
Abstract
Glial cell-mediated neuroinflammation and neuronal attrition are highly correlated with cognitive impairment in Alzheimer’s disease. YKL-40 is a secreted astrocytic glycoprotein that serves as a diagnostic biomarker of Alzheimer’s disease. High levels of YKL-40 are associated with either advanced Alzheimer’s disease or the normal aging process. However, the functional role of YKL-40 in Alzheimer’s disease development has not been firmly established. In a 5xFAD mouse model of Alzheimer’s disease, we observed increased YKL-40 expression in the cerebrospinal fluid of 7-month-old mice and was correlated with activated astrocytes. In primary astrocytes, Aβ1-42 upregulated YKL-40 in a dose-dependent manner and was correlated with PI3-K signaling pathway activation. Furthermore, primary neurons treated with YKL-40 and/or Aβ1-42 resulted in significant synaptic degeneration, reduced dendritic complexity, and impaired electrical parameters. More importantly, astrocyte-specific knockout of YKL-40 over a period of 7 days in symptomatic 5xFAD mice could effectively reduce amyloid plaque deposition in multiple brain regions. This was also associated with attenuated glial activation, reduced neuronal attrition, and restored memory function. These biological phenotypes could be explained by enhanced uptake of Aβ1-42 peptides, increased rate of Aβ1-42 degradation and acidification of lysosomal compartment in YKL-40 knockout astrocytes. Our results provide new insights into the role of YKL-40 in Alzheimer’s disease pathogenesis and demonstrate the potential of targeting this soluble biomarker to alleviate cognitive defects in symptomatic Alzheimer’s disease patients.
Similar content being viewed by others

Introduction
Alzheimer’s disease (AD) is the most common form of dementia, characterized by progressive attrition of neuronal subsets resulting in impaired neuroplasticity and a continuum of clinical features ranging from mild cognitive impairment to complete cognitive and physical incapacitation [1, 2]. Amyloid plaques and tau-associated neurofibrillary tangles are two key diagnostic hallmarks of AD [3,4,5]. However, previous attempts to restore cognitive deficits in AD patients by targeting amyloid-beta (Aβ) have not been effective. The recently FDA-approved Aβ-directed monoclonal antibody drug Lecanemab is the latest in a long line of therapeutics that aimed to reduce the presumptive neurotoxic effects of Aβ [6].
Neuroinflammation plays a prominent role in AD progression [7,8,9]. In early AD, Aβ-producing neurons may initiate neuroinflammation by attracting microglia and stimulating the production of pro-inflammatory cytokines and chemokines that aggravate AD progression. In late AD, fully activated microglia surrounding amyloid plaques exert their phagocytic functions to clear toxic Aβ aggregates. Thus, neuroinflammatory processes can have detrimental or beneficial roles in AD disease progression.
YKL-40, a secreted chitin-binding lectin belonging to glycosyl hydrolase family 18, has been identified as a biomarker for advanced AD. High YKL-40 levels in cerebrospinal fluid (CSF) have been observed in early-stage AD patients [10], and are associated with acute and chronic neuroinflammation [11]. The central nervous system (CNS) YKL-40 appears to be derived from astrocytes and rare white matter neurons rather than infiltrating macrophages [10, 12]. YKL-40 transcription in astrocytes is stimulated through STAT3 and RelB/p50 transcriptional complexes by IL-1β and IL-6 released from macrophages [13]. However, the regulation and the biological function of YKL-40 in AD have not been firmly defined. Pathologically, elevated YKL-40 expression is associated with cortical thinning, hippocampal atrophy, and cognitive decline [14]. Mouse models of AD exhibit increased expression of chitinase-3 like 1 (Chi3l1), a mouse homologue of human YKL-40 [15]. Chi3l1 was mainly expressed in astrocytes in the mouse brain, and an increased number of Chi3l1-positive reactive astrocytes was reported in advanced AD and associated with IBA1-positive microglia [16]. More recently, the characterization of a YKL-40-knockout mouse line revealed that YKL-40 suppressed glial cell-mediated phagocytosis and that YKL-40 expression is regulated by the circadian clock [17]. However, how YKL-40 regulates the Aβ processing and degradation are still unclear.
Toxic Aβ peptides are cleared in AD brains by multiple mechanisms. They could be taken up by phagocytosis or endocytosis through binding to lipoprotein receptor-related protein 1 (LRP-1) on astrocytes [18], triggering receptor expressed on myeloid cells 2 (TREM-2) on macrophages [19], or formyl peptide receptors (FPR) on microglia [20, 21]. Alternatively, Aβ peptides could be degraded by multiple proteases in the cytoplasm or extracellular space including neprilysin [22], cathepsin B [23], and α1-antichymotrypsin [24]. Autophagy, which has been implicated in the degradation of aggregated proteins, has also been shown to play a role in Aβ clearance in both microglia and astrocytes [25]. Finally, the ability of astrocytes to send out extensive processes to blood vessels allows the clearance of Aβ from the brain to the blood circulation [26]. The functional role of YKL-40 in Aβ processing and degradation has not been clearly defined. In this study, we report a significant enhancement in memory functions associated with astrocyte-specific knockout of YKL-40 and uncovered endocytic genes that may explain these biological effects.
Materials and methods
Mice
B6SJL-Tg(APPSwFlLon, PSEN1*M146L*L286V)6799Vas/Mmjax (stock#034840; 5xFAD hereafter) [27, 28], and B6;FVB-Tg(Aldhl11-cre/ERT2)1Khakh/J (stock#029655; Aldh hereafter) were purchased from the Jackson Laboratory (Bar Harbor, Maine). A YKL-40/Chi3l1 conditional knockout mouse strain, B6-Chil1tm1amc/Cyagen was generated by a commercial source (Cyagen, Inc.) (Chi3l1fl hereafter). A knockout cassette carrying a Neor gene was placed between exon 5 and 6 with flox sites flanking exon 5. Cre-mediated recombination event will eliminate exon 5 in the knockout allele. The above three mouse strains were maintained at the Chinese University of Hong Kong. Separately, a conventional YKL-40/Chi3l1 mouse line, C57BL/6 J-Chil1em1cyagen (Chi3l1−/− hereafter), was also purchased from Cyagen, Inc. and maintained in the animal center of Weifang Medical University. All mouse strains were backcrossed into a C57BL/6 J genetic background for at least 5 generations. To generate various YKL-40/Chi3l1 knockout in 5xFAD experimental groups, double heterozygous Aldh;Chi3l1+/fl and 5xFAD;Chi3l1+/fl mice were crossed to derive all experimental groups.
Primary cultures
For astrocytes, cortices of postnatal day 0 to 1 (P0-P1) pups were collected and triturated 3 times and resuspended with 2 ml of DMEM + 1% Pen/Strep + 5% fetal bovine serum (FBS, Gibco) and plated onto two 60-mm dishes coated with poly-D-lysine (PDL) (Sigma) and incubated at 37 °C, 5% CO2. For primary neurons, cortices or hippocampi from embryonic day 17.5 (E17.5) mice were triturated in ice-cold dissociating medium (DMEM + 1% Pen/Strep + 1% Antimycin/Antibiotic–Antimycotic, CAISSON Labs, ABL02-10) and 2 × 106 cells were seeded onto PDL coated 6-well dish in neurobasal medium (Gibco) + 1% Pen/Strep + 1% Antimycin/Antibiotic–Antimycotic + 2% B-27 Supplement (Gibco) + 0.25% L-Glutamine (200 mM) (Gibco). One day later, all wells were treated with 1 mM AraC (Sigma Aldrich). Half of the medium was changed every 3–4 days (without AraC).
Mouse dissection
Mice were anesthetized with ketamine/xylazine and head was fixed on a stereotaxic apparatus. Mouse skin was incised longitudinally to expose the dura over the cisterna magna. Then, the siphon glass tube was pushed into the cisterna magna and a total of 15–20 μl CSF was collected for each mouse. Next, the thoracic cavity was exposed, and blood was collected using a syringe through the right atrium and was immediately transferred into an EDTA treated 1.5 ml Eppendorf tube.
Mouse YKL-40/Chi3l1 ELISA immunoassay
Mouse Chitinase 3-like 1 Quantikine ELISA kit (R & D, MC3L10) was used to determine YKL-40/Chi3l1 expression levels in CSF and plasma of 4-, 7-, 9-, and 16-month-old WT and 5xFAD mice according to the manufacturer’s instructions. A microplate reader was used to determine the OD 450-nm values within 30 min. All standards, control, and samples were assayed in duplicates. The relative CSF and plasma YKL-40/Chi3l1 expression were normalized with total protein measured by the BCA kit.
Morris water maze
Male mice from 6.5-month- to 7.5-month-old were tested. Mice went through 1 day of cued training, 4 days of acquisition training, and a final day of probe trial. 5 trials were created on Day 0 to train mice to reach a visible platform. This was followed by 4 days of acquisition training with mice released at different release points to locate the submerged platform. Probe Trial was conducted on Day 5 to evaluate spatial memory ability. Only 1 trial that lasted for 60 secs was created for each mouse with no platform zone, and with the same release direction. EthoVision XT 11.5 software was used to track the number of entries, escape latency, swimming speed, and total path length. The tank was divided into 4 quadrants and was calibrated in the computer software to create physical distance information from pixel-based information.
Western blotting analysis
Cells were solubilized in RIPA buffer and diluted with 5X Loading buffer plus 2-mercaptoethanol and boiled. Approximately 5–10 μg protein samples and PageRuler Pre-stained Protein Ladder (Thermo Fisher Scientific) were resolved on SDS-PAGE gel. Following transfer onto Nitrocellulose membrane (Bio-Rad), the membrane was incubated in blocking solution (5% milk) and probed with primary antibodies in 5% milk at 4 °C overnight. Bound antibodies were detected with peroxidase-conjugated secondary antibodies (in 5% milk) for at least 1 hour (h) at room temperature. The membranes were developed by Enhanced Chemiluminescence. The membranes were exposed to autoradiography film in a dark room or directly detected using ChemiDoc Touch Imaging System (Bio-Rad).
Immunofluorescence staining
Each well of primary neurons in 12-well plates was fixed with 1 ml of 4% paraformaldehyde (PFA) (Sigma-Aldrich) at room temperature for 20 min. The neurons were permeabilized and blocked with 5% goat serum + 0.05% Triton X-100 in PBS for 60 min at room temperature. Primary antibodies were diluted to working concentration with 5% goat serum + 0.05% Triton X-100 in PBS and incubated at 4 °C, overnight. All neurons were probed with Alexa-conjugated secondary antibodies in 5% goat serum + 0.05% Triton X-100 in PBS for 1 h, washed and stained with DAPI (1 μg/mL) (Sigma-Aldrich) in PBS for 5–10 min at room temperature. Stained neurons were mounted with VECTASHIELD Mounting Medium (VECTOR) covered by coverslips and observed using Olympus SP8 Confocal System. Captured images were analyzed using ImageJ Fiji software. Primary astrocytes were cultured in 8-well chambers (3 × 104 per well) for 24 h. Cells were fixed with 4% paraformaldehyde solution, blocked with 3% BSA and 0.2% Triton X-100 at room temperature for 1 h, and then incubated with primary antibody overnight at 4 °C. After washing, the cells were incubated with the secondary antibodies at room temperature for 1 h. The nuclei were then stained with DAPI and images were acquired by confocal microscopy.
MTS/PMS cell viability assay
Primary neurons were seeded at 104 cells/well in 96-well plates coated with 10 mg/mL PDL. Neurons were treated with different doses of Aβ and YKL-40 for 48 h and 72 h. 3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) powder (Promega) was dissolved in PBS (2 mg/mL), pH6.0 ~ 6.5. PMS powder (Sigma) was also dissolved in PBS at a concentration of 0.92 mg/mL. The MTS / PMS solution was mixed at a ratio of 20 (MTS):1(PMS). Next, MTS/PMS solution was mixed with phenol red-free DMEM medium (Gibco) at the ratio of 1:5 and added to neurons. After incubating at 37 °C for 4 h, the OD values were measured at 490 nm on a spectrophotometer (Bio-Rad).
Drugs treatment
Primary astrocytes were treated with 1 mM tamoxifen (Sigma-Aldrich) for three consecutive days. For in vivo experiments, mice were intraperitoneally injected with 100 µl tamoxifen (20 mg/mL in corn oil) for 7 consecutive days. Lyophilized Aβ1-42 peptide powder (AnaSpec) was dissolved in 80 μl 1% NH4OH, then immediately diluted with 1X PBS to a concentration of 1 mg/mL (221.5 mM). After vortexing, large aggregates were removed by centrifugation (14,000 × g at RT for 15 min), and preparations were stored at − 20 °C. 1% NH4OH/PBS solution was used to dilute the Aβ stock to a working concentration.
Microelectrode array recoding (MEA)
Primary cortical neurons were harvested from E17.5 mice and seeded on microelectrode arrays (MEAs) (Multichannel System) for 24 days. The neuronal spontaneous electric activity was recorded beginning at DIV14 using an MEA2100 MultiChannel Systems on a heated stage at 37 °C. Neurons exhibited mature spikes (around DIV18) were exposed to 10 μM Aβ, 400 ng/mL YKL-40, and 10 μM Aβ + 400 ng/mL YKL-40, respectively. MEA recordings were measured at DIV14 (baseline), DIV19 (24 h after drug treatment), and DIV21 (72 h after drug treatment). The spikes were normalized to the pre-treatment activity of each array.
Quantitative RNA sequencing and data analysis
Primary astrocytes isolated from wild-type mice and YKL-40 knockout mice were cultured in DMEM with 10% FBS and lysed in 1 ml TRIZOL™ Reagent (Invitrogen). Total RNA was extracted following the manufacturer’s instructions. Then, RNA samples were subjected to quantitative RNA sequencing by Genesky (Genesky Biotechnologies Inc., Shanghai). DEGs with threshold values of log2 fold-change (FC) > 1 and p-value less than 0.05 were captured and presented in the volcano map. For functional enrichment analysis, all DEGs were mapped to the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, and significantly enriched KEGG pathways were searched. Furthermore, a gene set enrichment analysis (GSEA) (Broad Institute) tool kit was used to obtain the enrichment score for defined gene sets, such as GO gene sets and KEGG pathway gene sets.
Aβ uptake and degradation by astrocytes
Primary astrocytes were seeded in 96-well culture plates (2 × 104 per well) with 10% FBS-containing medium for 24 h. The cells were treated with 0.1 μM HiLyte™ Fluor 555-labeled Aβ42 (AnaSpec) for 24 h. The fluorescence intensity was determined at 540 nm excitation and 570 nm emission using a fluorescence microplate reader (Molecular Device, SpectraMax Gemini EM).
Intracellular lysosomal pH detection
The intracellular pH of astrocytes was detected using the ratiometric probe LysoSensor™ Yellow/Blue DND-160 (Invitrogen, L7545). After treatment with 5 μM Aβ1-42 for 96 h, astrocytes were rinsed with PBS and dye was added according to the manufacturer's protocol, followed by intracellular pH measurement with a fluorescence microplate reader. Fluorescence intensity was detected at EX = 385 nm EM = 540 nm and EX = 330 nm EM = 440 nm.
Flow cytometry
Primary astrocytes isolated from wild-type mice and YKL-40 knockout mice were prepared in a 6-well plate (1 × 105 cells per well) 24 h before the assay. The cells were incubated with 0.1 μM Aβ42-555 or APC Beads (BD, 661620) for 24 h at 37 °C. After incubation, the cells were washed three times with cold PBS and then suspended in PBS. The cell suspension was gravity filtered through 35 μm nylon mesh prior and then introduced to a flow cytometer. The autofluorescence of the cells incubated without Aβ42-555 or APC Beads was used as a control.
Data analysis
The mean surface area, integrated density, and synapse density of confocal images were measured by ImageJ Fiji software. GraphPad Prism software (Version 9.0, San Diego, CA, USA) was used to analyze the statistical significance and was used to construct various graphs. Neuronal spikes were identified and analyzed using Signal Processing Toolbox in MATLAB (2020a; MathWorks, California, United States)(see codes for spike analysis in Supplemental Materials section).
For MEA data analysis, recordings were performed for 5 min using threshold crossing methods at a 250000 Hz sampling rate. The raw data were filtered at 300 Hz high pass cut-off frequency using MC_Rack software. Thresholds were set at 3 times the standard deviation noise level of the individual channels. The pre-spike and post-spike durations were both set to 2 ms. Only channels that showed 10 or more spikes and low baseline noise (< 40 V) were included for analysis. We called electrodes that met these criteria “active electrodes”. All active channels of each drug and time point were pooled together for their respective statistical analyses. Significant differences between drugs at a given time point were tested by analysis of variance (ANOVA) followed by Dunnett’s multiple comparison post hoc test with a significance threshold of α = 0.05. Data were presented as mean ± SEM, n = 100–120 neurons from three independent experiments.
For Sholl Analysis, the image stacks of GFAP and MAP2 staining were reconstructed as binarized representations, and the Sholl method of concentric circles was used to determine astrocyte/neuron ramification [29]. Each cell was manually tracked to exclude adjacent cells by Simple Neurite Tracing (SNT) and analyzed by selecting the center of its soma and then running the Sholl analysis procedure, which counts the number of intersections at circles of increasing radii from the center. The maximal ring with an intersecting process (max distance), the mean number of intersections (branching complexity) for all circles, and the ramification index were calculated for each cell. Astrocytes: start radius (μm): 0.552; step size (μm): 1.664; samples per radius: 2. Neurons: start radius (μm): 3; step size (μm): 3; samples per radius: 2. Data were shown as mean ± SEM of 70–75 astrocytes from five independent experiments and 50–70 neurons from four independent experiments. GraphPad Prism software (Version 9.0, San Diego, CA, USA) was used to analyze statistical significance and graph constructions.
Statistical analysis
Quantitative data were represented as mean ± SEM. Differences between groups were tested with one-way ANOVA with Tukey’s post hoc comparisons, two-way ANOVA followed by Tukey’s multiple comparisons, and an unpaired t-test. Mann–Whitney test and Kruskal–Wallis test were used for non-parametric data when appropriate. Significance levels were defined as follows: *p < 0.05, **p < 0.01, ***p < 0.001.
Results
Increased astrocytic YKL-40 expression in 5xFAD mice
5xFAD mouse strain was selected to unravel the role of YKL-40 in AD. The 5xFAD mouse strain expresses human APP and PSEN1 transgenes with a total of five AD-linked mutations [30,31,32]. Immunostaining revealed extensive Aβ plaques in 5xFAD mice, even in the presymptomatic phase at 4 months old (Additional file 1: Fig. S1A). As expected, the plaque numbers in the dentatae gyrus (DG) region were significantly increased in 7- and 9-month-old 5xFAD mice (Additional file 1: Fig. S1B-C). Co-staining of DG and CA1 regions revealed extensive overlapping signals of GFAP and YKL-40 of 5xFAD mice (Fig. 1A). The YKL-40 levels showed a progressive increase with age from 4 to 9 months old (Fig. 1B, C). Furthermore, GFAP-positive reactive glial cells were detected in close proximity to Aβ-positive senile plaques (Additional file 1: Fig. S1B). Similar observations were detected in cortex, CA3, and thalamus (not shown). Further co-staining also revealed YKL-40-positive cells in the immediate vicinity of Aβ deposits (Additional file 1: Fig. S1D). These results imply an association between YKL-40 expression in astrocytes and amyloid plaque deposition. Because YKL-40 is a prognostic biomarker for AD, CSF and plasma were collected from 4-, 7-, 9-, and 16-month-old wild-type and 5xFAD mice. Using ELISA, we observed a significant (~ threefold) increase in soluble YKL-40 in the CSF of 5xFAD mice from 7 months onward (Fig. 1D). In WT mice, this increasing trend was not statistically significant. While similar increases in soluble YKL-40 were also detected in 5xFAD mice plasma, the ~ 1.5-fold increase was relatively modest compared with that observed in CSF (Fig. 1E). Next, primary astrocytes were treated with Aβ1-42 to mimic the in vivo condition of 5xFAD. Aβ1-42 induced a dose-dependent upregulation of intracellular and secreted YKL-40 in the conditioned medium (CM) of WT astrocytes at protein (Fig. 1F–H) and mRNA (Fig. 1I) levels.

Elevated Astrocytic YKL-40 expression in 5xFAD mice. A Representative confocal images (Mag. 10X) of dentate gyrus region from 4-month (m), 7 m, and 9 m 5xFAD mouse brains immuno-stained with DAPI (blue signal), anti-YKL-40 (green signal), and anti-GFAP (red signal) antibodies. Scale bar, 100 μm. Mean intensity (B) and mean surface area (C) of YKL-40 signal in DG region were quantified from 4 m-, 7 m-, and 9 m- WT and 5xFAD mice. 4 m (n = 6), 7 m (n = 6), 9 m (n = 4). Data are mean ± SEM. One-way ANOVA with Tukey’s post hoc comparisons. *p < 0.05, **p < 0.01, ***p < 0.001. D YKL-40 expression level in cerebral spinal fluid (CSF) extracted from WT and 5xFAD mice. E YKL-40 expression level in plasma extracted from WT and 5xFAD mice. YKL-40 protein was measured by ELISA and data was normalized by total protein. Data are mean ± SEM of 6 to 17 animals. One-way ANOVA with Tukey’s post hoc comparisons. *p < 0.05, **p < 0.01, ***p < 0.001 compared to WT-4 m. F Western blotting analysis of YKL-40 expression level from astrocyte lysates and conditioned medium (CM) treated with 0, 50, 200, and 1000 nM Aβ1-42 for 72 h. GAPDH was used as an internal control. The relative YKL-40 expression level in CM was normalized with total protein. G-H Quantification of relative fold change of YKL-40. n = 6. Data are mean ± SEM. One-way ANOVA with Tukey’s post hoc comparisons. *p < 0.05, ***p < 0.001 compared to control. I. Relative Chi3l1 gene expression from primary astrocytes treated with 0, 50, 200, and 1000 nM Aβ1-42 for 72 h. n = 3 to 6 from independent experiments. Data are mean ± SEM. J. Western blotting analysis of PI3-K pathway expression from astrocytes lysates treated with 0, 50 nM, 200 nM, 1000 nM Aβ1-42 for 72 h. GAPDH was used as internal control
Alterations in the PI3-K pathway have been shown to impair neurotransmission and long-term plasticity [33], and we have previously demonstrated a transcriptional link between PTEN and YKL-40 [34]. Primary astrocytes treated with Aβ1-42 induced activations of multiple components of the PI3-K pathway. In particular, both AKT and S6 were robustly activated by ~ 30-fold while both mTOR and p70 S6K were modestly activated by 2- to threefold (Fig. 1J and Additional file 1: Fig. S1E). In contrast, two members of the pro-inflammatory signaling pathway, JAK2 and STAT3, were not significantly activated (data not shown). Thus, the ability of Aβ1-42 to increase YKL-40 expression appears to be correlated with PI3-K activation state in primary mouse astrocytes.
YKL-40 induces neurotoxicity in primary neurons
To examine whether YKL-40 modulates the neurotoxic effects of Aβ1-42 in neuronal cells, primary cortical and hippocampal neurons were cultured from E17.5 embryos. Treatments with increasing amounts of either Aβ1-42 (1–10 μM) or YKL-40 (100–400 ng/mL) resulted in significant dose- and time-dependent loss of cell viability by as much as 30% (Fig. 2A-D and Additional file 1: Fig. S2A). Combined treatments with two dose combinations (10 μM Aβ1-42 + 100 ng/mL YKL-40 or 10 μM Aβ1-42 + 400 ng/mL YKL-40) resulted in further decreases in cell viability by 40% (Fig. 2A–D and Additional file 1: Fig. S2A).

YKL-40 induces neuronal toxicity and dendritic degradation. Primary neurons were established from E17.5 wild type mice. A–D MTS assay was used to determine cell viability of DIV14 primary cultured cortex (Cor) & hippocampus (Hip) neurons treated with Aβ1-42 and/or YKL-40 for 48 or 72 h. Data are mean ± SEM. n = 4 to 12. One-way ANOVA with Tukey’s post hoc comparisons. *p < 0.05, **p < 0.01, ***p < 0.001 compared to control. E Confocal images (Mag. 10X) of primary neurons immuno-stained with an anti-MAP2 (cyan signal) antibody. Scale bars, 100 μm. F MAP2 mean surface area was quantified. Data are mean ± SEM. One-way ANOVA with Tukey’s post hoc comparisons. *p < 0.05, **p < 0.01, ***p < 0.001 compared to control. G–I Sholl Analysis (Image J) of dendritic intersection number from tracing images. Data are mean ± SEM of 50 to 70 neurons. Two-way ANOVA with Tukey’s post hoc comparisons. **p < 0.01, ***p < 0.001 compared to control
To better visualize the neuronal morphologies, treated cells were immuno-stained with anti-MAP2 antibody (Fig. 2E). Aβ1-42 or YKL-40, added either alone or in combination, caused clear neurite retraction and decreased branching, as reflected in the reductions in mean intensity and mean surface area by 50 to 60% (Fig. 2F). Individual neurons were traced using the Simple Neurite Tracing plugin, and the dendritic complexity was analyzed using Sholl analysis. As shown in Fig. 2G–I and Additional file 1: Fig. S2B–H, Aβ1-42 and YKL-40 added alone or in combination significantly decreased neuronal complexity, reducing the intersection number, soma perimeter, dendritic length, axon length, axon node numbers, dendrite node, and dendrite end. However, no synergistic effect between the two compounds was observed.
Disturbed spontaneous electric activity and synaptic degradation in Aβ1-42- and YKL-40-treated primary neurons
To explore whether Aβ1-42 and YKL-40 could alter the spontaneous firing activity of cortical neurons, neuronal activities were recorded using a 60-channel microelectrode array (MEA). As shown in Fig. 3A, four electrodes from each group were used to record the electrical activities of DIV14 to DIV21 neurons. As expected, the neuronal spontaneous spike rate in the control group progressively increased from DIV14 to DIV21 (Fig. 3A). However, when Aβ1-42 and YKL-40 were added alone or in combination, this spontaneous electrical parameter was significantly suppressed in DIV19 and DIV21 (Fig. 3A). In terms of spike amplitude, the spontaneous firing activity plateaued around DIV19 in the control group (Fig. 3B). Treatment with Aβ1-42 and/or YKL-40 caused suppression to baseline levels in DIV21 (Fig. 3B). For spike numbers per second, while a significant increase was observed in DIV21 in the control group, this enhancement was not detected in groups treated with Aβ1-42 or YKL-40 either alone or in combination. In fact, there were significant decreases in DIV19 neurons following treatment with Aβ1-42 or with a combination of Aβ1-42 + YKL-40; as well as DIV21 neurons following treatment with YKL-40 alone. (Fig. 3C).

Impaired neuronal electric activities of Aβ1-42 and/or YKL-40 treated primary neurons. A Representative raw spike traces from four active electrode versus time after exposure to 10 μM Aβ, 400 ng/mL YKL-40, and 10 μM Aβ + 400 ng/mL YKL-40 in DIV18 cultures. B–C Quantified data showing time-dependent mean amplitude and spike rate for MEA neuronal cultures. Data are mean ± SEM of 100–120 neurons from three independent experiments. One-way ANOVA with Tukey’s post hoc comparisons. *p < 0.05, **p < 0.01, ***p < 0.001. D Confocal images (Mag. 10X) of primary neurons immuno-stained with anti-MAP2 (cyan signal), anti-PSD95 (green signal), and anti-SYP (red signal) antibodies. Scale bars, 100 μm. E–F Mean intensity and surface area were quantified. Mean surface area (n = 12), Density (n = 8) with at least 100 neurons included. Data are mean ± SEM. One-way ANOVA with Tukey’s post hoc comparisons. *p < 0.05, **p < 0.01
The adverse effects exerted by YKL-40 or Aβ1-42 on the electrical properties of neurons could be caused by biochemical or structural defects in synapses. We explored whether the addition of YKL-40 influenced synaptic morphology. Synaptic density was measured using anti-SYP and anti-PSD-95 co-staining of primary cortical neurons at DIV14 (Fig. 3D). As shown in Fig. 3E, F, significant reductions in synaptic density by 1.2- to 1.6-fold were observed in neurons treated with either 400 ng/mL YKL-40 or 10 μM Aβ1-42 alone. However, no further decline was observed when YKL-40 was added simultaneously with Aβ1-42, suggesting that Aβ1-42 treatment and YKL-40 expression may share a common mechanism for inducing neurodegeneration.
YKL-40 knockout reduces Aβ plaques and neuronal loss in 5xFAD mice
To explore the role of YKL-40 in 5xFAD, the Chi3l1 (YKL-40) gene was conditionally deleted in astrocytes of 5xFAD mice. A transgenic mouse line, Aldh, that allows pan-astrocyte inducible knockout of genes was used (Additional file 1: Fig. S3A)[35]. The exon 5 of Chi3l1 was deleted by tamoxifen injection one week prior to the experiment (Additional file 1: Fig. S3B, E). WT (Aldh;Chi3l1+/+) and heterozygous (Aldh;Chi3l1+/fl) mice had detectable YKL-40 expression in GFAP+ cells in both DG and TH (thalamus) regions (Additional file 1: Fig. S3F-G). In contrast, YKL-40 knockout mice (Aldh;Chi3l1fl/fl) had drastically reduced YKL-40 expression in GFAP+ cells, suggesting effective knockout of this gene (Additional file 1: Fig. S3F–G). Next, we crossed Aldh;Chi3l1+/fl mice with 5xFAD;Chi3l1+/fl mice to yield six experimental groups: 5xFAD;Aldh;Chi3l1+/+, 5xFAD;Aldh;Chi3l1+/fl, 5xFAD;Aldh;Chi3l1fl/fl, 5xFAD, WT;Aldh;Chi3l1fl/fl, and WT (Additional file 1: Fig. S3C). As expected, immunofluorescence analysis revealed the loss of YKL-40 expression in GFAP+ cells of 5xFAD;Aldh;Chi3l1+/fl (heterozygous knockout) and 5xFAD;Aldh;Chi3l1fl/fl (homozygous knockout) DG regions (Fig. 4A).

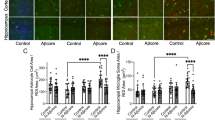
Knockout of YKL-40 in astrocytes reduces Aβ deposition in 5xFAD mice brain. All mice were IP injected with 20 mg/mL tamoxifen in corn oil (100 µl per mice/per day) for 7 consecutive days before experiments. Experiments were carried out 7 days after last tamoxifen injection. A Confocal images (Mag. 10X) of dentate gyrus region of 5xFAD;Aldh;Chi3l1+/+; 5xFAD;Aldh;Chi3l1+/fl; 5xFAD;Aldh;Chi3l1fl/fl mouse brain immuno-stained with DAPI (blue signal), anti-YKL-40 (green signal), and anti-GFAP (red signal) antibodies. Scale bars, 100 μm. B Parallel brain sections were co-stained with DAPI (blue signal), anti-Aβ (green signal), and anti-GFAP (red signal) antibodies. C High magnification image of Dentate Gyrus regions. Quantification of plaque number from Aβ signals and mean surface area. Data are mean ± SEM. One-way ANOVA with Tukey’s post hoc comparisons. *p < 0.05, **p < 0.01, ***p < 0.001 compared to 5xFAD;Aldh;Chi3l1+/+
To determine the effect of YKL-40 knockout on Aβ1-42 accumulation in 5xFAD mice, immunofluorescence analysis was performed on brain tissues from 7-month-old WT and YKL-40-deficient mice. Significant global reductions in Aβ plaque were observed in both 5xFAD;Aldh;Chi3l1+/fl 5xFAD;Aldh;Chi3l1fl/fl mice (Fig. 4B). The plaque numbers and mean surface area were reduced by 66% and 58%, respectively in the DG regions of 5xFAD;Aldh;Chi3l1fl/fl mice (Fig. 4C). Similar observations were detected in motor cortex + retrosplenial cortex (MOC), cortex (CTX), Cornu Ammonis 3 (CA3), and thalamus (TH) regions (Additional file 1: Fig. S4A, B).
NeuN is a marker of postmitotic neurons and loss of NeuN-positive cells is observed in mouse models of AD [36]. Immunostaining showed significant increased in NeuN immunoreactivity in 5xFAD;Aldh;Chi3l1fl/fl knockout mice to a level similar to the WT (Fig. 5A, B). In addition, significant increases in p-AMAPR in both cortical and hippocampal regions were observed in YKL-40 knockout 5xFAD mice compared with control 5xFAD mice (Fig. 5C, D). As a major excitatory neurotransmitter receptor in CNS, disturbed AMPAR surface diffusion is thought to be a critical mechanism underlying glutamatergic synaptic plasticity [37]. However, YKL-40-depletion in 5xFAD mice failed to alter the expression of several synaptic markers, such as PSD95 and synaptophysin (SYP) (Fig. 5C). In fact, YKL-40 knockout only mildly increased SYP expression in the hippocampal lysates of 5xFAD mice (Fig. 5C).

Depletion of YKL-40 in astrocytes prevents neuronal loss in the brain of 5xFAD mice. All mice were IP injected with 20 mg/mL tamoxifen in corn oil (100 µl per mice/per day) for 7 consecutive days before experiments. Experiments were conducted 7 days after last tamoxifen injection. A Confocal images (Mag. 10X) of dentate gyrus region 7-m mice brain immuno-stained with DAPI (blue signal), anti-NeuN (green signal), and anti-GFAP (red signal) antibodies. Scale bars, 100 μm. B Mean NeuN + density were quantified. Data are mean ± SEM of 7–9 brains (2–3 slices for each brain), then analysed by One-way ANOVA with Tukey’s post hoc comparisons. ***p < 0.001, compared to 5xFAD mice. DG: dentate gyrus. C Western blotting analysis of neuronal proteins in lysates isolated from cortex (Cor) and hippocampus (Hip). GAPDH was used as an internal control. D Western results were quantified for cortical (Cor) and hippocampal (Hip) samples. Data are mean ± SEM. n = 7. One-way ANOVA with Tukey’s post hoc comparisons. *p < 0.05, **p < 0.01, ***p < 0.001 compared to WT mice
Furthermore, we marked microglia and astrocytes with IBA1 and GFAP, respectively, and significant decreases in glial activation in multiple brain regions of YKL-40 knockout 5xFAD mice were observed via confocal microscopy (Additional file 1: Fig. S5A–D). Western blotting analysis of hippocampal and cortical lysates revealed elevated IBA1 and GFAP expression in 5xFAD mice compared with WT mice (Additional file 1: Fig. S5E–H). However, a significant decrease in IBA1 expression was only observed in the cortex of YKL-40 knockout 5xFAD mice (Additional file 1: Fig. S5G). This discrepancy may be due to the heterogeneity of cell types in bulk tissue sample analyses.
YKL-40 astrocyte-conditional knockout rescues cognitive deficits in 5xFAD mice
Morris water maze tests were conducted to examine whether the knockout of YKL-40 in astrocytes could rescue the memory deficits of 5xFAD mice. A significant decrease in escape latency over four days of training was defined as a strong learning ability in this spatial learning test. Memory deficits were defined by a reduced number of entries plus shortened latency to find the platform on day 5 (probe trial). No differences in escape latency between groups were observed on day 0 and day 1 (Fig. 6A, B). After four days of training, the escape latency of 5xFAD;Aldh;Chi3l1fl/fl mice was significantly shortened compared with that of 5xFAD mice (Fig. 6C). As expected, WT, WT;Aldh;Chi3l1fl/fl, and 5xFAD;Aldh;Chi3l1fl/fl mice showed greater decreases in escape latencies over four days of training ( Fig. 6C,I). On day 5, mice were allowed to explore the tank for 60 s following release from a fixed location (Fig. 6H). No differences in the total distance traveled and mean velocity were detected among the six groups (Fig. 6D, E). Deletion of YKL-40 increased the number of entries and reduced the escape latency of 5xFAD mice (Fig. 6F, G). No differences were observed between WT and WT;Aldh;Chi3l1fl/fl mice (Fig. 6F, G). Taken together, the observed improvements in spatial memory and learning ability of YKL-40 knockout 5xFAD mice support the detrimental effects of YKL-40 in the Aβ deposition process.

Knockout of YKL-40 in astrocytes attenuates cognitive deficits of 5xFAD mice. All mice were IP injected with 20 mg/mL tamoxifen in corn oil (100 µl per mice/per day) for 7 consecutive days before experiments. Experiments were carried out 7 days after last tamoxifen injection. Spatial memory and learning ability were tested using Morris Water Maze. A-C Quantified escape latency at day 0 (visible platform), day 1, and day 4. D–G Quantified total length, mean speed, escape latency, and number of entries at day 5 (probe trial). Data are mean ± SEM. One-way ANOVA with Tukey’s post hoc comparisons. *p < 0.05, **p < 0.01, ***p < 0.001. H Schematic representation of Morris water maze. I Quantified escape latency from day 1 to day 4 (training trials). Data are mean ± SEM. n = 23 ~ 25. One-way ANOVA with Tukey’s post hoc comparisons. *p < 0.05, **p < 0.01, ***p < 0.001
YKL-40 knockout enhances endocytosis through acidification of lysozyme
To unravel the underlying mechanism responsible for the elevated Aβ plaque clearance following YKL-40 knockout, we compared the transcriptomes of primary astrocytes from WT and Chi3l1−/− mice. Transcriptomic analysis revealed 294 up-regulated and 64 down-regulated genes with multiple enriched KEGG terms including phagosome (Additional file 1: Fig. S6A, B). GSEA analysis further identified genes involved in phagocytic vesicles and lysosome signaling (Additional file 1: Fig. S6C, D). We first tested if the endocytic pathway was required for Aβ uptake and retention in primary astrocytes using a panel of endocytic inhibitors. As shown in Fig. 7A, chlorpromazine (CPZ) and genistein, which inhibit clathrin-mediated and caveolae-mediated endocytosis, respectively, could significantly suppress the accumulation of fluorescence-labelled Aβ1-42–555 peptide. To examine if Aβ uptake was altered by YKL-40 knockout, primary astrocytes were exposed to Aβ1-42–555 peptide and the relative fraction of cells labelled positive were counted. There was a ~ twofold increase in Aβ1-42–555-positive cells in Chi3l1−/− astrocytes compared to WT (Fig. 7B). Similar results were obtained by using the flow cytometry method (Fig. 7C). However, the addition of exogenous YKL-40 failed to restore homeostatic Aβ levels in Chi3l1−/− cells suggesting that YKL-40 may act intracellularly (Fig. 7C). Furthermore, Aβ1-42 in Chi3l1−/− cells was mainly localized in the lysosomes (Fig. 7D) and there was a ~ 2.5-fold more lysosomal-associated Aβ1-42 detected in Chi3l1−/− than WT astrocytes (Fig. 7E). When lysosomal-associated Aβ1-42 was monitored over a period of 96 h, we observed that the rate of clearance was greater in Chi3l1−/− astrocytes than in WT (Fig. 7F). We further monitored the lysosomal pH of astrocytes using LysoSensor dyes. While WT and Chi3l1−/− astrocytes have similar lysosomal pH, treatment with Aβ1-42 for 96 h significantly increased the acidity of Chi3l1−/− astrocytes by twofold when compared with WT (Fig. 7G). Taken together, these results imply that high level of YKL-40 in AD patients can promote neurotoxicity by acting directly at the level of Aβ uptake and degradation in astrocytes.

Knockout of YKL-40 enhances Aβ induced receptor mediated endocytosis and promotes Aβ lysosomal degradation in primary astrocytes. A Astrocytes were pre-treated with indicated endocytosis inhibitors for 6 h and then incubated with fluorescent labeled Aβ1-42 at a concentration of 0.1 µM for 24 h, the fluorescence intensity of Aβ1-42 up-taken by astrocytes was measured using plate reader (Bars, SD; n = 5; ns, not significant; ***p < 0.001). B Astrocytes were treated with fluorescent labeled Aβ1-42 (0.1 µM) for 24 h. The percentage of Aβ1-42 positive cells was analyzed using image J. Data are mean ± SD; n = 10, *p < 0.05. C Astrocytes were exposed fluorescent labeled Aβ1-42 (0.1 µM) together with or without recombinant YKL-40 protein (250 ng/mL) for 24 h, and the Aβ1-42 positive cells were analyzed with flow cytometry. Data are mean ± SD; n = 3; **p < 0.01, ***p < 0.001. D The sub-cellular localization of Aβ42-555 peptide in WT and YKL-40 KO astrocytes 24 h after addition was revealed by immunostaining with lysosomal protein LAMP1 (green signal), and Aβ1-42 (red signal). E Data are quantified as percentage of Aβ1-42 colocalized with lysosome was analyzed using image J. Data are mean ± SD; n = 10, **p < 0.01. F Astrocytes were pre-treated with fluorescent labeled Aβ1-42 (0.1 µM) for 24 h, and the levels of Aβ1-42 contained in cells after treatment were measured using plate reader every 24 h. Data are mean ± SD; n = 6, **p < 0.01. G Astrocytes were treated with Aβ1-42 (5 µM) for 96 h, the pH of lysosomes were measured using LysoSensor™ Yellow/Blue DND-160. Data are ± SD; n = 6, ***p < 0.001
Discussion
The present study revealed several major insights into the role of YKL-40 in AD. First, we demonstrated that YKL-40 levels in CSF are elevated with disease progression in 5xFAD mice. Second, YKL-40 could induce neurotoxicity, astrogliosis, microglia accumulation, and suppressed spontaneous electrical activities. More importantly, we have demonstrated drastic Aβ clearance in an astrocyte-specific knockout of YKL-40 and provided in vivo evidence of the rescue of memory deficits in this mouse line. Finally, our study established a transcriptional link between YKL-40 and the endocytic pathways, and thus providing a mechanistic basis for the rescued memory deficits seen in YKL-40 knockout mice.
Studies on YKL-40 have reported opposite biological functions in different disease pathologies. YKL-40 has been reported to be neuroprotective and/or anti-inflammatory in traumatic brain injury [38], experimental autoimmune encephalomyelitis [39], and bacterial infection [40]; whereas others have observed attenuated cell viability and retracted neurites in primary cortical neurons treated with YKL-40 [41, 42]. More consistent with the present study, it has been reported that a YKL-40 chemical inhibitor, K284-6111, could suppress Aβ deposition and restore memory function in 12 months old Tg2576 mice [43]. Similar to our findings, the therapeutic effects were immediately observed following a short treatment of only 28 days. The target for YKL-40 appears to be the ERK-dependent PTX3 pathway [43]. Separately, YKL-40 knockout in an APP/PS1 model was shown to impede amyloid accumulation [17]. They also demonstrated that YKL-40 was regulated by the circadian clock and could suppress the abilities of astrocytes or microglia to phagocytose Aβ. Similarly, our 5xFAD mouse model with the astrocyte-specific knockout of YKL-40 also demonstrated reduced amyloid plaque accumulation, reactive astrocytes, microglia and preserved synaptic structures. More critically, our study has gone further to demonstrate that YKL-40 knockout in astrocytes of 5xFAD mice rescued memory deficits.
The interaction between astrocytes and microglia plays a critical role in Aβ-induced neuroinflammation and plaque clearance. The study by Lananna et. al. indicated that YKL-40 knockout in APP/PS1 model did not alter the abundance of microglia cells but the levels of the lysosomal marker, CD68, were increased. In contrast, we observed a significant reduction in total IBA1-staining intensity in our knockout model to a level similar to wild-type mice. While we have not evaluated the functional states of microglia in our model, these differences may be due to the differences in the AD mouse models being used. Indeed, the ability of microglia cells to clear Aβ is still controversial and appears to be stage-specific [44,45,46].
The Morris water maze tests conducted using 7-month-old 5xFAD;Aldh;Chi3l1fl/fl transgenic mice clearly demonstrated the rescue of cognitive impairment. Although the heterozygous loss of YKL-40 resulted in a 50% decrease in Aβ plaque deposition, no significant improvement in the Morris water maze test results was observed. The fact that YKL-40 was knockout just two weeks prior to the Morris water maze tests implies that symptomatic relief could be attainable even at the full onset of AD. This rapidity in plaque clearance and restoring memory functions suggests that YKL-40 may play a pivotal role in Aβ processing and clearance in 7-month-old 5XFAD mice. We also observed a greater number of NeuN + cells in the DG region of 5xFAD;Aldh;Chi3l1fl/fl mice when compared to 5xFAD;Aldh;Chi3l1+/+ control. This is unlikely due to neurogenesis, but is more likely to be the result of reduced neuronal loss. Consistently, YKL-40 deletion moderately restored synaptophysin expression and p-AMAPR/AMPAR levels in 5xFAD;Aldh;Chi3l1fl/fl mice compared with 5xFAD mice. Various studies have supported that decreased p-AMPAR Ser845 of GluA1 could lead to dysregulated synaptic plasticity and memory deficits. We speculate that the deletion of YKL-40 might restore GluA1 Ser845 phosphorylation and enhance AMPAR channel opening in 5xFAD;Aldh;Chi3l1fl/fl mice. Because AMPAR function is highly related to long-term potentiation (LTP) [47, 48], future studies should investigate whether YKL-40/Chi3l1-induced AMPAR dysfunction and cognitive impairment are associated with altered LTP function.
Based on our RNA-Seq analysis, the observed enhanced clearance and degradation of Aβ in 5xFAD;Aldh;Chi3l1fl/fl mice could be caused by the upregulation of genes involved in Aβ uptake and processing. For example, TLR2 has been shown to bind aggregated Aβ42 and inhibition of TLR2 in murine microglia abolished pro-inflammatory and phagocytotic responses following fibrillary Aβ exposure [49, 50]. Similarly, activation of TLR9 led to the amelioration of amyloid burden in the Tg2576 AD transgenic mouse model [51]. The greater rate of Aβ degradation observed in 5xFAD;Aldh;Chi3l1fl/fl mice could be attributed to elevated expression of members of the cathepsin family of lysosomal proteases, including Cathepsin B, C, D, H, K, and S. Among them, cathepsin D (CTSD) mediates the degradation of both Aβ and tau in vitro and Ctsd knockout mice have 3- to fourfold increase in cerebral Aβ burden [52]. On the contrary, cathepsin S and B (CTSS and CTSB) are risk factor genes for AD as they produce toxic Aβ peptides through their β-secretase activities [53, 54]. Besides, the addition of exogenous YKL-40 to YKL-40 KO astrocytes did not reverse the enhanced Aβ uptake raising the possibility that either astrocytes lack YKL-40 receptors or the effect of YKL-40 on Aβ uptake acts through intracellular targets. Currently, there are 6 known receptors for YKL-40 including AGER, CD44, IL13RA2, LGALS3, PTGDR2, and TMEM219 [14]. A detailed expression analysis would be required to address the relative abundance of these receptors in mouse astrocytes. For potential intracellular targets for YKL-40, we have recently demonstrated the nuclear localization of YKL-40 in human glioblastoma cell lines [55]. Whether YKL-40 has direct transcriptional activity on gene expression could be an area of future investigation.
Conclusion
The present study provides mechanistic insights supporting a functional role for YKL-40 in aggravating AD disease pathologies. There is strong evidence that elevated YKL-40 expression in CSF is associated with increasing Aβ burden. Extrapolate the data from astrocyte-specific knockout of YKL-40, it is concluded that YKL-40 is a risk gene for AD. First, it is toxic to neuronal cells by reducing cell viability, electrical activities, and dendritic processes. Second, it impedes Aβ uptake and subsequent degradation by astrocytes through transcriptional downregulation of genes involved in endocytosis, phagocytosis, and lysosomal functions. All these exacerbate the accumulation of Aβ in AD brains and provide credence for future development of therapeutics that target this molecule.
Availability of data and materials
All data generated or analysed during this study are included in this published article [and its Additional files].
Abbreviations
- Aβ:
-
Amyloid-beta
- AD:
-
Alzheimer’s disease
- BACE1:
-
β-Site amyloid precursor protein cleaving enzyme 1
- CA3:
-
Cornu Ammonis 3
- CSF:
-
Cerebrospinal fluid
- Chi3l1 :
-
Chitinase-3 like 1
- CTX:
-
Cortex
- DIV:
-
Days in vitro
- DG:
-
Dentatae gyrus
- MOC:
-
Motor cortex + retrosplenial cortex
- TH:
-
Thalamus
- MEA:
-
Microelectrode array
- PI3-K:
-
Phosphoinositide 3-kinase
- PDL:
-
Poly-D-lysine; and S6K, S6 kinase
References
Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chetelat G, Teunissen CE, et al. Alzheimer’s disease. Lancet. 2021;397(10284):1577–90.
As A. 2021 Alzheimer’s disease facts and figures. Alzheimers Dementia. 2021;17(3):327–406.
Leng FD, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol. 2021;17(3):157–72.
Lane CA, Hardy J, Schott JM. Alzheimer’s disease. Eur J Neurol. 2018;25(1):59–70.
Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell. 2019;179(2):312–39.
Jeremic D, Navarro-Lopez JD, Jimenez-Diaz L. Efficacy and Safety of anti-amyloid-beta monoclonal antibodies in current Alzheimer’s disease phase III clinical trials: a systematic review and interactive web app-based meta-analysiS. Ageing Res Rev. 2023;90:102012.
Cuello AC. Early and Late CNS Inflammation in Alzheimer’s Disease: Two Extremes of a Continuum? Trends Pharmacol Sci. 2017;38(11):956–66.
Gratuze M, Leyns CEG, Holtzman DM. New insights into the role of TREM2 in Alzheimer’s disease. Mol Neurodegener. 2018;13(1):66.
Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 Variants in Alzheimer’s disease. N Engl J Med. 2013;368(2):117–27.
Craig-Schapiro R, Perrin RJ, Roe CM, Xiong C, Carter D, Cairns NJ, et al. YKL-40: a novel prognostic fluid biomarker for preclinical Alzheimer’s disease. Biol Psychiatry. 2010;68(10):903–12.
Bonneh-Barkay D, Wang G, Starkey A, Hamilton RL, Wiley CA. In vivo CHI3L1 (YKL-40) expression in astrocytes in acute and chronic neurological diseases. J Neuroinflammation. 2010;7:34.
Querol-Vilaseca M, Colom-Cadena M, Pegueroles J, San Martin-Paniello C, Clarimon J, Belbin O, et al. YKL-40 (Chitinase 3-like I) is expressed in a subset of astrocytes in Alzheimer’s disease and other tauopathies. J Neuroinflammation. 2017;14(1):118.
Bhardwaj R, Yester JW, Singh SK, Biswas DD, Surace MJ, Waters MR, et al. RelB/p50 complexes regulate cytokine-induced YKL-40 expression. J Immunol. 2015;194(6):2862–70.
Connolly K, Lehoux M, O’Rourke R, Assetta B, Erdemir GA, Elias JA, et al. Potential role of chitinase-3-like protein 1 (CHI3L1/YKL-40) in neurodegeneration and Alzheimer’s disease. Alzheimers Dement. 2023;19(1):9–24.
Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflamm. 2006;3:27.
Moreno-Rodriguez M, Perez SE, Nadeem M, Malek-Ahmadi M, Mufson EJ. Frontal cortex chitinase and pentraxin neuroinflammatory alterations during the progression of Alzheimer’s disease. J Neuroinflamm. 2020;17(1):58.
Lananna BV, McKee CA, King MW, Del-Aguila JL, Dimitry JM, Farias FHG, et al. Chi3l1/YKL-40 is controlled by the astrocyte circadian clock and regulates neuroinflammation and Alzheimer’s disease pathogenesis. Sci Transl Med. 2020;12(574):eaax3519.
Deane R, Bell RD, Sagare A, Zlokovic BV. Clearance of amyloid-beta peptide across the blood-brain barrier: implication for therapies in Alzheimer’s disease. CNS Neurol Disord Drug Targets. 2009;8(1):16–30.
Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell. 2015;160(6):1061–71.
Brandenburg LO, Konrad M, Wruck CJ, Koch T, Lucius R, Pufe T. Functional and physical interactions between formyl-peptide-receptors and scavenger receptor MARCO and their involvement in amyloid beta 1–42-induced signal transduction in glial cells. J Neurochem. 2010;113(3):749–60.
Slowik A, Merres J, Elfgen A, Jansen S, Mohr F, Wruck CJ, et al. Involvement of formyl peptide receptors in receptor for advanced glycation end products (RAGE)–and amyloid beta 1–42-induced signal transduction in glial cells. Mol Neurodegener. 2012;7:55.
Fukami S, Watanabe K, Iwata N, Haraoka J, Lu B, Gerard NP, et al. Abeta-degrading endopeptidase, neprilysin, in mouse brain: synaptic and axonal localization inversely correlating with Abeta pathology. Neurosci Res. 2002;43(1):39–56.
Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9(8):857–65.
Abraham CR. Reactive astrocytes and alpha1-antichymotrypsin in Alzheimer’s disease. Neurobiol Aging. 2001;22(6):931–6.
Pomilio C, Pavia P, Gorojod RM, Vinuesa A, Alaimo A, Galvan V, et al. Glial alterations from early to late stages in a model of Alzheimer’s disease: Evidence of autophagy involvement in Abeta internalization. Hippocampus. 2016;26(2):194–210.
Morris AW, Carare RO, Schreiber S, Hawkes CA. The cerebrovascular basement membrane: role in the clearance of beta-amyloid and cerebral amyloid angiopathy. Front Aging Neurosci. 2014;6:251.
Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416(6880):535–9.
Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26(40):10129–40.
Le Douce J, Maugard M, Veran J, Matos M, Jego P, Vigneron PA, et al. Impairment of glycolysis-derived l-serine production in astrocytes contributes to cognitive deficits in Alzheimer’s disease. Cell Metab. 2020;31(3):503-17 e8.
Elder GA, Sosa MAG, De Gasperi R. Transgenic mouse models of Alzheimer’s disease. Mt Sinai J Med. 2010;77(1):69–81.
Hall AM, Roberson ED. Mouse models of Alzheimer’s disease. Brain Res Bull. 2012;88(1):3–12.
Jullienne A, Trinh MV, Obenaus A. Neuroimaging of mouse models of Alzheimer’s disease. Biomedicines. 2022. https://doi.org/10.3390/biomedicines10020305.
Knafo S, Sanchez-Puelles C, Palomer E, Delgado I, Draffin JE, Mingo J, et al. PTEN recruitment controls synaptic and cognitive function in Alzheimer’s models. Nat Neurosci. 2016;19(3):443–53.
Wang YB, Wong CW, Yan MF, Li LS, Liu T, Or PMY, et al. Differential regulation of the pro-inflammatory biomarker, YKL-40/CHI3L1, by PTEN/phosphoinositide 3-kinase and JAK2/STAT3 pathways in glioblastoma. Cancer Lett. 2018;429:54–65.
Srinivasan R, Lu TY, Chai H, Xu J, Huang BS, Golshani P, et al. New transgenic mouse lines for selectively targeting astrocytes and studying calcium signals in astrocyte processes In Situ and In Vivo. Neuron. 2016;92(6):1181–95.
Zhang J, Wu N, Wang S, Yao Z, Xiao F, Lu J, Chen B. Neuronal loss and microgliosis are restricted to the core of abeta deposits in mouse models of Alzheimer’s disease. Aging Cell. 2021;20(6): e13380.
Henley JM, Wilkinson KA. AMPA receptor trafficking and the mechanisms underlying synaptic plasticity and cognitive aging. Dialogues Clin Neurosci. 2022;15(1):11–27.
Wiley CA, Bonneh-Barkay D, Dixon CE, Lesniak A, Wang GJ, Bissel SJ, Kochanek PM. Role for mammalian chitinase 3-like protein 1 in traumatic brain injury. Neuropathology. 2015;35(2):95–106.
Bonneh-Barkay D, Wang GJ, LaFramboise WA, Wiley CA, Bissel SJ. Exacerbation of experimental autoimmune encephalomyelitis in the absence of breast regression protein 39/Chitinase 3-like 1. J Neuropathol Exp Neurol. 2012;71(11):948–58.
Dela Cruz CS, Liu W, He CH, Jacoby A, Gomitzky A, Ma B, et al. Chitinase 3-like-1 promotes Streptococcus pneumoniae killing and augments host tolerance to lung antibacterial responses. Cell Host Microbe. 2012;12(1):34–46.
Huang C, Huang B, Bi FF, Yan LH, Tong JB, Huang JF, et al. Profiling the genes affected by pathogenic TDP-43 in astrocytes. J Neurochem. 2014;129(6):932–9.
Matute-Blanch C, Calvo-Barreiro L, Carballo-Carbajal I, Gonzalo R, Sanchez A, Vila M, et al. Chitinase 3-like 1 is neurotoxic in primary cultured neurons. Sci Rep. 2020;10(1):7118.
Ham HJ, Lee YS, Yun J, Son DJ, Lee HP, Han SB, Hong JT. K284–6111 alleviates memory impairment and neuroinflammation in Tg2576 mice by inhibition of Chitinase-3-like 1 regulating ERK-dependent PTX3 pathway. J Neuroinflamm. 2020. https://doi.org/10.1186/s12974-020-02022-w.
Grathwohl SA, Kalin RE, Bolmont T, Prokop S, Winkelmann G, Kaeser SA, et al. Formation and maintenance of Alzheimer’s disease beta-amyloid plaques in the absence of microglia. Nat Neurosci. 2009;12(11):1361–3.
Lee CY, Landreth GE. The role of microglia in amyloid clearance from the AD brain. J Neural Transm. 2010;117(8):949–60.
Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci. 2008;28(33):8354–60.
Sheng M, Kim MJ. Postsynaptic signaling and plasticity mechanisms. Science. 2002;298(5594):776–80.
Kerchner GA, Nicoll RA. Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat Rev Neurosci. 2008;9(11):813–25.
Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B, et al. TLR2 is a primary receptor for Alzheimer’s amyloid beta peptide to trigger neuroinflammatory activation. J Immunol. 2012;188(3):1098–107.
Vollmar P, Kullmann JS, Thilo B, Claussen MC, Rothhammer V, Jacobi H, et al. Active immunization with amyloid-beta 1–42 impairs memory performance through TLR2/4-dependent activation of the innate immune system. J Immunol. 2010;185(10):6338–47.
Scholtzova H, Kascsak RJ, Bates KA, Boutajangout A, Kerr DJ, Meeker HC, et al. Induction of toll-like receptor 9 signaling as a method for ameliorating Alzheimer’s disease-related pathology. J Neurosci. 2009;29(6):1846–54.
Suire CN, Abdul-Hay SO, Sahara T, Kang D, Brizuela MK, Saftig P, et al. Cathepsin D regulates cerebral Abeta42/40 ratios via differential degradation of Abeta42 and Abeta40. Alzheimers Res Ther. 2020;12(1):80.
Hook G, Reinheckel T, Ni J, Wu Z, Kindy M, Peters C, Hook V. Cathepsin B gene knockout improves behavioral deficits and reduces pathology in models of neurologic disorders. Pharmacol Rev. 2022;74(3):600–29.
Schechter I, Ziv E, Cathepsins S. B and L with aminopeptidases display beta-secretase activity associated with the pathogenesis of Alzheimer’s disease. Biol Chem. 2011;392(6):555–69.
Shi M, Ge Q, Wang X, Diao W, Yang B, Sun S, et al. Functional analysis of the short splicing variant encoded by CHI3L1/YKL-40 in glioblastoma. Front Oncol. 2022;12: 910728.
Acknowledgements
The authors thank the Core laboratory staff at the School of Biomedical Sciences for their technical supports. We thank Prof. Raymond Chang and Prof. Jacque Ip for their constructive comments of our experimental results.
Funding
This study was supported by a General Research Fund grant (#14123718) to AMC from the University Grants Committee of Hong Kong, and a General Program Grant (ZR2020MH146) to YW from the Shandong Provincial Natural Science Foundation.
Author information
Authors and Affiliations
Contributions
XYZ conceptualization, performed cell-based and animal experiments, investigation, data analysis, wrote the original draft; SKKC supervised cell-based and animal experiments; MS performed cell-based and animal experiments, investigation, data analysis; PMY Or: maintenance of mouse colonies; ZL supervised behavioral tests; JYHL supervised MEA experiments; WLHH conducted cell-based and biochemical experiments; TL conducted cell-based and biochemical experiments; KL performed animal experiments; JAR provided resources and supervised MEA experiments; AMC and YW conceptualization, project administration, supervision, analyzed the data, wrote and edited the manuscript and secured funding.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All animal works were approved by the Hong Kong Department of Health and conducted in accordance with the Animal Experimentations and Ethics Committee of the Chinese University of Hong Kong (No.18-104-GRF) and by Animal Ethics Committee of Weifang Medical University (No. 2019SDL055).
Consent for publication
Not applicable.
Competing interests
All authors do not have any competing interests to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Age-dependent amyloid-beta plaques deposition in 5xFAD mouse brains and Aβ-induced signaling in primary astrocytes. A. Confocal images (Mag. 10X) of stitched 4m (presymptomatic), 7m (symptomatic), and 9m (advanced) 5xFAD mouse brains immuno-stained with DAPI (blue signal), anti-Aβ (green signal), and anti-GFAP (red signal) antibodies. Scale bars, 2000 μm. B. Higher magnifications of dentate gyrus (DG) region of 7m brains are shown. C. The mean plaque surface area and plaque numbers in DG region were quantified using ImageJ software. 4m (n=8), 7m (n=8), 9m (n=6). Data are mean ± SEM. One-way ANOVA with Tukey’s post hoc comparisons. *p < 0.05, **p < 0.01,***p < 0.001. D. Parallel immunofluorescence analysis similar to panel (B) was performed with Thioflavin T (ThT, green) and anti-YKL-40 (red). E. Quantification of data from Fig. 1J showing relative phosphorylation levels of AKT, P70S6, mTOR, and S6 following Aβ treatment. Data are mean ± SEM. One-way ANOVA with Tukey’s post hoc comparisons. ns, not significant, *p < 0.05, **p < 0.01. Fig. S2. Neuronal damages induced by Aβ1-42 (Aβ) and YKL-40 treatment. Primary neurons were derived from E17.5 wild type embryos. A. Brightfield photomicrographs taken from DIV14 treated with 10 μM Aβ, 400 ng/ml YKL-40, and 10 μM Aβ+400 ng/ml YKL-40 for 72 hrs. Scale bars, 200 μm. B. Binary figures of MAP2 immuno-stained confocal images, and their reconstructed Simple Neurite Tracing (SNT, Plugin in ImageJ software) tracing images following a 72 hrs exposure to the indicated concentrations of Aβ and YKL-40. C-H. Sholl Analysis (plugin from ImageJ) was performed from tracing images. Quantitative data for soma perimeter, dendritic length, axon length and nodes, dendritic nodes and ends are shown. Data are mean ± SEM of 50 to 70 neurons. One-way ANOVA with Tukey’s post hoc comparisons. ***p < 0.001 compared to control. Fig. S3. Generation of astrocyte-specific YKL-40/Chi3l1 conditional knockout mice. A. Genotyping of Aldhl11-cre/ERT2. Tails of one month old pups from a typical litter were processed for DNA extraction and PCR analysis was carried out using the following primer pairs: Aldhl1 and Aldhl2; Aldhl3 and Aldhl4. B. Knockout strategy of YKL-40/Chi3l1. C. Mating strategy used to generate the six experimental groups for this study. See Materials and Methods section for detailed descriptions. D. Tails of one month old pups from a typical litter were processed for DNA extraction and PCR analysis was carried out using the following primer pair: mChil1_flox_F and mChil1_flox_R. Wild-type (+/+) and heterozygous (+/- or +/fl) littermates are indicated. Abbreviations, Ma, markers; C, control. E. Primary astrocytes were treated without (-) or with (+) 1 μM tamoxifen solution. The 546-basepair (bp) knockout specific PCR product is indicated. F. Confocal images (Mag. 10X) of dentate gyrus (DG) and (G) thalamus (TH) regions of 3.5-month-old Aldh;Chi3l1+/+, Aldh;Chi3l1+/fl, and Aldh;Chi3l1fl/fl mouse brains immuno-stained with DAPI (blue signal), anti-YKL-40 (green signal), and anti-GFAP (red signal) antibodies. Scale bars, 100 μm. All mice were IP injected with 20 mg/ml tamoxifen in corn oil (100 μl per mice/per day) for 7 consecutive days before brains were harvested. Fig. S4. Knockout of YKL-40 in astrocytes reduces Aβ deposition in 5xFAD mice brain. All mice were IP injected with 20 mg/ml tamoxifen in corn oil (100 µl per mice/per day) for 7 consecutive days before experiments. Experiments were carried out 7 days after last tamoxifen injection. A. Confocal images (Mag. 10X) of motor cortex + metrosplenial cortex (MOC), mortex (CTX), Cornu Ammonis 3 (CA3), and thalamus (TH) regions of 5xFAD;Aldh;Chi3l1+/+; 5xFAD;Aldh;Chi3l1+/fl; 5xFAD;Aldh;Chi3l1fl/fl mouse brain immuno-stained with DAPI (blue signal), anti-Aβ (green signal), and anti-GFAP (red signal) antibodies. Scale bars, 100 μm. B. Quantification of plaque number from Aβ signals and mean surface area. Data are mean ± SEM. One-way ANOVA with Tukey’s post hoc comparisons. *p < 0.05,**p < 0.01, ***p < 0.001 compared to 5xFAD;Aldh;Chi3l1+/+. Fig. S5. YKL-40 depletion in astrocytes reduces glial activation. All mice were IP injected with 20 mg/ml tamoxifen in corn oil (100 μl per mice/per day) for 7 consecutive days before experiments. Experiments were conducted 7 days after last tamoxifen injection. A-B. Confocal images (Mag. 10X) of 7m 5xFAD mice brain immuno-stained with DAPI (blue signal), anti-IBA1 (green signal), and anti-GFAP (red signal) antibodies. Scale bar, 100 μm. C–D. Mean surface area was quantified. n=2 to 4. Data are mean ± SEM. E–F. Western blotting analysis of GFAP, and IBA1 expression from cortex (Cor) and hippocampal (Hip) lysates. GAPDH was used as an internal control. G-H. Quantification results of Western blot analysis are shown. Data are mean ± SEM. One-way ANOVA with Tukey’s post hoc comparisons.*p < 0.05, **p < 0.01,***p < 0.001. Fig. S6. Transcriptomic analysis of YKL-40 knockout astrocytes. (A) The volcano plot shows the 258 differentially expressed genes (294 up-regulated and 64 down-regulated, Foldchange>2, p<0.05) between WT and Chi3l1-/- (KO) in primary astrocytes by RNA-seq. (B) KEGG enrichment analysis of all DEGs showed that KO-associated with phagosome signaling pathway. GSEA analysis showed that genes associated with (C) phagocytic vesicles and (D) lysosome signaling were significantly up-regulated in KO astrocytes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zeng, X., Cheung, S.K.K., Shi, M. et al. Astrocyte-specific knockout of YKL-40/Chi3l1 reduces Aβ burden and restores memory functions in 5xFAD mice. J Neuroinflammation 20, 290 (2023). https://doi.org/10.1186/s12974-023-02970-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-023-02970-z