaxis is impaired in monocytes from patients with generalized anxiety disorder [105], suggesting that future therapeutic interventions aim at modulating monocytes motility toward the CNS.
The study by live two-photon imaging observed that the patrolling monocytes climbed to Aβ+ on the lumen wall of vein, internalized Aβ and circulated back into the bloodstream, hinting that monocytes can clear vascular Aβ of AD [106]. In this year, Uekawa et al. reported that brain border-associated macrophages (BAM) through the Aβ-binding innate immunity receptor CD36 lead to cognitive impairment. The absence of CD36 in BAM also declines brain Aβ40 without affecting plaques in the brain, and enhanced the vascular clearance of exogenous Aβ [107]. It has been evidenced that macrophages of AD patients have interrelated defects in the transcriptome, glycome, Aβ phagocytosis, and Aβ degradation [108].
As the major innate immune cells, peripheral macrophages are the chief phagocytes in the periphery [109] and their role of infiltrating into the CNS and association with Aβ deposit have been contentious. Macrophages can attract other cells migrating to injured or infected tissues via releasing signal molecules [109]. Since the similarity in phenotype between the macrophages and microglia, it is difficult to fully differentially distinguish them [110, 111], thus, most studies on AD focus on either a single cell or a joint analysis of both. Wisniewski et al. analyzed these two types of cells using an electron microscopy and considered that macrophages were more effective to eliminate Aβ plaques than microglia [112]. In response to Aβ stimulation, macrophages tend to produce less inflammatory cytokines than microglia [113]. In year 2020, Reed-Geaghan et al. reported that Aβ plaque was surrounded by microglia rather than infiltrating macrophages [114], speculating that microglia are more impactful in clearing Aβ plaque than macrophages. A new subset of macrophages has been found within the CNS in the border region areas, including the pia mater, perivascular space and choroid plexus and are replaced by circulating monocytes [115,116,117].
In short, these data showed that the study on the role of monocytes themselves and the effect of Aβ on circulating monocytes in AD are limited and is still unclear. More studies are needed to determine the direct role of circulating monocytes in AD and impact of Aβ on circulating monocytes.
Effect on NK cells
As innate immune cells, NK cells represent approximately 15% of peripheral blood lymphocytes and are a subpopulation of cytotoxic lymphocytes. NK cells are divided into several NK cell subsets according to the differential expression of some phenotypical and functional markers. NK cells have an immunomodulatory role and accompanied by high secretion of cytokines and chemokines without pre-stimulation, such as interferon-γ (IFN-γ) [118], and also can kill other cells. The increased ratio of cytotoxic NK cells in the periphery was considered as a preclinical sign of AD and one of the Aβ neuropathological mechanism [119].
Importantly, NK cells can also stimulate macrophages contributing to chronic inflammation in the CNS of AD [118, 120]. During the aging process, the cytotoxic activity of NK cells in healthy people is impaired obviously compared to young people [70]. However, the role of NK cells in AD has always been disputed. Araga et al. reported a lowered cytotoxic function of NK cells in AD patients compared to healthy subjects [121, 122]. But Solerte et al. obtained an opposite result, which showed significantly enhanced levels of TNF-α and IFN-γ, as well as higher cytotoxic capacity of NK cells in AD when compared to healthy elderly subjects [123, 124]. In mild AD patients, no change in NK cell activation capacity was observed through comparing the expression of CD107a, a marker for granular release, and levels of granzyme B and IFN-γ [125], whether NK cells are altered in severe AD patients is not clear. In Rag2–/–/Il2rγ–/–deficient mice crossed with 5xFAD mice, NK cells, T cells, and B cells do not develop accompanied by raising Aβ levels [126]. However, in Rag2–/– mice crossed with PSAPP mice without T and B cells with NK cell-sufficient, there was a reduce Aβ plaques [127], which implies that NK cells may be more involved in clearing Aβ plaques compared to T and B cells, however, this warrants further investigation in such mice.
Similar to neutrophils, NK cells can also infiltrate the brain of the APP/PS1 mice causing Aβ pathology [115, 128, 129], however, neutrophils not only infiltrate the brain of three mouse models of AD (APP/PS1, 5 × FAD and 3 × Tg-AD mice) [130, 131], but also infiltrate the brains of AD patients. Up to now, the research on the infiltration of NK cells into the AD brain is limited. Using single-cell RNA (scRNA) sequencing data of sorted NKs (from datasets GSE181279 and GSE142853) illustrated the landscape of immune cells and immunity-related genes characteristics from the peripheral blood mononuclear cells in AD to confirm NK infiltration in the AD brain, which showed that the peripheral NK cells may infiltrate the brain and contribute to neuroinflammation in AD patients [129]. However, further in vivo studies are required to validate the CNS infiltration of peripheral NK cells and to investigate their role in AD. Meanwhile, the studies proposed that signal transducer and activator of transcription 3 (STAT3) may play a critical role in NK activation and infiltration into the brain in AD [129]. Due to the fact that NK cells do not require prior activation to secrete cytokines and chemokines to play their role, we speculate that circulating NK cells do not need the peripheral Aβ stimulation directly. Whether NK cells infiltration is needed Aβ as a potent chemoattractant for attracting them entry into the brain, it remains unclear. However, it is certain, NK cells are early responders who can receive the signals from target organs and quickly coordinate local inflammation upon arrival and involved the pathological changes of AD.
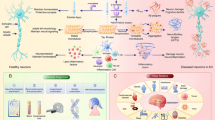
The role of the peripheral blood Aβ and peripheral innate immune cells in AD and its animal models is presented in Fig. 1.

The role of the peripheral blood Aβ and innate immune cells in AD and its animal models. In human peripheral blood, a large amount of Aβ is derived from circulating platelets. And the liver is another origin of brain Aβ deposits and it is also a major organ responsible for cleaning up circulating Aβ in the blood. Clearing brain-derived Aβ is through transporting to the periphery. LRP-1 of Aβ receptor expression on hepatocytes of the aging animals can regulate Aβ clearance ability. Innate immune cells of the peripheral blood, including neutrophils, NK cells, monocytes and macrophages infiltrate into the CNS in AD and its animal models under Aβ chemotaxis, as well as produce inflammatory cytokines, such as IL-17 and IFN-γ. Aβ42 triggered LFA-1 regulates neutrophil extravasation into the brain. Meanwhile circulating activated platelets produce both Aβ and proinflammatory mediators, which could amplify peripheral inflammation and endothelial senescence, leading to change the permeability of the BBB and contributing to Aβ across BBB into brain, as well as accelerating Aβ deposition in the brain and enhancing Aβ level in peripheral blood. In the CNS, infiltrated peripheral innate immune cells and inflammatory mediators can stimulate astrocytes and microglia activation to produce inflammatory cytokines, which promote brain Aβ deposits and induce AD pathology. Finally, this leads to neuronal cell death, synaptic degradation and inflammation as well as gliosis, further exacerbating neurodegeneration and ultimate causing dementia
Conclusion
The peripheral blood Aβ is a potent chemoattractant for the peripheral innate immune cells infiltration into brain of AD, which is a crucial step to cause pathological changes in AD. There are communications between peripheral and the CNS in AD through several pathways, although the directionality and timing of these pathways are poorly understood.
In the early stage of disease, the peripheral Aβ is involved in the pathogenesis of AD through activating innate immune cells and promoting them to secretion of inflammatory cytokines and molecules leading to enhancing the BBB permeability or damage BBB. In the late stage, the peripheral Aβ may activate the peripheral and central inflammatory processes by affecting the proliferation and differentiation of innate immune cells. The recruitment of the peripheral innate immune cells may lead to increased production of proinflammatory cytokines by microglia, promoting the recruitment of more peripheral innate immune cells to move to Aβ plaques of brain. The peripheral innate immune cells could participate in engulfing and clearing the Aβ plaques in AD. Colocalization of innate immune cells with Aβ plaques is now a well-recognized neuropathological feature of AD. Based on the facts that (1) Aβ deposition in the brain may potentially correlate to the level in the peripheral circulatory system, and (2) anti-Aβ mabs therapy in AD is reducing blood Aβ levels through the peripheral sink mechanism of action, thus promotes CNS amyloid transforming to produce soluble monomers, across BBB to restore the decreased blood Aβ levels, we considered that the peripheral blood Aβ contributing to AD pathology is via impacting the peripheral innate immune cells partly. These give us a better understanding of the effects of the peripheral blood Aβ on the peripheral innate immune cells in AD pathology. However, due to insufficient study in this area, further investigation is necessary to understand the relationship between the peripheral Aβ and the peripheral innate immune cells in AD.
Availability of data and materials
Not applicable.
References
Alexiou A, Chatzichronis S, Ashraf GM. Chapter 23 - Prediction of Alzheimer's disease. In: Martin CR, Preedy VR, editors. Diagnosis and management in dementia. Academic Press; 2020. p. 365–78. https://doi.org/10.1016/B978-0-12-815854-8.00023-9.
Povova J, et al. Epidemiological of and risk factors for Alzheimer’s disease: a review. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2012;156:108–14. https://doi.org/10.5507/bp.2012.055.
2020 Alzheimer’s disease facts and figures. Alzheimers Dement; 2020. https://doi.org/10.1002/alz.12068.
Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell. 2019;179:312–39. https://doi.org/10.1016/j.cell.2019.09.001.
Uddin MS, et al. Emerging proof of protein misfolding and interactions in multifactorial Alzheimer’s disease. Curr Top Med Chem. 2020;20:2380–90. https://doi.org/10.2174/1568026620666200601161703.
Scheltens P, et al. Alzheimer’s disease. Lancet. 2021;397:1577–90. https://doi.org/10.1016/s0140-6736(20)32205-4.
Alexander GC, Emerson S, Kesselheim AS. Evaluation of aducanumab for Alzheimer disease: scientific evidence and regulatory review involving efficacy, safety, and futility. JAMA. 2021;325:1717–8. https://doi.org/10.1001/jama.2021.3854.
Zhu F, Li C, Chu F, Tian X, Zhu J. Target dysbiosis of gut microbes as a future therapeutic manipulation in Alzheimer’s disease. Front Aging Neurosci. 2020;12:544235. https://doi.org/10.3389/fnagi.2020.544235.
Tolar M, Abushakra S, Sabbagh M. The path forward in Alzheimer’s disease therapeutics: reevaluating the amyloid cascade hypothesis. Alzheimers Dement. 2020;16:1553–60. https://doi.org/10.1016/j.jalz.2019.09.075.
Veitch DP, et al. Understanding disease progression and improving Alzheimer’s disease clinical trials: recent highlights from the Alzheimer’s Disease Neuroimaging Initiative. Alzheimers Dement. 2019;15:106–52. https://doi.org/10.1016/j.jalz.2018.08.005.
Imbimbo BP, et al. Solanezumab for the treatment of mild-to-moderate Alzheimer’s disease. Expert Rev Clin Immunol. 2012;8:135–49. https://doi.org/10.1586/eci.11.93.
Zhang Y, Lee DH. Sink hypothesis and therapeutic strategies for attenuating Abeta levels. Neuroscientist. 2011;17:163–73. https://doi.org/10.1177/1073858410381532.
Shibata M, et al. Clearance of Alzheimer’s amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106:1489–99. https://doi.org/10.1172/jci10498.
Deane R, et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–44. https://doi.org/10.1016/j.neuron.2004.07.017.
DeMattos RB, et al. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2001;98:8850–5. https://doi.org/10.1073/pnas.151261398.
DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM. Brain to plasma amyloid-beta efflux: a measure of brain amyloid burden in a mouse model of Alzheimer’s disease. Science. 2002;295:2264–7. https://doi.org/10.1126/science.1067568.
Logovinsky V, et al. Safety and tolerability of BAN2401–a clinical study in Alzheimer’s disease with a protofibril selective Aβ antibody. Alzheimers Res Ther. 2016;8:14. https://doi.org/10.1186/s13195-016-0181-2.
FDA, T. U. S. FDA Converts novel Alzheimer’s disease treatment to traditional approval. The U. S. Food and drug administration. https://www.fda.gov/news-events/press-announcements/fda-converts-novel-alzheimers-disease-treatment-traditional-approval. Accessed 06 July 2023.
FDA, T. U. S. FDA grants full approval to Leqembi, opening up coverage of Alzheimer’s drug by Medicare. https://endpts.com/alzheimers-drug-leqembi-gets-full-approval-from-fda-allowing-medicare-coverage/. Accessed 06 July 2023.
Walker KA, Ficek BN, Westbrook R. Understanding the role of systemic inflammation in Alzheimer’s disease. ACS Chem Neurosci. 2019;10:3340–2. https://doi.org/10.1021/acschemneuro.9b00333.
Shi M, et al. Role of adaptive immune and impacts of risk factors on adaptive immune in Alzheimer’s disease: are immunotherapies effective or off-target? Neuroscientist. 2022;28:254–70. https://doi.org/10.1177/1073858420987224.
Hou JH, et al. Association of peripheral immunity with cognition, neuroimaging, and Alzheimer’s pathology. Alzheimers Res Ther. 2022;14:29. https://doi.org/10.1186/s13195-022-00968-y.
Bettcher BM, Tansey MG, Dorothée G, Heneka MT. Peripheral and central immune system crosstalk in Alzheimer disease - a research prospectus. Nat Rev Neurol. 2021;17:689–701. https://doi.org/10.1038/s41582-021-00549-x.
Chen M, Inestrosa NC, Ross GS, Fernandez HL. Platelets are the primary source of amyloid beta-peptide in human blood. Biochem Biophys Res Commun. 1995;213:96–103. https://doi.org/10.1006/bbrc.1995.2103.
Li QX, et al. Secretion of Alzheimer’s disease Abeta amyloid peptide by activated human platelets. Lab Invest. 1998;78:461–9.
Skovronsky DM, Lee VM, Praticò D. Amyloid precursor protein and amyloid beta peptide in human platelets. Role of cyclooxygenase and protein kinase C. J Biol Chem. 2001;276:17036–43. https://doi.org/10.1074/jbc.M006285200.
Canobbio I, Catricalà S, Balduini C, Torti M. Calmodulin regulates the non-amyloidogenic metabolism of amyloid precursor protein in platelets. Biochim Biophys Acta. 2011;1813:500–6. https://doi.org/10.1016/j.bbamcr.2010.12.002.
Canobbio I, Abubaker AA, Visconte C, Torti M, Pula G. Role of amyloid peptides in vascular dysfunction and platelet dysregulation in Alzheimer’s disease. Front Cell Neurosci. 2015;9:65. https://doi.org/10.3389/fncel.2015.00065.
Chen L, et al. Abnormal platelet amyloid-β precursor protein metabolism in SAMP8 mice: evidence for peripheral marker in Alzheimer’s disease. J Cell Physiol. 2019;234:23528–36. https://doi.org/10.1002/jcp.28921.
Matsubara E, et al. Soluble Abeta homeostasis in AD and DS: impairment of anti-amyloidogenic protection by lipoproteins. Neurobiol Aging. 2004;25:833–41. https://doi.org/10.1016/j.neurobiolaging.2003.10.004.
Wu T, Chen L, Zhou L, Xu J, Guo K. Platelets transport β-amyloid from the peripheral blood into the brain by destroying the blood-brain barrier to accelerate the process of Alzheimer’s disease in mouse models. Aging (Albany NY). 2021;13:7644–59. https://doi.org/10.18632/aging.202662.
Bassendine MF, Taylor-Robinson SD, Fertleman M, Khan M, Neely D. Is Alzheimer’s disease a liver disease of the brain? J Alzheimers Dis. 2020;75:1–14. https://doi.org/10.3233/jad-190848.
Zheng H, et al. Tissue-specific metabolomics analysis identifies the liver as a major organ of metabolic disorders in amyloid precursor protein/presenilin 1 mice of Alzheimer’s disease. J Proteome Res. 2019;18:1218–27. https://doi.org/10.1021/acs.jproteome.8b00847.
Cheng Y, et al. Physiological β-amyloid clearance by the liver and its therapeutic potential for Alzheimer’s disease. Acta Neuropathol. 2023;145:717–31. https://doi.org/10.1007/s00401-023-02559-z.
Xiang Y, et al. Physiological amyloid-beta clearance in the periphery and its therapeutic potential for Alzheimer’s disease. Acta Neuropathol. 2015;130:487–99. https://doi.org/10.1007/s00401-015-1477-1.
Li C, Liu K, Zhu J, Zhu F. The effects of high plasma levels of Aβ(1–42) on mononuclear macrophage in mouse models of Alzheimer’s disease. Immun Ageing. 2023;20:39. https://doi.org/10.1186/s12979-023-00366-4.
Jakhmola-Mani R, Islam A, Katare DP. Liver-brain axis in sporadic Alzheimer’s disease: role of ten signature genes in a mouse model. CNS Neurol Disord Drug Targets. 2021;20:871–85. https://doi.org/10.2174/1871527319666201209111006.
Huang Z, Lin HWK, Zhang Q, Zong X. Targeting Alzheimer’s disease: the critical crosstalk between the liver and brain. Nutrients. 2022. https://doi.org/10.3390/nu14204298.
Kitazume S, et al. Soluble amyloid precursor protein 770 is released from inflamed endothelial cells and activated platelets: a novel biomarker for acute coronary syndrome. J Biol Chem. 2012;287:40817–25. https://doi.org/10.1074/jbc.M112.398578.
Kuo YM, et al. Elevated abeta42 in skeletal muscle of Alzheimer disease patients suggests peripheral alterations of AbetaPP metabolism. Am J Pathol. 2000;156:797–805. https://doi.org/10.1016/s0002-9440(10)64947-4.
Van Nostrand WE, Melchor JP. Disruption of pathologic amyloid beta-protein fibril assembly on the surface of cultured human cerebrovascular smooth muscle cells. Amyloid. 2001;8(Suppl 1):20–7.
Chen L, et al. Studies on APP metabolism related to age-associated mitochondrial dysfunction in APP/PS1 transgenic mice. Aging (Albany NY). 2019;11:10242–51. https://doi.org/10.18632/aging.102451.
D’Alonzo ZJ, et al. Peripheral metabolism of lipoprotein-amyloid beta as a risk factor for Alzheimer’s disease: potential interactive effects of APOE genotype with dietary fats. Genes Nutr. 2023;18:2. https://doi.org/10.1186/s12263-023-00722-5.
Biere AL, et al. Amyloid beta-peptide is transported on lipoproteins and albumin in human plasma. J Biol Chem. 1996;271:32916–22. https://doi.org/10.1074/jbc.271.51.32916.
Maarouf CL, et al. Impaired hepatic amyloid-beta degradation in Alzheimer’s disease. PLoS ONE. 2018;13:e0203659. https://doi.org/10.1371/journal.pone.0203659.
Lam V, et al. Synthesis of human amyloid restricted to liver results in an Alzheimer disease-like neurodegenerative phenotype. PLoS Biol. 2021;19:e3001358. https://doi.org/10.1371/journal.pbio.3001358.
Deane R, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907–13. https://doi.org/10.1038/nm890.
Deane R, Wu Z, Zlokovic BV. RAGE (yin) versus LRP (yang) balance regulates alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke. 2004;35:2628–31. https://doi.org/10.1161/01.STR.0000143452.85382.d1.
Sutcliffe JG, Hedlund PB, Thomas EA, Bloom FE, Hilbush BS. Peripheral reduction of β-amyloid is sufficient to reduce brain β-amyloid: implications for Alzheimer’s disease. J Neurosci Res. 2011;89:808–14. https://doi.org/10.1002/jnr.22603.
Xu J, et al. Mitochondrial dysfunction in platelets and hippocampi of senescence-accelerated mice. J Bioenerg Biomembr. 2007;39:195–202. https://doi.org/10.1007/s10863-007-9077-y.
Shi C, et al. Effects of ageing and Alzheimer’s disease on mitochondrial function of human platelets. Exp Gerontol. 2008;43:589–94. https://doi.org/10.1016/j.exger.2008.02.004.
Thomas T, Nadackal TG, Thomas K. Aspirin and non-steroidal anti-inflammatory drugs inhibit amyloid-beta aggregation. NeuroReport. 2001;12:3263–7. https://doi.org/10.1097/00001756-200110290-00024.
Harris JR. In vitro fibrillogenesis of the amyloid beta 1–42 peptide: cholesterol potentiation and aspirin inhibition. Micron. 2002;33:609–26. https://doi.org/10.1016/s0968-4328(02)00029-x.
Casoli T, et al. Release of beta-amyloid from high-density platelets: implications for Alzheimer’s disease pathology. Ann N Y Acad Sci. 2007;1096:170–8. https://doi.org/10.1196/annals.1397.082.
Medeiros R, et al. Aspirin-triggered lipoxin A4 stimulates alternative activation of microglia and reduces Alzheimer disease-like pathology in mice. Am J Pathol. 2013;182:1780–9. https://doi.org/10.1016/j.ajpath.2013.01.051.
Doost Mohammadpour J, et al. Non-selective NSAIDs improve the amyloid-β-mediated suppression of memory and synaptic plasticity. Pharmacol Biochem Behav. 2015;132:33–41. https://doi.org/10.1016/j.pbb.2015.02.012.
Sepúlveda C, et al. The cAMP/PKA pathway inhibits beta-amyloid peptide release from human platelets. Neuroscience. 2019;397:159–71. https://doi.org/10.1016/j.neuroscience.2018.11.025.
Bu XL, et al. Blood-derived amyloid-β protein induces Alzheimer’s disease pathologies. Mol Psychiatry. 2018;23:1948–56. https://doi.org/10.1038/mp.2017.204.
Ryan J, et al. Randomized placebo-controlled trial of the effects of aspirin on dementia and cognitive decline. Neurology. 2020;95:e320–31. https://doi.org/10.1212/wnl.0000000000009277.
Stevenson-Hoare J, et al. Plasma biomarkers and genetics in the diagnosis and prediction of Alzheimer’s disease. Brain. 2023;146:690–9. https://doi.org/10.1093/brain/awac128.
Nakamura A, et al. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature. 2018;554:249–54. https://doi.org/10.1038/nature25456.
Aschenbrenner AJ, et al. Neurofilament light predicts decline in attention but not episodic memory in preclinical Alzheimer’s disease. J Alzheimers Dis. 2020;74:1119–29. https://doi.org/10.3233/jad-200018.
Chatterjee P, et al. Plasma glial fibrillary acidic protein is elevated in cognitively normal older adults at risk of Alzheimer’s disease. Transl Psychiatry. 2021;11:27. https://doi.org/10.1038/s41398-020-01137-1.
Scudellari M. Ageing research: blood to blood. Nature. 2015;517:426–9. https://doi.org/10.1038/517426a.
Dong X, Nao J, Shi J, Zheng D. Predictive value of routine peripheral blood biomarkers in Alzheimer’s disease. Front Aging Neurosci. 2019;11:332. https://doi.org/10.3389/fnagi.2019.00332.
Banks WA, Reed MJ, Logsdon AF, Rhea EM, Erickson MA. Healthy aging and the blood-brain barrier. Nat Aging. 2021;1:243–54. https://doi.org/10.1038/s43587-021-00043-5.
Nation DA, et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med. 2019;25:270–6. https://doi.org/10.1038/s41591-018-0297-y.
Bratton DL, Henson PM. Neutrophil clearance: when the party is over, clean-up begins. Trends Immunol. 2011;32:350–7. https://doi.org/10.1016/j.it.2011.04.009.
Tseng CW, Liu GY. Expanding roles of neutrophils in aging hosts. Curr Opin Immunol. 2014;29:43–8. https://doi.org/10.1016/j.coi.2014.03.009.
Vida C, et al. Impairment of several immune functions and redox state in blood cells of Alzheimer’s disease patients. Relevant role of neutrophils in oxidative stress. Front Immunol. 2017;8:1974. https://doi.org/10.3389/fimmu.2017.01974.
Baik SH, et al. Migration of neutrophils targeting amyloid plaques in Alzheimer’s disease mouse model. Neurobiol Aging. 2014;35:1286–92. https://doi.org/10.1016/j.neurobiolaging.2014.01.003.
Zenaro E, et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat Med. 2015;21:880–6. https://doi.org/10.1038/nm.3913.
Ying G, et al. Humanin, a newly identified neuroprotective factor, uses the G protein-coupled formylpeptide receptor-like-1 as a functional receptor. J Immunol. 2004;172:7078–85. https://doi.org/10.4049/jimmunol.172.11.7078.
Sayed A, et al. The neutrophil-to-lymphocyte ratio in Alzheimer’s disease: current understanding and potential applications. J Neuroimmunol. 2020;349:577398. https://doi.org/10.1016/j.jneuroim.2020.577398.
Kaneko R, et al. Increased neutrophils in inflammatory bowel disease accelerate the accumulation of amyloid plaques in the mouse model of Alzheimer’s disease. Inflamm Regen. 2023;43:20. https://doi.org/10.1186/s41232-023-00257-7.
Bawa KK, et al. A peripheral neutrophil-related inflammatory factor predicts a decline in executive function in mild Alzheimer’s disease. J Neuroinflammation. 2020;17:84. https://doi.org/10.1186/s12974-020-01750-3.
Kong Y, et al. PET imaging of neutrophils infiltration in Alzheimer’s disease transgenic mice. Front Neurol. 2020;11:523798. https://doi.org/10.3389/fneur.2020.523798.
Smyth LCD, et al. Neutrophil-vascular interactions drive myeloperoxidase accumulation in the brain in Alzheimer’s disease. Acta Neuropathol Commun. 2022;10:38. https://doi.org/10.1186/s40478-022-01347-2.
Song L, Yang YT, Guo Q, Zhao XM. Cellular transcriptional alterations of peripheral blood in Alzheimer’s disease. BMC Med. 2022;20:266. https://doi.org/10.1186/s12916-022-02472-4.
Sas AR, et al. A new neutrophil subset promotes CNS neuron survival and axon regeneration. Nat Immunol. 2020;21:1496–505. https://doi.org/10.1038/s41590-020-00813-0.
Mason HD, McGavern DB. How the immune system shapes neurodegenerative diseases. Trends Neurosci. 2022;45:733–48. https://doi.org/10.1016/j.tins.2022.08.001.
Jorfi M, Maaser-Hecker A, Tanzi RE. The neuroimmune axis of Alzheimer’s disease. Genome Med. 2023;15:6. https://doi.org/10.1186/s13073-023-01155-w.
Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14:133–50. https://doi.org/10.1038/nrneurol.2017.188.
Herisson F, et al. Direct vascular channels connect skull bone marrow and the brain surface enabling myeloid cell migration. Nat Neurosci. 2018;21:1209–17. https://doi.org/10.1038/s41593-018-0213-2.
Shechter R, et al. Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity. 2013;38:555–69. https://doi.org/10.1016/j.immuni.2013.02.012.
Da Mesquita S, et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature. 2018;560:185–91. https://doi.org/10.1038/s41586-018-0368-8.
Louveau A, et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat Neurosci. 2018;21:1380–91. https://doi.org/10.1038/s41593-018-0227-9.
Kuyumcu ME, et al. The evaluation of neutrophil-lymphocyte ratio in Alzheimer’s disease. Dement Geriatr Cogn Disord. 2012;34:69–74. https://doi.org/10.1159/000341583.
Mehta NH, et al. Peripheral immune cell imbalance is associated with cortical beta-amyloid deposition and longitudinal cognitive decline. Sci Rep. 2023;13:8847. https://doi.org/10.1038/s41598-023-34012-2.
Dong Y, et al. Neutrophil hyperactivation correlates with Alzheimer’s disease progression. Ann Neurol. 2018;83:387–405. https://doi.org/10.1002/ana.25159.
Huang X, et al. Leukocyte surface biomarkers implicate deficits of innate immunity in sporadic Alzheimer’s disease. Alzheimers Dement. 2023;19:2084–94. https://doi.org/10.1002/alz.12813.
Liu ZH, et al. Improving blood monocyte energy metabolism enhances its ability to phagocytose amyloid-β and prevents Alzheimer’s disease-type pathology and cognitive deficits. Neurosci Bull. 2023. https://doi.org/10.1007/s12264-023-01077-y.
Town T, et al. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14:681–7. https://doi.org/10.1038/nm1781.
Fiala M, et al. Ineffective phagocytosis of amyloid-beta by macrophages of Alzheimer’s disease patients. J Alzheimers Dis. 2005;7:221–32. https://doi.org/10.3233/jad-2005-7304. (discussion 255–262).
Malm TM, et al. Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to beta-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol Dis. 2005;18:134–42. https://doi.org/10.1016/j.nbd.2004.09.009.
Baruch K, et al. PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer’s disease. Nat Med. 2016;22:135–7. https://doi.org/10.1038/nm.4022.
Yin H, et al. Loss of the m6A methyltransferase METTL3 in monocyte-derived macrophages ameliorates Alzheimer’s disease pathology in mice. PLoS Biol. 2023;21:e3002017. https://doi.org/10.1371/journal.pbio.3002017.
Danziger R, et al. The effects of enhancing angiotensin converting enzyme in myelomonocytes on ameliorating Alzheimer’s-related disease and preserving cognition. Front Physiol. 2023;14:1179315. https://doi.org/10.3389/fphys.2023.1179315.
El Khoury J, et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–8. https://doi.org/10.1038/nm1555.
Naert G, Rivest S. CC chemokine receptor 2 deficiency aggravates cognitive impairments and amyloid pathology in a transgenic mouse model of Alzheimer’s disease. J Neurosci. 2011;31:6208–20. https://doi.org/10.1523/jneurosci.0299-11.2011.
Varvel NH, et al. Replacement of brain-resident myeloid cells does not alter cerebral amyloid-β deposition in mouse models of Alzheimer’s disease. J Exp Med. 2015;212:1803–9. https://doi.org/10.1084/jem.20150478.
Prokop S, et al. Impact of peripheral myeloid cells on amyloid-β pathology in Alzheimer’s disease-like mice. J Exp Med. 2015;212:1811–8. https://doi.org/10.1084/jem.20150479.
Eckfeld C, et al. TIMP-1 is a novel ligand of Amyloid Precursor Protein and triggers a proinflammatory phenotype in human monocytes. J Cell Biol. 2023. https://doi.org/10.1083/jcb.202206095.
Conti E, et al. TSPO modulates oligomeric amyloid-β-induced monocyte chemotaxis: relevance for neuroinflammation in Alzheimer’s disease. J Alzheimers Dis. 2023;95:549–59. https://doi.org/10.3233/jad-230239.
Sacerdote P, Panerai AE, Frattola L, Ferrarese C. Benzodiazepine-induced chemotaxis is impaired in monocytes from patients with generalized anxiety disorder. Psychoneuroendocrinology. 1999;24:243–9. https://doi.org/10.1016/s0306-4530(98)00079-1.
Michaud JP, Bellavance MA, Préfontaine P, Rivest S. Real-time in vivo imaging reveals the ability of monocytes to clear vascular amyloid beta. Cell Rep. 2013;5:646–53. https://doi.org/10.1016/j.celrep.2013.10.010.
Uekawa K, et al. Border-associated macrophages promote cerebral amyloid angiopathy and cognitive impairment through vascular oxidative stress. Mol Neurodegener. 2023;18:73. https://doi.org/10.1186/s13024-023-00660-1.
Dover M, et al. Polyunsaturated fatty acids mend macrophage transcriptome, glycome, and phenotype in the patients with neurodegenerative diseases, including Alzheimer’s disease. J Alzheimers Dis. 2023;91:245–61. https://doi.org/10.3233/jad-220764.
Ma WT, Gao F, Gu K, Chen DK. The role of monocytes and macrophages in autoimmune diseases: a comprehensive review. Front Immunol. 2019;10:1140. https://doi.org/10.3389/fimmu.2019.01140.
Bennett ML, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A. 2016;113:E1738-1746. https://doi.org/10.1073/pnas.1525528113.
Jordão MJC, et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science. 2019. https://doi.org/10.1126/science.aat7554.
Wisniewski HM, Barcikowska M, Kida E. Phagocytosis of beta/A4 amyloid fibrils of the neuritic neocortical plaques. Acta Neuropathol. 1991;81:588–90. https://doi.org/10.1007/bf00310142.
Rawji KS, et al. Immunosenescence of microglia and macrophages: impact on the ageing central nervous system. Brain. 2016;139:653–61. https://doi.org/10.1093/brain/awv395.
Reed-Geaghan EG, Croxford AL, Becher B, Landreth GE. Plaque-associated myeloid cells derive from resident microglia in an Alzheimer’s disease model. J Exp Med. 2020. https://doi.org/10.1084/jem.20191374.
Villacampa N, Heneka MT. Microglia: You’ll Never Walk Alone! Immunity. 2018;48:195–7. https://doi.org/10.1016/j.immuni.2018.02.009.
Wyatt-Johnson SK, Brutkiewicz RR. The complexity of microglial interactions with innate and adaptive immune cells in Alzheimer’s disease. Front Aging Neurosci. 2020;12:592359. https://doi.org/10.3389/fnagi.2020.592359.
Mrdjen D, et al. High-dimensional single-cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity. 2018;48:599. https://doi.org/10.1016/j.immuni.2018.02.014.
Prinz M, Priller J. The role of peripheral immune cells in the CNS in steady state and disease. Nat Neurosci. 2017;20:136–44. https://doi.org/10.1038/nn.4475.
Yasuno F, et al. Mutual effect of cerebral amyloid β and peripheral lymphocytes in cognitively normal older individuals. Int J Geriatr Psychiatry. 2017;32:e93–9. https://doi.org/10.1002/gps.4660.
Radde, R. et al. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep 7, 940–946 (2006). https://doi.org:https://doi.org/10.1038/sj.embor.7400784
Araga S, et al. Natural killer cell activity in patients with dementia of the Alzheimer type. Arch Neurol. 1990;47:380–1. https://doi.org/10.1001/archneur.1990.00530040020007.
Araga S, Kagimoto H, Funamoto K, Takahashi K. Reduced natural killer cell activity in patients with dementia of the Alzheimer type. Acta Neurol Scand. 1991;84:259–63. https://doi.org/10.1111/j.1600-0404.1991.tb04948.x.
Solerte SB, et al. Increased natural killer cell cytotoxicity in Alzheimer’s disease may involve protein kinase C dysregulation. Neurobiol Aging. 1998;19:191–9. https://doi.org/10.1016/s0197-4580(98)00050-5.
Solerte SB, Cravello L, Ferrari E, Fioravanti M. Overproduction of IFN-gamma and TNF-alpha from natural killer (NK) cells is associated with abnormal NK reactivity and cognitive derangement in Alzheimer’s disease. Ann N Y Acad Sci. 2000;917:331–40. https://doi.org/10.1111/j.1749-6632.2000.tb05399.x.
Le Page A, et al. NK cells are activated in amnestic mild cognitive impairment but not in mild Alzheimer’s disease patients. J Alzheimers Dis. 2015;46:93–107. https://doi.org/10.3233/jad-143054.
Marsh SE, et al. The adaptive immune system restrains Alzheimer’s disease pathogenesis by modulating microglial function. Proc Natl Acad Sci U S A. 2016;113:E1316-1325. https://doi.org/10.1073/pnas.1525466113.
Späni C, et al. Reduced β-amyloid pathology in an APP transgenic mouse model of Alzheimer’s disease lacking functional B and T cells. Acta Neuropathol Commun. 2015;3:71. https://doi.org/10.1186/s40478-015-0251-x.
Zhang Y, et al. Depletion of NK cells improves cognitive function in the Alzheimer disease mouse model. J Immunol. 2020;205:502–10. https://doi.org/10.4049/jimmunol.2000037.
Liu Z, Li H, Pan S. Discovery and validation of key biomarkers based on immune infiltrates in Alzheimer’s disease. Front Genet. 2021;12:658323. https://doi.org/10.3389/fgene.2021.658323.
Wang J. Neutrophils in tissue injury and repair. Cell Tissue Res. 2018;371:531–9. https://doi.org/10.1007/s00441-017-2785-7.
Webers A, Heneka MT, Gleeson PA. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol Cell Biol. 2020;98:28–41. https://doi.org/10.1111/imcb.12301.
Acknowledgements
This work was supported by the project of The First hospital, Jilin University in Changchun city, Jilin Province and Sanming Project of Medicine in Shenzhen city (SZSM201801014), Guangdong Province of China.
Funding
This study was supported by Department of Finance of Jilin Province (JLSWSRCZX2021-098); Sanming Project of Medicine in Shenzen Municipality (SZSM201801014); Project of The First hospital, Jilin University.
Author information
Authors and Affiliations
Contributions
MS and FC prepared the manuscript and figure; JZ and FZ conceived and finalized the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors of the manuscript have read and agreed to its content and are accountable for all aspects of the accuracy and integrity of the manuscript in accordance with ICMJE criteria. All authors confirm that the article is original, has not already been published in a journal, and is not currently under consideration by another journal. All authors agree to the terms of the BioMed Central Copyright and License Agreement.
Competing interests
The authors have no competing interests to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shi, M., Chu, F., Zhu, F. et al. Peripheral blood amyloid-β involved in the pathogenesis of Alzheimer’s disease via impacting on peripheral innate immune cells. J Neuroinflammation 21, 5 (2024). https://doi.org/10.1186/s12974-023-03003-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-023-03003-5