
Abstract
Background
Perinatal infection/inflammation is associated with a high risk for neurological injury and neurodevelopmental impairment after birth. Despite a growing preclinical evidence base, anti-inflammatory interventions have not been established in clinical practice, partly because of the range of potential targets. We therefore systematically reviewed preclinical studies of immunomodulation to improve neurological outcomes in the perinatal brain and assessed their therapeutic potential.
Methods
We reviewed relevant studies published from January 2012 to July 2023 using PubMed, Medline (OvidSP) and EMBASE databases. Studies were assessed for risk of bias using the SYRCLE risk of bias assessment tool (PROSPERO; registration number CRD42023395690).
Results
Forty preclinical publications using 12 models of perinatal neuroinflammation were identified and divided into 59 individual studies. Twenty-seven anti-inflammatory agents in 19 categories were investigated. Forty-five (76%) of 59 studies reported neuroprotection, from all 19 categories of therapeutics. Notably, 10/10 (100%) studies investigating anti-interleukin (IL)-1 therapies reported improved outcome, whereas half of the studies using corticosteroids (5/10; 50%) reported no improvement or worse outcomes with treatment. Most studies (49/59, 83%) did not control core body temperature (a known potential confounder), and 25 of 59 studies (42%) did not report the sex of subjects. Many studies did not clearly state whether they controlled for potential study bias.
Conclusion
Anti-inflammatory therapies are promising candidates for treatment or even prevention of perinatal brain injury. Our analysis highlights key knowledge gaps and opportunities to improve preclinical study design that must be addressed to support clinical translation.
Similar content being viewed by others

Introduction
Perinatal inflammation is highly associated with neonatal mortality and morbidity, including neurodevelopmental disorders such as vision and hearing impairments, learning difficulties, autism spectrum disorder, behavioural hyperactivity, schizophrenia and cerebral palsy (CP) [1,2,3]. Of particular concern, the risk of CP is increased several-fold in both preterm and term infants exposed to perinatal inflammation (odds ratio: 2.5–9.3) [4,5,6]. The cumulative lifetime economic cost of CP in the USA was estimated to be over USD 11.5 billion in 2003 [3]. More recent evidence indicates that the cost of disability associated with perinatal brain injury continues to rise, and that prevention of such injury would substantially reduce the socio-economic burden on affected individuals, their families and society [7].
The only commonly used treatment for targeting inflammation, namely corticosteroids (glucocorticoids), may exacerbate brain injury and increase the risk of cerebral palsy [8]. Magnesium sulphate for preterm neuroprotection, currently recommended for maternal administration when preterm labour is expected before 30 weeks of gestation, may in part act through inhibition of the NF-κB inflammatory pathway [9, 10]. However, recent follow-up studies to school age suggest it does not significantly improve longer-term neurodevelopmental outcomes compared to placebo [11, 12], although these studies are relatively small due to incomplete follow-up. Conversely, both small and large animal studies suggest that therapeutic hypothermia is not neuroprotective after exposure to perinatal infection/inflammation at term [13,14,15,16]. Collectively, these data suggest that current therapeutics aimed at improving neurodevelopmental outcomes in preterm and term infants are at best partially effective, and that development of targeted anti-inflammatory treatments is an important area of unmet medical need [17, 18].
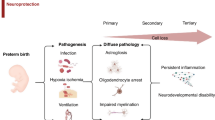
There is strong evidence that chronic inflammation related to perinatal infection and hypoxia–ischaemia can independently or synergistically cause inflammation in the fetus and neonate [19, 20]. In recent cohort studies, long-term neurodevelopmental disturbances were associated with chronic systemic inflammation and diffuse injury in the white matter tracts in both term and preterm infants [2, 6, 21,22,23,24]. As previously described, both systemic and central nervous system inflammation are strongly associated with cell death, dysmaturation and disturbed neuronal and oligodendrocyte development and reductions in brain growth [25,26,27,28]. These disturbances in white and grey matter development at the cellular level likely underpin altered brain microstructure, reduced white and grey matter volumes [29, 30] and long-term behavioural and intellectual disabilities after exposure to perinatal inflammation.
Despite this strong preclinical evidence that exposure to inflammation does trigger brain injury, and encouraging preclinical studies, no anti-inflammatory interventions have been shown to prevent clinical perinatal brain injury. In part this reflects confusion about the most appropriate drug targets and lack of clarity on the most appropriate preclinical studies to provide a foundation for safety and efficacy trials in humans. In this systemic review we aimed to evaluate the rigour of preclinical studies undertaken in the last 10 years that investigated potential immunomodulatory therapeutics to reduce perinatal inflammation-induced brain injury. A secondary aim was to determine the current knowledge gaps for clinical translation of the identified therapeutics.
Analysis strategy
Search method
This systematic review was conducted according to the Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) guidelines [31] (Additional file 1: Table S1 and Additional file 2: Table S2). The protocol was developed and registered with the International Prospective Register of Systematic Reviews (PROSPERO; registration number CRD42023395690).
Searches were conducted using Pubmed, Medline (OvidSP) and EMBASE databases for publications between January 2012 and July 2023. The following search terms: (preterm brain injury OR perinatal encephalopathy OR neonatal encephalopathy) AND (anti-inflammatory) were utilised. Other sources used to identify studies included relevant manuscripts and reviews. Reviews, conference abstracts, and articles written in a language other than English or for which no translation was available were excluded. Search results for both databases were collated, and duplicate articles were manually removed. Abstracts were identified and screened by an unbiased investigator (SBK) and duplicated by another investigator (NTT).
Selection criteria
Studies were deemed eligible if they met the following criteria: (1) conducted in an in vivo model of preterm/term equivalent age; (2) intervention possesses immunomodulatory or antimicrobial effects, or exclusively impacts immune activation (Table 3); (3) clear histological (based on the assessment of tissue inflammation and injury) and/or functional outcomes are reported; and (4) comparison to a vehicle control group is made. Studies were excluded if they: (1) were conducted in vitro; (2) did not meet the age criteria (i.e. were conducted in adult/paediatric equivalent subjects); (3) tested drugs reported to have therapeutic impacts beyond immunomodulation; (4) did not report outcomes relating to neuroinflammation and related brain injury, or (5) did not include appropriate control groups. In vitro studies were excluded from this analysis due to their limited ability to capture complex interactions between systemic immune activation and brain pathophysiology.
Data extraction
Studies were grouped by therapeutic agent and then further subdivided by species, age, type of insult to induce inflammation/injury, treatment and dosing regimen, extent of temperature monitoring, subject sex and main study outcomes (pathological/functional) and outcome (protection/no protection). The (SYstematic Review Centre for Laboratory animal Experimentation) SYRCLE risk of bias tool, described below, was used to evaluate the potential for individual study bias.
Studies were assessed according to the extent of temperature control, whether the insult and treatment were randomised, whether investigators were blinded to the intervention during histological and or functional assessments, and whether males and females were included in the analysis.
Studies were defined as being neuroprotective if there was a statistically significant improvement (P < 0.05) in brain histopathology and/or functional outcomes in the insult group that received treatment compared to the insult group that received vehicle/placebo.
Risk of bias
A risk of bias assessment for the selected studies was conducted using the SYRCLE Risk of Bias (RoB) tool [32]. The SYRCLE’s RoB tool assesses the quality of animal studies (e.g. randomisation and blinding procedures in study design) to critically appraise the preclinical research methodology. The 10 RoB assessment domains were scored as either “yes” for low risk of bias, “no” for high risk of bias, or “unclear” if the experimental methods did not explicitly address the domain assessment (Table 3).
Results
We identified 808 relevant records. After excluding reviews, duplicates, and records for which the full text was not available, we screened a total of 764 records and excluded 724 for one or more of the following reasons: ex vivo studies, inappropriate developmental age, brain histology and functional outcomes were not examined, or the therapeutic under investigation did not explicitly affect the immune system. A total of 40 publications investigating 19 categories of therapeutic were included in this analysis (Figs. 1 and 2). Publications that used more than one model of injury or showed different outcomes based on different treatment regimens (e.g. different drug dose and timing of drug delivery) were further subdivided into individual studies. The original 40 publications were thereby subdivided into 59 individual studies, which are summarised in Table 1.

Flowchart illustrating the number of papers identified through database searching and other relevant sources, the number of full text articles screened, assessed, and excluded, and the final number of original papers surveyed. Publications that used more than one paradigm of encephalopathy or multiple treatment regimens were further subdivided if outcomes differed according to experimental paradigm or treatment regimen. After subdividing these publications there was a total of 59 individual studies. The studies are summarised in Table 1

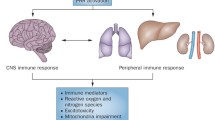
Outline of systemic and central nervous system inflammatory responses targeted by the immune-based therapeutics identified in this systematic review. Created with BioRender.com
Preclinical models of neuroinflammation
Fetal or neonatal rodents (rats or mice) were the predominant species used (n = 47 studies). Eight studies used fetal rodents from embryonic days 15–20, broadly corresponding to the neural development of human infants at < 22 weeks of gestation [33, 34]. The 35 postnatal rodent studies ranged from postnatal days (P) 0–11. Eight studies used rodents between P 0–6, which is broadly comparable to human brain development at 22–32 weeks of gestation. Fifteen studies used rodents at P7, which is comparable to the preterm human brain at approximately 30–34 weeks. Sixteen studies used rodents at P 9–11, which is broadly comparable to human brain development at term [33, 34]. There were 12 large animal studies: six studies used fetal sheep at 0.7 of gestation, which is comparable to the preterm human brain at approximately 30 weeks of gestation [35, 36]. One used term neonatal piglets (postnatal day 1) and 5 used fetal sheep at 0.8–0.9 of term gestation; these ages are comparable to neural maturation in the term human brain [35, 36].
Fourteen individual methods of causing inflammatory injury were identified (Fig. 3A). Studies were divided into three categories: inflammation initiated by pathogen-associated molecular patterns (infection-related inflammation, n = 17), inflammation initiated without pathogen-associated molecular patterns (non-infection related inflammation, n = 35), and combined infection- and not infection-related inflammation (n = 7). Twenty-three studies provoked neuroinflammation using the Rice–Vannucci model of carotid artery ligation followed by a period of moderate hypoxia. One study used neonatal hypoxia [37], and two studies used bilateral carotid artery occlusion [38, 39]. Five studies used umbilical cord occlusion in fetal sheep [40,41,42,43,44], one study used spontaneous fetal growth restriction in neonatal piglets [45] and 2 studies induced fetal inflammation by injecting IL-1β between the fetal membranes [46]. Eleven studies induced inflammation using the Gram-negative bacterial cell wall component lipopolysaccharide (LPS); four administered LPS maternally (using either intrauterine or intraperitoneal injection) [46,47,48,49,50] and seven infused LPS directly to the fetus or newborn using either single intracerebral, intracisternal or intraperitoneal injection to the neonate [51,52,53,54] or repeated fetal i.v. LPS infusions [55, 56]. Five studies used intracisternal injection of live S. pneumoniae to the newborn [57]. Seven studies combined either intraperitoneal injection of live S. epidermidis (n = 2) [58] or LPS (n = 5) with neonatal hypoxia–ischaemia [50, 59,60,61].

A Number of studies (n) which promoted inflammation using infection related, non-infection related and combined infection and non-infection related techniques and whether they showed the intervention to be neuroprotective (white) or not neuroprotective (black). B The number of studies (n) that showed neuroprotective outcomes (white) versus the number of studies that were not protective (black) for each therapy
Therapeutic doses, regimens, outcomes, and survival times
Twenty anti-inflammatory/immunomodulatory therapies in 17 categories were investigated. A description of each therapy, a summary of the number of studies that reported neuroprotection vs. no protection for each therapy are outlined in Table 2 and Fig. 3B, respectively.
Nine studies started the intervention before the insult (Fig. 4A), and 50 studies administered the intervention after the insult. For the latter approach, most studies started treatment either within the first hour (n = 20/50, 40%) or between 1 and 6 h after the insult (n = 21/50, 42%) (Fig. 4B). Only 4 studies started treatment between 1 and 3 d after the insult, of which 3 reported neuroprotection and 1 reported increased injury after treatment (Fig. 4B). The treatment dose, regimen and survival times (Fig. 4C) varied markedly. The main outcomes for each therapy are described below and in Table 1, in order of least-to-most effective, according to the proportion of studies that reported no improvement or deleterious outcomes vs. those that showed improved outcomes, as indicated by brain histopathology or behavioural assessment.

A The number of studies (n) that showed neuroprotection (white) or no protection (black) after administering the treatment at ≤ 1 h, 4 h, 14 h, 2 d or 3 d before the insult. B The number of studies (n) that showed neuroprotection (white) or no protection (black) after administering the treatment at < 1 h, 1–6 h, 18 h, 1 d, 2 d, or 3 d after the insult. C The number of studies (n) that showed neuroprotection (white) or no protection (black) stratified by survival time after the insult
One study administered the anti-fungal treatment fluconazole (Table 2) to the fetus 2 days after exposure to intra-amniotic Candida albicans and showed increased neuroinflammation and oligodendrocyte loss (P < 0.05, Kruskal–Wallis with Dunnett’s post hoc test) [61] Table 1).
Ten studies tested corticosteroids (hydrocortisone, dexamethasone or betamethasone, Table 2). The less potent corticosteroid, hydrocortisone, in a dose of 10 µg given intracerebroventricularly 2 h after HI was associated with reduced infarct size at 2 days (P < 0.05, one-way ANOVA with Newman–Keuls post hoc) [62]. Similarly, reduced infarct size after 2 days was seen with 300 µg given intranasally 2 h after HI. However, protective or injurious effects were not seen with lower or higher intranasal doses (50–1000 µg) (P < 0.05, one-way ANOVA with Newman–Keuls post hoc) [62] (Table 1). Repeated i.p. doses of dexamethasone (range: 0.1–0.5 mg/kg) given 4 days before HI were associated with increased neuronal cell death after 1 day recovery (P < 0.05) [63]. A single intracerebral injection of dexamethasone or betamethasone given 1 h before LPS was associated with improved histological and behavioural outcomes (P < 0.05, one-way ANOVA with Tukey’s post hoc) [54]. However, a single intranasal dose of dexamethasone (0.1 µg) given 2 h after HI was not associated with improved outcomes after 2 days [62].
In preterm fetal sheep, a single 12 mg i.m. dose of maternal dexamethasone given either 4 h before or 15 min after global HI was associated with increased electrographic seizures (P < 0.05, repeated measures ANOVA with Fisher’s LSD post hoc) and increased white and grey matter injury after 7 days (P < 0.05, three-way ANOVA) [43, 44] (Table 1). Similarly, maternal betamethasone (11.2 mg at 48 and 24 h before preterm birth) was associated with increased inflammation, oxidative stress and vascular extravasation in neonatal lambs exposed to high tidal volume ventilation (P < 0.05, two-way ANOVA with Holm–Sidak post hoc test) [64] (Table 1).
Four studies investigated giving repeated doses of inter-alpha inhibitor proteins (a serine protease inhibitor; Table 2) (30 mg/kg i.p.) to the neonate. Two showed no improvement in histology (at 3 days) and behavioural outcomes (at ~ 16 weeks), respectively, with treatment started from one to six hours after HI (P < 0.05, one-way ANOVA) [65, 66]. In contrast, two studies showed reduced tissue loss (P < 0.05, one-way ANOVA with Fisher’s LSD post hoc) and improved memory at 3 days and ~ 13 weeks (P < 0.05, repeated measures ANOVA with Tukey’s post hoc), respectively, when treatment was started within the first hour after HI, although improved outcomes were only seen in male offspring [65, 66] (Table 1).
Two studies used the complement inhibitor RLS0071 (Table 2). Both studies gave single or repeated doses of 10 mg/kg i.p., starting 1 h after HI. One showed no improvement in histological outcomes after 2 days (P < 0.05, paired T-test and ANOVA) [67]. One showed reduced cortical infarct area 2 days after HI when complement inhibitor was combined with therapeutic hypothermia (compared to hypothermia alone) (P < 0.05, paired T test and ANOVA) [67] (Table 1).
Two studies used a toll-like receptor 7 (TLR7) agonist (gardiquimod (GDQ), Table 2) at a dose of 1.8 mg/kg via fetal intracerebroventricular infusion from 1 h after global HI. Improved neuronal and oligodendrocyte survival were seen 3 days after treatment (P < 0.05, two-way ANOVA with Fisher’s LSD post hoc) [41], whereas there was delayed onset of epileptiform discharges and no overall histological improvement after 7 days recovery (*P < 0.05, repeated measures ANOVA with Fisher’s LSD post hoc) [42] (Table 1).
Eight, studies tested antibiotics for induced bacterial infection. One study used a single i.p. dose of 15 mg/kg vancomycin (Table 2) given 2 min after neonatal S. epidermidis inoculation combined with HI. Treatment was associated with attenuated brain tissue loss 9 days later (P < 0.05, Kruskal–Wallis test with Dunn’s post hoc) [58]. In the same animal model, combining pentoxifylline (40 mg/kg i.p.) with vancomycin did not augment vancomycin-induced protection (P > 0.05, Kruskal–Wallis test with Dunn’s post hoc) [58] (Table 1). One study used 10 mg/kg doxycycline i.p. given 1 h after HI and showed reduced lesion size and neuronal loss after 42 days (P < 0.05, Mann–Whitney U test) [68]. One study administered ceftriaxone (Table 2) at a dose of 100 mg/kg i.p. 18 h after intracisternal S. pneumonia inoculation. Treatment was associated with increased neuronal loss after 42 h (P < 0.05, Mann–Whitney test), and reduced learning and memory after 3 weeks (P < 0.05, two-way ANOVA) [57] (Table 1). One study used 100 mg/kg ceftriaxone i.p. combined with the non-bacteriolytic antibiotic daptomycin 10 mg/kg s.c. given 18 h after intracisternal S. pneumonia inoculation and showed reduced cortical necrosis after 42 h (P < 0.05, Mann–Whitney test) [57] (Table 1). Three studies combined ceftriaxone i.p. with daptomycin 10 mg/kg s.c. and 2 doses of the matrix metalloprotease inhibitor trocade (75 mg/kg) given 24 h apart starting at 18 h after intracisternal S. pneumonia inoculation. This treatment regimen was associated with reduced hippocampal apoptosis and cortical necrosis after 42 h (P < 0.05, Mann–Whitney test), and improved hearing, learning and memory at 3 weeks (P < 0.05, two-way ANOVA) [57] (Table 1).
Three studies used a single dose of a nuclear factor kappa B (NF-κB) inhibitor (Tat-NBD, Table 2) delivered intranasally to the neonate at a dose of 1.4 mg/kg 10 min after the insult. Two showed reduced tissue loss after 7 days in rat pups exposed to a combination of HI and LPS and one showed no improvement in histological outcomes in pups exposed to HI alone (P < 0.05, unpaired t-test or one-way ANOVA with Newman–Keuls post hoc) [59] (Table 1).
Four studies tested fingolimod (FTY720, Table 2), a sphingosine-1-phosphate receptor modulator [69]. Of these, two gave it antenatally to the mother, as a single dose of 1 mg/kg i.p. immediately or 30 min after maternal LPS-exposure and showed improved histological outcomes (reduced markers of inflammation in the white matter and cortex) after 6- and 4-h recovery, respectively (P < 0.05, Mann–Whitney test) [47, 48]. Two studies gave fingolimod to the neonate via single or repeated doses (0.3–1 mg/kg, i.p.). The 1 mg/kg dose was associated with worse histological outcomes (increased cortical tissue loss) compared to vehicle 7 days after HI (P < 0.05, unpaired t-test.), whereas 0.3 mg/kg was associated with reduced total seizure duration and improved behavioural outcomes at 7 weeks after HI (P < 0.05, two-way ANOVA with Tukey’s post hoc) [37, 70] (Table 1).
One study used repeated doses of a glycogen synthase kinase 3 β (GSK3β) inhibitor (SB216763, Table 2) at 10 mg/kg i.p. to the neonate from 14 h before the insult and showed reduced tissue loss at 7 days after HI (P < 0.05, one-way ANOVA with Holm–Sidak’s post hoc) [71] (Table 1).
One study gave an innate defence regulator protein 1018 (IDR-1018, Table 2) in a single dose (8 µg/g i.p.) to the neonate at 3 h after LPS + HI and showed reduced white and grey matter tissue loss 7 days after treatment (P < 0.05, t test) [60] (Table 1).
One study used single intracisternal infusion of lipoxin A4 (Table 2) at a dose of 10 mg/kg starting 1 h after HI and showed reduced infarct area and improved motor function and cognition at 24 h and 3 weeks, respectively (P < 0.05, one-way ANOVA with Tukey’s post hoc) [72] (Table 1).
Recombinant human IL-35 was administered i.v. to the neonate at the time of HI and 1 day later, reduced infarct volume was shown 2 days after treatment (P < 0.05, one-way ANOVA with Tukey’s post hoc) [73] (Table 1).
Two studies administered a single dose of a melanocortin receptor 1 agonist (BMS-470539, Table 2) intranasally at 1 h after HI [74]. The concentrations tested ranged from 50 µg/kg to 500 µg/kg, with survival times between 2 days and 4 weeks. Outcomes were dose dependent; 50 µg/kg did not improve outcomes, whereas 500 µg/kg and 160 µg/kg reduced infarct area and improved sensorimotor function at 2 days and 4 weeks, respectively (P < 0.05, one-way ANOVA or Student t-test with Tukey’s post hoc) (Table 1).
Three studies used cyclooxygenase 2 (COX2) inhibitors (ibuprofen and celecoxib; Table 2) administered to the neonate via single or repeated doses of 10–20 mg/kg from 5 min to 2 h (i.p.) after LPS exposure, or 1 day (oral) after delivery in a model of spontaneous growth restriction. One showed improved histological outcomes (reduced inflammation and improved white and grey matter integrity) and motor function after one day (P < 0.05 one-way ANOVA with Student–Newman–Keuls post hoc) [53], one showed reduced inflammation in the frontal cortex after 10 days (P < 0.05, t-test) [75], and one showed reduced white matter gliosis, improved myelination and neuronal survival after three days (P < 0.05, two-way ANOVA with Holm–Sidak post hoc) [45] (Table 1).
Three studies used either granulocyte (G-CSF, Table 2) or colony stimulating factor 1 (CSF-1/M-CSF, Table 2). When G-CSF was given as a single i.p. 50 µg/kg dose intraperitoneally at 1 h after HI, improved blood brain barrier integrity and reduced inflammation were reported after 2 days recovery (P < 0.05, one-way ANOVA with Tukey’s post hoc) [76]. CSF-1 was given via repeated doses of 80 µg/kg intranasally at 1 and 24 h after HI. Reduced and sensorimotor and cognitive function were shown after 2 days and 4 weeks recovery, respectively (P < 0.05, one-way ANOVA with Tukey’s post hoc) [77] (Table 1).
Etanercept, a soluble TNF receptor (Table 2) that inhibits TNF activity, was administered directly to the fetus or neonate in three studies. A single i.p. dose (5 mg/kg) was associated with improved white matter integrity 1 day after hypoxia–ischaemia (HI) (P < 0.05, ANOVA with Bonferroni post hoc) [52]. One study gave repeated doses of etanercept i.v. to fetal sheep (5 mg/kg) starting immediately after LPS exposure and one study administered it to the fetal sheep brain via repeated intracerebroventricular infusions (1 mg) starting from 3 days after HI. Both showed improved reduced neuroinflammation and reduced white matter injury (*P < 0.05, two-way ANOVA with Fisher’s LSD post hoc) and or reduced suppression of electroencephalogram power (repeated measures ANOVA with Fisher’s LSD post hoc) at 10 days and 3 weeks [40, 56] (Table 1).
For studies targeting IL-1, eight studies gave IL-1 receptor antagonists (anakinra or 101.10, Table 2) at doses of 1 to 13 mg/kg. Three treated prophylactically, i.e. starting before the insult, and used single dosing in fetal mice. All showed improved histological (reduced markers of neurotoxicity and improved microvascular integrity) (P < 0.05, Kruskal–Wallis one-way ANOVA) and functional outcomes (improved visual evoked potentials) (P < 0.05, Kruskal–Wallis one-way ANOVA with Dunn’s post-test) in the offspring when assessed at 4–6 h, and 15–30 days after the insult, respectively [46, 49] (Table 1). Three studies treated the neonate directly using repeated doses started immediately after the insult in postnatal mice exposed to maternal LPS and or neonatal hypoxia. All showed improved histological (P < 0.05, ANOVA with Newman–Keuls post hoc test) and functional outcomes (P < 0.05, unpaired t-test with Welch correction) 40 days later [50] (Table 1). One study gave three doses of anakinra between 5 min and 22 h after the insult to LPS-treated male rat pups and showed improved histological and MRI outcomes 1 day later (P < 0.05, Kruskal–Wallis tests with Dunn’s multiple comparisons) [51]. One study gave anakinra 1 h after progressive repeated LPS exposure in fetal sheep and showed both improved histological (P < 0.05, two-way ANOVA with Fisher’s LSD) and functional (improved electroencephalogram power, P < 0.05 two-way ANOVA with repeated measures) after 4 days [55]. Two studies gave one or two doses of 5.1 to 7.7 mg/kg to the fetus of a mouse anti-ovine IL-1β monoclonal antibody (Table 2) starting 15 min after the insult. Both showed improved histological outcomes (blood brain barrier penetration and reduced grey matter apoptosis) after 1-day recovery (P < 0.05, P < 0.05, one-way ANOVA with FSD post hoc) [38, 39] (Table 1).
Temperature monitoring
Ten out of 59 studies (16%) reported monitoring core temperature during the study (Table 1, Fig. 5A). Of these, 5 reported maintaining core temperature during the insult (HI) but not during recovery. One study reported temporal core temperature data throughout the experimental period [58]. Twenty-three studies reported maintaining ambient air temperature (range: 28–38 ℃) during the study period, 18/23 (78%) reported neuroprotection. Twenty five studies did not report temperature monitoring as part of their study protocol, however 11/25 studies (44%) were conducted in fetal sheep, where fetal core temperature is maintained in utero between 39.0 and 39.5 ℃ by the intrauterine environment [78, 79]. An overview of type of temperature control for the studies included can be seen in Fig. 5A.

A The number of studies (n) that showed neuroprotection (white) or no protection (black) and monitored ambient temperature, core temperature, or did not report temperature monitoring. B The number of studies (n) that showed neuroprotection (white) or no protection (black) which reported outcomes in both males and females (♂ + ♀), males only (♂), females only (♀), or did not report the sex of the subjects
Subject sex
Forty-one out of 59 studies (69%) reported outcomes in both sexes, but 11 of these studies did not report numbers or ratios of males and females (Table 1, Fig. 5B). Four out of 59 studies reported outcomes in males only [51, 63, 66]. Of these, 2 reported improved outcomes, one reported no improvement and one reported worse outcomes with treatment. Fifteen out of 59 studies (25%) did not report the sex of the subjects, of these, 14/15 studies showed improved outcomes (Fig. 5B).
Study bias
The SYRCLE RoB tool [32] was used to measure risk of study bias (Table 3). Thirty out of 40 papers stated that allocation to groups was random, although only two papers gave specific details relating to how the randomisation was performed [47, 74]. Nine out of 40 papers reported the baseline characteristics of the groups analysed. No studies explicitly reported randomly housing animals during the experiment or noted whether the caregivers and examiners were blinded to treatment groups. Seventeen out of the 40 papers (42%) did not report blinding of the assessor/s during the analysis, while one paper reported conducting a random outcome assessment [65]. Seventeen out of 40 papers (42%) did not address incomplete outcome data and were therefore at risk of attrition bias. All papers appeared to be free from selective outcome reporting (Table 3).
Discussion
Perinatal inflammation is a major cause of neurodevelopmental impairments in preterm and term infants [25, 26]. Developing effective therapeutic interventions for the ‘at risk’ fetus or neonate requires that we improve our understanding of the pathophysiological mechanisms that lead to neurodevelopmental impairments, identify therapeutic targets, and test pharmacological interventions in a translational research pipeline that incorporates high quality small and large animal trials. In this systematic review, we set out to identify which immunomodulatory interventions have been trialled between 2012 and 2023 for inflammation-induced brain injury and determine key knowledge gaps in the literature that need to be addressed in animal studies before progressing potential therapies into human trials for perinatal neuroprotection.
Modelling perinatal infection/inflammation
There is compelling evidence that both mild and moderate-to-severe HIE and infection/inflammation are highly associated with microgliosis and activation of distinct inflammatory pathways in the peripheral and central nervous system, as previously reviewed [19, 26, 80]. Most of the studies surveyed here (59%) used models of ‘non-infection’ related inflammation (hypoxia with or without ischaemia). A few studies modelled ‘infection’ related inflammation (28%) or combined ‘infection and non-infection’ related insults (12%). None tested interventions in the setting of Gram-positive infection, such as mycoplasmas (e.g. Ureaplasma spp.), which are among the most common bacterial isolates in pregnancies complicated by chorioamnionitis (fetal infection/inflammation), preterm birth [81], and neurodevelopmental impairment. For example, amniotic fluid cultures that are positive for Ureaplasma urealyticum are associated with a higher risk of adverse psychomotor development, abnormal neurological outcome and a higher risk of cerebral palsy at 2 years of age compared to patients with negative amniotic fluid cultures [82].
None of the studies surveyed used polymicrobial models of inflammation. There is emerging evidence that multiple bacteria and viruses reside in the placenta and amniotic fluid, raising the possibility that, at least in some cases, there may be a polymicrobial aetiology to perinatal infection/inflammation-induced impairments in brain development [83,84,85,86,87]. This concept is supported by studies in animal models that show combining viral and bacterial inflammation in pregnant mice is associated with increased rates of preterm birth, tissue inflammation and necrosis relative to either inflammatory stimulus alone [88, 89]. Furthermore, few studies modelled repeated fetal or neonatal infection/inflammation. Repeated infections occur in approximately two thirds of preterm infants ≤ 30 weeks of gestation and are associated with an increased risk of white matter abnormalities and mortality [90, 91]. Another consideration is that none of the studies surveyed tested immunomodulators in models of viral infection. This highlights another important knowledge gap given the strong association between congenital infections with viruses, such as cytomegalovirus herpes simplex virus type 1 and severe acute respiratory syndrome coronavirus 2, and long-term neurological sequelae [92,93,94,95].
Controlling for iatrogenic hypo/hyperthermia
Most publications (n = 47/59, 79%) used neonatal rodents. Rigorous studies in neonatal rodents offer many advantages for neuroprotection research, as previously highlighted [96]. However, due to their small body mass relative to surface area, lack of subcutaneous fat, naked skin and limited shivering response, neonatal rats produce less heat and lose more body heat than adults [96, 97]. These factors make them functionally poikilothermic and susceptible to rapid changes in body and brain temperature during changes in environmental temperature [98]. Small changes in body temperature are known to affect neurological outcomes in animal and human studies [96, 99, 100]. Furthermore, as previously reviewed, neuroprotective effects of various pharmacological interventions, including anaesthetics, can be confounded by drug-induced hypothermia mediated by increased heat loss [100]. Conversely, neuroprotection can be masked by delayed hyperthermia [101, 102]. Thus, care is required to ensure that iatrogenic changes in body temperature do not occur to ensure that outcomes are not confounded by unappreciated changes in body temperature or environmental conditions.
Of concern, only 16% of studies published since 2012 measured core temperature; half of these studies measured core temperature during the insult, and one explicitly reported temperature data after treatment [58]. Most of the studies measured environmental temperature which ranged from 28–38 ℃ (− 4 to 0 ℃ below core temperature). We identified 4 studies that used maternal LPS exposure to model antenatal infection/inflammation, all reported modest improvements in neurological outcomes, but none monitored maternal body temperature. Two of these studies administered fingolimod, a peripheral vasodilator [103], to the mother. One study did not state whether temperature was maintained, the other reported maintaining ambient temperature between 21 and 22 ℃. This is an important consideration since maternal LPS exposure is commonly associated with pyrexia. Intrapartum fever is associated with adverse neonatal outcomes and increased risk of cerebral palsy and neonatal encephalopathy [104], likely mediated by a combination of increased release of oxygen free radicals and excitatory neurotransmitters, enhanced glutamate toxicity on neurons and glia, blood brain barrier dysfunction and proteolysis [105]. Thus, it is not possible to know whether neuroprotective effects of fingolimod were mediated by iatrogenic hypothermia in the pregnant dams or direct anti-inflammatory effects of fingolimod.
Of the 25 studies that did not report controlling body temperature, 10 were conducted in fetal sheep. A major advantage of testing potential neuroprotectants in fetal sheep is that their body temperature is regulated by the pregnant ewe and therefore unless the ewe is febrile, fetal core temperature is highly stable [78, 79]. Collectively these observations highlight the need for animal studies to improve core temperature monitoring throughout the experimental period to ensure that outcomes of preclinical drug trials are not confounded by fluctuations in maternal, fetal or neonatal body temperature.
Limitations of current immunomodulatory therapies: corticosteroids and antibiotics
Currently there are no clinically proven treatments to prevent infection/inflammation related brain injury. Of the immunomodulatory interventions identified in this systematic review corticosteroids and antibiotics are among the most routinely used interventions in perinatal medicine. In our analysis, the corticosteroids dexamethasone and betamethasone showed the least promising outcomes, with 5/10 (50%) of studies reporting either no improvement or deleterious effects. Indeed, in human studies corticosteroids have been associated with exacerbation of perinatal brain injury, including increased risk of both intraventricular haemorrhage, cerebral palsy and hyperactivity in childhood [8, 106]. The potential for corticosteroids to cause deleterious effects in the perinatal brain are postulated to relate to the stage of neurodevelopment at the time of exposure, the dose and duration of exposure relative to the timing of the insult [107], and their potential to cause hyperglycaemia, which animal and human studies have shown to augment encephalopathy after HI [44, 108]. Furthermore, meta-analysis suggests that prophylactic antibiotics given to women at risk of preterm labour with ruptured membranes are associated with an increased risk of neonatal death and disability [109]. These observations are supported by animal studies, for example treating pregnant rabbits with antibiotics 24 h after intrauterine E. coli administration was associated with improved survival but increased white matter cell death [110]. The mechanisms for this are unclear, however it is possible that bacterial lysis promotes the release of bacterial fragments that augment inflammation-induced injury.
Consistent with this hypothesis, we identified two studies in this review that showed increased injury with stand-alone antibiotic or anti-fungal treatments [57, 61]. By contrast, combining antibiotics with the matrix metalloproteinase-9 inhibitor trocade was associated with improved outcomes, suggesting that in cases of fetal or neonatal infection combining antibiotics with an anti-inflammatory intervention could be a more effective approach [57]. Conversely, another study showed that combining antibiotics with the phosphodiesterase inhibitor pentoxifylline did not augment vancomycin-induced protection against Gram-positive bacterial infection, indicating that targeting the right anti-inflammatory mechanism/s to augment antimicrobial treatment is an important consideration [58]. In this analysis anti-cytokine therapies, particularly those targeting the primary effector cytokine IL-1, were most associated with improved outcomes in models of both infection related inflammation and non-infection related inflammation. This raises the possibility that use of anti-cytokine therapies alone or as an adjuvant to antibiotic therapy could be an effective approach to prevent or mitigate inflammation-induced injury in the perinatal brain.
Who are we treating and when are we treating them?
A key translational consideration for testing potential neuroprotectants is who and when to treat. A minority of therapeutics (8/20; 40%) identified in this review were tested across multiple preclinical models of infection related, non-infection related or combined inflammation. Almost half of the studies (29/59; 46%) started the intervention before or immediately after the insult (within 60 min). Whilst this approach provides useful insight into the early pathophysiology of injury, it unlikely to be practical for clinical translation. Clinically, it is difficult to identify fetuses who are at risk of injury since the positive predictive value of fetal heart rate monitoring and biophysical profiling for predicting adverse neurodevelopmental outcomes is low [111, 112]. Similarly, early neonatal cranial ultrasound is not reliable at detecting ongoing diffuse white matter injury. Instead, its validity has been shown in the setting of advanced severe cystic white matter injury, which is now less common than diffuse non-cystic injury [113,114,115]. Diffusion magnetic resonance imaging (MRI) has been shown both in preclinical models and in preterm infants to accurately detect acute white matter injury [116,117,118,119,120,121]. However, it is not feasible to systematically screen all high-risk infants with diffusion MRI in the first few days after birth. Twenty-one out of 59 studies (35%) started the intervention between 1 and 6 h, and 9 studies (15%) started treatment between 18 h and 3 days after the insult. Ideally, pharmacological interventions need to be administered around the time of bulk cell death/injury, which primarily occurs within hours to days after the insult. As well, there is evidence that chronic inflammation makes a contribution to the sub-acute and chronic phases of injury, which develop several days to weeks after the initial insult [9, 122,123,124]. This suggests that delayed use of immunomodulatory interventions, alone or in combination with interventions that target other pathways of cell damage or repair (e.g. antioxidants, trophic factors, stem cells or stem cell secretomes), could be an effective strategy to mitigate delayed or tertiary brain injury. Ultimately, this raises the need to identify biomarkers of evolving brain injury to facilitate early treatment [125,126,127], along with understanding the therapeutic window of opportunity for potential interventions in carefully designed animal trials to progress promising therapies from the animal lab to the bedside.
Assessment of long-term functional and histological outcomes
Another important limitation of the studies identified in this review is that most studies (61%) used survival times of ≤ 7 days, and less than half (22/59 studies; 37%) assessed functional outcomes. Indeed 40% of studies used survival times of hours to 2 days after the insult. Short survival times provide important information about acute histological and functional outcomes, but it is well established that injury evolves many days–weeks after the insult [9, 123, 128] and that functional and histological outcomes are sometimes discordant [96]. Twenty-one out of 59 studies (35%) assessed outcomes beyond 1 week, most (18/21 studies; 85%) reported neuroprotection, however none reported measuring core temperature during treatment or beyond the initial insult. Thus, assessment of histological and functional outcomes in studies beyond the first few hours to days after the insult is an important consideration for future animal trials designed to evaluate the efficacy of potential therapeutics.
Controlling for potential effects of subject sex on neurological outcomes
Most studies (40/59) reported using subjects of both sexes in the experimental design, however 11 of these studies did not report numbers or ratios of males and females. The remaining studies either did not report the sex of the subjects or tested interventions in males only. Studies investigating the impact of infectious and non-infectious insults have reported sexual dimorphisms in the severity and evolution of immune responses [129], perinatal brain injury [130, 131] and responses to treatment [132, 133]. Four of the 59 studies only used male subjects in their experimental protocol. In addition, only eight studies accounted for sex in outcome reporting. Of these, two stated that a post hoc analysis was performed to assess sex differences between the groups [46]. The remaining five studies reported sex differences as primary outcomes [37, 62, 65], and showed a bias towards neuroprotective effects in males. It remains unclear whether similar differences exist in human trials [11, 134, 135]. Overall, these data raise the need for greater emphasis on evaluating the impact of sex in future animal studies.
Risk of bias
To evaluate study bias, we used the SYRCLE risk of bias assessment tool. No studies reported random housing of animals. This is a particularly important consideration for small animal (rodent) studies. For example, there is compelling evidence that differences in light exposure, which may vary with respect to rack location, can affect reproduction and behaviour [136, 137]. Additionally, ambient temperature can vary with respect to position of the cage with ambient temperature in the top cage being up to 5℃ higher than the bottom cage [138]. Seventeen out of 40 papers (42%) did not report blinding of examiners during outcome assessments, and 17/40 papers (42%) did not state whether incomplete outcome data were addressed. Only 9/40 papers (23%) reported baseline characteristics, raising the possibility that potential confounders (e.g. unequal distributions of sex, body weight, relevant physiological parameters) may not have been addressed in the analysis. Collectively these data highlight possible inconsistencies in the quality of the data surveyed. We cannot definitively conclude that the methodological issues identified in our analysis affected the outcomes of the studies. Nevertheless, if this critical information is not reported or accounted for in publications, it is difficult to assess the significance of past and future studies in a meaningful way.
Conclusion
There is an important unmet need to identify and develop effective immunomodulatory interventions for the prevention of perinatal brain injury. Despite many successful preclinical trials, there are no immunomodulatory treatments for perinatal neuroprotection in clinical practice. In this systematic review, we examined preclinical publications between 2012 and 2023 and highlight opportunities to improve the way that preclinical animal trials are designed, carried out and reported to help overcome the ‘translational block’ and close the gap between animal studies and human trials for perinatal neuroprotection. Future studies should evaluate potential therapies in diverse preclinical models that replicate relevant disease pathophysiology, control for iatrogenic changes in temperature that may occur as part of the experimental insult or treatment, address pragmatic treatment regimens that are conducive to clinical application, control for potential effects of subject sex on outcomes, assess long-term functional and histological outcomes, and follow relevant guidelines that mitigate study bias.
Availability of data and materials
The datasets used during the current study are available from the corresponding author upon reasonable request.
References
Fleischmann C, Reichert F, Cassini A, Horner R, Harder T, Markwart R, Tröndle M, Savova Y, Kissoon N, Schlattmann P, et al. Global incidence and mortality of neonatal sepsis: a systematic review and meta-analysis. Arch Dis Child. 2021;106:745.
Wu YW, Colford JM Jr. Chorioamnionitis as a risk factor for cerebral palsy: a meta-analysis. JAMA. 2000;284:1417–24.
Honeycutt A, Dunlap L, Chen H, Al Homsi G, Grosse S, Schendel DE. Economic costs associated with mental retardation, cerebral palsy, hearing loss, and vision impairment–United States, 2003. MMWR Morb Mortal Wkly Rep. 2004;53:57–9.
Grether JK, Nelson KB. Maternal infection and cerebral palsy in infants of normal birth weight.[Erratum appears in JAMA 1998 Jan 14;279(2):118]. JAMA. 1998;1997(278):207–11.
Soraisham AS, Trevenen C, Wood S, Singhal N, Sauve R. Histological chorioamnionitis and neurodevelopmental outcome in preterm infants. J Perinatol. 2013;33:70–5.
Wu YW, Escobar GJ, Grether JK, Croen LA, Greene JD, Newman TB. Chorioamnionitis and cerebral palsy in term and near-term infants. JAMA. 2003;290:2677–84.
Shih STF, Tonmukayakul U, Imms C, Reddihough D, Graham HK, Cox L, Carter R. Economic evaluation and cost of interventions for cerebral palsy: a systematic review. Dev Med Child Neurol. 2018;60:543–58.
Barrington KJ. The adverse neuro-developmental effects of postnatal steroids in the preterm infant: a systematic review of RCTs. BMC Pediatr. 2001;1:1.
Galinsky R, Dhillon SK, Kelly SB, Wassink G, Davidson JO, Lear CA, van den Heuij LG, Bennet L, Gunn AJ. Magnesium sulphate reduces tertiary gliosis but does not improve EEG recovery or white or grey matter cell survival after asphyxia in preterm fetal sheep. J Physiol. 2023;601:1999.
Sugimoto J, Romani AM, Valentin-Torres AM, Luciano AA, Ramirez Kitchen CM, Funderburg N, Mesiano S, Bernstein HB. Magnesium decreases inflammatory cytokine production: a novel innate immunomodulatory mechanism. J Immunol. 2012;188:6338–46.
Doyle LW, Anderson PJ, Haslam R, Lee KJ, Crowther C, Australasian Collaborative Trial of Magnesium Sulphate Study G. School-age outcomes of very preterm infants after antenatal treatment with magnesium sulfate vs placebo. JAMA. 2014;312:1105–13.
Chollat C, Enser M, Houivet E, Provost D, Benichou J, Marpeau L, Marret S. School-age outcomes following a randomized controlled trial of magnesium sulfate for neuroprotection of preterm infants. J Pediatr. 2014;165:398-400.e393.
Falck M, Osredkar D, Maes E, Flatebø T, Wood TR, Sabir H, Thoresen M. Hypothermic neuronal rescue from infection-sensitised hypoxic-ischaemic brain injury is pathogen dependent. Dev Neurosci. 2017;39:238–47.
Osredkar D, Sabir H, Falck M, Wood T, Maes E, Flatebø T, Puchades M, Thoresen M. Hypothermia does not reverse cellular responses caused by lipopolysaccharide in neonatal hypoxic-ischaemic brain injury. Dev Neurosci. 2015;37:390–7.
Osredkar D, Thoresen M, Maes E, Flatebø T, Elstad M, Sabir H. Hypothermia is not neuroprotective after infection-sensitized neonatal hypoxic-ischemic brain injury. Resuscitation. 2014;85:567–72.
Martinello KA, Meehan C, Avdic-Belltheus A, Lingam I, Mutshiya T, Yang Q, Akin MA, Price D, Sokolska M, Bainbridge A, et al. Hypothermia is not therapeutic in a neonatal piglet model of inflammation-sensitized hypoxia-ischemia. Pediatr Res. 2022;91:1416–27.
Kelly SB, Green E, Hunt RW, Nold-Petry CA, Gunn AJ, Nold MF, Galinsky R. Interleukin-1: an important target for perinatal neuroprotection? Neural Regen Res. 2023;18:47–50.
Green EA, Garrick SP, Peterson B, Berger PJ, Galinsky R, Hunt RW, Cho SX, Bourke JE, Nold MF, Nold-Petry CA. The role of the interleukin-1 family in complications of prematurity. Int J Mol Sci. 2023;24:2795.
Galinsky R, Lear CA, Dean JM, Wassink G, Dhillon SK, Fraser M, Davidson JO, Bennet L, Gunn AJ. Complex interactions between hypoxia-ischemia and inflammation in preterm brain injury. Dev Med Child Neurol. 2018;60:126–33.
Bui CB, Pang MA, Sehgal A, Theda C, Lao JC, Berger PJ, Nold MF, Nold-Petry CA. Pulmonary hypertension associated with bronchopulmonary dysplasia in preterm infants. J Reprod Immunol. 2017;124:21–9.
O’Muircheartaigh J, Robinson EC, Pietsch M, Wolfers T, Aljabar P, Grande LC, Teixeira R, Bozek J, Schuh A, Makropoulos A, et al. Modelling brain development to detect white matter injury in term and preterm born neonates. Brain. 2020;143:467–79.
Leviton A, Paneth N, Reuss ML, Susser M, Allred EN, Dammann O, Kuban K, Van Marter LJ, Pagano M, Hegyi T, et al. Maternal infection, fetal inflammatory response, and brain damage in very low birth weight infants. Developmental Epidemiology Network Investigators. Pediatric Res. 1999;46:566–75.
O’Shea TM, Allred EN, Kuban KC, Dammann O, Paneth N, Fichorova R, Hirtz D, Leviton A. Extremely Low Gestational Age Newborn Study I: elevated concentrations of inflammation-related proteins in postnatal blood predict severe developmental delay at 2 years of age in extremely preterm infants. J Pediatr. 2012;160(395–401): e394.
O’Shea TM, Shah B, Allred EN, Fichorova RN, Kuban KC, Dammann O, Leviton A, Investigators ES. Inflammation-initiating illnesses, inflammation-related proteins, and cognitive impairment in extremely preterm infants. Brain Behav Immun. 2013;29:104–12.
Galinsky R, Davidson JO, Dean JM, Green CR, Bennet L, Gunn AJ. Glia and hemichannels: key mediators of perinatal encephalopathy. Neural Regen Res. 2018;13:181–9.
Hagberg H, Mallard C, Ferriero DM, Vannucci SJ, Levison SW, Vexler ZS, Gressens P. The role of inflammation in perinatal brain injury. Nat Rev Neurol. 2015;11:192–208.
Kelly SB, Dean JM, Zahra VA, Dudink I, Thiel A, Polglase GR, Miller SL, Hooper SB, Bennet L, Gunn AJ, Galinsky R. Progressive inflammation reduces high-frequency EEG activity and cortical dendritic arborisation in late gestation fetal sheep. J Neuroinflamm. 2023;20:124.
Prasad JD, van de Looij Y, Gunn KC, Ranchhod SM, White PB, Berry MJ, Bennet L, Sizonenko SV, Gunn AJ, Dean JM. Long-term coordinated microstructural disruptions of the developing neocortex and subcortical white matter after early postnatal systemic inflammation. Brain Behav Immun. 2021;94:338–56.
Thompson DK, Kelly CE, Chen J, Beare R, Alexander B, Seal ML, Lee KJ, Matthews LG, Anderson PJ, Doyle LW, et al. Characterisation of brain volume and microstructure at term-equivalent age in infants born across the gestational age spectrum. Neuroimage Clin. 2019;21: 101630.
Erdei C, Bell KA, Garvey AA, Blaschke C, Belfort MB, Inder TE. Novel metrics to characterize temporal lobe of very preterm infants on term-equivalent brain MRI. Pediatr Res. 2023;94:979l.
Page MJ, Moher D, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, Shamseer L, Tetzlaff JM, Akl EA, Brennan SE, et al. PRISMA 2020 explanation and elaboration: updated guidance and exemplars for reporting systematic reviews. BMJ. 2021;372: n160.
Hooijmans CR, Rovers MM, de Vries RB, Leenaars M, Ritskes-Hoitinga M, Langendam MW. SYRCLE’s risk of bias tool for animal studies. BMC Med Res Methodol. 2014;14:43.
Hagberg H, Peebles D, Mallard C. Models of white matter injury: comparison of infectious, hypoxic-ischemic, and excitotoxic insults. Ment Retard Dev Disabil Res Rev. 2002;8:30–8.
Patel SD, Pierce L, Ciardiello AJ, Vannucci SJ. Neonatal encephalopathy: pre-clinical studies in neuroprotection. Biochem Soc Trans. 2014;42:564–8.
Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Hum Dev. 1979;3:79–83.
Barlow RM. The foetal sheep: morphogenesis of the nervous system and histochemical aspects of myelination. J Comp Neurol. 1969;135:249–62.
Najafian SA, Farbood Y, Sarkaki A, Ghafouri S. FTY720 administration following hypoxia-induced neonatal seizure reverse cognitive impairments and severity of seizures in male and female adult rats: the role of inflammation. Neurosci Lett. 2021;748: 135675.
Chen X, Hovanesian V, Naqvi S, Lim YP, Tucker R, Donahue JE, Stopa EG, Stonestreet BS. Systemic infusions of anti-interleukin-1β neutralizing antibodies reduce short-term brain injury after cerebral ischemia in the ovine fetus. Brain Behav Immun. 2018;67:24–35.
Patra A, Chen X, Sadowska GB, Zhang J, Lim YP, Padbury JF, Banks WA, Stonestreet BS. Neutralizing anti-interleukin-1β antibodies reduce ischemia-related interleukin-1β transport across the blood-brain barrier in fetal sheep. Neuroscience. 2017;346:113–25.
Lear CA, Lear BA, Davidson JO, Sae-Jiw J, Lloyd JM, Dhillon SK, Gunn AJ, Bennet L. Tumour necrosis factor blockade after asphyxia in foetal sheep ameliorates cystic white matter injury. Brain. 2022;146:1453.
Cho KHT, Wassink G, Galinsky R, Xu B, Mathai S, Dhillon SK, van den Heuij LG, Davidson JO, Weaver-Mikaere L, Bennet L, et al. Protective effects of delayed intraventricular TLR7 agonist administration on cerebral white and gray matter following asphyxia in the preterm fetal sheep. Sci Rep. 2019;9:9562.
Cho KHT, Fraser M, Xu B, Dean JM, Gunn AJ, Bennet L. Induction of tertiary phase epileptiform discharges after postasphyxial infusion of a toll-like receptor 7 agonist in preterm fetal sheep. Int J Mol Sci. 2021;22:6593.
Koome ME, Davidson JO, Drury PP, Mathai S, Booth LC, Gunn AJ, Bennet L. Antenatal dexamethasone after asphyxia increases neural injury in preterm fetal sheep. PLoS ONE. 2013;8: e77480.
Lear CA, Davidson JO, Mackay GR, Drury PP, Galinsky R, Quaedackers JS, Gunn AJ, Bennet L. Antenatal dexamethasone before asphyxia promotes cystic neural injury in preterm fetal sheep by inducing hyperglycemia. J Cereb Blood Flow Metab. 2018;38:706–18.
Wixey JA, Sukumar KR, Pretorius R, Lee KM, Colditz PB, Bjorkman ST, Chand KK. Ibuprofen treatment reduces the neuroinflammatory response and associated neuronal and white matter impairment in the growth restricted newborn. Front Physiol. 2019;10:541.
Nadeau-Vallée M, Chin PY, Belarbi L, Brien M, Pundir S, Berryer MH, Beaudry-Richard A, Madaan A, Sharkey DJ, Lupien-Meilleur A, et al. Antenatal suppression of IL-1 protects against inflammation-induced fetal injury and improves neonatal and developmental outcomes in mice. J Immunol. 2017;198:2047–62.
Yavuz A, Sezik M, Ozmen O, Asci H. Fingolimod against endotoxin-induced fetal brain injury in a rat model. J Obstet Gynaecol Res. 2017;43:1708–13.
Yalcin SE, Sezik M, Yavuz A, Savran M, Asci H, Ozmen O. Combined use of magnesium sulfate and fingolimod for antenatal neuroprotection against inflammation-mediated experimental preterm brain injury in a rat model. Fetal Pediatr Pathol. 2022;41:603–15.
Leitner K, Al Shammary M, McLane M, Johnston MV, Elovitz MA, Burd I. IL-1 receptor blockade prevents fetal cortical brain injury but not preterm birth in a mouse model of inflammation-induced preterm birth and perinatal brain injury. Am J Reprod Immunol. 2014;71:418–26.
Girard S, Sébire H, Brochu ME, Briota S, Sarret P, Sébire G. Postnatal administration of IL-1Ra exerts neuroprotective effects following perinatal inflammation and/or hypoxic-ischemic injuries. Brain Behav Immun. 2012;26:1331–9.
Pierre WC, Akakpo L, Londono I, Pouliot P, Chemtob S, Lesage F, Lodygensky GA. Assessing therapeutic response non-invasively in a neonatal rat model of acute inflammatory white matter injury using high-field MRI. Brain Behav Immun. 2019;81:348–60.
Shin SH, Kim EK, Lee KY, Kim HS. TNF-α antagonist attenuates systemic lipopolysaccharide-induced brain white matter injury in neonatal rats. BMC Neurosci. 2019;20:45.
Fan LW, Kaizaki A, Tien LT, Pang Y, Tanaka S, Numazawa S, Bhatt AJ, Cai Z. Celecoxib attenuates systemic lipopolysaccharide-induced brain inflammation and white matter injury in the neonatal rats. Neuroscience. 2013;240:27–38.
Pang Y, Fan LW, Zheng B, Campbell LR, Cai Z, Rhodes PG. Dexamethasone and betamethasone protect against lipopolysaccharide-induced brain damage in neonatal rats. Pediatr Res. 2012;71:552–8.
Kelly SB, Stojanovska V, Zahra VA, Moxham A, Miller SL, Moss TJM, Hooper SB, Nold MF, Nold-Petry CA, Dean JM, et al. Interleukin-1 blockade attenuates white matter inflammation and oligodendrocyte loss after progressive systemic lipopolysaccharide exposure in near-term fetal sheep. J Neuroinflamm. 2021;18:189.
Galinsky R, Dhillon SK, Dean JM, Davidson JO, Lear CA, Wassink G, Nott F, Kelly SB, Fraser M, Yuill C, et al. Tumor necrosis factor inhibition attenuates white matter gliosis after systemic inflammation in preterm fetal sheep. J Neuroinflammation. 2020;17:92.
Muri L, Grandgirard D, Buri M, Perny M, Leib SL. Combined effect of non-bacteriolytic antibiotic and inhibition of matrix metalloproteinases prevents brain injury and preserves learning, memory and hearing function in experimental paediatric pneumococcal meningitis. J Neuroinflammation. 2018;15:233.
Lai JCY, Svedin P, Ek CJ, Mottahedin A, Wang X, Levy O, Currie A, Strunk T, Mallard C. Vancomycin is protective in a neonatal mouse model of Staphylococcus epidermidis-potentiated hypoxic-ischemic brain injury. Antimicrob Agents Chemother. 2020;64:10–128.
Yang D, Sun YY, Lin X, Baumann JM, Dunn RS, Lindquist DM, Kuan CY. Intranasal delivery of cell-penetrating anti-NF-κB peptides (Tat-NBD) alleviates infection-sensitized hypoxic-ischemic brain injury. Exp Neurol. 2013;247:447–55.
Bolouri H, Sävman K, Wang W, Thomas A, Maurer N, Dullaghan E, Fjell CD, Ek CJ, Hagberg H, Hancock RE, et al. Innate defense regulator peptide 1018 protects against perinatal brain injury. Ann Neurol. 2014;75:395–410.
Ophelders DR, Gussenhoven R, Lammens M, Küsters B, Kemp MW, Newnham JP, Payne MS, Kallapur SG, Jobe AH, Zimmermann LJ, et al. Neuroinflammation and structural injury of the fetal ovine brain following intra-amniotic Candida albicans exposure. J Neuroinflammation. 2016;13:29.
Harding B, Conception K, Li Y, Zhang L. Glucocorticoids protect neonatal rat brain in model of hypoxic-ischemic encephalopathy (HIE). Int J Mol Sci. 2016;18:17.
Chang KH, Yeh CM, Yeh CY, Huang CC, Hsu KS. Neonatal dexamethasone treatment exacerbates hypoxic-ischemic brain injury. Mol Brain. 2013;6:18.
Stojanovska V, Barton SK, Tolcos M, Gill AW, Kluckow M, Miller SL, Zahra V, Hooper SB, Galinsky R, Polglase GR. The effect of antenatal betamethasone on white matter inflammation and injury in fetal sheep and ventilated preterm lambs. Dev Neurosci. 2018;40:497–507.
Chen X, Nakada S, Donahue JE, Chen RH, Tucker R, Qiu J, Lim YP, Stopa EG, Stonestreet BS. Neuroprotective effects of inter-alpha inhibitor proteins after hypoxic-ischemic brain injury in neonatal rats. Exp Neurol. 2019;317:244–59.
Bradford A, Hernandez M, Kearney E, Theriault L, Lim YP, Stonestreet BS, Threlkeld SW. Effects of juvenile or adolescent working memory experience and inter-alpha inhibitor protein treatment after neonatal hypoxia-ischemia. Brain Sci. 2020;10:999.
Kumar P, Hair P, Cunnion K, Krishna N, Bass T. Classical complement pathway inhibition reduces brain damage in a hypoxic ischemic encephalopathy animal model. PLoS ONE. 2021;16: e0257960.
Widerøe M, Havnes MB, Morken TS, Skranes J, Goa PE, Brubakk AM. Doxycycline treatment in a neonatal rat model of hypoxia-ischemia reduces cerebral tissue and white matter injury: a longitudinal magnetic resonance imaging study. Eur J Neurosci. 2012;36:2006–16.
Mao-Draayer Y, Sarazin J, Fox D, Schiopu E. The sphingosine-1-phosphate receptor: a novel therapeutic target for multiple sclerosis and other autoimmune diseases. Clin Immunol. 2017;175:10–5.
Herz J, Köster C, Crasmöller M, Abberger H, Hansen W, Felderhoff-Müser U, Bendix I. Peripheral T cell depletion by FTY720 exacerbates hypoxic-ischemic brain injury in neonatal mice. Front Immunol. 2018;9:1696.
D’Angelo B, Ek CJ, Sun Y, Zhu C, Sandberg M, Mallard C. GSK3β inhibition protects the immature brain from hypoxic-ischaemic insult via reduced STAT3 signalling. Neuropharmacology. 2016;101:13–23.
Zhu JJ, Yu BY, Fu CC, He MZ, Zhu JH, Chen BW, Zheng YH, Chen SQ, Fu XQ, Li PJ, Lin ZL. LXA4 protects against hypoxic-ischemic damage in neonatal rats by reducing the inflammatory response via the IκB/NF-κB pathway. Int Immunopharmacol. 2020;89: 107095.
Liu G, Li M, Qian S, Yu L, Qian L, Feng X. Interleukin-35 exhibits protective effects in a rat model of hypoxic-ischemic encephalopathy through the inhibition of microglia-mediated inflammation. Transl Pediatr. 2022;11:651–62.
Yu S, Doycheva DM, Gamdzyk M, Yang Y, Lenahan C, Li G, Li D, Lian L, Tang J, Lu J, Zhang JH. Activation of MC1R with BMS-470539 attenuates neuroinflammation via cAMP/PKA/Nurr1 pathway after neonatal hypoxic-ischemic brain injury in rats. J Neuroinflamm. 2021;18:26.
Wixey JA, Reinebrant HE, Buller KM. Post-insult ibuprofen treatment attenuates damage to the serotonergic system after hypoxia-ischemia in the immature rat brain. J Neuropathol Exp Neurol. 2012;71:1137–48.
Li L, McBride DW, Doycheva D, Dixon BJ, Krafft PR, Zhang JH, Tang J. G-CSF attenuates neuroinflammation and stabilizes the blood-brain barrier via the PI3K/Akt/GSK-3β signaling pathway following neonatal hypoxia-ischemia in rats. Exp Neurol. 2015;272:135–44.
Hu X, Li S, Doycheva DM, Huang L, Lenahan C, Liu R, Huang J, Xie S, Tang J, Zuo G, Zhang JH. Rh-CSF1 attenuates neuroinflammation via the CSF1R/PLCG2/PKCε pathway in a rat model of neonatal HIE. J Neuroinflamm. 2020;17:182.
Galinsky R, Draghi V, Wassink G, Davidson JO, Drury PP, Lear CA, Gunn AJ, Bennet L. Magnesium sulfate reduces EEG activity but is not neuroprotective after asphyxia in preterm fetal sheep. J Cereb Blood Flow Metab. 2017;37:1362–73.
Wassink G, Davidson JO, Crisostomo A, Zhou KQ, Galinsky R, Dhillon SK, Lear CA, Bennet L, Gunn AJ. Recombinant erythropoietin does not augment hypothermic white matter protection after global cerebral ischaemia in near-term fetal sheep. Brain Commun. 2021;3: fcab172.
Robertson SA, Hutchinson MR, Rice KC, Chin PY, Moldenhauer LM, Stark MJ, Olson DM, Keelan JA. Targeting Toll-like receptor-4 to tackle preterm birth and fetal inflammatory injury. Clin Transl Immunol. 2020;9: e1121.
Viscardi RM. Ureaplasma species: role in diseases of prematurity. Clin Perinatol. 2010;37:393–409.
Berger A, Witt A, Haiden N, Kaider A, Klebermasz K, Fuiko R, Langgartner M, Pollak A. Intrauterine infection with Ureaplasma species is associated with adverse neuromotor outcome at 1 and 2 years adjusted age in preterm infants. J Perinat Med. 2009;37:72–8.
Silasi M, Cardenas I, Kwon JY, Racicot K, Aldo P, Mor G. Viral infections during pregnancy. Am J Reprod Immunol. 2015;73:199–213.
Gervasi MT, Romero R, Bracalente G, Chaiworapongsa T, Erez O, Dong Z, Hassan SS, Yeo L, Yoon BH, Mor G, et al. Viral invasion of the amniotic cavity (VIAC) in the midtrimester of pregnancy. J Matern Fetal Neonatal Med. 2012;25:2002–13.
DiGiulio DB. Diversity of microbes in amniotic fluid. Semin Fetal Neonatal Med. 2012;17:2–11.
DiGiulio DB, Romero R, Amogan HP, Kusanovic JP, Bik EM, Gotsch F, Kim CJ, Erez O, Edwin S, Relman DA. Microbial prevalence, diversity and abundance in amniotic fluid during preterm labor: a molecular and culture-based investigation. PLoS ONE [Electronic Resource]. 2008;3: e3056.
DiGiulio DB, Romero R, Kusanovic JP, Gomez R, Kim CJ, Seok KS, Gotsch F, Mazaki-Tovi S, Vaisbuch E, Sanders K, et al. Prevalence and diversity of microbes in the amniotic fluid, the fetal inflammatory response, and pregnancy outcome in women with preterm pre-labor rupture of membranes. Am J Reprod Immunol. 2010;64:38–57.
Cardenas I, Mor G, Aldo P, Lang SM, Stabach P, Sharp A, Romero R, Mazaki-Tovi S, Gervasi M, Means RE. Placental viral infection sensitizes to endotoxin-induced pre-term labor: a double hit hypothesis. Am J Reprod Immunol. 2011;65:110–7.
Cardenas I, Means RE, Aldo P, Koga K, Lang SM, Booth CJ, Manzur A, Oyarzun E, Romero R, Mor G. Viral infection of the placenta leads to fetal inflammation and sensitization to bacterial products predisposing to preterm labor. J Immunol. 2010;185:1248–57.
Rand KM, Austin NC, Inder TE, Bora S, Woodward LJ. Neonatal infection and later neurodevelopmental risk in the very preterm infant. J Pediatr. 2016;170:97–104.
Tsai MH, Chu SM, Lee CW, Hsu JF, Huang HR, Chiang MC, Fu RH, Lien R, Huang YC. Recurrent late-onset sepsis in the neonatal intensive care unit: incidence, clinical characteristics and risk factors. Clin Microbiol Infect. 2014;20:O928-935.
Zhang L, Li Z, Han X, Du H, Cao Y, Liu Y, Wang W. Association between congenital cytomegalovirus infection and brain injury in neonates: a meta-analysis of cohort studies. Behav Neurol. 2021;2021:9603660.
Qiao H, Guo M, Shang J, Zhao W, Wang Z, Liu N, Li B, Zhou Y, Wu Y, Chen P. Herpes simplex virus type 1 infection leads to neurodevelopmental disorder-associated neuropathological changes. PLoS Pathog. 2020;16: e1008899.
Yu W, Hu X, Cao B. Viral infections during pregnancy: the big challenge threatening maternal and fetal health. Matern Fetal Med. 2022;4:72–86.
Hessami K, Norooznezhad AH, Monteiro S, Barrozo ER, Abdolmaleki AS, Arian SE, Zargarzadeh N, Shekerdemian LS, Aagaard KM, Shamshirsaz AA. COVID-19 pandemic and infant neurodevelopmental impairment: a systematic review and meta-analysis. JAMA Netw Open. 2022;5: e2238941.
Galinsky R, Dean JM, Lear CA, Davidson JO, Dhillon S, Wassink G, Bennet L, Gunn AJ. In the era of therapeutic hypothermia, how well do studies of perinatal neuroprotection control temperature? Dev Neurosci. 2017;39:7–22.
Taylor PM. Oxygen consumption in new-born rats. J Physiol. 1960;154:153–68.
Hahn P, Koldovský O. Prenatal and postnatal development of metabolism and its regulation in mammals. Cesk Fysiol. 1966;215:437–79.
DeBow SB, Clark DL, MacLellan CL, Colbourne F. Incomplete assessment of experimental cytoprotectants in rodent ischemia studies. Can J Neurol Sci. 2003;30:368–74.
Galinsky R, Bennet L, Groenendaal F, Lear CA, Tan S, van Bel F, Juul SE, Robertson NJ, Mallard C, Gunn AJ. Magnesium is not consistently neuroprotective for perinatal hypoxia-ischemia in term-equivalent models in preclinical studies: a systematic review. Dev Neurosci. 2014;36:73–82.
Busto R, Dietrich WD, Globus MY, Valdes I, Scheinberg P, Ginsberg MD. Small differences in intraischemic brain temperature critically determine the extent of ischemic neuronal injury. J Cereb Blood Flow Metab. 1987;7:729–38.
Minamisawa H, Smith ML, Siesjo BK. The effect of mild hyperthermia and hypothermia on brain damage following 5, 10, and 15 minutes of forebrain ischemia. Ann Neurol. 1990;28:26–33.
Camm J, Hla T, Bakshi R, Brinkmann V. Cardiac and vascular effects of fingolimod: mechanistic basis and clinical implications. Am Heart J. 2014;168:632–44.
Gunn AJ, Bennet L. Is temperature important in delivery room resuscitation? Semin Neonatol. 2001;6:241–9.
Gunn AJ, Thoresen M. Hypothermic neuroprotection. NeuroRx. 2006;3:154–69.
French NP, Hagan R, Evans SF, Mullan A, Newnham JP. Repeated antenatal corticosteroids: effects on cerebral palsy and childhood behavior. Am J Obstet Gynecol. 2004;190:588–95.
Malaeb SN, Stonestreet BS. Steroids and injury to the developing brain: net harm or net benefit? Clin Perinatol. 2014;41:191–208.
Tam EWY, Kamino D, Shatil AS, Chau V, Moore AM, Brant R, Widjaja E. Hyperglycemia associated with acute brain injury in neonatal encephalopathy. Neuroimage Clin. 2021;32: 102835.
Flenady V, Hawley G, Stock OM, Kenyon S, Badawi N. Prophylactic antibiotics for inhibiting preterm labour with intact membranes. 2013. Cochrane Database Syst Rev. https://doi.org/10.1002/14651858.CD000246.pub2.
Debillon T, Gras-Leguen C, Vérielle V, Winer N, Caillon J, Rozé JC, Gressens P. Intrauterine infection induces programmed cell death in rabbit periventricular white matter. Pediatr Res. 2000;47:736–42.
Lalor JG, Fawole B, Alfirevic Z, Devane D. Biophysical profile for fetal assessment in high risk pregnancies. Cochrane Database Syst Rev. 2008;2008: Cd000038.
Nelson KB, Dambrosia JM, Ting TY, Grether JK. Uncertain value of electronic fetal monitoring in predicting cerebral palsy. N Engl J Med. 1996;334:613–8.
Inder TE, Wells SJ, Mogridge NB, Spencer C, Volpe JJ. Defining the nature of the cerebral abnormalities in the premature infant: a qualitative magnetic resonance imaging study. J Pediatr. 2003;143:171–9.
Maalouf EF, Duggan PJ, Counsell SJ, Rutherford MA, Cowan F, Azzopardi D, Edwards AD. Comparison of findings on cranial ultrasound and magnetic resonance imaging in preterm infants. Pediatrics. 2001;107:719–27.
Mirmiran M, Barnes PD, Keller K, Constantinou JC, Fleisher BE, Hintz SR, Ariagno RL. Neonatal brain magnetic resonance imaging before discharge is better than serial cranial ultrasound in predicting cerebral palsy in very low birth weight preterm infants. Pediatrics. 2004;114:992–8.
Lodygensky GA, Kunz N, Perroud E, Somm E, Mlynarik V, Hüppi PS, Gruetter R, Sizonenko SV. Definition and quantification of acute inflammatory white matter injury in the immature brain by MRI/MRS at high magnetic field. Pediatr Res. 2014;75:415–23.
Galinsky R, van de Looij Y, Mitchell N, Dean JM, Dhillon SK, Yamaguchi K, Lear CA, Wassink G, Davidson JO, Nott F, et al. Magnetic resonance imaging correlates of white matter gliosis and injury in preterm fetal sheep exposed to progressive systemic inflammation. Int J Mol Sci. 2020;21:8891.
Lodygensky GA, West T, Stump M, Holtzman DM, Inder TE, Neil JJ. In vivo MRI analysis of an inflammatory injury in the developing brain. Brain Behav Immun. 2010;24:759–67.
Inder T, Huppi PS, Zientara GP, Maier SE, Jolesz FA, di Salvo D, Robertson R, Barnes PD, Volpe JJ. Early detection of periventricular leukomalacia by diffusion-weighted magnetic resonance imaging techniques. J Pediatr. 1999;134:631–4.
Chau V, Brant R, Poskitt KJ, Tam EW, Synnes A, Miller SP. Postnatal infection is associated with widespread abnormalities of brain development in premature newborns. Pediatr Res. 2012;71:274–9.
Glass HC, Bonifacio SL, Chau V, Glidden D, Poskitt K, Barkovich AJ, Ferriero DM, Miller SP. Recurrent postnatal infections are associated with progressive white matter injury in premature infants. Pediatrics. 2008;122:299–305.
Fleiss B, Gressens P. Tertiary mechanisms of brain damage: a new hope for treatment of cerebral palsy? Lancet Neurol. 2012;11:556–66.
Lear BA, Lear CA, Davidson JO, Sae-Jiw J, Lloyd JM, Gunn AJ, Bennet L. Tertiary cystic white matter injury as a potential phenomenon after hypoxia-ischaemia in preterm f sheep. Brain Commun. 2021;3: fcab024.
Chakkarapani AA, Aly H, Benders M, Cotten CM, El-Dib M, Gressens P, Hagberg H, Sabir H, Wintermark P, Robertson NJ. Therapies for neonatal encephalopathy: targeting the latent, secondary and tertiary phases of evolving brain injury. Semin Fetal Neonatal Med. 2021;26: 101256.
Shi Z, Luo K, Deol S, Tan S. A systematic review of noninflammatory cerebrospinal fluid biomarkers for clinical outcome in neonates with perinatal hypoxic brain injury that could be biologically significant. J Neurosci Res. 2022;100:2154–73.
Pavlidis E, Lloyd RO, Boylan GB. EEG - a valuable biomarker of brain injury in preterm infants. Dev Neurosci. 2017;39:23–35.
Pavel AM, Mathieson SR, Livingstone V, O’Toole JM, Pressler RM, de Vries LS, Rennie JM, Mitra S, Dempsey EM, Murray DM, et al. Heart rate variability analysis for the prediction of EEG grade in infants with hypoxic ischaemic encephalopathy within the first 12 h of birth. Front Pediatr. 2022;10:1016211.
Geddes R, Vannucci RC, Vannucci SJ. Delayed cerebral atrophy following moderate hypoxia-ischemia in the immature rat. Dev Neurosci. 2001;23:180–5.
Smith PLP, Mottahedin A, Svedin P, Mohn CJ, Hagberg H, Ek J, Mallard C. Peripheral myeloid cells contribute to brain injury in male neonatal mice. J Neuroinflamm. 2018;15:301.
Smith AL, Alexander M, Rosenkrantz TS, Sadek ML, Fitch RH. Sex differences in behavioral outcome following neonatal hypoxia ischemia: insights from a clinical meta-analysis and a rodent model of induced hypoxic ischemic brain injury. Exp Neurol. 2014;254:54–67.
Ardalan M, Chumak T, Vexler Z, Mallard C. Sex-dependent effects of perinatal inflammation on the brain: implication for neuro-psychiatric disorders. Int J Mol Sci. 2019;20:2270.
Bennet L, Galinsky R, Draghi V, Lear CA, Davidson JO, Unsworth CP, Gunn AJ. Time and sex dependent effects of magnesium sulphate on post-asphyxial seizures in preterm fetal sheep. J Physiol. 2018;596:6079–92.
Galinsky R, Dhillon SK, Lear CA, Yamaguchi K, Wassink G, Gunn AJ, Bennet L. Magnesium sulfate and sex differences in cardiovascular and neural adaptations during normoxia and asphyxia in preterm fetal sheep. Am J Physiol Regul Integr Comp Physiol. 2018;315:R205-r217.
Vohr BR, Allan WC, Westerveld M, Schneider KC, Katz KH, Makuch RW, Ment LR. School-age outcomes of very low birth weight infants in the indomethacin intraventricular hemorrhage prevention trial. Pediatrics. 2003;111:e340-346.
Wyatt JS, Gluckman PD, Liu PY, Azzopardi D, Ballard R, Edwards AD, Ferriero DM, Polin RA, Robertson CM, Thoresen M, et al. Determinants of outcomes after head cooling for neonatal encephalopathy. Pediatrics. 2007;119:912–21.
Donnelly H, Saibaba P. Light intensity and the oestrous cycle in albino and normally pigmented mice. Lab Anim. 1993;27:385–90.
Vanderschuren LJ, Niesink RJ, Spruijt BM, Van Ree JM. Influence of environmental factors on social play behavior of juvenile rats. Physiol Behav. 1995;58:119–23.
Clough G. Environmental effects on animals used in biomedical research. Biol Rev Camb Philos Soc. 1982;57:487–523.
Funding
This study was supported by project grants from the National Health and Medical Research Council of Australia (R.G.; 1164954), the Cerebral Palsy Alliance, Harold and Cora Brennen Benevolent Trust and the Victorian Government’s Operational Infrastructure Support Program.
Author information
Authors and Affiliations
Contributions
Sharmony B. Kelly and Robert Galinsky conceptualised and designed the study. Sharmony B. Kelly, Nhi T Tran and Robert Galinsky undertook formal analysis and interpretation of the data. Sharmony B. Kelly and Robert Galinsky prepared the figures. Robert Galinsky provided overall oversight of the research. All authors critically reviewed the manuscript and approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supplementary table 1.
Additional file 2.
Supplementary table 2.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kelly, S.B., Tran, N.T., Polglase, G.R. et al. A systematic review of immune-based interventions for perinatal neuroprotection: closing the gap between animal studies and human trials. J Neuroinflammation 20, 241 (2023). https://doi.org/10.1186/s12974-023-02911-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-023-02911-w