
Abstract
Background
Repetitive mild traumatic brain injury (mTBI) can result in chronic visual dysfunction. G-protein receptor 110 (GPR110, ADGRF1) is the target receptor of N-docosahexaenoylethanolamine (synaptamide) mediating the anti-neuroinflammatory function of synaptamide. In this study, we evaluated the effect of an endogenous and a synthetic ligand of GPR110, synaptamide and (4Z,7Z,10Z,13Z,16Z,19Z)-N-(2-hydroxy-2-methylpropyl) docosa-4,7,10,13,16,19-hexaenamide (dimethylsynaptamide, A8), on the mTBI-induced long-term optic tract histopathology and visual dysfunction using Closed-Head Impact Model of Engineered Rotational Acceleration (CHIMERA), a clinically relevant model of mTBI.
Methods
The brain injury in wild-type (WT) and GPR110 knockout (KO) mice was induced by CHIMERA applied daily for 3 days, and GPR110 ligands were intraperitoneally injected immediately following each impact. The expression of GPR110 and proinflammatory mediator tumor necrosis factor (TNF) in the brain was measured by using real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR) in an acute phase. Chronic inflammatory responses in the optic tract and visual dysfunction were assessed by immunostaining for Iba-1 and GFAP and visual evoked potential (VEP), respectively. The effect of GPR110 ligands in vitro was evaluated by the cyclic adenosine monophosphate (cAMP) production in primary microglia isolated from adult WT or KO mouse brains.
Results
CHIMERA injury acutely upregulated the GPR110 and TNF gene level in mouse brain. Repetitive CHIMERA (rCHIMERA) increased the GFAP and Iba-1 immunostaining of glia cells and silver staining of degenerating axons in the optic tract with significant reduction of N1 amplitude of visual evoked potential at up to 3.5 months after injury. Both GPR110 ligands dose- and GPR110-dependently increased cAMP in cultured primary microglia with A8, a ligand with improved stability, being more effective than synaptamide. Intraperitoneal injection of A8 at 1 mg/kg or synaptamide at 5 mg/kg significantly reduced the acute expression of TNF mRNA in the brain and ameliorated chronic optic tract microgliosis, astrogliosis, and axonal degeneration as well as visual deficit caused by injury in WT but not in GPR110 KO mice.
Conclusion
Our data demonstrate that ligand-induced activation of the GPR110/cAMP system upregulated after injury ameliorates the long-term optic tract histopathology and visual impairment caused by rCHIMERA. Based on the anti-inflammatory nature of GPR110 activation, we suggest that GPR110 ligands may have therapeutic potential for chronic visual dysfunction associated with mTBI.
Similar content being viewed by others

Introduction
The white matter of brain, mainly composed of myelinated axons that relay a coordinating communication of grey matter of brain, is known to be susceptible to the impact of the acceleration/deceleration forces [1,2,3]. The optic tract is particularly vulnerable, and the disruption of the visual process can be detected even in mild TBI (mTBI) [4,5,6], especially in repeated mild close head brain injury such as blast-related injury [7]. According to the report from DVBIC (the Defense and Veterans Brain Injury Center), approximately 74% of the service members in Iraq and Afghanistan wars diagnosed with TBI since 2000 has experienced visual impairments that adversely affect the cognitive performance and quality of individual life. We and others have reported that repetitive mTBI in animal models produces profound neuropathological changes in the optic tract and visual deficit at acute and chronic phases [6, 8,9,10,11]. Although the visual deficit is an obvious confounding factor for motor, sensory, cognitive, and neuropsychiatric symptoms of TBI, there has been little effort for developing effective strategy to improve the TBI-linked visual deficit.
Synaptamide (N-docosahexaenoylethanolamin) is a metabolite of docosahexaenoic acid, an omega-3 fatty acid enriched in the brain [12]. Synaptamide is an endogenous ligand of GPR110 (ADGRF1) that belongs to the adhesion G-protein coupled receptor (GPCR) group VI family [13]. By binding to the GPCR-autoproteolysis-inducing domain of GPR110, synaptamide triggers the downstream cAMP/PKA/CREB signaling pathway [13, 14] and stimulates neurite outgrowth and synaptogenesis in mouse cortical neuron and induces neuronal differentiation of mouse neural stem cells at nanomolar concentrations [13, 15]. In an animal model of neuroinflammation induced by lipopolysaccharide (LPS), GPR110-dependent anti-inflammatory effects of synaptamide have also been observed [16, 17], suggesting GPR110 as a potential therapeutic target for neuroinflammation. In addition, (4Z,7Z,10Z,13Z,16Z,19Z)-N-(2-hydroxy-2-methylpropyl) docosa-4,7,10,13,16,19-hexaenamide (dimethylsynaptamide, A8), a chemical analogue of synaptamide with improved stability, was recently shown to be an effective GPR110 ligand that can activate cAMP signaling for axon regeneration [18]. Closed-head impact model of engineered rotational acceleration (CHIMERA) is a suitable animal model to study the long-term pathophysiology of TBI [9, 19], and repetitive CHIMERA (rCHIMERA) can produce significant visual deficit along with the glia activation and axon degeneration in the optic tract [11]. In this study, we investigated the therapeutic potential of GPR110 ligands in repetitive mTBI based on the optic tract histopathology and visual deficit induced by rCHIMERA and anti-inflammatory effects of GPR110/cAMP signaling.
Materials and methods
Experimental animals
Mice were housed in the animal facility of the National Institute on Alcohol Abuse and Alcoholism (NIAAA) with free access to standard food and water under a 12-h light–dark cycle. GPR110 heterozygous mice on C57BL/6J genetic background were generated by the Knockout Mouse Project (KOMP) Repository (MMRRC_046507-UCD). Heterozygous GPR110 male and female mice were bred to produce GPR110 knockout offspring. The WT littermates were used as controls. Adult male and female mice at an age of 2–4 months were used for all experiments. GPR110 KO mice developed normally and showed no visible abnormal phototype at this age. All animal experiments were conducted according to the NIH guidelines for the health and use of laboratory animals (LMS-HK-13).
rCHIMERA injury and treatments
The mild closed traumatic brain injury was induced by rCHIMERA (rCHI) as previously described [9]. Briefly, mice were anesthetized with 5% isoflurane in oxygen (1 L/min) for 3 min and placed in a supine position on the CHIMERA apparatus with the head lying flat on the hole of the base plate. An impact of 0.55 J of energy was delivered to the mouse head by a computer-controlled pneumatic air pressure. An impact was given daily for three consecutive days at a 24-h interval, and immediately following each impact, mice were intraperitoneally injected with vehicle, synaptamide, oleoylethanolamide (OEA), or A8 that was synthesized as previously reported [18]. The mice were returned to the home cage and allowed to recover, and the samples were collected according to the experimental timeline shown in Scheme 1. WT and GPR110 KO mice were randomly divided into four groups: (1) Sham, (2) rCHI + V, (3) rCHI + SYN, and (4) rCHI + A8. The solution of synaptamide and A8 for the intraperitoneal (i.p.) injection was freshly prepared from the DMSO stock by diluting with mixture of N,N-dimethylacetamide (DMAC, Sigma-Aldrich, St. Louis, MO, USA, cat# 271012-1L): Solutol H15 (Sigma-Aldrich, St. Louis, MO, USA, cat# 42966 ): 1 × PBS (1:1:2). The investigator who performed the experiment was blinded with respect to the compound identity for treatments until all data analyses were completed.

The experimental scheme
Quantitative real-time reverse transcription polymerase chain reaction (RT-PCR)
Mice were anesthetized with isoflurane and transcardially perfused with 0.1 M phosphate buffer (pH 7.4). The cerebral cortex was rapidly dissected, and total RNA was extracted by Trizol reagent (Thermo Fisher Scientific, cat# 15596026, MA, USA), reverse transcribed to cDNA with High Capacity cDNA Reverse Transcription kit (Applied Biosystem, cat# 4368814, CA, USA). Quantitative RT-PCR was performed using TaqMan™ probes (Thermo Fisher Scientific) run on the QuantStudio 3 Real-Time PCR system (Applied Biosystems by Thermo Fisher Scientific). cDNA samples were reacted with the TaqMan™ probe for GPR110 (Assay ID: Mm00505409_m1, cat# 4331182) or TNF (Assay ID: Mm00443258_m1, cat# 4331182) using TaqMan™ Fast Advanced Master Mix (Thermo Fisher Scientific, cat# 4444557) in the presence of the TaqMan™ probe for HPRT (hypoxanthine guanine phosphoribosyl transferase) (Assay ID: Mm00446968_m1, cat# 4448490) which was used as an internal control. Data were analyzed using comparative Ct method. The relative mRNA level of target gene normalization to HPRT was calculated as 2-ΔΔCt value.
Immunofluorescence and silver staining
The animals were deeply anesthetized with isoflurane and transcardially perfused with 0.1 M phosphate buffer (pH 7.4) and fixed with 4% paraformaldehyde. The brains were carefully removed and post-fixed in 4% paraformaldehyde solution for overnight at 4 °C and subsequently transferred into 30% sucrose solution at 4 °C. The brains were embedded with O.C.T. compound medium (VWR, PA, USA, cat# 25608-930) and frozen on dry ice and stored at – 80 °C. Coronal sections (25 μm thickness) were sliced by Leica Cryostat (Leica Biosystems Inc., IL, USA) and stored in cryoprotective solution at – 20 °C. Three sections per mouse (approximately bregma 1.68 to − 2.2 mm) were selected for staining.
The immunofluorescence double staining for Iba-1 and GFAP was conducted according to the procedure previously described [9]. The sections were incubated with Iba-1 (Wako Chemicals, VA, USA, cat# 019-19741) and GFAP antibodies (Novus Biologicals, CO, USA, cat# NBP1-05198) at 4 °C for overnight and Alexa fluor 488-conjugated F (ab’)2 fragment goat anti-rabbit IgG and sheep anti-chicken IgG (Jackson Immuno-Research labs, PA, USA, cat# 703-035-155) at RT for 1 h. After washing, the sections were mounted on the slides and covered with mounting medium containing DAPI (Vector laboratories, CA, USA, cat# H-1500). Immunofluorescence images were captured by an Olympus 1 × 81 microscopic system. The quantification of GFAP and Iba-1 expression was performed by measuring the fluorescence intensity (per μm2) using Metamorph software (Molecular Devices Inc., CA, USA).
The silver staining was performed using FD NeuroSilver™ Kit (FD NeuroTechnologies, Inc, MD, USA, cat# PK301A) according to the manufacturer's protocol. The 20×-magnified images of the optic tract were acquired in bright field with an Olympus 1 × 81 microscopic system. The staining intensity of images was quantified using Image J (NIH, MD, USA).
Visual evoked potential and electroretinogram
The visual evoked potential (VEP) and electroretinogram (ERG) were recorded with the Epsion Visual Electrophysiology System (Diagnosys, LLC, MA, USA). All procedures were performed under dim red light after the mice were acclimated in the room at least for 1 h. Mice were anesthetized with an intraperitoneal injection of ketamine and xylazine cocktail solution (100 mg/kg ketamine + 10 mg/kg xylazine). Each pupil of the mouse was dilated with a drop of 2.5% phenylephrine hydrochloride solution before being placed on a heated platform of color dome. A reference electrode was placed in the lower lip of the mouse and a ground electrode placed on the tail. For VEP measurements, the active electrode was subcutaneously inserted in the middle of the two ears. Both eyes were stimulated by the flash stimuli of white light (6500 K) at an intensity of 3.0 cd s/m2 repeatedly with each set including 100 sweeps. Three sets of readings were recorded and averaged to obtain the amplitude and latency (implicit time) of the N1 component (the first negative peak: P1–N1) as described earlier [11]. For ERG recording, a drop of topical petrolatum ophthalmic ointment was applied to the corneal surface of one eye on which a gold wire electrode was placed with the other eye covered. A light-adapted (photopic) protocol was used for ERG measurement as described earlier [20]. After testing, the mice were transferred to the home cage and placed on a heat pad to recover. The investigator was blinded regarding the identity of the experimental groups to prevent bias.
Determination of the brain synaptamide and A8 level
Synaptamide and A8 were analyzed by reversed phase liquid chromatography coupled to high-resolution tandem mass spectrometry using a Thermo Scientific Q-Exactive mass spectrometer. Mice were intraperitoneally injected with a mixture of d4-synaptamide and A8. At 1, 2, and 24 h after injection, mice were perfused with 1× PBS, and the cerebral cortex was collected and homogenized in 500 μL of water/methanol (1:1) mixture containing 2 μM URB597 (a fatty acid amide hydrolase inhibitor) and 50 μg/mL butyl hydroxytoluene (BHT) (Sigma-Aldrich, St. Louis, MO, USA, cat# W218405). Protein concentration of the homogenate was determined by the bicinchoninic acid protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA, cat# 23225). A mixture of deuterated internal standards of d4-anandamide and d6-A8 was added to 300 μL of homogenate, which was then brought to BHT-methanol/water (7:3) and centrifuged for 20 min at 4 °C. Supernatants were loaded onto a Strata-X polymeric C18 reverse-phase SPE cartridge (33 μm, 30 mg/mL, Phenomenex, Torrance, CA, USA) that was wetted with BHT-methanol and equilibrated with water. After washing with water, samples were eluted with 2.5 mL BHT-methanol into glass tubes, dried under N2, and resuspended in a small volume of BHT-methanol. Separation was made on an Eclipse C18 HPLC column (1.8 μm, 2.1 mm × 50 mm, Agilent Technologies, Santa Clara, CA, USA) using a tertiary gradient consisting of water (A), methanol (B), and acetonitrile (Avantor, Radnor Township, PA, USA) (C), with all solvents containing 0.01% acetic acid (Thermo Scientific). After pre-equilibration of column with A/B (60%/40%), 5 μL extract was injected, and the solvent composition was linearly changed to A/B/C (36.3%/15%/48.7%) in 5 min, followed by a linear gradient to A/B/C (13.5%/68.4%/18.1%) over 22 min. The mass transitions of 376.3 to 66.085, 400.3 to 72.081, 352.3 to 66.085, and 406.4 to 78.118 were used to detect d4-synaptamide, analog 8, d4-anandamide, and d6-A8, respectively. Quantitation of d4-synaptamide and A8 was made using d4-anandamide and d6-A8 as the respective internal standard. Results were normalized to protein amount and presented as fmol per microgram protein.
Determination of cAMP in primary microglia
The brain tissue from GPR110-WT or KO mice at 4 months of age were dissociated into single cells using the Adult Brain Dissociation Kit (Miltenyi Biotec, Gaithersburg, MD, USA, cat#130-107-677) according to the manufacturer’s instruction. To isolate the microglia, the single-cell suspension was incubated with CD11b Microbeads for 30 min at 4 °C. The cells were resuspended in MACS buffer and passed through the LS column (Miltenyi Biotech). The column was washed 4 times with MACS buffer and then magnetically labeled CD11b-positive cells were flushed out of the columns twice using MACS buffer. Enriched CD11b-positive microglia cells were pelleted by centrifuging at 300 × g at 4 °C for 10 min and resuspended in DMEM media supplemented with 10% fetal bovine serum (FBS). Isolated CD11b-positive microglia cells were cultured in poly-D-lysin coated 384-well white/clear bottom plate for 7 days by changing 50% of DMEM media supplemented with 10% FBS every other day. On the day of the cAMP assay, media was changed to the stimulation media consisting of phenol red-free DMEM supplemented with 0.5 mM IBMX (Tocris, Minneapolis, MN, USA), a phosphodiesterase inhibitor. Microglia cells were treated with different concentrations of synaptamide or A8 complexed with 0.05% fatty acid-free BSA (Sigma-Aldrich) in the presence of 40 μM vitamin E for 10 min. Separately, microglia cells were treated with 10 μM Forskolin (Tocris, cas# 66575-29-9) for 10 min. The cAMP production was measured by a homogenous time-resolved fluorescence assay with a cAMP Gs Dynamic Kit (Cis Bio, Bedford, MA, USA, cat#62AM4PEB) using the FlexStaion 3 device (Molecular devices)
Statistical analysis
The data are presented as mean ± standard error of the mean (SEM). Statistical analyses were performed using GraphPad Prism (version 6.04, GraphPad Software Inc.) for one-way or two-way analysis of variance (ANOVA) followed by the Tukey’s post hoc test for multiple comparisons, and p < 0.05 was considered statistically significant.
Results
CHIMERA induces GPR110 gene expression in the brain
To investigate the possible role of GPR110 activation in improving TBI outcome, we first examined the GPR110 mRNA expression in adult mouse brain using quantitative RT-PCR at 1 and 24 h after single or multiple CHIMERA given daily for three consecutive days. Both single and multiple CHIMERA significantly upregulated GPR110 gene level at 1 and 24 h after injury compared to the sham group (F = 38.53, p < 0.0001, n = 3-4/group, Fig. 1), indicating that gpr110 expression responds quickly to the brain injury.

Induction of GPR110 mRNA by single or multiple CHIMERA. The GPR110 mRNA in brain measured by RT-PCR using TaqMan probes shows upregulation at 1 h and 24 h after single (CHIMERA) or repeated CHIMERA applied daily for 3 days at a 24-h interval (rCHIMERA). The data are expressed as mean ± SEM (n = 3–4) and are representative of three independent experiments. **p < 0.05; ****p < 0.0001 vs. Sham
GPR110 ligands dose-dependently stimulate cAMP production in primary microglia
A methylated analogue of synaptamide, (4Z,7Z,10Z,13Z,16Z,19Z)-N-(2-hydroxy-2-methylpropyl)docosa-4,7,10,13,16,19-hexaenamide (A8, NCGC00248435) (Fig. 2A), was previously shown to be resistant to hydrolysis by fatty acid amide hydrolase (FAAH) and to produce cAMP more effectively than synaptamide in cortical neurons [18]. Since rCHIMERA induces microglia activation, we examined the capability of this ligand to stimulate cAMP production in primary microglia (Fig. 2B) where GPR110 was shown to be expressed [17]. As shown in cortical neurons [18], A8 dose-dependently increased microglial production of cAMP more effectively than synaptamide with EC50 of 0.79 nM. This increase was GPR110-dependent as the microglia from GPR110 KO mice did not respond to A8 while forskolin raised the cAMP level in both preparations (Fig. 2C).

Dose-dependent effect of GPR110 ligands on cAMP production in mouse primary microglia cells. A The chemical structure of GPR110 ligands, synaptamide and A8. B The concentration-dependent production of cAMP in primary microglia stimulated by GPR110 ligands. The EC50 values of synaptamide and A8 are 1.27 and 0.79 nM. Microglia isolated from adult GPR110-WT and KO mice were treated with varying concentrations of synaptamide or A8 for 10 min. The fold change data are presented as mean ± SEM from a representative experiment out of three independent experiments performed in triplicates. C The cAMP production in primary microglia after stimulation with forskolin. No difference was found in the cAMP production from WT and KO microglia. The microglia cells were treated with 10 μM forskolin for 10 min and cAMP production was measured in triplicate. The data is representative of three independent experiments
Determination of treatment dose and ligand stability in vivo
To determine the proper dose of GPR110 ligands for in vivo treatment, we examined their effects on the expression of an inflammation marker TNF after rCHIMERA. We chose the time point at 2 h after rCHIMERA because the maximum expression of TNF mRNA occurred at this time although it remained elevated throughout the duration of experiment up to 24 h after injury (F = 12.61, p < 0.001, n = 3–4/group, Fig. 3A). Based on the previous finding that synaptamide at 5 mg/kg significantly suppressed LPS-induced neuroinflammation [16, 17] as well as the effectiveness of A8 in microglia (Fig. 2C), we examined the dose range of 1–5 mg/kg for synaptamide and 0.1–1 mg/kg for A8 (Fig. 3B). We found that the brain mRNA level of TNF was significantly upregulated at 2 h after injury (F = 5.1, p < 0.001, 14.7 ± 2.27-fold, p < 0.001 vs. Sham). Intraperitoneal administration of A8 or synaptamide immediately following each CHIMERA dose-dependently suppressed the TNF expression. Compared with the vehicle-treated injured group (rCHI + V), synaptamide at 5 mg/kg or A8 at 1 mg/kg significantly suppressed TNF expression (p < 0.05 and p < 0.05 vs. rCHI + V for 1 mg/kg A8 and 5 mg/kg synaptamide, respectively). Synaptamide at 2 mg/kg or A8 at 0.1 and 0.5 mg/kg also showed the trend to reduce the TNF mRNA expression although the reduction was not statistically significant. The time–course of synaptamide and A8 in the brain was examined by injecting d4-synaptamide at 5 mg/kg and A8 at 1 mg/kg (Fig. 3C). Both d4-synaptamide and A8 were detected in the brain cortex obtained after transcardiac perfusion, but their level was less than 1% of the initial amounts injected at all time points. At 1 and 2 h after injection, the stable analogue A8 was found in the brain cortex at a significantly higher level (0.71 ± 0.09 and 0.67 ± 0.02 fmol/μg protein at 1 and 2 h, respectively) than synaptamide (0.3 ± 0.07 and 0.13 ± 0.04 fmol/μg protein at 1 and 2 h, respectively), even though the injected dose of A8 was 5-fold less compared with synaptamide. Both compounds were no longer detectable in the brain cortex after 24 h, indicating rapid metabolism and clearance of these compounds. Considering these results, we selected 5 mg/kg of synaptamide and 1 mg/kg of A8 for treatment for in vivo experiments.

Dose-dependent effect of synaptamide and A8 on TNF mRNA expression and their stability in vivo. A The mRNA level of TNF in the mouse cortex quantified by qRT-PCR using TaqMan probes at 2, 4, and 24 h after rCHIMERA. The TNF mRNA level from injured mice was upregulated at all time points compared with Sham control (***p < 0.001, *p < 0.05 vs. Sham) with the most pronounced increase occurring at 2 h after the last injury (*p < 0.05 vs. rCHI-4 h or rCHI-24 h). The data are expressed as mean ± SEM (n = 4). Each dot symbol represents an individual animal within each group. B Dose-dependent effects of GPR110 ligands in cortical TNF mRNA level evaluated at 2 h after the last injury. Mice were intraperitoneally (i.p.) injected with varying doses of synaptamide (SYN, 1, 2, and 5 mg/kg) or A8 (0.1, 0.5, 1 mg/kg) immediately following each injury. The increase in the mRNA level of TNF after rCHI (rCHI + V, **p < 0.001 vs. Sham) was significantly suppressed by A8 at 1 mg/kg and synaptamide at 5 mg/kg (#p < 0.05 vs. rCHI + V). The data are expressed as mean ± SEM (n = 4–5). Each dot symbol represents an individual animal within each group. C The time course of A8 and d4-synaptamide detected in the mouse brain. A8 (1 mg/kg) and d4-synaptamide (5 mg/kg) intraperitoneally injected were detected by tandem mass spectrometry. The A8 level in mouse cortex is significantly higher than synaptamide at 1 h and 2 h after intraperitoneal injection. The data are expressed as mean ± SEM (n = 3). **p < 0.001, ***p < 0.001
Synaptamide and A8 suppress rCHIMERA-induced gliosis in a GPR110-dependent manner
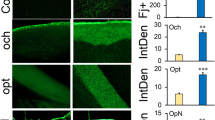
A number of studies have been reported that repetitive mild TBI results in microglia and astrocyte activation in white matter [21,22,23,24]. We examined the effect of GPR110 ligands on the expression of GFAP (an astrocyte marker) and Iba-1 (a microglia marker) in the optic tract (OT) at 24 h (Fig. S1) and 7 days after injury by rCHIMERA (Fig. 4). At 24 h after injury, synaptamide significantly suppressed the Iba-1 (F = 43.63, p < 0.0001 vs. rCHI + V) and GFAP expression in OT (F = 14.5, p < 0.0001vs. rCHI + V) but N-oleoylethanolamine (OEA), an inactive lipid control, showed no effect, indicating that the suppression of glia cell activation was a specific effect of synaptamide (Fig. S1). Similarly, significant increases in the expression of Iba-1 (Fig. 4A, B) and GFAP (Fig. 4C, D) were observed in the OT from both WT and GPR110 KO mice at 7 days after injury (Iba-1: F = 34.11, p < 0.0001, GFAP: F = 15.38, p < 0.0001 vs. Sham-WT). Treating WT injured mice with synaptamide or A8 significantly reduced both Iba-1(rCHI + SYN vs. rCHI + V, p < 0.001; rCHI + A8 vs. rCHI + V, p < 0.01) and GFAP expression (rCHI + SYN vs. rCHI + V, p < 0.01; rCHI + A8 vs. rCHI + V, p < 0.05); however, this effect was not observed in GPR110 KO injured mice (Fig. 4B and D). A8 and synaptamide also produced similar effects, reducing glia activation in the corpus callosum at 7 days after injury (Fig. S2). To determine the long-term effects, we examined the expression of Iba-1 and GFAP in the OT at 3.5 months after injury (Fig. 5). Compared with the injured group with vehicle treatment (rCHI + V), synaptamide or A8 treatment significantly suppressed Iba-1 (rCHI + SYN vs. rCHI + V, p < 0.05; rCHI + A8 vs. rCHI + V, p < 0.001) and GFAP expression (rCHI + SYN vs. rCHI + V, p < 0.05; rCHI + A8 vs. rCHI + V, p < 0.001) in the OT of injured WT mice; however, injured GPR110 KO mice did not respond to the treatments (Fig. 5B and D). These results indicate that synaptamide and A8 inhibit gliosis in the optic tract in a GPR110-dependent manner.

GPR110-dependent amelioration of rCHIMERA-induced glia cell activation by synaptamide and A8. A, C Representative immune-fluorescence micrographic images of Iba-1 (A) and GFAP (C) in the optic tract from WT and GPR110 KO mice at 1 week after injury (rCHI). WT and GPR110 KO mice were injected with A8 (1 mg/kg, i.p.) or synaptamide (5 mg/kg, i.p.) after each CHIMERA, and brains were collected for immunostaining at 7 days after the last injury. B, D Quantitative analysis showing significant suppression of Iba-1 (B) and GFAP expression (D) by the treatment with synaptamide or A8 compared to the vehicle-treated group (rCHI + V) in WT but not in GPR110 KO mice after rCHIMERA. The optic tract region (OT) is outlined with dashed lines. The data are expressed as mean ± SEM (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001 vs. Sham-WT

GPR110-dependent attenuation of rCHIMERA-induced chronic gliosis by synaptamide and A8. A, C. Representative immune-fluorescence micrographic images of Iba-1 (A) and GFAP (C) in the optic tract from WT and GPR110 KO mice at 3.5 months after injury (rCHI). WT and GPR110 KO mice were injected with A8 (1 mg/kg, i.p.) or synaptamide (5 mg/kg, i.p.) after each CHIMERA, and brains were collected for immunostaining at 3.5 months after the last injury. B, D Quantitative analysis showing significant suppression of Iba-1 (B) and GFAP expression (D) by the treatment with synaptamide or A8 compared to the vehicle-treated group (rCHI + V) in WT but not in GPR110 KO mice after rCHIMERA. The optic tract region (OT) is outlined with dashed lines. The data are expressed as mean ± SEM (n = 3). **p < 0.01, ****p < 0.0001 vs. Sham-WT
Synaptamide and A8 attenuate rCHIMERA-induced axonal damage in a GPR110-dependent manner
It is well established that degenerating axons have a high affinity for silver ions [25]. Using silver staining, we examined the axonal damage in the OT from WT and KO mice at 3.5 months after injury. The pronounced increase in silver staining was observed in the OT from both rCHIMERA-injured WT and GPR110 KO mice (F = 44.48, p < 0.0001 vs. Sham). The silver staining intensity of OT from the injured mice treated with synaptamide or A8 was significantly reduced compared with the injured group treated with vehicle only (rCHI + SYN vs. rCHI + V, p < 0.01; rCHI + A8 vs. rCHI + V, p < 0.05). However, no such effect was observed in GPR110 KO mice after treatment with synaptamide or A8 (Fig. 6).

GPR110-dependent decrease in chronic axonal damage after treatment with synaptamide or A8. A Representative micrographic images of silver-stained axons in the optic tract from WT and GPR110 KO mice. WT and GPR110 KO mice were injected with synaptamide (5 mg/kg, i.p.) or A8 (1 mg/kg, i.p.) after each CHIMERA, and brains were collected for silver staining at 3.5 months after the last injury. B Quantitative analysis showing significant suppression of silver staining in the optic tract by the treatment with synaptamide or A8 compared to the vehicle-treated group (rCHI + V) in WT but not in GPR110 KO mice after rCHIMERA. The optic tract region (OT) is outlined with dashed lines. The data are expressed as mean ± SEM (n = 4). ***p < 0.001, ****p < 0.0001 vs. Sham-WT
GPR110 activation improved rCHIMERA-induced visual deficit
Since rCHIMERA-induced upregulation of gliosis and axonal damage observed in the OT may be associated with visual dysfunction, we evaluated the visual evoke potential (VEP) along with the electroretinogram (ERG). At 2 weeks after injury, rCHIMERA significantly decreased N1 amplitude of VEP compared with the Sham group regardless of the genotype (F = 43.77, p < 0.0001; p < 0.001 vs. Sham for WT; p < 0.001vs. Sham for KO). The treatment with the GPR110 ligand A8 ameliorated this reduction in WT (p < 0.05 vs. rCHI + V) but not in GPR110 KO mice (Fig. S3A, B). Neither rCHIMERA nor GPR110 ligand affected the ERG as indicated by the unaltered amplitude and latency of a and b waves measured at 2 weeks after injury (Fig. S3C-E). A prolonged impact of rCHIMERA was also observed as the N1 amplitude was significantly reduced compared with the Sham group regardless of the genotype (F = 60.74, p < 0.0001; WT-rCHI + V: 21.88 ± 3.37 vs. Sham-WT: 59.61 ± 2.1, p < 0.01; KO-rCHI + V: 21.50 ± 1.57 vs. Sham-KO: 60.02 ± 1.98, p < 0.001) at 3 months post injury (Fig. 7). The N1 amplitude was significantly improved after the intraperitoneal injection of synaptamide (37.55 ± 5.20 μV, p < 0.05 vs. rCHI + V) or A8 (34.16 ± 2.65 μV, p < 0.01vs. rCHI + V) compared to the vehicle treatment after injury (21.88 ± 3.37 μV) in WT but not in GPR110 KO mice (Fig. 7A, B). The N1 latency was not significantly altered either by rCHIMERA or treatments with GPR110 ligands (Fig. 7C).

GPR110-dependent improvement of visual function impaired by rCHIMERA by synaptamide and A8. A Average trace of VEP evaluated at 3 months post injury. Full-field flash VEP was elicited at a constant intensity of 3.0 cd·s/m2 with the active electrode subcutaneously inserted in the middle of the two ears. B, C Quantitative analysis of the VEP response. N1 amplitude (B) and latency (C) indicates that treatment with synaptamide or A8 increased the N1 amplitude in WT but not in GPR110 KO injured mice without affecting N1 latency. No significant difference in the N1 amplitude or latency in sham animals was observed between two genotypes. The data are expressed as mean ± SEM (n = 7–12). Each dot symbol represents each animal per group. ***p < 0.001, ****p < 0.0001 vs. Sham-WT
Discussion
Numerous preclinical studies have searched for effective intervention for the long-lasting complications of repetitive mild traumatic brain injury. However, there are few successful candidates that can be translated into the clinic [26, 27]. In this study, we explored the therapeutic potential of GPR110 ligands, synaptamide and A8, in a clinically relevant mouse model of close head injury based on the optic tract neuropathology and visual dysfunction as the injury outcome. We found that these compounds produce GPR110-dependent amelioration of chronic neuropathology and vision deficit caused by rCHIMERA.
GPR110 is highly expressed in the neural stem cells, kidney, and developing brain but GPR110 expression is markedly diminished in the adult brain (Lee et al, 2016). In some cancer conditions such as prostate, liver, and breast cancer, upregulated expression of GPR110 has been reported [28,29,30]. Significant upregulation of GPR110 has also been demonstrated in adult neural tissues in response to injury [18] or LPS stimulation [17]. Likewise, the GPR110 gene level in adult mouse brains rapidly increased after single or multiple CHIMERA in our closed head injury model (Fig. 1).
G protein-coupled receptors (GPCRs) have been the targets for potential therapeutic agents in diverse fields [31, 32]. Many adhesion GPCRs are involved in cellular adhesion and signaling in immunology and neurology as well as developmental biology [33, 34]. GPR110 is the target receptor of synaptamide [13] that ameliorates LPS-induced neuroinflammation through the cAMP/PKA/CREB singling pathway in vivo and in cultured microglia where GPR110 is expressed [16, 17]. Recently, synaptamide has been shown to reduce inflammation and cognitive impairment in animal models of neuroinflammation and TBI [35, 36]. In LPS-induced neuroinflammation, synaptamide was shown to reduce the expression of proinflammatory cytokines/chemokines without affecting anti-inflammatory/pro-resolving cytokines [16]. Although the role of rapid induction of GPR110 after injury is not clear, the anti-inflammatory nature of GPR110 signaling may help attenuate uncontrolled inflammatory signals as part of the neuroprotective responses to injuries.
Neuroinflammation is an important mechanism underlying the pathogenesis of traumatic brain injury [37, 38], while microglia are potent immune effector cells producing and releasing proinflammatory and cytotoxic mediators in response to brain injury [23] Therefore, the GPR110-dependent microglial production of cAMP (Fig. 2), a well-established regulator of immune responses [39], is likely an important contributing mechanism to the effectiveness of GPR110 ligands on rCHIMERA-induced optic tract gliosis (Figs. 4 and 5, S1-S2) and visual dysfunction observed in this study (Fig. 7, S3). In addition, GPR110 ligands may have activated GPR110/cAMP signaling in other cellular components such as neurons and suppressed axonal damage (Fig. 6), also contributing to the improved injury outcome.
A8, a chemical analogue of synaptamide with improved stability, has been recently described as a better ligand to GPR110 compared with the endogenous ligand synaptamide [18]. Indeed, A8 was more effective for GPR110-dependent cAMP production in microglia (Fig. 2) and suppression of the inflammatory signal after brain injury caused by rCHIMERA (Fig. 3). Both A8 and synaptamide were detected in the brain after intraperitoneal injection indicating that they passed the brain–blood barrier and were delivered to the brain. The detected level of A8 was significantly higher compared to synaptamide although 5-fold less A8 was injected than synaptamide, confirming improved in vivo stability of A8. Enhanced in vivo stability and biological effectiveness of A8 suggest that A8 may have better translational potential than synaptamide.
Persistent gliosis caused by repetitive mild TBI is often associated with a functional deficit in animals and humans [22, 40,41,42]. The rCHIMERA-induced chronic gliosis and axonal degeneration in the optic tract [11, 19] are accompanied by visual impairment [11]. The vulnerability of the optic tract to mild TBI can be utilized for the evaluation of drug candidates for therapeutic potential in mild repetitive TBI. Such strategy led to the current demonstration of anti-inflammatory GPR110 ligands as effective agents in ameliorating the chronic optic tract histopathology and visual dysfunction. The positive effect of GPR110 ligands observed in the optic tract may occur similarly in other cerebral white matter tracts including the corpus callosum, internal capsule, and corticospinal tracts that are known to be disrupted significantly after TBI [43]. Consequently, these GPR110 ligands may find further applications to other brain functions impaired by mild repetitive TBI, particularly memory and executive function that require intact white matter tracts.
Conclusions
Our results demonstrate that synaptamide and A8 attenuate optic tract histopathology and visual impairment caused by rCHIMERA by activating the GPR110/cAMP system that is upregulated after injury. This study provides new insight for the translational potential of targeting GPR110 using its ligands for improving the chronic outcome after repeated mild TBI.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- A8:
-
(4Z,7Z,10Z,13Z,16Z,19Z)-N-(2-hydroxy-2-methylpropyl) docosa-4,7,10,13,16,19-hexaenamide
- ANOVA:
-
Analysis of variance
- cAMP :
-
Cyclic adenosine monophosphate
- CHIMERA:
-
Closed-Head Impact Model of Engineered Rotational Acceleration
- ERG:
-
Electroretinogram
- FBS:
-
Fetal bovine serum
- GPCR:
-
G-protein coupled receptor
- GPR110:
-
G-protein receptor 110
- KO:
-
Knockout
- LPS:
-
Lipopolysaccharide
- mTBI:
-
Mild traumatic brain injury
- qRT-PCR:
-
Quantitative reverse transcription polymerase chain reaction
- rCHIMERA:
-
Repetitive CHIMERA
- SEM:
-
Standard error of the mean
- Synaptamide:
-
N-Docosahexaenoylethanolamine
- TNF:
-
Tumor necrosis factor
- VEP:
-
Visual evoked potential
- WT:
-
Wild-type
References
Strain JF, Didehbani N, Spence J, Conover H, Bartz EK, Mansinghani S, et al. White matter changes and confrontation naming in retired aging National Football League athletes. J Neurotrauma. 2017;34(2):372–9. https://doi.org/10.1089/neu.2016.4446.
Lancaster MA, Olson DV, McCrea MA, Nelson LD, LaRoche AA, Muftuler LT. Acute white matter changes following sport-related concussion: a serial diffusion tensor and diffusion kurtosis tensor imaging study. Hum Brain Mapp. 2016;37(11):3821–34. https://doi.org/10.1002/hbm.23278.
Champagne AA, Peponoulas E, Terem I, Ross A, Tayebi M, Chen Y, et al. Novel strain analysis informs about injury susceptibility of the corpus callosum to repeated impacts. Brain Commun. 2019;1:fcz021.
Sen N. An insight into the vision impairment following traumatic brain injury. Neurochem Int. 2017;111:103–7. https://doi.org/10.1016/j.neuint.2017.01.019.
Armstrong RA. Visual problems associated with traumatic brain injury. Clin Exp Optom. 2018;101(6):716–26. https://doi.org/10.1111/cxo.12670.
Wright WG, Tierney RT, McDevitt J. Visual-vestibular processing deficits in mild traumatic brain injury. J Vestib Res. 2017;27(1):27–37. https://doi.org/10.3233/VES-170607.
Gilmore CS, Camchong J, Davenport ND, Nelson NW, Kardon RH, Lim KO, et al. Deficits in visual system functional connectivity after blast-related mild TBI are associated with injury severity and executive dysfunction. Brain Behav. 2016;6:e00454.
Goodrich GL, Martinsen GL, Flyg HM, Kirby J, Garvert DW, Tyler CW. Visual function, traumatic brain injury, and posttraumatic stress disorder. J Rehabil Res Dev. 2014;51(4):547–58. https://doi.org/10.1682/JRRD.2013.02.0049.
Chen H, Desai A, Kim HY. Repetitive closed-head impact model of engineered rotational acceleration induces long-term cognitive impairments with persistent astrogliosis and microgliosis in mice. J Neurotrauma. 2017;34(14):2291–302. https://doi.org/10.1089/neu.2016.4870.
Evanson NK, Guilhaume-Correa F, Herman JP, Goodman MD. Optic tract injury after closed head traumatic brain injury in mice: a model of indirect traumatic optic neuropathy. PLoS One. 2018;13(5):e0197346. https://doi.org/10.1371/journal.pone.0197346.
Desai A, Chen H, Kim HY. Multiple mild traumatic brain injuries lead to visual dysfunction in a mouse model. J Neurotrauma. 2020;37(2):286–94. https://doi.org/10.1089/neu.2019.6602.
Kim HY, Spector AA. N-Docosahexaenoylethanolamine: a neurotrophic and neuroprotective metabolite of docosahexaenoic acid. Mol Asp Med. 2018;64:34–44. https://doi.org/10.1016/j.mam.2018.03.004.
Lee JW, Huang BX, Kwon H, Rashid MA, Kharebava G, Desai A, et al. Orphan GPR110 (ADGRF1) targeted by N-docosahexaenoylethanolamine in development of neurons and cognitive function. Nat Commun. 2016;7(1):13123. https://doi.org/10.1038/ncomms13123.
Huang BX, Hu X, Kwon HS, Fu C, Lee JW, Southall N, et al. Synaptamide activates the adhesion GPCR GPR110 (ADGRF1) through GAIN domain binding. Commun Biol. 2020;3(1):109. https://doi.org/10.1038/s42003-020-0831-6.
Rashid MA, Katakura M, Kharebava G, Kevala K, Kim HY. N-Docosahexaenoylethanolamine is a potent neurogenic factor for neural stem cell differentiation. J Neurochem. 2013;125(6):869–84. https://doi.org/10.1111/jnc.12255.
Park T, Chen H, Kevala K, Lee JW, Kim HY. N-Docosahexaenoylethanolamine ameliorates LPS-induced neuroinflammation via cAMP/PKA-dependent signaling. J Neuroinflammation. 2016;13(1):284. https://doi.org/10.1186/s12974-016-0751-z.
Park T, Chen H, Kim HY. GPR110 (ADGRF1) mediates anti-inflammatory effects of N-docosahexaenoylethanolamine. J Neuroinflammation. 2019;16(1):225. https://doi.org/10.1186/s12974-019-1621-2.
Kwon H, Kevala K, Xin H, Patnaik S, Marugan J, Kim H-Y. Ligand-induced GPR110 activation facilitates axon growth after Injury. Int J Mol Sci. 2021;22:3386.
Namjoshi DR, Cheng WH, McInnes KA, Martens KM, Carr M, Wilkinson A, et al. Merging pathology with biomechanics using CHIMERA (Closed-Head Impact Model of Engineered Rotational Acceleration): a novel, surgery-free model of traumatic brain injury. Mol Neurodegener. 2014;9(1):55. https://doi.org/10.1186/1750-1326-9-55.
Benchorin G, Calton MA, Beaulieu MO, Vollrath D. Assessment of murine retinal function by electroretinography. Bio Protoc. 2017;7(7). https://doi.org/10.21769/BioProtoc.2218.
Wang G, Zhang J, Hu X, Zhang L, Mao L, Jiang X, et al. Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. J Cereb Blood Flow Metab. 2013;33(12):1864–74. https://doi.org/10.1038/jcbfm.2013.146.
Mouzon BC, Bachmeier C, Ferro A, Ojo JO, Crynen G, Acker CM, et al. Chronic neuropathological and neurobehavioral changes in a repetitive mild traumatic brain injury model. Ann Neurol. 2014;75(2):241–54. https://doi.org/10.1002/ana.24064.
Loane DJ, Kumar A. Microglia in the TBI brain: the good, the bad, and the dysregulated. Exp Neurol. 2016;275(Pt 3):316–27. https://doi.org/10.1016/j.expneurol.2015.08.018.
Sajja VS, Hlavac N, VandeVord PJ. Role of glia in memory deficits following traumatic brain injury: biomarkers of glia dysfunction. Front Integr Neurosci. 2016;10:7.
de Olmos JS, Beltramino CA, de Olmos de Lorenzo S. Use of an amino-cupric-silver technique for the detection of early and semiacute neuronal degeneration caused by neurotoxicants, hypoxia, and physical trauma. Neurotoxicol Teratol. 1994;16(6):545–61. https://doi.org/10.1016/0892-0362(94)90033-7.
Loane DJ, Faden AI. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci. 2010;31(12):596–604. https://doi.org/10.1016/j.tips.2010.09.005.
Schouten JW. Neuroprotection in traumatic brain injury: a complex struggle against the biology of nature. Curr Opin Crit Care. 2007;13(2):134–42. https://doi.org/10.1097/MCC.0b013e3280895d5c.
Lum AM, Wang BB, Beck-Engeser GB, Li L, Channa N, Wabl M. Orphan receptor GPR110, an oncogene overexpressed in lung and prostate cancer. BMC Cancer. 2010;10(1):40. https://doi.org/10.1186/1471-2407-10-40.
Ma B, Zhu J, Su J, Pan F, Ji Y, Luan L, et al. The role of GPR110 in lung cancer progression. Ann Transl Med. 2020;8(12):745. https://doi.org/10.21037/atm-20-3146.
Ma B, Zhu J, Tan J, Mao Y, Tang L, Shen C, et al. Gpr110 deficiency decelerates carcinogen-induced hepatocarcinogenesis via activation of the IL-6/STAT3 pathway. Am J Cancer Res. 2017;7(3):433–47.
Lagerström MC, Schiöth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov. 2008;7(4):339–57. https://doi.org/10.1038/nrd2518.
Purcell RH, Hall RA. Adhesion G protein-coupled receptors as drug targets. Annu Rev Pharmacol Toxicol. 2018;58(1):429–49. https://doi.org/10.1146/annurev-pharmtox-010617-052933.
Bjarnadóttir TK, Fredriksson R, Schiöth HB. The adhesion GPCRs: a unique family of G protein-coupled receptors with important roles in both central and peripheral tissues. Cell Mol Life Sci. 2007;64(16):2104–19. https://doi.org/10.1007/s00018-007-7067-1.
Yona S, Lin HH, Siu WO, Gordon S, Stacey M. Adhesion-GPCRs: emerging roles for novel receptors. Trends Biochem Sci. 2008;33(10):491–500. https://doi.org/10.1016/j.tibs.2008.07.005.
Tyrtyshnaia A, Bondar A, Konovalova S, Sultanov R, Manzhulo I. N -Docosahexanoylethanolamine reduces microglial activation and improves hippocampal plasticity in a murine model of neuroinflammation. Int J Mol Sci. 2020;21(24):9703. https://doi.org/10.3390/ijms21249703.
Ponomarenko Arina I, Tyrtyshnaia Anna A, Pislyagin Evgeny A, Dyuizen Inessa V, Sultanov Ruslan M, Manzhulo Igor V. N-docosahexaenoylethanolamine reduces neuroinflammation and cognitive impairment after mild traumatic brain injury in rats. Sci Rep. 2021;11(1):756. https://doi.org/10.1038/s41598-020-80818-9.
Corps KN, Roth TL, McGavern DB. Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol. 2015;72(3):355–62. https://doi.org/10.1001/jamaneurol.2014.3558.
Dinet VP, KG Badaut J. Brain–immune interactions and neuroinflammation after traumatic brain injury. Front Neurosci. 2019;13:1178.
Raker VKB, C.; Steinbrink, K. The cAMP pathway as therapeutic target in autoimmune and inflammatory diseases. Front Immunol. 2016;7:123.
Mannix R, Berglass J, Berkner J, Moleus P, Qiu J, Andrews N, et al. Chronic gliosis and behavioral deficits in mice following repetitive mild traumatic brain injury. J Neurosurg. 2014;121(6):1342–50. https://doi.org/10.3171/2014.7.JNS14272.
Tagge CA, Fisher AM, Minaeva OV, Gaudreau-Balderrama A, Moncaster JA, Zhang XL, et al. Concussion, microvascular injury, and early tauopathy in young athletes after impact head injury and an impact concussion mouse model. Brain. 2018;141(2):422–58. https://doi.org/10.1093/brain/awx350.
Vonder Haar C, Martens KM, Bashir A, McInnes KA, Cheng WH, Cheung H, et al. Repetitive closed-head impact model of engineered rotational acceleration (CHIMERA) injury in rats increases impulsivity, decreases dopaminergic innervation in the olfactory tubercle and generates white matter inflammation, tau phosphorylation and degeneration. Exp Neurol. 2019;317:87–99. https://doi.org/10.1016/j.expneurol.2019.02.012.
Rutgers DR, Toulgoat F, Cazejust J, Fillard P, Lasjaunias P, Ducreux D. White matter. abnormalities in mild traumatic brain injury: a diffusion tensor imaging study. Am J Neuroradiol. 2008;29(3):514–9. https://doi.org/10.3174/ajnr.A0856.
Acknowledgements
The authors thank Dr. Sam Patnaik at NCAT for providing A8 and d6-A8 and the NIAAA animal facility staff for their support with animal care.
Funding
This research was supported by the intramural program of National Institute of Alcohol Abuse and Alcoholism (NIAAA) in National Institutes of Health (NIH) and Center for Neuroscience and Regenerative Medicine (CNRM) with Henry M. Jackson Foundation for the Advancement of Military Medicine. The funding agencies did not have any role in the design and implementation of this study. Open Access funding provided by the National Institutes of Health (NIH).
Author information
Authors and Affiliations
Contributions
HC and HYK designed the experiments and wrote the manuscript; HC performed CHIMERA procedures, histopathological analysis, and VEP/ERG measurement; KK performed mass spectrometric analysis of synaptamide and A8 in brain; EA performed cAMP analysis in primary microglia cells; JM designed and provided A8 and d6-A8 with expert knowledge on their chemical properties. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Synaptamide-specific suppression of glia cell activation in optic tract (OT) at one day post-rCHIMERA. A. Representative micrographic images of Iba-1 and GFAP immunofluorescence. WT mice were injected with synaptamide or oleoylethanolamine (OEA) at 5 mg/kg (i.p.) after each CHIMERA, and brains were collected for immunostaining at 1 day after the last injury. B, C. Quantitative analysis of Iba-1 (B) and GFAP expression (B), showing that synaptamide suppressed Iba-1 and GFAP expression induced by rCHIMEA while oleoylethanolamine (OEA) had no effect. The synaptamide treatment without injury did not affect the GFAP and Iba-1 expression in the brain. The data are expressed as mean ± SEM (n=3). *p<0.05, **p<0.01, ****p<0.001 vs. Sham. Figure S2. GPR110-dependent inhibition of glia cell activation in corpus callosum by synaptamide and A8 at 7 days after injury. A, C. Representative micrographic images of Iba-1 (A) and GFAP (C) immunofluorescence in the corpus callosum (CC) from WT and GPR110 KO mice at 1 week after injury (rCHI). WT and GPR110 KO mice were injected with synaptamide (5 mg/kg, i.p.) or A8 (1 mg/kg, i.p.) after each CHIMERA, and brains were collected for immunostaining at 7 days after the last injury. B, D. Quantitative analysis showing significant suppression of Iba-1 (B) and GFAP expression (D) by the treatment with synaptamide or A8 compared to the vehicle-treated group (rCHI + V) in WT but not in GPR110 KO mice after rCHIMERA. The corpus callosum region (CC) is outlined with dashed lines. The data are expressed as mean ± SEM (n=3). *p<0.05, **p<0.01, ***p<0.001 vs. Sham-WT. Figure S3. Increases in N1 amplitude of VEP by A8 at 2 weeks after rCHIMERA. A: Average traces of VEP evaluated at 2 weeks post injury. Full-field flash VEP was elicited at a constant intensity of 3.0 cd·s/m2 with the active electrode subcutaneously inserted in the middle of the two ears. B, Quantitative analysis of the N1 amplitude and latency showing that A8 increased the N1 amplitude in WT but not in GPR110 KO injured mice without affecting N1 latency. No significant difference in the N1 amplitude or latency in sham animals was observed between two genotypes. The data are expressed as mean ± SEM (n=8-10). Each dot symbol represents each animal per group. ***p<0.001, ****p<0.0001 vs. Sham-WT. C. Average tracts of ERG at 2 weeks post injury obtained using a light-adapted (photopic) protocol. D, E. Quantitative analysis of a and b amplitude and latency showing that A8 or injury did not change a and b amplitude and latency for both WT and GPR110 KO mice. No significant difference in these ERG parameters was observed between two genotypes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, H., Kevala, K., Aflaki, E. et al. GPR110 ligands reduce chronic optic tract gliosis and visual deficit following repetitive mild traumatic brain injury in mice. J Neuroinflammation 18, 157 (2021). https://doi.org/10.1186/s12974-021-02195-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-021-02195-y