
Abstract
Background
In 2014, we first described novel autoantibodies to the inositol 1,4,5-trisphosphate receptor type 1 (ITPR1-IgG/anti-Sj) in patients with autoimmune cerebellar ataxia (ACA) in this journal. Here, we provide a review of the available literature on ITPR1-IgG/anti-Sj, covering clinical and paraclinical presentation, tumour association, serological findings, and immunopathogenesis.
Methods
Review of the peer-reviewed and PubMed-listed English language literature on ITPR1-IgG/anti-Sj. In addition, we provide an illustrative report on a new patient with ITPR1-IgG-associated encephalitis with cognitive decline and psychosis.
Results
So far, at least 31 patients with serum ITPR1-IgG/anti-Sj have been identified (clinical information available for 21). The most common manifestations were ACA, encephalopathy with seizures, myelopathy, and (radiculo)neuropathy, including autonomic neuropathy. In 45% of cases, an underlying tumour was present, making the condition a facultative paraneoplastic neurological disorder. The neurological syndrome preceded tumour diagnosis in all but one case. In most cases, immunotherapy had only moderate or no effect. The association of ITPR1-IgG/anti-Sj with manifestations other than ACA is corroborated by the case of a 48-year-old woman with high-titre ITPR1-IgG/anti-Sj antibodies and rapid cognitive decline, affecting memory, attention and executive function, and psychotic manifestations, including hallucinations, investigated here in detail. FDG-PET revealed right-temporal glucose hypermetabolism compatible with limbic encephalitis. Interestingly, ITPR1-IgG/anti-Sj mainly belonged to the IgG2 subclass in both serum and cerebrospinal fluid (CSF) in this and further patients, while it was predominantly IgG1 in other patients, including those with more severe outcome, and remained detectable over the entire course of disease. Immunotherapy with intravenous methylprednisolone, plasma exchange, and intravenous immunoglobulins, was repeatedly followed by partial or complete recovery. Long-term treatment with cyclophosphamide was paralleled by relative stabilization, although the patient noted clinical worsening at the end of each treatment cycle.
Conclusions
The spectrum of neurological manifestations associated with ITPR1 autoimmunity is broader than initially thought. Immunotherapy may be effective in some cases. Studies evaluating the frequency of ITPR1-IgG/anti-Sj in patients with cognitive decline and/or psychosis of unknown aetiology are warranted. Tumour screening is essential in patients presenting with ITPR1-IgG/anti-Sj.
Similar content being viewed by others


Introduction
In 2014, we described in this journal novel immunoglobulin G (IgG) autoantibodies against the inositol 1,4,5-trisphosphate receptor type 1 (ITPR1; also termed IP3R1 or InsP3R1) in patients with autoimmune cerebellar ataxia [1]. The target antigen was identified by us in 2010 and the antibody initially termed anti-Sj after the index sample code. When tested by immunohistochemistry on rodent or primate cerebellum sections, IgG from serum and cerebrospinal fluid (CSF) samples of ITPR1-IgG/anti-Sj-positive patients characteristically bind to dendrites and cell bodies of Purkinje cells (PC). ITPR1-IgG/anti-Sj-associated ataxia thus forms part of a broader spectrum of anti-PC antibody-related ataxias referred to collectively as ‘medusa head ataxias’, based on the specific immunohistochemical staining pattern [2,3,4]. Many of these antibodies target proteins involved in intracellular calcium homoeostasis, such as anti-Homer-3 [5], anti-PKCγ [6], anti-mGluR1 [7, 8], anti-GluR-delta2 [9, 10], anti-CARPVIII [11, 12] and, possibly, anti-ARHGAP26/anti-Ca [13,14,15]. However, ITPR1 is also expressed in other central and peripheral neurons (Fig. 1) [16, 17], and, accordingly, we and others subsequently described ITPR1-IgG/anti-Sj antibodies also in patients with paraneoplastic and non-paraneoplastic (radiculo)neuropathy, autonomic neuropathy, myelopathy, possible brainstem encephalitis and encephalopathy with seizures [16, 18]. While ITPR1 is usually considered an intrathecal antigen, surface localization of ITPR1 has been reported to occur in some neurons and other cell types [19,20,21,22,23,24,25].

(modified images from the Human Protein Atlas image database [40]; https://www.proteinatlas.org; licensed under the Creative Commons Attribution-ShareAlike 3.0 International License)
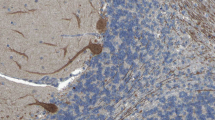
ITPR1 protein expression, as detected by immunohistochemistry, in the cerebellum (a), cerebral cortex (b), hippocampus (c), and lateral ventricle wall/basal ganglia (d), together with protein expression scores (e)
Here, we give a comprehensive review of all ITPR1-IgG/anti-Sj-seropositive cases published so far and discuss the immunopathophysiology of this rare syndrome. We include in the analysis a recent case in which ITPR1-IgG/anti-Sj was associated with progressive cognitive decline, affecting mainly short-term memory, executive dysfunction, attention deficits, a sleep disorder, and psychotic symptoms.
Methods
Literature review and case report
Cases were identified by a PubMed search including all English language articles on ITPR1-IgG/anti-Sj in patients with autoimmune encephalitis published between our first description of ITPR1-IgG in 2014 and 2022 using the following search term: (“inositol 1,4,5-trisphosphate receptor 1” OR ITPR1 OR IP3R1 OR InsP3R1) AND (antibody OR autoantibody OR antibodies OR autoantibodies OR “immunoglobulin G” OR IgG), and from the authors own files. Clinical and paraclinical data of a novel case of ITPR1-IgG/anti-Sj-associated autoimmune encephalitis were obtained retrospectively from the patient’s records. A descriptive statistical analysis of all data obtained was performed.
Antibody testing
Immunohistochemistry (IHC) was performed as previously described [1]. Briefly, 4-μm cryosections of primate cerebellum (Euroimmun, Lübeck, Germany) were incubated for 1 h with diluted patient and control serum or cerebrospinal fluid (CSF) samples, respectively. Bound patient serum and CSF IgG was detected using goat secondary antibodies to human IgG, IgM and IgA, respectively, labelled with fluorescein isothiocyanate (FITC) (Euroimmun).
Specificity of the patient’s antibody response for ITPR1 was further confirmed using a fixed cell-based assay (CBA) as previously described [16]. Briefly, murine full-length ITPR1 was expressed in human embryonic kidney (HEK) 293 cells. HEK293 cells were then grown on sterile cover glasses for 48 h and subsequently fixed with acetone. Cover glasses were then cut into millimetre-sized fragments (biochips), which were used as assay substrate. Biochips with mock-transfected HEK293 cells were used as control substrate. Both substrates were incubated with the patients’ serum samples or control samples in parallel. Binding of patient serum and CSF IgG was visualized using a goat secondary antibody to human IgG labelled with FITC (Euroimmun).
For IgG subclass analyses, unconjugated sheep anti-human IgG antibodies specific for IgG subclasses 1 to 4 (The Binding Site, Schwetzingen, Germany) were substituted for the FITC-labelled goat anti-human IgG antibody and an Alexa Fluor® 568-labelled donkey anti-sheep IgG secondary antibody (Invitrogen; absorbed against human IgG) was used to detect the IgG subclass-specific secondary antibodies, both in the IHC and in the CBA assay.
Results
Section A: Illustrative case report
We included in the analysis a new, so far unpublished case described in detail in the Appendix of this article. A summary can be found in Table 1. ITPR1-IgG/anti-Sj was associated with progressive cognitive decline in this patient, affecting mainly short-term memory, executive dysfunction, attention deficits, a sleep disorder, and psychotic symptoms. Immunotherapy with high-dose intravenous methylprednisolone (IVMP) given during periods of acute deterioration, either alone or with plasma exchange (PLEX) and intravenous immunoglobulins (IVIG), repeatedly resulted in significant recovery. Long-term therapy with cyclophosphamide led to some stabilization. However, several relapse-like end-of-dose episodes occurred, characterized by either worsening of pre-existing symptoms or occurrence of new symptoms. Clinical and paraclinical data covering a period of more than 2 years, including serological, CSF, magnetic resonance imaging (MRI), and fluorodeoxyglucose positron emission tomography (PET) findings as well as neuropsychological test results, are provided in the Appendix.
Section B: Review of the literature
Including the case described in Section A, 21 patients with serum antibodies to ITPR1 and central and peripheral neurological symptoms have been described in the literature so far (and at least 10 further patients have been identified in historic series of samples with anti-PC antibodies but not yet published due to the sparsity of available clinical data [4, 18]):
-
1.
In 2014, we reported on ITPR1-IgG/anti-Sj in a woman with a 10-year history of progressive ataxia of the upper limbs, dysarthria, and gaze disturbances. MRI revealed pontocerebellar atrophy [1]. A follow-up report on this patient was published in 2017 by Berzero et al. [26]. Although the patient was positive for a BRCA1 (breast cancer 1, early onset) gene mutation and had a history of breast cancer on her mother’s side of the family, no tumour was initially found despite extensive diagnostic work-up, including regular screening with whole-body 18FDG-PET. Steroid treatment, ten cycles of PLEX, and three cycles of intravenous immunoglobulin, all first given several years after disease onset, had no significant beneficial effect with regard to disease progression. The disease came to a halt only when the patient was already severely disabled [1, 26]. Eleven years after onset, detection of suspicious lymph nodes upon mammography led to a diagnosis of bifocal ductal grade I breast cancer. Of note, immunopathology revealed substantial ITPR1 expression in the tumour and the metastatic lymph nodes [18]. Two further ITPR1-IgG/anti-Sj-seropositive patients investigated in the original 2014 report had “cerebral ataxia” and a “chronic cerebellar syndrome responsive to immunotherapy”, respectively, but no additional clinical information was available [1].
-
2.
In 2016, we reported on serum ITPR1-IgG/anti-Sj in three male patients with (radiculo)polyneuropathy and, in one of them, neurogenic pain, consistent with the fact that ITPR1 has been shown to be expressed also in the anterior horn of the spinal cord, in the substantia gelatinosa, and in the motor, sensory (including the dorsal root ganglia) and autonomic peripheral nervous system. In two cases, the disease was associated with an ITPR1-expressing adenocarcinoma of the lung and multiple myeloma, respectively. Of note, ITPR1-IgG/anti-Sj serum titres declined after removal of the lung tumour, which was paralleled by clinical stabilization [16].
-
3.
In 2017, three further ITPR1-IgG/anti-Sj-seropositive patients (two of them female) with cerebellar ataxia were described [27], one of whom also developed dysautonomia. One patient was successively treated with steroids, glatiramer acetate, and interferon-β-1a for suspected MS, none of which was convincingly effective in slowing disease progression (tumour status unknown), while no data on treatment outcome were available for the remaining patients (no evidence of paraneoplastic aetiology).
-
4.
In 2018, a series of 11 (8 × female) patients with serum ITPR1-IgG/anti-Sj autoantibodies from the Mayo Clinic was published. Alfugham et al. [18] reported cerebellar ataxia in three cases, peripheral neuropathy in five (1 × autonomic), encephalitis with seizures in two, and myelopathy in two. Additional symptoms compatible with possible brainstem involvement included double vision and vertigo. In one patient, just as in the present case, visual blurring occurred. In five of the 11 patients (45%), ITPR1-IgG/anti-Sj was associated with cancer. In none of the seven patients receiving immunotherapy did the neurologic impairment improve significantly.
-
5.
In 2022, a Chinese patient with ITPR1-IgG/anti-Sj-positive cerebellar ataxia associated with an adenocarcinoma of the lung and multiple brain metastases was described [28]. A broad screening for other paraneoplastic and non-paraneoplastic anti-neural antibodies was negative.
In addition to the cases summarized above, five further patients with ITPR1-IgG/anti-Sj detected only in the CSF have been reported (2 × seronegative [29, 30], 2 × no serum available [18], 1 × no information provided [18]). However, as all previously published ITPR1-IgG/anti-Sj-positive patients had serum antibodies to ITPR1, and the specificity of the recombinant assays used to detect ITPR1-IgG/anti-Sj has not been formally evaluated for CSF, the clinical significance of this finding is unknown. Moreover, co-existing CSF IgG antibodies to N-methyl d-aspartate receptors (NMDAR) (2×) and glial fibrillary astrocytic protein (GFAP) (3×) [16, 29, 30] were reportedly present in the CSF of some of these patients. CSF GFAP-IgG and CSF NMDAR-IgG can also cause autoimmune encephalomyelitis or encephalitis, respectively, rendering it impossible to attribute these patients’ clinical and paraclinical features to either of these antibodies with sufficient certainty. In one of these patients [30], ITPR1-IgG was negative in the CSF on three occasions, including at disease onset, but reportedly temporarily positive once, and negative in the serum in four matched serum samples, ITPR1-specific binding of the patient’s IgG to brain tissue was not demonstrated, and the associated tumour did not express ITPR1, which in summary casts doubts on the diagnosis. Accordingly, these cases were not included in this literature review. Of note, the new patient described in detail in Section A of the present article was negative for NMDAR-IgG and GFAP-IgG, as determined in three consecutive paired CSF and serum samples.
Very recently, a patient with suspected ITPR1-IgG seropositivity and confabulations, amnesia, and motor memory loss has been reported [31]. However, binding of IgG to ITPR1-IgG transfected cells was found only at an extremely low titre (1:10), and no corresponding, ITPR1-specific tissue staining was apparently observed. In consequence, doubts remain regarding the diagnosis also in this case. Accordingly, the case was not included in the statistical analysis.
Epidemiology
Ethnic origin was not reported for most patients (5 × Caucasian, not reported in the remainder, which included one report from a Chinese group); 9 patients were seen at European institutions; 11 at a North American institution; and one at a Chinese institution. The male:female ratio was 1:1.5. Interestingly, however, 6/7 patients with peripheral neuropathy were male. The median age at onset was 64 years (range 13–83), with no significant difference between patients with peripheral and patients with central nervous system involvement (median 60 years vs. 65 years).
Of particular interest, one study aimed at prospectively evaluating the frequency of ITPR1-IgG/anti-Sj among specimens submitted for paraneoplastic autoantibody evaluation, reporting a frequency of 15/100,000 specimens/year (although it is unclear whether this includes three of the four patients reportedly positive only in the CSF). This would be 15 times the frequency of anti-Tr/DNER (delta/notch-like epidermal growth factor-related receptor) (1/100,000/year) but lower than that of anti-Hu/ANNA-1 (100/100,000/year), anti-Ri/ANNA-2 (30/100,000/year) and anti-Yo/PCA-1 (80/100,000/year), based on a very large sample (N = 52,000) [18].
Cerebellar ataxia
Symptoms of cerebellar ataxia, either alone (N = 2) or in combination with other symptoms (N = 9), were present in 11/21 (52%) ITPR1-IgG/anti-Sj-seropositive patients, including limb ataxia with dysmetria, dysdiadochokinesis, intention tremor, gait ataxia, dysarthria, and gaze disturbances (Table 2). Co-existing symptoms in patients with cerebellar ataxia included double vision, visual blurring, subacute hearing loss, mild bilateral lower limb weakness, dysautonomia (2×), REM sleep disturbances, vertigo (2×), dysphasia, agraphia, and cognitive decline and other neuropsychiatric symptoms (Table 2).
Cognitive decline and other neuropsychiatric symptoms
In the patient described in Section A, ITPR1-IgG/anti-Sj seropositivity was associated with rapidly progressive, severe cognitive decline, mainly affecting memory, attention and executive functions; optic hallucinations; and depression. A further patient originally presented with subacute pandysautonomia but developed dementia, encephalopathy and “electrographic seizures” 4 years later. MRI showed generalized brain atrophy, affecting particularly the temporal lobes [18]. The patient initially responded well to steroid treatment but the disease subsequently progressed. FDG-PET/CT suggested a lung tumour with mediastinal lymphadenopathy. ITPR1-IgG/anti-Sj was detected at a very high serum titre of 1:30,720 and a high CSF titre of 1:128. Of note, signs of possible limbic involvement (confusion, severe memory loss, suspected temporal lobe epilepsy) were also present in two ITPR1-IgG/anti-Sj-seropositive patients identified by us in 2016, one of whom also had cerebellar ataxia. As no detailed clinical data are available, the cases of these two patients have not yet been published (but were briefly mentioned in [16]). In another patient, who had breast and endometrial carcinomas, insomnia, anxiety and depression were reported, but no cognitive decline [18]. Severe depression of unknown cause developed also in an ITPR1-IgG/anti-Sj-seropositive patient reported in reference [16].
Sleep disorders
Sleep disorders have recently been described in association with a number of autoantibody-associated CNS disorders, including anti-IgLON5 [32, 33], anti-DPPX [34,35,36], anti-CASPR2 and anti-NMDAR encephalitis [37]. It should therefore not go unmentioned that the patient described in Section A reported hypersomnia at onset. When, later, psychomotor restlessness occurred, treatment with pipamperone was started (20 mg/day) in February 2020 and intensified (40 mg/day) in October 2020 because of increasing restlessness and hyposomnia. In January 2020 an attempt to withdraw pipamperone resulted in disruptions in sleep and inner unrest, and treatment was subsequently resumed at a reduced dose (20 mg/day). Sleep disorders were also present in at least two other previously described ITPR1-IgG/anti-Sj-seropositive patients, one with insomnia [18] and one with REM sleep disturbances [27].
Poly(radiculo)neuropathy
Electroneurography (ENG) indicated axonal and demyelinating polyneuropathy (PNP) in 2, axonal PNP in 1, and demyelinating PNP in 1 patient (ENG results not reported in 2 further patients). Both motor (at least 3 patients) and sensory (at least 6 patients) nerves were affected. The onset of PNP-associated symptoms was subacute in 4 and slowly progressive in 1 (no data in 2). Electromyography (EMG) corroborated the presence of PNP in all six cases with available data. In at least one case, clinical and electrophysiological findings suggested GBS [16]. In that patient, first severely delayed and missing F waves, which indicate damage to the proximal motor nerve axons or anterior horn α-motor neuron cell bodies, and A waves were noted, and, later, delayed motor nerve conduction velocities and decreased sensory nerve action potentials [16]. Interestingly, in this case also delayed motor neuron conduction velocity and distal motor latency of the facial nerve and dysautonomia was observed.
In another patient, also delayed and lost F waves were noted as well as A waves, decreased and lost compound muscle action potentials, missing sensory nerve action potentials and reduced sensory nerve conduction velocities in the upper and lower extremities and no conduction blocks. EMG suggested denervation [16].
Pain
Of note, most severe pain was present since onset in one of the patients with axonal and demyelinating peripheral neuropathy (ascending from the lower legs to the lumbar region) at an intensity that required treatment with tilidine, metamizole and gabapentin [16]. Pain was also a leading symptom a in second patient with EMG-confirmed symmetric diffuse axonal neuropathy [18]. We have previously pointed to the fact that ITPR1 is expressed in the sensory dorsal root ganglion, the trigeminal ganglia, and the substantia gelatinosa, in which C fibre axons synapse with neurons of the pain-transmitting lateral spinothalamic tract and damage to which can cause pain and hyperalgesia. Moreover, ITPR1 dysregulation was found to play a role in hyperalgesia and allodynia in animal studies [5].
Dysautonomia
Interestingly, the onset of symptoms was accompanied by diarrhoea lasting around 3 weeks in the new patient described in Section A. This is similar to a previously described ITPR1-IgG/anti-Sj-positive patient, in whom the onset of polyradiculoneuropathy was preceded by bowel and bladder dysfunction, which lasted several months [16]. That patient also exhibited pathological reduced heart frequency variability, a typical sign of autonomic neuropathy of the heart. Given that ITPR1 is expressed in the sympathetic ganglia as well as in the smooth muscle cells of the bowel and the bladder and that ITPR1-IgG/anti-Sj was shown to bind to smooth muscle cells in the enteric wall [1], a possible autoimmune pathogenesis cannot be excluded also in the present case. Diarrhoea is also a typical prodromal symptom in DPPX (dipeptidyl-aminopeptidase-like protein 6) syndrome, another novel antibody-related autoimmune disease of the CNS [34,35,36, 38], and paraneoplastic enteropathy is a well-recognized complication of cancer. As mentioned above, subacute dysautonomia (confirmed by thermoregulatory sweat test and autonomic reflex test) was also present in the sole previously reported patient with cognitive decline and serum ITPR1-IgG/anti-Sj antibodies [18]. Overall, dysautonomia has been reported in 4/19 (21%) ITPR1-IgG/anti-Sj-seropositive patients [16, 18, 27]. Studies evaluating the frequency of ITPR1-IgG/anti-Sj in patients with paraneoplastic or non-paraneoplastic autonomic enteropathy and other types of dysautonomia may thus be warranted.
Notably, repeated stroke episodes attributed to intermittent atrial fibrillation occurred in one patient with ITPR1-IgG/anti-Sj-associated dysautonomia [16]. ITPR1 is also present in human and murine cardiac myocytes [39,40,41] and has been implicated in modulating the propensity of the human myocardium to develop arrhythmias [39].
Further symptoms
In addition to the predominant cognitive deficits and neuropsychiatric symptoms, the patient described in Section A also developed transient symptoms of limb and gait ataxia, blurred vision for a period of around 6–8 weeks, and persisting tetraparesis. Interestingly, “weakness in extremities”, “subacute spastic paraparesis”, “extensive myelitis” and “blurred vision”, respectively, have been described in four further recently published ITPR1-IgG/anti-Sj-positive patients [18], again suggesting that neuronal damage or dysfunction in ITPR1-IgG/anti-Sj-associated disease may be more widespread than previously known.
CSF findings
Lumbar puncture was normal twice in our patient described in Section A, but on another occasion revealed blood–CSF barrier dysfunction (as indicated by an increased albumin CSF/serum quotient [QAlb]), which was also present in the only other patient with available data [16]. Including the present patient, CSF total protein, which strongly correlates with QAlb, was elevated in 4/7 seropositive patients. OCB were negative in 5/6 patients, including in 4 matched CSF/serum samples taken over a period of 4 months in one of them, and pleocytosis was present in 1/5. In one case, breast cancer cells were present in the CSF [18]; this highlights the need for detailed pathological examination of CSF cells in patients with PND.
Serological findings
All ITPR1-IgG/anti-Sj-seropositive patients analysed have been positive in recombinant CBA and have, in addition, shown the distinct binding to PC somata and dendrites described in the index patient when tested by immunohistochemistry using cerebellum tissue sections [1, 16, 18, 26, 27]. Reported CBA titres ranged between 1:320 and 1:245,760 (median 1:30,720; N = 13) in the serum and between 1:4 and 1:8192 (median 1:128; N = 5) in the CSF. IHC titres varied between 1:100 and 1:32,000 (median 1:3200; N = 7) in the serum; IHC CSF titres were reported only for two patients (1:250 and 1:1000, respectively). The CSF/serum ratio was reported for 6 patients (4 × CBA, 2 × IHC) and ranged between 0.004 and 0.033 (median 0.017; N = 6). While this could indicate intrathecal synthesis based on a normal CSF/serum ratio for total IgG of around 1/440, data on ITPR1-IgG-specific antibody indices (AI), which control for possible blood–CSF barrier dysfunction, were assessed only in two cases. In both cases the ITPR1-AI was below 4 (1 × based on IHC titres and on CBA titres, 1 × based on CBA titres), indicating an extrathecal origin of ITPR1-IgG [16]. In line with this finding, CSF-restricted OCB were absent in these two patients.
Radiological findings
Magnetic resonance imaging (MRI) showed cerebellar or pontocerebellar atrophy in 3 patients with ACA and available data; 1 additional patient showed several demyelinating supratentorial brain lesions (this patient had previously been considered to suffer from multiple sclerosis but steroids, interferon-beta and glatiramer acetate were all not effective); and 1 patient only ischaemic brain lesions. MRI was normal in 1. In three cases, MRI findings were not reported. Spinal cord lesions (longitudinally extensive in one) were noted in two patients, both of whom were diagnosed with myelopathy; in one of these, PNP was present in addition and MRI showed diffuse enhancement of the cervical nerve roots. Among patients with encephalitis/encephalopathy, a FDG-PET/CT revealed glucose hypermetabolism in the right medial temporal lobe (amygdala, parahippocampus) and in the basal ganglia was noted in 1; generalized brain atrophy on MRI (prominent in temporal lobes) in another; and subcortical right occipito-parietal T2-hyperintense lesions with gadolinium enhancement in a third patient.
EEG findings
EEG findings were not systematically reported. One patient with high-titre ITPR1-IgG serum antibodies developed a status epilepticus [18]; in a further high-titre patient (positive in both serum and CSF), “EEG-electrographic seizures” were documented [18]. In the patient reported in Section A, right parietal delta or theta activity and intermittent right temporal slowing was noted on several occasions, with intermittent normalization following immunotherapy. Of note, epileptic seizures have also been observed in mice lacking ITPR1 [42].
Tumour associations
In a substantial proportion of patients ITPR1-IgG/anti-Sj was found to be associated with tumours [1, 16, 18, 26, 28], some of which were demonstrated to express ITPR1 [16, 26]. ITPR1-IgG/anti-Sj-associated disease may thus be considered a facultative paraneoplastic neurological syndrome (PNS). Overall, a tumour was found in 9/20 (45%) of the ITPR1/anti-Sj-seropositive patients with available data (3 × breast, 1 × breast and endometrial, 3 × lung [including non-small lung cancer/adenocarcinoma in 2, not specified in 1], 1 × renal, 1 × multiple myeloma) [1, 16, 18, 26, 27]. However, this may underestimate the rate of paraneoplastic cases, given that (a) the tumour status was unknown in some patients, that (b) neurological manifestations may precede tumour diagnosis by many years in patients with paraneoplastic neurological disorder (PND) (as was indeed the case in one of the ITPR1-IgG/anti-Sj-seropositive index patients), and (c) that the underlying tumour not uncommonly remains occult in PND, often being found only upon autopsy. A positive CA125 test was noted in one additional case (see Section A), cervical dysplasia in another [18], and severe weight loss in a further one [18]. One additional case occurred 5 years after bone marrow transplantation for Fanconi anaemia, a genetic condition that confers a high risk of cancer [18]. According to the updated criteria for PNS [43], ITPR1-IgG/anti-Sj could be classified as “intermediate-risk antibody (associated with cancer in 30–70%)”.
In all but one case, the tumour was not known before onset of the neurological symptoms. One patient had a history of two cancers (breast and endometrial), but no information of tumour dissemination at the time of presentation was given [18]. Of 7 patients with a solid tumour and available data on tumour dissemination, five (71%) had metastases at the time of neurological presentation (3 × lymph nodes, 1 × bones and liver, 1 × brain). In one case, ITPR1-expressing breast cancer (associated with a germline mutation in the BCRA1 gene) was not found until 11 years after neurological onset, when it had already spread to the lymph nodes [26, 27].
Mastectomy and adjuvant tamoxifen led to tumour remission in the latter case, but the patient’s neurological status remained unchanged, probably reflecting early irreversible loss of PC, as also observed in other types of PND. In a patient with adenocarcinoma of the lung, removal of the tumour led to mild clinical improvement, but the patient was still unable to walk or stand [16]. No information on the effect of tumour removal is available for the remaining patients; however, two had died at last follow-up (1 × renal cancer with lymphadenopathy; 1 × metastatic breast cancer) [18].
Response to immunotherapy
Including the present case, the response to immunotherapy was reported for 10 patients (7 × PLEX, 7 × IVIG, 7 × IVMP/”IV steroids”/“steroids”, 1 × rituximab, 1 × glatiramer acetate and interferon beta-1a for suspected multiple sclerosis; 1 × cyclophosphamide). However, no significant effect was noted in most cases, with the exception of the patient reported in detail in the Appendix; the only other patient with cognitive decline, as mentioned above, who initially showed a good response to steroid treatment (but later progressed); and one patient with myelopathy in whom at least a short-lasting mild benefit from treatment with PLEX, IVIG and rituximab was found. As an important limitation, only very limited data on exact treatment regimens and, importantly, treatment duration were reported for most cases. Moreover, it cannot be excluded that the number of PLEX cycles was not sufficient to eliminate the antibodies in some cases; in one patient ITPR1-IgG/anti-Sj was still detectable at a titre of 1:1000 (CBA) 1 week after 7 cycles of PLEX [16]; no information on ITPR1-IgG/anti-Sj titres after PLEX treatment was reported for the remaining cases.
Four patients had died at the time of publication, but no information on the cause of death was reported, and no data was provided on the length of follow-up for most patients. One patient died 1 year after onset; at the time of neurological manifestation, he had already metastasis to the bones and liver from renal cell cancer; in another deceased patient, malignant breast cells were present in the CSF at the time of neurological onset. Eight patients were alive 2 years (mild dementia at last follow-up), 5 years (wheel-chair bound), at least 4 years (progressive with cognitive decline and seizures), at least 1.5 years (severe neurological sequelae), 23 months (PNP), 5 months (mild PNP, severe depression), 11.5 years (ACA, severely disabled), 5 months (ACA, no improvement), and 4 years (progressive ACA), respectively, after neurological onset.
Pathophysiology
It is still not known with certainty whether the antibody itself causes the symptoms present in patients with ITPR1 autoimmunity, since no passive transfer experiments, which would be required to demonstrate a direct pathogenic impact of ITPR1-IgG/anti-Sj, have been conducted so far. However, several lines of indirect evidence support the notion of an autoimmune pathogenesis at least in the case presented in Section A:
-
1.
ITPR1, the target antigen of ITPR1-IgG, is expressed not only in PC but also in neurons in the cerebral cortex, the hippocampal formation and amygdala, and the temporal cortex, which renders encephalitis/limbic encephalitis a plausible complication of anti-ITPR1 autoimmunity, as well as in the basal ganglia [16, 17] (Fig. 1).
-
2.
An FDG-PET scan performed during an episode of acute deterioration indeed demonstrated glucose hypermetabolism in the right medial temporal lobe (amygdala, parahippocampus), compatible with limbic encephalitis, and in the basal ganglia, and EEG repeatedly demonstrated theta and delta wave activity, including intermittent right-temporal slowing, during a relapse-like episode. Neuropsychologically, a diagnosis of a cognitive disorder consistent with right-temporal lobe damage was made.
-
3.
Treatment with high-dose IVMP or a combination of PLEX, IVMP and IVIG, repeatedly resulted in significant and prompt clinical improvement, sometimes with complete recovery, as demonstrated by neuropsychological testing and confirmed both by the patient herself and her caregiver. On two occasions, immunotherapy was followed also by EEG normalization.
-
4.
Similarly, immunosuppressive treatment with cyclophosphamide was paralleled by clinical improvement and relative stabilization of the disease course. Of note, the patient and her caregiver both reported that symptoms repeatedly worsened at the end of each cyclophosphamide cycle but soon improved after the next cyclophosphamide infusion, finally resulting in shortening of the treatment intervals.
-
5.
ITPR1-IgG/anti-Sj remained clearly positive over the entire course of disease, with serum titres ranging between 1:100 and 1:1000 (cut-off ≥ 1:10).
-
6.
ITPR1-IgG/anti-Sj was detectable also in the CSF during acute deterioration, although at low titres suggestive of passive diffusion from the peripheral blood into the CNS rather than intrathecal synthesis. In fact, clinically apparent infections (including respiratory infection, urinary tract infection and cholecystitis) or laboratory signs of inflammation, such as an increase in CRP levels or BSR, preceded or accompanied most episodes of clinical deterioration. Eleven of 13 CRP tests performed during the 1st year of disease yielded positive results (maximum 22.3 mg/l). This is of note, since peripheral infections may alter the blood–brain barrier (e.g., due to an increase in serum levels of proinflammatory cytokines, metalloproteinases, ROIs, etc.) and facilitate passive diffusion of serum IgG into the CNS. As mentioned above, acute deterioration was in fact associated with blood–CSF barrier disruption on one occasion (indicated by an elevated albumin CSF/serum ratio). Similarly, QAlb was also elevated twice in one of the only two other ITPR1-IgG-seropositive patients with published data [16]. Preceding infections are thought to trigger clinical attacks also in other autoantibody-associated disorders [44,45,46].
In this context, it is important to underline that the absence of total IgG OCB and of intrathecal ITPR1-IgG/anti-Sj synthesis, as suggested by a negative AI, does not per se argue against a pathogenetic role of the antibody. Rather, negative OCB and negative AIs are frequent findings in antibody-related autoimmune disorders of the CNS, including MOG-IgG-positive encephalomyelitis [45,46,47,48,49] and AQP4-IgG-positive neuromyelitis optica spectrum disorders [44, 50], two diseases in which a direct pathogenic impact of the antibody has been proven or is highly likely [51,52,53,54]. A negative ITPR1-IgG-AI, indicating an extrathecal origin of CSF ITPR1-IgG/anti-Sj, was reported also in the only other patient investigated so far [16].
-
7.
No co-existing anti-neural autoantibodies were present in addition to ITPR1-IgG/anti-Sj in the CSF and serum in our patient, despite very broad screening for such reactivities. Similarly, ITRP1-IgG/anti-Sj was absent in 80 neurological disease controls with autoantibody-related neurological syndromes or multiple sclerosis in three previous studies [1, 18, 27].
-
8.
ITPR1-IgG/anti-Sj has been shown to be absent in healthy individuals (N = 250) [16, 18, 27] as well as in patients with non-neurological disorders (N = 105) [18, 27] upon testing by cell-based assays and was absent in almost 52,000 samples from patients with neurological symptoms of putative autoimmune aetiology in the 2018 study by Alfugham et al. [18]. This renders accidental coincidence at least unlikely.
-
9.
Despite wide-ranging differential diagnostic endeavours, no other disorder that would better explain the patient’s symptoms was found.
It is of note that ITPR1-IgG/anti-Sj belonged exclusively to the IgG2 subclass in the present patient, in contrast to two of the index cases with ACA, which showed mainly IgG1 antibodies with only weak IgG2 and IgG3 reactivity (no data available from the third index patient) [1]. However, ITPR1-IgG/anti-Sj of exclusively the IgG2 subclass was also observed in two of the three patients with peripheral neuropathy previously reported by us [16].
Although complement activation by IgG1 and IgG3 is much stronger than that by IgG2, at least in humans (partly due to reduced binding of C1q to IgG2), IgG2 is still capable of triggering the classical complement pathway [55,56,57,58], putatively depending on antigen surface density [55,56,57], antibody concentration and hexamer formation [59]. IgG2 is also said to have limited Fc-receptor-mediated immune effector functions, because it binds only poorly to FcγRIIIa [60, 61]. However, myeloid cell-mediated antibody-dependent cellular cytotoxicity (ADCC) may be less affected than NK cell-mediated ADCC [57, 62].
Interestingly, all exclusively ITPR1-IgG2-positive patients (N = 3), the present case among them, were relatively mildly affected, while the exclusively [16] or mainly [1] ITPR1-IgG1-positive patients with available outcome data (N = 2) were severely disabled at most recent follow-up. IgG subclasses were not assessed in the remaining ITPR1-IgG/anti-Sj-seropositive patients reported so far [18, 26, 27]. IgG subclass analysis should be included in all future studies on ITPR1-IgG/anti-Sj, since differences in subclass distribution may be associated with pathogenetic and, in consequence, clinical and prognostic differences. Future studies should address the therapeutic and prognostic impact of differences in IgG subclass distribution.
ITPR1 is traditionally thought to be expressed mostly in the membrane of the smooth endoplasmic reticulum (ER) (and to a lesser degree on rough ER and the nuclear envelope). Therefore, it may not be accessible to serum or CSF ITPR1-IgG. If that is so, ITPR1-IgG/anti-Sj may just be a diagnostic marker, as is the case in other paraneoplastic neurological disorders, and T cell-mediated pathomechanisms may play a more important role. In a previous study, we demonstrated the presence of ITPR1-specific PBMCs in an ITPR1-IgG/anti-Sj-seropositive patient by use of a 3H-thymidine proliferation assay, including, besides ITPR1-specific B cells, CD4+ T cells and CD8+ CD45RO+ T cells [16].
On the other hand, surface localization of ITPR1 has indeed been reported to occur in some cell types, including neurons [19,20,21,22,23,24,25]. Moreover, uptake of human [63,64,65,66,67,68,69,70,71,72] as well as mouse [73] IgG autoantibodies targeting intracellular antigens by neurons has been described in numerous studies, as we have outlined elsewhere [2]. For example, incorporation of paraneoplastic anti-CDR2 and CDR2L antibodies (anti-Yo) has been reported to affect PC calcium homoeostasis [72] and to cause defective PC arborization [72] and PC cell death in cultured PCs [64]. Similarly, amphiphysin autoantibodies from patient sera transferred to rats were found to be internalized by neurons, to alter the function of inhibitory synapses in vivo [74], and to cause dose-dependent stiffness, with spasms resembling human stiff-person syndrome [74,75,76], as well as anxiety behaviour [77]. Autoantibodies to recoverin have been shown to induce caspase-dependent apoptosis after being taken up by retinal cells, probably via endocytosis [78]. A pathogenic effect has also been proposed for antibodies to the intracellular antigen GAD65 (glutamate decarboxylase 65) [79, 80]. Finally, endocytic uptake of autoantibodies has also been found for a subset of anti-DNA antibodies [81,82,83,84]. Of note, incorporation of IgG and IgM by PCs has been reported to occur independent of binding of the antibodies to PC surface antigens [65]. Taken together, these findings would imply that, at least with regard to some subsets of neurons, antigen surface expression may not be a prerequisite of antibody uptake and autoantibody-mediated functional or cellular cell damage [2]. ITPR1-IgG-related damage might then be mediated by disruption of Ca2+ homoeostasis via blocking or stimulation of ITPR1. This could result either in neuronal dysfunction (which would be compatible with the lack of radiological evidence for inflammatory brain damage or atrophy in the patient described in Section A and the partial—and once almost complete—recovery observed after immunotherapy in that patient) or, given that release of Ca2+ from the ER upon ITPR1 stimulation plays an important role in the induction of apoptosis [85,86,87], neuronal cell death. Further studies, including passive transfer experiments, are needed to shed more light on the pathogenetic relevance of ITPR1-IgG/anti-Sj.
Serum ITPR1-IgG/anti-Sj titres in the present patient were clearly positive (1:1000, 1:320, 1:100, respectively) but lower than those reported in two of the three index patients with cerebellar ataxia [1] and two of the three patients with neuropathy described in Ref. [2], all of whom were tested using the same methods as applied in the present case. However, no correlation of ITPR1-IgG/anti-Sj titres with the severity or type of clinical or oncological phenotype was found in a previous study [18], although more data from larger cohorts are certainly needed to draw definite conclusions.
Discussion
ITPR1-IgG/anti-Sj autoantibodies in patients with neurological symptoms have been first described only relatively recently [1]. This new reactivity was initially thought to be associated mainly with autoimmune cerebellar ataxia. However, as shown in this review, the spectrum of syndromes associated with ITPR1-IgG/anti-Sj is much broader. A substantial proportion of all published cases (~ 45%) occurred in tumour patients, which renders the condition a facultative paraneoplastic disorder and the antibodies potential tumour markers.
In addition to providing a general overview of the clinical and paraclinical features and summarizing the current knowledge on the pathophysiology of this new disorder, we here specifically point to the association of serum ITPR1-IgG/anti-Sj with dementia and psychosis by reporting on a patient in whom these antibodies were associated with rapidly progressive, severe cognitive decline, mainly affecting memory, attention and executive functions, optic hallucinations and depression.
While this is the only detailed report on that association, neuropsychiatric symptoms have been described in a few further patient positive for serum ITPR1-IgG/anti-Sj, including dementia and encephalopathy [18], confusion [16], memory loss [16], insomnia, anxiety and depression [18], as well as temporal lobe atrophy [18] and suspected temporal lobe epilepsy [16]. This provides a rationale for investigating ITPR1 autoimmunity in patients with psychiatric symptoms or dementia of unknown cause as well as in patients with suspected limbic encephalitis.
Little is known so far in terms of optimum treatment of patients with ITPR1-IgG/anti-Sj-associated autoimmunity. It is therefore an important observation that immunotherapy repeatedly resulted in significant clinical improvement and in relative stability in the long-term in the patient described here. On the other hand, many ITPR1-IgG/anti-Sj-positive patients showed no or only little response to immunotherapy, and the reasons for differences in treatment response are not well understood. It is therefore of interest that a milder course of disease was associated with the exclusive presence of ITPR1-IgG/anti-Sj antibodies of the IgG2 subclass in some patients, which warrants systematic evaluation of IgG subclasses in ITPR1 autoimmunity. Moreover, ITPR1-IgG/anti-Sj was still detectable shortly after 7 cycles of PLEX in the only patient with available data. This might indicate a need for prolonged PLEX treatment (as already standard of care in many non-neurological autoimmune diseases) in some patients. When planning PLEX, it is import to consider that ITPR1-IgG/anti-Sj titres are very high in some cases (up to 1:245,760).
Future studies should also pay attention to less well-documented symptoms such as dysautonomia, bladder and bowel disturbances and pain as well as to spinal cord and optic nerve involvement.
Conclusions
Our findings support the notion that the spectrum of symptoms associated with autoimmunity to ITPR1 in neurological patients is broader than initially thought. They provide a rationale for studies evaluating the frequency of ITPR1-IgG/anti-Sj in patients with cognitive decline and/or psychosis of unknown aetiology. In many cases, the presence of ITPR1-IgG was first suggested by IHC. This highlights the importance of screening sera and CSF from patients with suspected autoimmune encephalitis by means of tissue-based assays in order to recognize reactivities that may prompt a second level screening for ITPR1 or other rarer antigens by means of antigen-specific assays, considering that many laboratories routinely use only commercial assays (blot or CBA) for the most common anti-neuronal and anti-glial antibodies. Passive transfer experiments are needed to clarify whether the disease is directly caused by the antibody or rather by T-cell-mediated mechanisms. Future studies investigating treatment responses and outcome in ITPR1-related autoimmunity should systematically evaluate ITPR1-IgG/anti-Sj subclasses, given that the presence of exclusively IgG2 antibodies was associated with a milder course of disease in some cases compared to patients with predominantly IgG1 antibodies. Considering the high rate of paraneoplastic cases, tumour screening seems essential in patients presenting with ITPR1-IgG/anti-Sj-related autoimmune encephalitis or polyneuropathy.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Abbreviations
- ACA:
-
Autoimmune cerebellar ataxia
- ADCC:
-
Antibody-dependent cellular cytotoxicity
- AI:
-
Antibody index
- ART:
-
Autonomic response testing
- BSR:
-
Blood sedimentation rate
- CA125:
-
Cancer antigen 125
- CBA:
-
Cell-based assay
- CDT:
-
Clock-drawing test
- cMRI:
-
Cranial MRI
- CSF:
-
Cerebrospinal fluid
- CRP:
-
C-reactive protein
- CT:
-
Computed tomography
- CTD:
-
Connective tissue disorders
- CYCL:
-
Cyclophosphamide
- ER:
-
Endoplasmic reticulum
- EEG:
-
Electroencephalography
- EMG:
-
Electromyography
- FDG-PET:
-
2-Deoxy-2-[18F]fluoro-d-glucose positron emission tomography
- FITC:
-
Fluorescein isothiocyanate
- Gd:
-
Gadolinium
- GFAP:
-
Glial fibrillary astrocytic protein
- HEK293:
-
Human embryonic kidney 293 cells
- IgG/M/A:
-
Immunoglobulin G/M/A
- IHC:
-
Immunohistochemistry
- ITPR1:
-
Inositol 1,4,5-trisphosphate receptor 1
- IVIG:
-
Intravenous immunoglobulins
- IVMP:
-
Intravenous methylprednisolone
- LETM:
-
Longitudinally extensive transverse myelitis
- MMSE:
-
Mini-Mental Status Examination
- MRI:
-
Magnetic resonance imaging
- NMDAR:
-
N-Methyl d-aspartate receptors (NMDAR)
- OCB:
-
Oligoclonal bands
- PC:
-
Purkinje cells
- PET–CT:
-
Combined positron emission tomography and computed tomography
- PLEX:
-
Plasma exchange
- QAlb:
-
Albumin CSF/serum ratio
- REM:
-
Rapid eye movements
- TST:
-
Thermoregulatory sweat test
References
Jarius S, Scharf M, Begemann N, Stocker W, Probst C, Serysheva I, Nagel S, Graus F, Psimaras D, Wildemann B, Komorowski L. Antibodies to the inositol 1,4,5-trisphosphate receptor type 1 (ITPR1) in cerebellar ataxia. J Neuroinflamm. 2014;11:206.
Jarius S, Wildemann B. “Medusa head ataxia”: the expanding spectrum of Purkinje cell antibodies in autoimmune cerebellar ataxia. Part 3: Anti-Yo/CDR2, anti-Nb/AP3B2, PCA-2, anti-Tr/DNER, other antibodies, diagnostic pitfalls, summary and outlook. J Neuroinflamm. 2015;12:168.
Jarius S, Wildemann B. “Medusa head ataxia”: the expanding spectrum of Purkinje cell antibodies in autoimmune cerebellar ataxia. Part 2: Anti-PKC-gamma, anti-GluR-delta2, anti-Ca/ARHGAP26 and anti-VGCC. J Neuroinflamm. 2015;12:167.
Jarius S, Wildemann B. “Medusa-head ataxia”: the expanding spectrum of Purkinje cell antibodies in autoimmune cerebellar ataxia. Part 1: anti-mGluR1, anti-Homer-3, anti-Sj/ITPR1 and anti-CARP VIII. J Neuroinflamm. 2015;12:166.
Zuliani L, Sabater L, Saiz A, Baiges JJ, Giometto B, Graus F. Homer 3 autoimmunity in subacute idiopathic cerebellar ataxia. Neurology. 2007;68:239–40.
Sabater L, Bataller L, Carpentier AF, Aguirre-Cruz ML, Saiz A, Benyahia B, Dalmau J, Graus F. Protein kinase Cgamma autoimmunity in paraneoplastic cerebellar degeneration and non-small-cell lung cancer. J Neurol Neurosurg Psychiatry. 2006;77:1359–62.
Sillevis Smitt P, Kinoshita A, De Leeuw B, Moll W, Coesmans M, Jaarsma D, Henzen-Logmans S, Vecht C, De Zeeuw C, Sekiyama N, Nakanishi S, Shigemoto R. Paraneoplastic cerebellar ataxia due to autoantibodies against a glutamate receptor. N Engl J Med. 2000;342:21–7.
Coesmans M, Smitt PA, Linden DJ, Shigemoto R, Hirano T, Yamakawa Y, van Alphen AM, Luo C, van der Geest JN, Kros JM, et al. Mechanisms underlying cerebellar motor deficits due to mGluR1-autoantibodies. Ann Neurol. 2003;53:325–36.
Shiihara T, Kato M, Konno A, Takahashi Y, Hayasaka K. Acute cerebellar ataxia and consecutive cerebellitis produced by glutamate receptor delta2 autoantibody. Brain Dev. 2007;29:254–6.
Shimokaze T, Kato M, Yoshimura Y, Takahashi Y, Hayasaka K. A case of acute cerebellitis accompanied by autoantibodies against glutamate receptor delta2. Brain Dev. 2007;29:224–6.
Bataller L, Sabater L, Saiz A, Serra C, Claramonte B, Graus F. Carbonic anhydrase-related protein VIII: autoantigen in paraneoplastic cerebellar degeneration. Ann Neurol. 2004;56:575–9.
Hoftberger R, Sabater L, Velasco F, Ciordia R, Dalmau J, Graus F. Carbonic anhydrase-related protein VIII antibodies and paraneoplastic cerebellar degeneration. Neuropathol Appl Neurobiol. 2014;40:650–3.
Doss S, Nümann A, Ziegler A, Siebert E, Borowski K, Stöcker W, Prüss H, Wildemann B, Endres M, Jarius S. Anti-Ca/anti-ARHGAP26 antibodies associated with cerebellar atrophy and cognitive decline. J Neuroimmunol. 2014;267:102–4.
Jarius S, Martinez-Garcia P, Hernandez AL, Brase JC, Borowski K, Regula JU, Meinck HM, Stocker W, Wildemann B, Wandinger KP. Two new cases of anti-Ca (anti-ARHGAP26/GRAF) autoantibody-associated cerebellar ataxia. J Neuroinflamm. 2013;10:7.
Wallwitz U, Brock S, Schunck A, Wildemann B, Jarius S, Hoffmann F. From dizziness to severe ataxia and dysarthria: new cases of anti-Ca/ARHGAP26 autoantibody-associated cerebellar ataxia suggest a broad clinical spectrum. J Neuroimmunol. 2017;309:77–81.
Jarius S, Ringelstein M, Haas J, Serysheva II, Komorowski L, Fechner K, Wandinger KP, Albrecht P, Hefter H, Moser A, et al. Inositol 1,4,5-trisphosphate receptor type 1 autoantibodies in paraneoplastic and non-paraneoplastic peripheral neuropathy. J Neuroinflamm. 2016;13:278.
Uhlen M, Bjorling E, Agaton C, Szigyarto CA, Amini B, Andersen E, Andersson AC, Angelidou P, Asplund A, Asplund C, et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol Cell Proteomics. 2005;4:1920–32.
Alfugham N, Gadoth A, Lennon VA, Komorowski L, Scharf M, Hinson S, McKeon A, Pittock SJ. ITPR1 autoimmunity: frequency, neurologic phenotype, and cancer association. Neurol Neuroimmunol Neuroinflamm. 2018;5:e418.
Tanimura A, Tojyo Y, Turner RJ. Evidence that type I, II, and III inositol 1,4,5-trisphosphate receptors can occur as integral plasma membrane proteins. J Biol Chem. 2000;275:27488–93.
Lischka FW, Zviman MM, Teeter JH, Restrepo D. Characterization of inositol-1,4,5-trisphosphate-gated channels in the plasma membrane of rat olfactory neurons. Biophys J. 1999;76:1410–22.
Vermassen E, Parys JB, Mauger JP. Subcellular distribution of the inositol 1,4,5-trisphosphate receptors: functional relevance and molecular determinants. Biol Cell. 2004;96:3–17.
Taylor CW, Dellis O. Plasma membrane IP3 receptors. Biochem Soc Trans. 2006;34:910–2.
Dellis O, Dedos SG, Tovey SC, Taufiq Ur R, Dubel SJ, Taylor CW. Ca2+ entry through plasma membrane IP3 receptors. Science. 2006;313:229–33.
Cunningham AM, Ryugo DK, Sharp AH, Reed RR, Snyder SH, Ronnett GV. Neuronal inositol 1,4,5-trisphosphate receptor localized to the plasma membrane of olfactory cilia. Neuroscience. 1993;57:339–52.
Khan AA, Steiner JP, Klein MG, Schneider MF, Snyder SH. IP3 receptor: localization to plasma membrane of T cells and cocapping with the T cell receptor. Science. 1992;257:815–8.
Berzero G, Hacohen Y, Komorowski L, Scharf M, Dehais C, Leclercq D, Fourchotte V, Buecher B, Honnorat J, Graus F, et al. Paraneoplastic cerebellar degeneration associated with anti-ITPR1 antibodies. Neurol Neuroimmunol Neuroinflamm. 2017;4:e326.
Fouka P, Alexopoulos H, Chatzi I, Dedos SG, Samiotaki M, Panayotou G, Politis P, Tzioufas A, Dalakas MC. Antibodies to inositol 1,4,5-triphosphate receptor 1 in patients with cerebellar disease. Neurol Neuroimmunol Neuroinflamm. 2017;4:e306.
Lam K, Chi MS, Chan ELY, Ip WK, Chan EYT, Au EYL. Adenocarcinoma of the lung associated with antibodies to inositol 1,4,5-trisphosphate receptor 1 (ITPR1-IgG/anti-Sj) and cerebellar ataxia. Neuroimmunol Rep. 2022;2:100104.
Xu L, Xian W, Li J, Yao X, Long Y. Purkinje cell (PC) antibody positivity in a patient with autoimmune glial fibrillary acidic protein (GFAP) astrocytopathy. Int J Neurosci. 2020. https://doi.org/10.1080/00207454.2020.1860965.
Cirkel A, Wandinger KP, Ditz C, Leppert J, Hanker L, Cirkel C, Neumann A, Brocke J, Hoftberger R, Komorowski L, et al. Paraneoplastic encephalomyeloradiculits with multiple autoantibodies against ITPR-1, GFAP and MOG: case report and literature review. Neurol Res Pract. 2021;3:48.
Schiff JR, Fiorillo BP, Sadjadi R, Henry TL, Gruen JK, Gensler LM. Confabulation, amnesia and motor memory loss as a presentation of apparent ITPR1 antibody autoimmune encephalitis. BMJ Case Rep. 2021;14:e244316.
Sabater L, Gaig C, Gelpi E, Bataller L, Lewerenz J, Torres-Vega E, Contreras A, Giometto B, Compta Y, Embid C, et al. A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: a case series, characterisation of the antigen, and post-mortem study. Lancet Neurol. 2014;13:575–86.
Gruter T, Mollers FE, Tietz A, Dargvainiene J, Melzer N, Heidbreder A, Strippel C, Kraft A, Hoftberger R, Schoberl F, et al. Clinical, serological and genetic predictors of response to immunotherapy in anti-IgLON5 disease. Brain. 2022. https://doi.org/10.1093/brain/awac090.
Balint B, Jarius S, Nagel S, Haberkorn U, Probst C, Blocker IM, Bahtz R, Komorowski L, Stocker W, Kastrup A, et al. Progressive encephalomyelitis with rigidity and myoclonus: a new variant with DPPX antibodies. Neurology. 2014;82:1521–8.
Piepgras J, Holtje M, Michel K, Li Q, Otto C, Drenckhahn C, Probst C, Schemann M, Jarius S, Stocker W, et al. Anti-DPPX encephalitis: pathogenic effects of antibodies on gut and brain neurons. Neurology. 2015;85:890–7.
Stoeck K, Carstens PO, Jarius S, Raddatz D, Stocker W, Wildemann B, Schmidt J. Prednisolone and azathioprine are effective in DPPX antibody-positive autoimmune encephalitis. Neurol Neuroimmunol Neuroinflamm. 2015;2:e86.
Munoz-Lopetegi A, Graus F, Dalmau J, Santamaria J. Sleep disorders in autoimmune encephalitis. Lancet Neurol. 2020;19:1010–22.
Boronat A, Gelfand JM, Gresa-Arribas N, Jeong H-Y, Walsh M, Roberts K, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R, Graus F, et al. Encephalitis and antibodies to DPPX, a subunit of Kv4.2 potassium channels. Ann Neurol. 2012;73:120–8.
Signore S, Sorrentino A, Ferreira-Martins J, Kannappan R, Shafaie M, Del Ben F, Isobe K, Arranto C, Wybieralska E, Webster A, et al. Inositol 1, 4, 5-trisphosphate receptors and human left ventricular myocytes. Circulation. 2013;128:1286–97.
Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419.
Przyklenk K, Maynard M, Whittaker P. First molecular evidence that inositol trisphosphate signaling contributes to infarct size reduction with preconditioning. Am J Physiol Heart Circ Physiol. 2006;291:H2008-2012.
Matsumoto M, Nakagawa T, Inoue T, Nagata E, Tanaka K, Takano H, Minowa O, Kuno J, Sakakibara S, Yamada M, et al. Ataxia and epileptic seizures in mice lacking type 1 inositol 1,4,5-trisphosphate receptor. Nature. 1996;379:168–71.
Graus F, Vogrig A, Muniz-Castrillo S, Antoine JG, Desestret V, Dubey D, Giometto B, Irani SR, Joubert B, Leypoldt F, et al. Updated diagnostic criteria for paraneoplastic neurologic syndromes. Neurol Neuroimmunol Neuroinflamm. 2021;8:e1014.
Jarius S, Ruprecht K, Wildemann B, Kuempfel T, Ringelstein M, Geis C, Kleiter I, Kleinschnitz C, Berthele A, Brettschneider J, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflamm. 2012;9:14.
Jarius S, Kleiter I, Ruprecht K, Asgari N, Pitarokoili K, Borisow N, Hummert MW, Trebst C, Pache F, Winkelmann A, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 3: Brainstem involvement - frequency, presentation and outcome. J Neuroinflamm. 2016;13:281.
Jarius S, Lechner C, Wendel EM, Baumann M, Breu M, Schimmel M, Karenfort M, Marina AD, Merkenschlager A, Thiels C, et al. Cerebrospinal fluid findings in patients with myelin oligodendrocyte glycoprotein (MOG) antibodies. Part 2: Results from 108 lumbar punctures in 80 pediatric patients. J Neuroinflamm. 2020;17:262.
Jarius S, Pellkofer H, Siebert N, Korporal-Kuhnke M, Hummert MW, Ringelstein M, Rommer PS, Ayzenberg I, Ruprecht K, Klotz L, et al. Cerebrospinal fluid findings in patients with myelin oligodendrocyte glycoprotein (MOG) antibodies. Part 1: Results from 163 lumbar punctures in 100 adult patients. J Neuroinflamm. 2020;17:261.
Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, Pache F, Stich O, Beume LA, Hummert MW, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflamm. 2016;13:280.
Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, Pache F, Stich O, Beume LA, Hummert MW, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 1: Frequency, syndrome specificity, influence of disease activity, long-term course, association with AQP4-IgG, and origin. J Neuroinflamm. 2016;13:279.
Jarius S, Paul F, Franciotta D, Ruprecht K, Ringelstein M, Bergamaschi R, Rommer P, Kleiter I, Stich O, Reuss R, et al. Cerebrospinal fluid findings in aquaporin-4 antibody positive neuromyelitis optica: results from 211 lumbar punctures. J Neurol Sci. 2011;306:82–90.
Jarius S, Paul F, Weinshenker BG, Levy M, Kim HJ, Wildemann B. Neuromyelitis optica. Nat Rev Dis Primers. 2020;6:85.
Levy M, Wildemann B, Jarius S, Orellano B, Sasidharan S, Weber MS, Stuve O. Immunopathogenesis of neuromyelitis optica. Adv Immunol. 2014;121:213–42.
Jarius S, Paul F, Franciotta D, Waters P, Zipp F, Hohlfeld R, Vincent A, Wildemann B. Mechanisms of disease: aquaporin-4 antibodies in neuromyelitis optica. Nat Clin Pract Neurol. 2008;4:202–14.
Jarius S, Wildemann B. AQP4 antibodies in neuromyelitis optica: diagnostic and pathogenetic relevance. Nat Rev Neurol. 2010;6:383–92.
Garred P, Michaelsen TE, Aase A. The IgG subclass pattern of complement activation depends on epitope density and antibody and complement concentration. Scand J Immunol. 1989;30:379–82.
Lucisano Valim YM, Lachmann PJ. The effect of antibody isotype and antigenic epitope density on the complement-fixing activity of immune complexes: a systematic study using chimaeric anti-NIP antibodies with human Fc regions. Clin Exp Immunol. 1991;84:1–8.
Rosner T, Kahle S, Montenegro F, Matlung HL, Jansen JHM, Evers M, Beurskens F, Leusen JHW, van den Berg TK, Valerius T. Immune effector functions of human IgG2 antibodies against EGFR. Mol Cancer Ther. 2019;18:75–88.
Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 2014;5:520.
Diebolder CA, Beurskens FJ, de Jong RN, Koning RI, Strumane K, Lindorfer MA, Voorhorst M, Ugurlar D, Rosati S, Heck AJ, et al. Complement is activated by IgG hexamers assembled at the cell surface. Science. 2014;343:1260–3.
Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, Daeron M. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood. 2009;113:3716–25.
Bruggemann M, Williams GT, Bindon CI, Clark MR, Walker MR, Jefferis R, Waldmann H, Neuberger MS. Comparison of the effector functions of human immunoglobulins using a matched set of chimeric antibodies. J Exp Med. 1987;166:1351–61.
Schneider-Merck T, Lammerts van Bueren JJ, Berger S, Rossen K, van Berkel PH, Derer S, Beyer T, Lohse S, Bleeker WK, Peipp M, et al. Human IgG2 antibodies against epidermal growth factor receptor effectively trigger antibody-dependent cellular cytotoxicity but, in contrast to IgG1, only by cells of myeloid lineage. J Immunol. 2010;184:512–20.
Greenlee JE, Burns JB, Rose JW, Jaeckle KA, Clawson S. Uptake of systemically administered human anticerebellar antibody by rat Purkinje cells following blood-brain barrier disruption. Acta Neuropathol. 1995;89:341–5.
Greenlee JE, Clawson SA, Hill KE, Wood BL, Tsunoda I, Carlson NG. Purkinje cell death after uptake of anti-Yo antibodies in cerebellar slice cultures. J Neuropathol Exp Neurol. 2010;69:997–1007.
Hill KE, Clawson SA, Rose JW, Carlson NG, Greenlee JE. Cerebellar Purkinje cells incorporate immunoglobulins and immunotoxins in vitro: implications for human neurological disease and immunotherapeutics. J Neuroinflamm. 2009;6:31.
Fabian RH, Petroff G. Intraneuronal IgG in the central nervous system: uptake by retrograde axonal transport. Neurology. 1987;37:1780–4.
Fabian RH, Ritchie TC. Intraneuronal IgG in the central nervous system. J Neurol Sci. 1986;73:257–67.
Borges LF, Busis NA. Intraneuronal accumulation of myeloma proteins. Arch Neurol. 1985;42:690–4.
Graus F, Illa I, Agusti M, Ribalta T, Cruz-Sanchez F, Juarez C. Effect of intraventricular injection of an anti-Purkinje cell antibody (anti-Yo) in a guinea pig model. J Neurol Sci. 1991;106:82–7.
Martin-Garcia E, Mannara F, Gutierrez-Cuesta J, Sabater L, Dalmau J, Maldonado R, Graus F. Intrathecal injection of P/Q type voltage-gated calcium channel antibodies from paraneoplastic cerebellar degeneration cause ataxia in mice. J Neuroimmunol. 2013;261:53–9.
Tanaka K, Tanaka M, Igarashi S, Onodera O, Miyatake T, Tsuji S. Trial to establish an animal model of paraneoplastic cerebellar degeneration with anti-Yo antibody. 2. Passive transfer of murine mononuclear cells activated with recombinant Yo protein to paraneoplastic cerebellar degeneration lymphocytes in severe combined immunodeficiency mice. Clin Neurol Neurosurg. 1995;97:101–5.
Schubert M, Panja D, Haugen M, Bramham CR, Vedeler CA. Paraneoplastic CDR2 and CDR2L antibodies affect Purkinje cell calcium homeostasis. Acta Neuropathol. 2014;128:835–52.
Sakai K, Gofuku M, Kitagawa Y, Ogasawara T, Hirose G. Induction of anti-Purkinje cell antibodies in vivo by immunizing with a recombinant 52-kDa paraneoplastic cerebellar degeneration-associated protein. J Neuroimmunol. 1995;60:135–41.
Geis C, Weishaupt A, Hallermann S, Grunewald B, Wessig C, Wultsch T, Reif A, Byts N, Beck M, Jablonka S, et al. Stiff person syndrome-associated autoantibodies to amphiphysin mediate reduced GABAergic inhibition. Brain. 2010;133:3166–80.
Sommer C, Weishaupt A, Brinkhoff J, Biko L, Wessig C, Gold R, Toyka KV. Paraneoplastic stiff-person syndrome: passive transfer to rats by means of IgG antibodies to amphiphysin. Lancet. 2005;365:1406–11.
Vincent A. Successful “passive transfer” of paraneoplastic stiff person syndrome with antibodies to an intracellular antigen. Brain. 2010;133:3164–5.
Geis C, Grunewald B, Weishaupt A, Wultsch T, Toyka KV, Reif A, Sommer C. Human IgG directed against amphiphysin induces anxiety behavior in a rat model after intrathecal passive transfer. J Neural Transm. 2012;119:981–5.
Shiraga S, Adamus G. Mechanism of CAR syndrome: anti-recoverin antibodies are the inducers of retinal cell apoptotic death via the caspase 9- and caspase 3-dependent pathway. J Neuroimmunol. 2002;132:72–82.
Hansen N, Grunewald B, Weishaupt A, Colaco MN, Toyka KV, Sommer C, Geis C. Human Stiff person syndrome IgG-containing high-titer anti-GAD65 autoantibodies induce motor dysfunction in rats. Exp Neurol. 2013;239:202–9.
Geis C, Weishaupt A, Grunewald B, Wultsch T, Reif A, Gerlach M, Dirkx R, Solimena M, Perani D, Heckmann M, et al. Human stiff-person syndrome IgG induces anxious behavior in rats. PLoS ONE. 2011;6:e16775.
Golan TD, Gharavi AE, Elkon KB. Penetration of autoantibodies into living epithelial cells. J Invest Dermatol. 1993;100:316–22.
Zack DJ, Stempniak M, Wong AL, Taylor C, Weisbart RH. Mechanisms of cellular penetration and nuclear localization of an anti-double strand DNA autoantibody. J Immunol. 1996;157:2082–8.
Yanase K, Smith RM, Puccetti A, Jarett L, Madaio MP. Receptor-mediated cellular entry of nuclear localizing anti-DNA antibodies via myosin 1. J Clin Invest. 1997;100:25–31.
Anderson NE, Budde-Steffen C, Wiley RG, Thurman L, Rosenblum MK, Nadeau SE, Posner JB. A variant of the anti-Purkinje cell antibody in a patient with paraneoplastic cerebellar degeneration. Neurology. 1988;38:1018–26.
Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–65.
Boehning D, Patterson RL, Sedaghat L, Glebova NO, Kurosaki T, Snyder SH. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat Cell Biol. 2003;5:1051–61.
Boehning D, Patterson RL, Snyder SH. Apoptosis and calcium: new roles for cytochrome c and inositol 1,4,5-trisphosphate. Cell Cycle. 2004;3:252–4.
Kalbe E, Kessler J, Calabrese P, Smith R, Passmore AP, Brand M, Bullock R. DemTect: a new, sensitive cognitive screening test to support the diagnosis of mild cognitive impairment and early dementia. Int J Geriatr Psychiatry. 2004;19:136–43.
Franciotta D, Columba-Cabezas S, Andreoni L, Ravaglia S, Jarius S, Romagnolo S, Tavazzi E, Bergamaschi R, Zardini E, Aloisi F, Marchioni E. Oligoclonal IgG band patterns in inflammatory demyelinating human and mouse diseases. J Neuroimmunol. 2008;200:125–8.
Reiber H. Cerebrospinal fluid—physiology, analysis and interpretation of protein patterns for diagnosis of neurological diseases. Mult Scler. 1998;4:99–107.
Jarius S, Steinmeyer F, Knobel A, Streitberger K, Hotter B, Horn S, Heuer H, Schreiber SJ, Wilhelm T, Trefzer U, et al. GABAB receptor antibodies in paraneoplastic cerebellar ataxia. J Neuroimmunol. 2013;256:94–6.
Mundiyanapurath S, Jarius S, Probst C, Stocker W, Wildemann B, Bosel J. GABA-B-receptor antibodies in paraneoplastic brainstem encephalitis. J Neuroimmunol. 2013;259:88–91.
Hahn S, Trendelenburg G, Scharf M, Denno Y, Brakopp S, Teegen B, Probst C, Wandinger KP, Buttmann M, Haarmann A, et al. Identification of the flotillin-1/2 heterocomplex as a target of autoantibodies in bona fide multiple sclerosis. J Neuroinflammation. 2017;14:123.
Sillevis Smitt P, Kinoshita A, De Leeuw B, Moll W, Coesmans M, Jaarsma D, Henzen-Logmans S, Vecht C, De Zeeuw C, Sekiyama N, et al. Paraneoplastic cerebellar ataxia due to autoantibodies against a glutamate receptor. N Engl J Med. 2000;342:21–7.
Acknowledgements
The authors are grateful to Mrs. Katharina Mattes and Mrs. Anna Eschlbeck (Neuroimmunology Group, University Hospital Heidelberg) and to the Nikon Imaging Center at the University of Heidelberg for excellent technical assistance. For the publication fee, we acknowledge financial support by Deutsche Forschungsgemeinschaft within the funding programme “Open Access Publikationskosten” as well as by Heidelberg University.
Funding
Open Access funding enabled and organized by Projekt DEAL. There was no specific funding for this study.
Author information
Authors and Affiliations
Contributions
SJ collected the data, performed the literature research, conducted the antibody tests, and wrote the manuscript. LK was involved in the development of the cell-based assay. CR, SB, HYC and CG were involved in patient care and provided clinical and paraclinical data. All authors were involved in revising the manuscript for intellectual content. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
The patient gave written informed consent for publication of this report.
Competing interests
S.J., S.B., H.Y.C., C.G., J.H., B.W. and C.R. report no conflicts of interest. L.K. is an employee of Euroimmun AG, Lübeck, Germany.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Appendix
Appendix
In this appendix, we report an ITPR1-IgG/anti-Sj-positive patient with progressive cognitive decline, affecting mainly short-term memory, executive dysfunction, attention deficits, a sleep disorder, and psychotic symptoms. Most previous cases have been described only very briefly. Given the rarity and the relative novelty of the disease, we decided to give a comprehensive description of the patient’s clinical and paraclinical findings to draw a more vivid ‘real-life’ picture of the disorder than statistical analyses alone can provide. Moreover, only detailed case descriptions allow evaluation of treatment responses and outcomes in a meaningful way.
Initial presentation and serological findings
A 48-year-old saleswoman with no pre-existing conditions except for euthyroid struma (treated by thyroidectomy more than 30 years earlier), cholecystolithiasis, type 2 diabetes and hypertension and no history of drug abuse first noticed increasing forgetfulness in summer 2019. At the end of September 2019, her family noted rapidly progressive short-term memory deficits interfering with everyday activities (e.g., missed appointments, forgetting to switch off the oven and iron), confused actions (cleaning of the floor with oil instead of water; inability to switch on the computer; dressing wet after showering) with a tendency to trivialize the errors made, and other personality changes (more quiescent and secluded, uncommunicative and taciturn, increased need for sleep; emotional blunting, less empathetic, increased irritability; at times sudden and impulsive agitation, newly occurring buying frenzy), which led to referral to a neurological hospital a month later.
On admission at the end of October 2019 the patient was fully conscious, oriented to person, time, place, and situation, and cooperative, and no focal neurological deficits were present except for bilateral dysdiadochokinesis. However, flat affect, increased irritability and perseverations were noted, and neuropsychological testing revealed severe cognitive impairments affecting concentration/attentional control, attention span, shared attention, short-term memory, working memory, long-term memory, processing speed, action planning and problem/error recognition (with constant perseveration of previous errors), leading to a diagnosis of ‘dysexecutive dementia’ (Mini-Mental Status Examination [MMSE] 27/30 [0/3 terms recalled], clock-drawing test [CDT] 3/7, DemTect [88] 9/18). The results of contrast-enhanced cranial MRI carried out a few days prior to admission were normal. Lumbar puncture performed on the day of admission showed a normal cell count, negative oligoclonal bands (OCB) [89], no qualitative evidence of intrathecal IgG, IgA or IgM synthesis, a normal albumin CSF/serum ratio and normal CSF total protein, CSF L-lactate and CSF glucose levels. However, serological screening for paraneoplastic and non-paraneoplastic antibodies demonstrated strong anti-neuronal serum reactivity characterized by distinct staining of the dense network of PC neurites in the molecular layer of the cerebellum, the PC somata, the main PC dendrite branches and the PC axons passing through the granular layer and cerebellar white matter (Fig. 2). An identical binding pattern was found with primate (1:320), rat (1:320) and mouse cerebellum (1:100) sections. The presence of serum autoantibodies to ITRP1 was demonstrated by use of a cell-based assay (CBA) employing ITPR1-transfected and mock-transfected HEK293 cells as test substrates [1, 16] (cells obtained from Euroimmun, Lübeck, Germany) (1:1000) (Fig. 2). The antibodies belonged exclusively to the IgG2 subclass; neither antibodies of the IgG1, IgG3 or IgG4 subclasses nor IgM or IgA antibodies to ITPR1 were detected by the immunohistochemical assay or by the CBA. A matched CSF sample was positive for ITPR1-IgG/anti-Sj in the CBA when tested undiluted, but was negative when tested at dilutions that would suggest intrathecal synthesis (as indicated by an antibody index [AI] ≥ 4), taking into account Qlim [90]. The findings of routine laboratory tests were normal except for mildly elevated serum C-reactive protein (CRP) (7 mg/l; normal: < 5) and blood sedimentation rate (22 mm/h). Electroencephalography (EEG) showed intermittent right-sided frontoparietal delta wave activity. Broad screening for other anti-neural autoantibodies (anti-Hu, -Yo, -Ri, -ANNA-3, -Tr/DNER, -Ma2/Ta, -amphiphysin, -GAD65, -NMDAR, -CASPR2, -LGI1, -AMPAR, -CV2/CRMP5, -SOX1, -mGluR1, -mGluR5, -GABA-A-R, GABA-B-R [91, 92], -Homer-3, -CARPVIII, -GluR-delta2, -ARHGAP26, -DPPX, -Zic4, -recoverin, -titin, -neurochondrin, -flotillin [93], -IgLON5, -glycine receptor, -AQP4, -MOG, -myelin), for systemic or organ-specific rheumatological autoantibodies and for anti-thyroidal autoantibodies (anti-TPO, anti-thyroglobulin) was negative, as was testing for neurolues, neuro-HIV, Creutzfeldt–Jakob disease, neurosarcoidosis and neuroborreliosis. Immunohistochemistry did not reveal evidence for additional anti-neural antibodies, irrespective whether primate, murine or rat CNS tissue was used. Testing of neuronal cell cultures, which is available at some research institutions, was not performed. Later the same day she left the ward in a disoriented state and was subsequently referred to a psychiatric hospital for risk of self-harm and impaired impulse control. There, besides marked impairment of short- and intermediate-term (but not biographic) memory and attention, also a significant loss in the capacity to judge and discern as well as behavioural abnormalities (e.g., ‘reading’ books holding them upside down) were documented (MMSE 23/30). CRP remained elevated (7.7 mg/l; 9.3 mg/l). A second (non-contrast-enhanced) MRI performed 3 weeks after the patient’s first admission was normal as well, as was a second lumbar puncture (0 cells/µl, no OCB, normal albumin CSF/serum ratio, negative HSV PCR and VZV PCR, negative measles virus AI). EEG showed right-sided frontoparietal delta waves. Considering the serological findings, a diagnosis of possible autoimmune encephalitis was made and treatment with high-dose IVMP (5 × 1 g/day) was commenced. This was followed by marked clinical improvement with almost complete resolution of the patient’s cognitive and psychiatric symptoms (MMSE 29/30). After discharge at the end of November 2019, she went back to work in her previous job, complaining only of mild residual cognitive and physical fatigue. According to her husband, she was “her old self again”.

Binding of ITPR1-IgG/anti-Sj to cerebellar sections (a–c; a: staining of the Purkinje cell layer and the molecular layer; b: staining of axonal cross sections in the cerebellar white matter) and to ITPR1-transfected HEK293 cells (d) but not to mock-transfected HEK293 cells (e). Green fluorescence (FITC) indicates binding of patient IgG; red fluorescence (AF568) indicates binding of patient IgG of the IgG2 subclass; blue fluorescence corresponds to cell nuclei stained by 4′,6-diamidino-2-phenylindole. Note that the vessel-associated staining present in panel (c) is caused by non-specific cross-reactivity of the detection antibody with primate IgG that has penetrated the vessel walls post mortem during preparation of the brain specimen, was therefore present also after incubation of the sections with PBS instead of serum (not shown), and does not indicate the presence of anti-vessel antibodies in our patient; no such cross-reactivity was seen when using another, whole-IgG-specific secondary antibody (a, b)
Repeat neuropsychological testing in mid-January 2020 confirmed marked and sustained improvement (MMSE 29/30 [3/3 terms recalled], CDT 7/7, DemTect 16/18; normalized affect and executive functions with restored problem recognition and solution capacity; only mild residual attention deficit). Cranial MRI and CSF analysis (no pleocytosis, OCB, intrathecal immunoglobulin synthesis, or blood–brain barrier dysfunction) were again normal. Routine laboratory tests were unremarkable except for an increase in CRP levels (13 mg/l; normal: < 5). EEG showed right-sided parieto-occipital theta wave activity. Serological testing again revealed high-titre ITRP1-IgG serum autoantibodies (CBA 1:1000; IHC 1:1000 on rat tissue, 1:320 on primate tissue, 1:100 on mouse tissue); as at first testing, only IgG2 antibodies were present. Incubation with undiluted CSF now resulted in slightly more distinct binding of IgG (in particular IgG2) to the ITPR1-transfected cells, but again no evidence for intrathecal ITPR1-IgG/anti-Sj synthesis was found. Again, a broad spectrum of other anti-neural antibodies was tested in both serum and CSF (anti-Hu, -Yo, -Ri, -ANNA-3, -Tr/DNER, -Ma2/Ta, -amphiphysin, -GAD65, -NMDAR, -CASPR2, -LGI1, -AMPAR, -CV2/CRMP5, -SOX1, -mGluR1, -mGluR5, -GABA-A-R, -GABA-B-R, -Homer-3, -CARPVIII, -GluR-delta2, -ARHGAP26, -DPPX, -Zic4, -recoverin, -titin, -neurochondrin, -flotillin, -IgLON5, -glycine receptor, -AQP4, -MOG, -myelin) but all results were negative. High-dose IVMP therapy (3 × 1 g/day) was repeated, followed by oral tapering, with the intention of eliminating the mild residual deficits and achieving long-term stability.
First relapse
Around 2 weeks later, however, while still being treated with oral prednisolone (60 mg/day), the patient contracted a respiratory infection, which was treated with amoxicillin. This was followed by a severe relapse a further 10 days later in February 2020, characterized by a marked decline in cognitive function (MMSE 24/30, CDT 1/7, DemTect 8/16), again particularly affecting executive functions, short-term memory (markedly impaired with false-positive contents/confabulation) and attention, increased distractibility, psychomotor restlessness, disorientation, wandering, confusion (attempts to empty a washing basket with nothing in it), impaired impulse control/disinhibition, hypergraphia, logorrhoea, perseverations and, for the first time, optic hallucinations (handing over of non-existent letters). A neuropsychological diagnosis of “right-temporally accentuated cognitive disorder” was made. CRP and blood sedimentation rate were not elevated at the time of readmission. Lumbar puncture was again normal (no pleocytosis, normal cytology, no OCB, no malignant cells), as was MRI of the brain and cervical spinal cord. Notably, however, cranial FDG-PET revealed elevated cerebral glucose metabolism involving the right medial temporal lobe (amygdala, parahippocampal gyrus) and the right striatum, especially the putamen. EEG again showed right-sided parieto-occipital theta wave activity. Broad screening for classical paraneoplastic and non-paraneoplastic anti-neural autoantibodies (anti-Hu, -Yo, -Ri, -Ma2/Ta, -NMDAR, -AMPAR, -LG1, -CASPR2, -GABABR, -DPPX, -GAD65/67, -amphiphysin, -SOX1, -GluR-delta2) and CTD-related antibodies was again unremarkable, but ITPR1-IgG/anti-Sj was still detectable in the serum at a CBA titre of 1:1000 and also in the CSF at a titre of 1:4. Assuming an ITPR1-IgG/anti-Sj-related pathogenesis, the patient was then treated with 5 cycles of PLEX (removed plasma from the first cycle tested positive for ITPR1-IgG/anti-Sj at a CBA titre of 1:320), followed by a third pulse of high-dose IVMP (5 × 1 g/day; with oral tapering until July). This resulted (after an initial IVMP-induced psychotic episode) in significant improvement of the patient’s cognitive status, especially of attention, memory and psychomotor activity. Due to the absence of complete recovery, treatment with intravenous immunoglobulins (IVIG) (4 × 35 g/day) and anti-psychotic therapy with risperidone, pipamperone and lorazepam was added, resulting in improved impulse control and a reduction in psychomotor restlessness. One month later, neuropsychological testing confirmed marked improvement, with mild residual impairment of attention and processing speed (MMSE 28/30, CDT 3/7, DemTect 14/18). EEG was now normal.
New symptoms
While a high proportion (9/20; 45%) of the ITPR1-IgG/anti-Sj-seropositive patients reported so far had acute cerebellar ataxia, no signs compatible with cerebellar involvement were noted in the present case during the first 10 months after onset, except for dysdiadochokinesis, which was apparent already at first presentation. At the end of March 2020, however, bilaterally pathological knee–heel (still present in July 2020) and finger–nose tests were found, and the patient developed an unsteady gait and mild tetraparesis (upper extremities: 4/5, lower extremities: 4/5; apparent also on arm and leg extension tests), prompting her to use a rollator for long walks. Cranial MRI performed in October 2020 and July 2021 again showed no cerebellar atrophy or any signs of cerebellitis. Serum ITPR1-IgG/anti-Sj was positive at a CBA titre of 1:100. At the most recent follow-up in August 2021, the patient’s gait instability and limb ataxia had resolved but she still used a rollator due to paraparesis. In April to May 2021, the patient in addition experienced an episode of blurred vision in both eyes lasting for 6–8 weeks, followed by complete recovery; no VEP was performed.
Second relapse
It was decided that regular PLEX treatments should be carried out. However, the next cycle of PLEX had to be interrupted after the first treatment due to a severe allergic reaction. Long-term therapy with cyclophosphamide was initiated instead, starting at the beginning of May 2020 (350 mg/m2/day for 3 days). This was paralleled by further improvement of the patient’s cognitive test results (MMSE 30/30, CDT 7/7, DemTect 15/18), although some impairment of attention, memory and processing speed persisted. Serum ITPR1-IgG/anti-Sj was detected at a CBA titre of 1:100 immediately after the first cyclophosphamide cycle.
In October 2020, around a month after the third cyclophosphamide cycle, another relapse-like episode occurred, with increasing attention and memory deficits, disorientation, psychomotor restlessness and formal thought disorders, associated with increased CRP (13 mg/l; normal < 5) but no leucocytosis or fever. EEG showed intermittent right-temporal slowing but no typical epileptiform discharges. Cranial MRI was unremarkable. Treatment with IVMP (5 × 1 g/day, with oral tapering), risperidone, and lorazepam was followed by significant neurocognitive improvement and complete psychiatric recovery at re-evaluation in November as well as EEG normalization.
Four further cycles of cyclophosphamide were given in November 2020 and in January, April and June 2021; these were paralleled by relative stability. The patient herself noted a mild increase in symptom severity shortly before each cyclophosphamide cycle. She also underlined at that time that she felt the previous IVMP therapies had been “highly effective”. Serum ITPR1-IgG/anti-Sj remained detectable at a CBA titre of 1:320 (November 2020).
Third (mild) relapse
In mid-July 2021 the patient was re-admitted with a mild depressive disorder associated with a mild subjective decline in short-term memory and executive functions (with no change observed upon neuropsychological testing). Lumbar puncture showed mild blood–CSF barrier dysfunction and, accordingly, slightly elevated CSF IgG and IgM levels, but no qualitative or quantitative evidence of intrathecal immunoglobulin synthesis. Brain MRI with gadolinium enhancement was normal. Treatment with IVMP (3 × 1 g/day) and sertraline resulted in prompt resolution of the patient’s symptoms. ITPR1-IgG/anti-Sj was still detectable in August at a titre of 1:1000 (CBA). In September 2021, it was planned to end the treatment with cyclophosphamide to avoid potential side effects of long-term use and to commence treatment with azathioprine.
Dementia/neurodegeneration markers
CSF beta-amyloid 1–40 levels were elevated on two occasions in November 2019 but normal in January 2020; CSF beta-amyloid 1–42 levels were normal when tested at first admission and in November 2019, as well as in January and February 2020; the CSF beta-amyloid 1–42/1–40 ratio was determined three times and was always normal. CSF tau protein levels were mildly elevated in February 2020 (368 ng/l) but normal in November 2019 and January 2020; phospho-tau levels were elevated in November 2019 (78 ng/l) but normal at first presentation and again in January and February 2020.
Tumour screening
ITPR1-IgG/anti-Sj-related autoimmunity has been reported to occur in a paraneoplastic context in some patients, but no tumour has yet been identified in the present case (last follow-up: August 2021). No M gradient was present; serum immune fixation and CSF isoelectric fixation, total serum IgG, IgA and IgM values, serum free kappa (k) and lambda (l) light chains, and the k/l quotient were all normal. In January 2020, tumour markers were tested. While the serum levels of CEA, CA19-9, SCC and NSE were normal, CA125 serum levels were markedly elevated (111 U/ml, normal: < 35; 88 U/ml in February 2020; 35 U/ml in August 2021). CT performed in November 2019 had shown bilaterally small nodular abnormalities in the lung (< 6 mm) as well as cystic structures at the isthmus uteri, both considered to be of unknown significance. Whole-body FDG-PET–CT (February 2020) was unremarkable in that regard, as were thoracic and abdominal follow-up CT (April 2021), pelvic MRI (February 2020), gynaecological examinations (November 2019 and February 2020), sonography of the abdomen (October 2019, February and April 2020; suggestive of chronic cholecystitis), and several chest X-rays [94].
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jarius, S., Bräuninger, S., Chung, HY. et al. Inositol 1,4,5-trisphosphate receptor type 1 autoantibody (ITPR1-IgG/anti-Sj)-associated autoimmune cerebellar ataxia, encephalitis and peripheral neuropathy: review of the literature. J Neuroinflammation 19, 196 (2022). https://doi.org/10.1186/s12974-022-02545-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-022-02545-4