
Abstract
Background
Aβ1-42 peptide abnormal production is associated with the development and maintenance of neuroinflammation and oxidative stress in brains from Alzheimer disease (AD) patients. Suppression of neuroinflammation may then represent a suitable therapeutic target in AD. We evaluated the efficacy of IFNβ1a in attenuating cognitive impairment and inflammation in an animal model of AD.
Methods
A rat model of AD was obtained by intra-hippocampal injection of Aβ1-42 peptide (23 μg/2 μl). After 6 days, 3.6 μg of IFNβ1a was given subcutaneously (s.c.) for 12 days. Using the novel object recognition (NOR) test, we evaluated changes in cognitive function. Measurement of pro-inflammatory or anti-inflammatory cytokines, reactive oxygen species (ROS), and SOD activity levels was performed in the hippocampus. Data were evaluated by one-way ANOVA with Fisher’s Protected Least Significant Difference (PLSD) test.
Results
We showed that treatment with IFNβ1a was able to reverse memory impairment and to counteract microglia activation and upregulation of pro-inflammatory cytokines (IL-6, IL-1β) in the hippocampus of Aβ1-42-injected rats. The anti-inflammatory cytokine IL-10, significantly reduced in the Aβ1-42 animals, recovered to control levels following IFNβ1a treatment. IFNβ1a also reduced ROS and lipids peroxidation and increased SOD1 protein levels in the hippocampus of Aβ1-42-injected rats.
Conclusion
This study shows that IFNβ1a is able to reverse the inflammatory and cognitive effects of intra-hippocampal Aβ1-42 in the rat. Given the role played by inflammation in AD pathogenesis and the established efficacy of IFNβ1a in the treatment of inflammatory diseases of the central nervous system such as multiple sclerosis, its use may be a viable strategy to inhibit the pro-inflammatory cytokine and oxidative stress cascade associated with Aβ deposition in the hippocampus of AD patients.
Similar content being viewed by others


Background
Alzheimer disease (AD), an age-dependent neurodegenerative disorder and the most common cause of dementia, is a multifactorial disease with a complex interplay of genetics and environmental factors [1, 2]. The pathological hallmarks of AD include the deposition of extracellular neuritic plaques of β-amyloid (Aβ) peptide and the formation of intracellular neurofibrillary tangles in the brain [3]. Aβ peptides are neurotoxic and may cause neurodegenerative changes, including apoptosis, oxidative stress, and neuroinflammation. Neuroinflammation has been widely recognized as a possible pathological contributor to AD, usually including activation of glial cells, such as microglia and astrocytes [4], and release of cytotoxic compounds, e.g., cytokines and ROS, able to cause neuronal damage and death [3, 5]. Since suppression of neuroinflammation could represent an interesting therapeutic target for AD, several approaches have been tested to smolder inflammatory processes by using anti-inflammatory drugs [5,6,7,8] or other drugs with anti-inflammatory effects in AD models [9]. However, although all anti-inflammatory strategies tried so far in AD patients have not achieved satisfactory results, indicating the need for a better understanding of the role of the immune system in cerebral proteinopathies and how to modulate it [10], intervention with drugs modulating pro-inflammatory cytokine production is still considered a potentially useful strategy to slow down the disease course of this dreadful disease.
Interferons (IFNs) are a super-family of cytokine proteins that play an important role in the host immune response to infections and immune-mediated diseases [11]. Interferon-β1a (IFNβ1a) is the prototypical regulatory cytokine with anti-inflammatory properties largely used in the past two decades to slow down pathological and clinical features of central nervous system (CNS) immune-mediated diseases, such as multiple sclerosis [12,13,14,15,16,17] and its animal model experimental autoimmune encephalomyelitis [18, 19]. IFNβ-1a displays several cellular and humoral immune effects, including inhibition of pro-inflammatory cytokine (IL-6, IL-1β, TNF-α, IFN-γ) and downregulation of glial cells and oxidative stress [20]. Interestingly, IFNβ1a significantly prevented cognitive decline in a large cohort of patients with multiple sclerosis, thus suggesting that modulation of neuroinflammatory pathways may prevent cognitive decline in humans [21, 22], as well as cortical atrophy associated with cognitive impairment in patients with multiple sclerosis [23]. We also preliminarily evaluated the safety and efficacy of IFNβ1a in subjects affected by mild-to-moderate AD in a phase 2a study mainly aimed to evaluate the safety of the drug in this elderly population [24], and although not statistically significant, we observed a reduction in disease progression during follow-up as measured by the AD Physical Self-Maintenance Scale.
Taken together, these human studies support the hypothesis that IFNβ1a could attenuate the inflammatory response in AD and led us to look for an experimental confirmation by assessing the efficacy of IFNβ1a in attenuating inflammation in an animal model of AD obtained by intra-hippocampal injection of Aβ1-42 peptide. Intra-hippocampal injection of Aβ1-42 peptide is considered a suitable animal model of AD [25] with several pathological and behavioral features of AD patients, including cognitive impairment and inflammatory reactivity [26]. Therefore, we first evaluated in rats the cognitive impairment induced by intra-hippocampal injection of Aβ1-42 peptide and the therapeutic effects of IFNβ1a treatment. The anti-inflammatory effects of IFNβ1a were then assessed by examining in post-mortem rat brains several inflammation markers, such as pro- and anti-inflammatory cytokines, and oxidative stress responses.
Methods
Animals
Adult female Wistar rats (3 months old) were used. Rats were housed in a specific pathogen-free environment, three per polypropylene cages in controlled temperature (23 ± 2 °C), humidity (50–55%), and light (12-h light/dark cycle), with access to food and water ad libitum. Procedures involving animal were carried out in accordance with the Italian institutional guidelines (D. LGS. no. 26, GU n.61, March 2014). All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. No other methods to perform the described experiments (3Rs) were found.
Experimental design
For this study, we used the following experimental groups: (1) a control group treated with saline only (control); (2) a sham-operated control group and treated with saline (sham); (3) an Aβ1-42 peptide-treated group; (4) an interferon-β1a (IFNβ1a)-treated group; and (5) an Aβ1-42 peptide + IFNβ1a-treated group. Rats received two bilateral intra-hippocampal injections of 23 μg/2 μl of Aβ1-42 peptide dissolved in 0.9% physiological saline solution. IFNβ1a was given subcutaneously (s.c.) at a dose of 3.6 μg (1 M Units, Rebif, Merk Serono, London) in a volume of 0.1 ml of 0.9% of saline solution. We used female rats since in AD disproportionately the female/male ratio is 2:1, although the biological basis of these sex-based differences in AD onset and progression remains elusive [27, 28].
Aβ1-42 peptide preparation and toxicity
Aβ1-42 protein was produced according to Carrotta et al. [29]. For the kinetics of aggregation, the sample of Aβ1-42 protein was loaded in a 96 black multi-well and added with 8 μM of thioflavin-T. The multi-well was read to the plate reader every 30 s at 450–485 nm wavelength for 8 h at 37 °C. After the 8 h of incubation, the formation of Aβ1-42 protein aggregates was also evaluated at the fluorescence microscope (Leica Microsystems, Heidelberg, Germany). In addition, the mean size of the Aβ1-42 plates was measured by fluorescence microscopy software (Leica QFluoro V1.1 software, Heidelberg, Germany).
LAN5 cells were cultured with RPMI 1640 medium (Celbio srl, Milan, Italy) supplemented with 10% fetal bovine serum (Gibco-Invitrogen, Milan, Italy) and 1% antibiotics (50 mg mL − 1 penicillin and 50 mg ml − 1 streptomycin). Cells were maintained in a humidified 5% CO2 atmosphere at 37 ± 0.1 °C. For dose-effect studies of Aβ1-42 toxicity, cells were treated with 50, 75, and 100 μM of Aβ1-42 for 24 h, and thereafter, their viability was evaluated by MTS assay ([3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulphophenyl)-2H-tetrazolium]; Promega Italia, S.r.l., Milan, Italy) and morphological analyses. After 24 h of cell treatment with Aβ1-42, 20 μl of the MTS solution was added to each well for 3 h at 37 °C, 5% CO2. The absorbance was read at 490 nm on the Microplate reader (WallacVictor 2 1420 Multilabel Counter) (PerkinElmer, Inc. Monza, Italy).
Aβ1-42 intra-hippocampal injection
The Aβ1-42 peptide intra-hippocampal injection was performed as described by Mudò et al. [30]. Shortly, rats were anesthetized with mixture of 1:1 of Zolazepam + tiletamine 15 mg/kg b.w. (Zoletil, Virbac) and xilazine 9 mg/kg b.w. (Nerfasin, ATI, Italy), placed in a David Kopf stereotaxic apparatus, and received two bilateral intra-hippocampal injections of 23 μg/2 μl of Aβ1-42 peptide, using the following stereotaxic coordinates from the Bregma, according to Paxinos and Watson (1998): first injection AP = − 3.6, L = 2, and V = 4.5; second injection AP = − 4.2, L = 2.4, and V = 4.5. The sham group was intra-hippocampal-injected with 2 μl of 0.9% physiological saline. Injections were performed by 30-gauge injector cannula that was connected by a piece of polyethylene tube to the 10 μl Hamilton syringe. Each injection was performed over 3 min, and following injection, the needle remained in the target location for 3 min to avoid Aβ1-42 peptide reflux along the needle tract and to achieve a proper diffusion of the drug. After surgery, each rat was treated with penicillin (100,000 U/i.m.) to prevent infection.
Behavioral testing
Using the novel object recognition (NOR) test, we evaluated changes in cognitive function of the experimental animals. Rats were tested in an open field Plexiglas square box, in a mean light intensity (100 lx) illuminated chamber. All experimental groups were subjected to a 5-min training session when they were presented two identical non-toxic objects (i.e., two metal cans) placed against a wall in the open field arena. To prevent coercion to explore the objects, rats were released against the opposite wall with the back to the objects. The time spent on exploring each object was recorded using ANY MAZE Video Tracking System (Ugo Basile, Italy); a 2-cm2 area surrounding the objects was defined such that nose entries were recorded as time exploring the objects. After the training session, animals were placed in their home cage for a 24-h retention interval. Then, they were returned to the arena containing two objects: one was identical to the familiar one but previously unused (to prevent olfactory cues and the necessity to wash objects during experimentation) and the other was a novel object (metal, glass, or hard plastic items). Time spent on exploring each object was recorded along 5-min session. Objects were randomized and counterbalanced across animals. The objects and arena were thoroughly cleaned at the end of each experimental session. The recognition index (RI), which is the time spent on investigating the novel object, divided by the total amount of exploration time of the novel (TN) and familiar objects (TF), [RI = TN/(TN + TF)], is a measure of novel object recognition and the main index of retention. If RI percentage is higher than 50%, it indicates more time spent on inquiring into the novel object, whereas less than 50% indicates that time was prevailingly spent on exploring the familiar object, and 50% indicates a null preference.
Immunohistochemistry
The rats under deep anesthesia were perfused through the aorta with 0.9% saline and brain dissected. The left side of the brain was used for histological investigations and the right side for molecular analysis. Immuno-histochemical investigation was performed as described by Di Liberto et al. [31]. The left side of brain was fixed with 4% of paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) for 2 days and then immersed in the sucrose 10% solution for 1 day and in sucrose 20% for 2 days. Subsequently, brains were frozen in cooled isopentane and 20-μm-thick coronal sections at hippocampal level were prepared and processed for immunohistochemistry as free-floating sections. Sections were washed for 5 min in 0.1 M PBS and incubated for 15 min with BSA (5 mg/ml) and Triton X-100 (0.3%) in PBS 0.1 M. Mouse monoclonal antibody anti-glial fibrillary acidic protein (anti-GFAP; diluted 1:400; MAB360 Chemicon) or rabbit anti-ionized calcium-binding adapter molecules-1 (anti-Iba-1; diluted 1:300 Wako Catalog No. 019-19741) was added to sections that were then incubated at 4 °C overnight. After two washing steps with PBS for 5 min, the sections were incubated at RT for 1 h with specific Cy2-conjugated secondary antibodies diluted 1:250 (711225152 and 115-165-003; Jackson Immuno Research, West Grove, PA, USA). Following two washing steps with PBS, the sections were counterstained by incubation for 10 min in 0.5 mg/ml of the fluorescent nuclear dye Hoechst 33258 (bisbenzimide, Sigma–Aldrich, Seelze, Germany). Following a short washing with PBS, sections were coverslipped in a glycerol-based medium and slides were examined under a fluorescence microscope (DMRBE, Leica Microsystems, Wetzlar, Germany).
Western blotting
Rats were sacrificed at the end of experimental procedures by an overdose of anesthesia, and the hippocampi were rapidly removed from their brain, collected, and stored at − 70 °C for later use. Dissected hippocampal tissue was homogenized in cold radio-immunoprecipitation assay (RIPA) buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% Triton, SDS 0.1%), supplemented with protease inhibitor cocktail (Sigma-Aldrich P8340) and phosphatase inhibitor cocktail (Sigma–Aldrich P5726). Samples were sonicated (30 pulsations/min), quantified by the Lowry method [32], and stored at − 80 °C. Western blotting was performed as previously described by Frinchi et al. [33]. Protein samples (50 μg per lane) and molecular weight marker (161-0376 BIO-RAD) were run on 10% or 12% polyacrylamide gel and electrophoretically transferred onto nitrocellulose membrane (RPN303E, Hybond-C-extra, GE Healthcare Europe GmbH). The membranes were incubated for 1 h in blocking buffer, 1x TBS, 0.1% Tween-20, and 5% w/v nonfat dry milk, with gentle shaking overnight at − 4 °C with specific antibody in blocking buffer. For detection of superoxide dismutase-1 (SOD1) and superoxide dismutase-2 (SOD2), the following antibodies were used: rabbit polyclonal anti-SOD1 1:1000 (Sc-11407 Santa Cruz Biotechnology); mouse anti-SOD2 1:500 (SOD2; sc-137254, Santa Cruz Biotechnology). For detection of GFAP and Iba-1, the following antibodies were used: mouse monoclonal antibody anti-GFAP 1:2000 (MAB360 Chemicon), rabbit anti-Iba-1 1:1000 (Wako Catalog No. 019-19741). The day after, the membranes were washed three times for 10 min with TBS/T and then incubated for 1 h at room temperature with goat anti-rabbit IgG (sc-2004 Santa Cruz Biotechnology) or goat anti-mouse IgG (Sc.7076 Cell Signaling Technology) horseradish peroxidase-conjugated diluted 1:10000. After three washings with TBS-T, immune complexes were visualized with a chemiluminescence reagent (RPN2236, GE Healthcare Europe GmbH) according to the manufacturer’s instructions. The Hyperfilm (ECL-films 28906837, GE Healthcare Europe GmbH) were developed using Kodak developer and fixer (catalog no. 1900984 and 1902485, Kodak GBX, Eastman Kodak). For the normalization of quantitative evaluation of bands, each membrane was stripped at 65 °C for 30 min in buffer containing NaCl 137 mM, TrisHCl 20 mM pH 7.6, and β-mercaptoethanol 0.01%. After two washings with TBST, the membranes were reprobed with an anti-β-actin antibody (sc-47778, Santa Cruz Biotechnology). The densitometric evaluation of bands was performed by measuring the optical density (O.D.) using the Image J software (Rasband WS, ImageJ, U.S. National Institutes of Health, Bethesda, Maryland, USA, https://imagej.nih.gov/ij/, 1997–2018).
Measurement of pro-inflammatory or anti-inflammatory cytokines by ELISA assay
Concentrations of interleukin-1β (IL-1β), interleukin-6 (IL-6), interleukin-10 (IL-10), and transforming growth factor-beta1 (TGF-β1) were measured in the hippocampus homogenates (20 mg of tissue sample) using enzyme-linked immunosorbent assay (ELISA) kits for rat (Cloud-Clone Corp, Wuhan, Hubei) according to the manufacturers’ protocols and as reported by Zizzo et al. [34].
Reactive oxygen species analysis
To assess reactive oxygen species (ROS) generation by fluorimeter analysis, 10 mg of tissue from rat hippocampus was weighed and suspended on 1000 μl of PBS1X with 10 μ of protease inhibitors (Amersham Life Science, Munich, Germany). Samples were then incubated with 1 mM dichlorofluorescein diacetate (DCFH-DA) for 10 min at room temperature in the dark. The conversion of non-fluorescent DCFH-DA to the highly fluorescent compound 20,70-dichlorofluorescein (DCF) by esterase activity was used to monitor the presence of peroxides due to the oxidative burst in the brain [34]. The samples were analyzed by fluorimeter (Microplate reader WallacVictor 2 - 1420 Multilabel Counter; PerkinElmer, Inc.), using the excitation filter set at 485 nm and the emission filter set at 530 nm. Relative ROS production was expressed as the change in fluorescence of the experimental groups compared with that of the control group (100%).
SOD activity levels
The hippocampus of rats was homogenized in PBS with protease inhibitors (Amersham Life Science, Munich, Germany). To remove insoluble material, tissue lysates were sonicated and centrifuged (14,000 rpm, at 4 °C, for 30 min). In the supernatant, total proteins were quantified by the Lowry method [32]. Volume corresponding to 50 μg of protein was used for total SOD enzymatic activity measurement, by using the SOD assay kit (Sigma–Aldrich) according to manufacturer’s instructions [34]. Absorbance was measured by using the iMark™ Microplate Absorbance Reader at 450 nm.
Lipid peroxidation assay
The lipid peroxidation assay kit (Sigma Aldrich) was used to detect the concentration of malondialdehyde (MDA), a final product of lipids peroxidation. Ten milligrams of hippocampal tissues was homogenized in 300 μl of MDA lysis buffer (supplied in the kit), and colorimetric reaction with thiobarbituric acid (TBA) was read on an iMark™ Microplate Absorbance Reader at 532 nm, according to manufacturer’s instructions [34].
Cell counting
The number of Iba-1 and GFAP-positive cells was estimated by counts made by systematic sampling of brain sections, every third section of total 10 sections, in the hippocampal region of rat brain. All counts were made in four rats for each group and were carried out double-blindly. Labeled cells were evaluated using ImageJ software (Cell Counter plugin; Rasband, W.S., ImageJ, U.S. National Institutes of Health, Bethesda, Maryland, USA, http://imagej.nih.gov/ij/, 1997–2018). The cell count values obtained from three to five random fields per section, in 10 sections examined, were expressed as means ± SEM values per square millimeter of tissue.
Cortisol levels
Rats under anesthesia were sacrificed between 11:00 and 12:00 am, and blood was taken by intracardiac puncture and collected in tubes coated with EDTA. Blood samples were centrifuged at 4000×g at 4 °C for 10 min, and the supernatant was stored at − 80 °C. The plasma levels of cortisol were measured using an automated electrochemiluminescence immunoassay (Roche Diagnostics Elecsys Cortisol II assays and COBAS E801), and values were expressed in nanograms per milliliter. The minimum level of detection for assays of cortisol was 0.15 ng/ml [35].
Statistical analysis
Data analysis was performed using the GraphPad Prism 6 software (GraphPad Software, Inc., La Jolla, CA, USA). Results are presented as mean ± SE, and in some cases are expressed as arbitrary units, with controls equal to 1, or as percentage of control. For the novel object recognition task, the parameter chosen to assess rats’ ability to discriminate novelty from familiar features was the recognition index (RI) and was calculated using the following formula: [RI = TN/(TN + TF)]. Statistical evaluations were performed by one-way ANOVA, followed by Fisher’s Protected Least Significant Difference (PLSD) test, with the exclusion of behavioral data for which we used the Tukey’s multiple comparison test. Differences in P value less than 0.05 were considered statistically significant.
Results
AD rat model
Aβ1-42 oligomers aggregation and toxicity
Aβ1-42 oligomers, prepared as reported in Carrotta et al. [29], were aggregated by incubation for 8 h at 37 °C. The results of aggregation kinetics of Aβ1-42 oligomers are shown in Fig. 1a–c. The Aβ1-42 oligomers aggregation was also evaluated by fluorescence microscope, and the Aβ1-42 plate’s mean size was measured by fluorescence microscopy software; results are shown in Fig. 1d. For Aβ1-42 cell toxicity, LAN5 cells were treated with 50, 75, and 100 μM of Aβ1-42 for 24 h and the cell viability was evaluated using MTS assay; results are shown in Fig. 1e, f. Based on the present aggregation and toxicity data of Aβ1-42 oligomers and on data of neurotoxicity dependent on the types and sizes of Aβ1-42 oligomers [36, 37], it was decided to inject Aβ1-42 oligomers aggregates, formed after 8 h of incubation and at concentration of 75 μM, in the dorsal hippocampus.

Aβ1-42 oligomers aggregation and toxicity. a–c Aggregation kinetics of Aβ1-42 oligomers. d Fluorescence imaging of Aβ1-42 plates. e, f Aβ1-42 cell toxicity in LAN5 cells treated with 50, 75, and 100 μM of Aβ1-42 for 24 h: cell viability (e) and dose-effect of cell morphological changes (f). Scale: upper panels 100 μm; lower panel 50 μm
Aβ1-42 intra-hippocampal injection and IFNβ1a treatment
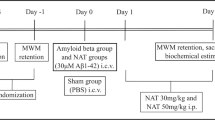
In Fig. 2a, the stereotaxic position of injection site in the dorsal hippocampus is shown. Fig. 2b shows the scheme of treatment performed. The amount of Aβ1-42 protein aggregates injected was 23 μg/2 μl according to toxicity data of previous experimental models [38, 39]. The scheme of dose and time of IFNβ1a treatment was based on previous work using a similar experimental rat model [20] or a rat model of autoimmune encephalomyelitis [40,41,42].

Aβ1-42 intra-hippocampal injection and IFNβ1a treatment. a Stereotaxic position of Aβ1-42 injection in the dorsal hippocampus. b Scheme of Aβ1-42 and IFNβ1a treatment performed
IFNβ1a treatment rescues cognitive performances impaired by intra-hippocampal injection of Aβ1-42 peptide
Using the NOR test, we evaluated changes in cognitive function induced by intra-hippocampal injection of Aβ1-42 peptide. At scheduled time (Fig. 2b), rats were tested in an open field arena in order to assess declarative memory as assessed by the recognition index of novel objects from familiar ones. When rats were trained with two identical objects, a one-way ANOVA did not show (Fig. 3a) any statistical variation in the RI% (F(4,35) = 0.6122; p = 0.6566) among the experimental groups. Twenty-four hours after the training, rats’ preference toward a novel object was evaluated. A one-way ANOVA showed (Fig. 3b) a significant effect of treatment (F(4,35) = 5.971; p < 0.001). The post hoc analysis conducted by Tukey’s multiple comparison test showed a significant reduction of RI% in the Aβ1-42 group as compared to the control (p < 0.01), to Aβ1-42 + IFNβ1a (p < 0.01), and to IFNβ1a (p < 0.05) groups (Fig. 3b). In the Aβ1-42 + IFNβ1a group, the treatment with IFNβ1 fully counteracted the RI% reduction observed in the Aβ1-42 group. In the IFNβ1a and sham groups, the RI% did not change as compared to control.

Cognitive evaluation by NOR test. Rats (n = 8 per group) exposed to the training with two identical objects did not show any statistical variation in the RI% (F(4,35) 0.6122; p = 0.6566) among the experimental groups (a). Twenty-four hours after the training was the evaluation of rats’ preference toward novel object. The results of one-way ANOVA showed b a significant effect of treatment (F(4,35) = 5.971; p < 0.001). The post hoc analysis conducted by Tukey’s multiple comparison test showed a significant reduction of RI% in the Aβ1-42 group with respect to control (p < 0.01), Aβ1-42 + IFNβ1a (p < 0.01), and IFNβ1a (p < 0.05) groups (b). In the Aβ1-42 + IFNβ1a group, the treatment with IFNβ1a full counteracted the RI% reduction observed in the Aβ1-42 group. In the IFNβ1a and sham groups, the RI% did not change as compared to control. *P < 0.05, **P < 0.01
Body weight and cortisol levels
Rats’ body weight was measured at the beginning and the end of the experimental period (Fig. 2b). Two-way repeated ANOVA measurements showed no significant differences in body weight (Fig. 4a) both among the various experimental groups and in each group at the end of the experimental period as compared to the beginning of the experiment.

Body weight and cortisol levels. Two-way repeated measurement ANOVA showed not significant differences in the body weight, both among the experimental groups and each group at the end of experimental period as compared to starting body weight (a). One-way ANOVA did not show significant change in cortisol level among the experimental groups (b)
Although corticosterone is considered the main glucocorticoid involved in the regulation of stress responses in rodents, we preferred to measure cortisol in consideration of procedure availability in our laboratory and of good correlation between serum cortisol and corticosterone [43]. The results are reported in Fig. 4b: one-way ANOVA did not show significant change in cortisol level among experimental groups.
IFNβ1a effects on glial cell activation by Aβ1-42 protein injection in the hippocampus
Since one of the features of AD pathology is activation of microglia and astrocytes induced by Aβ deposits, we analyzed by immunohistochemistry and Western blot Iba-1 and GFAP markers for microglia and astrocyte activation. As shown in Fig. 5a, b, the count of Iba-1-positive cells was significantly increased in the hippocampus of the Aβ1-42-treated group (F(4,22) = 26.97, p < 0.0001) as compared to the control (p < 0.0001) and sham groups (p < 0.001). However, in the Aβ1-42 + IFNβ1a group, the treatment with IFNβ1a significantly counteracted the effect of Aβ1-42 injection as shown by the Iba-1-positive cell number significantly reduced as compared to the Aβ1-42 group (p = 0.02). However, the cell number in the Aβ1-42 + IFNβ1a group was still significantly increased when compared to the control group (p < 0.0001) but not compared to the sham group (Fig. 5a, b). The sham group, but not the IFNβ1a group, showed a significant increase of Iba-1-positive cell number as compared to the control group (p < 0.001). Quantitative Western blot analyses of Iba-1 protein levels clearly showed a significant increase in the Aβ1-42 group as compared to both control (p < 0.05) and sham (p < 0.05) groups. In the Aβ1-42 + IFNβ1a group, the treatment with IFNβ1a counteracted the Aβ1-42 effect on Iba-1 protein levels. Indeed, Aβ1-42 + IFNβ1a Iba-1 protein levels are not significant when compared to the Aβ1-42 group, but they are also not significant when compared both to control and sham groups (Fig. 5c). This means that treatment with IFNβ1a in the Aβ1-42 + IFNβ1a group counteracted the increase of Iba-1 protein levels induced by Aβ1-42, as shown by its loss of significance with respect to controls, although it does not bring them back to the levels detected in the control and sham groups (Fig. 5c). Iba-1 protein levels in the IFNβ1a and sham groups did not show significant difference as compared to the control group.

IFNβ1a effects on microglial cell activation by Aβ1-42. a Iba-1 immunohistochemistry in the hippocampus. b Count of Iba-1-positive cells and c quantitative Western blot analyses of Iba-1 protein levels. b The count of Iba-1-positive cells showed a significant increase of cell number in the hippocampus region of the Aβ1-42-treated group (n = 6) and treatment with IFNβ1a (n = 6) significantly counteract this effect in the Aβ1-42+ IFNβ1a group (n = 6) as compared to sham group but not to the control group. c Similarly, quantitative Western blot analyses of Iba-1 protein levels showed that in the Aβ1-42 group, Iba-1 protein levels were significantly increased and treatment with IFNβ1a significantly counteract this effect in the Aβ1-42 + IFNβ1a group. Scale: 100 μm. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
The immunohistochemistry analysis of GFAP marker showed a not significant trend toward an increase in the astrocytes numb in all experimental groups as compared to control group (Fig. 6a, b). By contrast, the quantitative Western blot analyses of GFAP protein levels showed that in the Aβ1-42 and in the Aβ1-42 + IFNβ1a groups, GFAP protein levels were significantly increased (both p < 0.0001) as compared to the control group and in the Aβ1-42 + IFNβ1a group as compared to the sham group (Fig. 6c). Surprisingly, the treatment with IFNβ1a alone significantly reduced (p < 0.01) the GFAP protein levels as compared to the control group. The cell number of the sham group was not significantly different from the control group.

IFNβ1a effects on astroglial cell activation by Aβ1-42. a GFAP immunohistochemistry in the hippocampus. b Count of GFAP positive cells and c quantitative Western blot analyses of GFAP protein levels. b Count of astrocytes number showed no significant changes in all experimental groups. c By contrast, the quantitative Western blot analyses of GFAP protein levels revealed that in the Aβ1-42 group (n = 6), GFAP protein levels were significantly increased and this increase was not counteracted by treatment with IFNβ1a (n = 6) in the Aβ1-42 + IFNβ1a group (n = 6). Note IFNβ1a alone significantly reduced the GFAP protein levels as compared to the control group. Scale: 100 μm *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
IFNβ1a inhibits pro-inflammatory cytokines increase induced by Aβ1-42 protein injection in the hippocampus
We also analyzed by ELISA the hippocampal levels of pro-inflammatory cytokines, IL-1β and IL-6, and anti-inflammatory cytokines, IL-10 and TGF-β1. As shown in Fig. 7a, b, we found that both IL-1β and IL-6 levels were significantly increased in the Aβ1-42 group as compared to the control (p < 0.0001 and p < 0.01 respectively) and sham (p < 0.0001 and p < 0.001 respectively) groups. In the Aβ1-42 + IFNβ1a group, the treatment with IFNβ1a counteracted this Aβ1-42 effect on IL-1β and IL-6 levels (p < 0.0001 and p < 0.01 respectively), bringing them back to the levels of control and sham groups. In the IFNβ1a group, the treatment with IFNβ1a did not change the IL-1β levels as compared to the control, whereas it produced a substantial reduction of IL-6 levels as compared to the control and sham groups (p < 0.05).

IFNβ1a effects on inflammatory cytokines in the hippocampus. a, b Hippocampal levels of pro-inflammatory cytokines, IL-1β and IL-6. Note that IL-1β and IL-6 levels were significantly increased in the Aβ1-42 group (n = 5 respectively) and this increase was counteracted by treatment with IFNβ1 (n = 5 respectively) in the Aβ1-42 + IFNβ1a group (n = 5 respectively). c, d Hippocampal levels of anti-inflammatory cytokines, IL-10 and TGF-β1. Note IL-10 and TGF-β1 levels showed significant reduction in the Aβ1-42 group (n = 5 respectively) and following IFNβ1a (n = 5, respectively) treatment in the Aβ1-42 + IFNβ1a group (n = 5, respectively) were recovered to control (n = 5 and n = 6, respectively) levels. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
In contrast to hippocampal upregulation of IL-1 and IL-6 levels, the anti-inflammatory cytokine IL-10 was found significantly reduced in the Aβ1-42 group as compared to the control (p < 0.001) and sham (p < 0.001) groups (Fig. 7c). Following IFNβ1a treatment in the Aβ1-42 + IFNβ1a group, the IL-10 levels recovered to control levels, strengthening the anti-inflammatory property of IFNβ1a. The TGF-β1 levels were significantly reduced in the IFNβ1a group as compared to control or sham group (p < 0.01 and p < 0.001 respectively) and Aβ1-42 group as compared to sham (p < 0.05) group (Fig. 7d), whereas surprisingly in the Aβ1-42 + IFNβ1a group, TGF-β1 levels recovered to the control group levels. In the sham group, we did not find significant changes in both IL-10 and TGF-β1 levels as compared to the control group.
IFNβ1a treatment effects on ROS levels and SOD1 or SOD2 proteins and activity levels
SOD1 levels (Fig. 8a) were increased in the Aβ1-42 + IFNβ1a group as compared to the control (p < 0.05) and sham (p < 0.05) groups, whereas SOD2 levels (Fig. 8b) were significantly reduced in the same group as compared to the control (p < 0.05) and sham (p < 0.01) groups. The SOD2 levels were significantly lower in the IFNβ1a group as compared to the sham group (p < 0.05) but not to the control group, suggesting that IFNβ1a per se may negatively regulate the SOD2 levels. In the sham group, we did not find significant changes in both SOD1 and SOD2 levels as compared to the control group.

IFNβ1a effects on ROS and lipid peroxidation levels and SOD1 or SOD2 proteins and activity levels in the hippocampus. a, b The levels of SOD1 showed significant increase in the Aβ1-42 + IFNβ1a group (n = 5), whereas those of SOD2 appear significantly reduced in the same group (n = 5). c Total SOD activity levels showed significant decrease in Aβ1-42 group (n = 5), and this decrease was counteracted in the Aβ1-42 + IFNβ1a group (n = 5) by treatment with IFNβ1a. d ROS levels were significantly increased in the Aβ1-42 group (n = 5), and this increase was counteracted by treatment with IFNβ1a (n = 5). e The analysis of lipid peroxidation levels, measured as concentration of MDA, revealed a significant increase of MDA levels in the Aβ1-42 group (n = 5) that was completely blocked by IFNβ1a treatment in the Aβ1-42 + IFNβ1a group (n = 5). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Concerning the SOD activity (Fig. 8c), we found that SOD activity levels were significantly decreased in the Aβ1-42 group as compared to control (p < 0.01). In the Aβ1-42 + IFNβ1a group, the treatment with IFNβ1a counteracted the decrease of SOD activity level bringing it back to the control level. In the IFNβ1a group, the treatment with IFNβ1a did not change the SOD activity levels as compared to the control and sham groups. In the sham group, we did not find significant changes in SOD activity levels as compared to the control group.
Concerning the oxidative stress analysis (Fig. 8d), we found that ROS levels were significantly increased in the Aβ1-42 group as compared to control (p < 0.0001) and sham (p < 0.01) groups. In Aβ1-42+ IFNβ1a group, the treatment with IFNβ1a counteracted the increase of ROS levels bringing them back to the control level. ROS levels were substantially similar in the IFNβ1a, control, and sham groups.
Lipid peroxidation is the degradation of lipids that occurs as a result of oxidative damage, typically by reactive oxygen species, resulting in a well-defined chain reaction with the production of end products such as MDA. According to previous data [44] showing that Aβ1-42 injection induces ROS levels, we analyzed the level of lipid peroxidation in the hippocampus by measuring the concentration of MDA. The analysis (Fig. 8e) revealed a significant increase of MDA levels in the Aβ1-42 group as compared to control (p < 0.0001) and sham (p < 0.01) groups. This increase of MDA levels was positively correlated n with the increased levels of ROS. In the Aβ1-42 + IFNβ1a group, the treatment with IFNβ1a completely blocked the increase of MDA levels observed in the Aβ1-42 group. Interestingly, in the IFNβ1a group, the MDA levels were found significantly reduced as compared to control (p < 0.01) and sham (p < 0.001) groups. However, the MDA levels were found also significantly increased in the sham as compared to control group (p < 0.05).
Discussion
In our rat model of AD, the administration of Aβ1-42 oligomer aggregates in the dorsal hippocampus led to neuroinflammation, via the activation of glial immune system, and memory impairment as evaluated by the NOR test. The short treatment with IFNβ1a was able to reverse memory impairment and to suppress microglia activation and the upregulation of pro-inflammatory cytokine (IL-6, IL-1β) levels and oxidative stress in the hippocampus. All together, these data suggest a protective effect of IFNβ1a against Aβ1-42-induced functional alterations in the hippocampus of our rat AD model.
AD is characterized by an insidious clinical onset, by a progressive cognitive and memory decline, and by accumulation of Aβ plaques and neurofibrillary tangles leading to loss of neurons in the hippocampus and in the cerebral cortex [1,2,3]. In a previous pilot study, we evaluated the safety and efficacy of IFNβ1a in subjects affected by mild-to-moderate AD and detected a reduction in disease progression and improvements in the instrumental activities of daily living and physical self-maintenance scales [24]. Apart for this limited human study, IFNβ1a had never been previously tested in AD, unlike its long and widespread use in patients with multiple sclerosis, where it has been clearly shown that subcutaneous IFNβ1a protects against cognitive decline [15, 21, 22, 41, 45]. Although most of its pleiotropic effects occur in the peripheral immune system, a direct anti-inflammatory effect of IFNβ1a within the brain has also been proposed [23]. We actually found an anti-inflammatory effect of IFNβ1a in the CNS of experimental rats that reasonably explains the recovery from memory impairment caused by hippocampal Aβ1-42 peptide injection. Improvement of memory deficit in AD animal model after treatments targeting inflammation has already been reported [46, 47]. However, it has been reported that microglia of adult mice following chronic exposure to IFNβ1a express an aging-like phenotype and negatively affect learning abilities [48, 49].
Histological studies of brains from individuals with AD have revealed a direct relationship between Aβ1-42 peptide abnormal production and the development and/or maintenance of neuro-inflammation and oxidative stress [5, 50]. Accordingly, exposure of the brain to Aβ1-42 peptide causes inflammation, by activating microglia as well as astrocytes, and promotes the production of cytotoxic molecules, such as pro-inflammatory cytokines and reactive oxygen species, that contribute to dysfunction, injury, and ultimately neuronal loss [3, 51,52,53,54,55]. In addition, the release of inflammatory mediators may in turn increase Aβ1-42 peptide production, which may further contribute to plaque formation and progression to neuronal loss [3, 52]. Besides this consolidated evidence of the close relationship between plaques and activated glial cells, neuro-inflammation is increasingly believed to be an early player in the pathological cascade leading to AD rather than a mere consequence.
Although its role is still debated, the recognition of an inflammatory contribution to AD pathogenesis has led to therapeutic attempts using several anti-inflammatory agents [9]. Therapeutic approaches using phenols, phytoestrogens, neuro-steroids, and other natural phytochemicals have been explored in AD and experimental models, with some promising results such as cognitive improvements and attenuation of neuro-inflammation. Several non-steroidal anti-inflammatory drugs have also been tested in the attempt to prevent the onset or to slow down progression of AD [1, 2, 56, 57]. Flavonoids have been tested in AD models and seem to be able to reduce AD severity by modulating the production of microglia pro-inflammatory cytokines (TNF-α and IL-1β) or by reducing their Aβ-induced cytokine production [3]. In the present work, we tested IFNβ1a in an AD rat model and showed that its anti-inflammatory ability is mainly associated to the block of IL-1β and Il-6 upregulation levels in the hippocampus. Deposition of Aβ peptide may also activate astrocytes inducing astrogliosis with release of pro-inflammatory agents, such as IL-1, IL-6, and IL-10, and oxidative stress with production of reactive oxygen and nitrogen species [58, 59]. Changes in astrocyte function have been observed in brains from individuals with AD, as well as in AD in vitro and in vivo animal models [60]. However, in the present work, we could not observe a reduction of astrocytes activation in the Aβ1-42 group treated with IFNβ1a, as evaluated by the GFAP-positive cell number and GFAP protein levels.
IL-1β, synthesized and released by both activated microglia and astrocytes, is considered to be a major pro-inflammatory cytokine in the brain and play a key role in the progression of AD [61]. Similarly, IL-6 is also a pro-inflammatory cytokine mainly produced by activated microglia and when increased in brain of AD and AD animal models [5, 52] may impair cognitive processes, such as spatial learning and memory [62, 63], and stimulate the synthesis of Aβ precursor protein. Indeed, active microglia constitute the core immune system in the brain and release pro-inflammatory cytokines and free radicals that may elicit neurodegenerative processes, and since the pro-inflammatory mediators, IL-1β and Il-6, are produced primarily by microglia in the brain, the IFNβ1a inhibition of IL-1β and IL-6 in the hippocampus of Aβ1-42-treated group suggests a direct effect on microglial cells. This is supported by our observation of a reduction of microglial activation in the Aβ1-42 group treated with IFNβ1a, as reflected by the decrease in number of microglia cells and Iba-1 protein levels. Previously, following chronic exposure to IFNβ1a, Deczkowska et al. [48] reported modification in microglia morphology and expression of genes present in aged microglia, suggesting that microglia can contribute to the pro-inflammatory response of the brain in AD or aging, thereby exacerbating cognitive loss and disease pathology. In our rat model, we did not observe alterations in the microglia or astrocytes morphology, probably because the treatment time with IFNβ1a was shorter compared to those given by Deczkowska et al. [48]. In addition, in the present study, we did not explore gene expressed in aged microglia.
In contrast to hippocampal upregulation of IL-1 and IL-6 levels, the anti-inflammatory cytokines, IL-10 and TGF-β1, were significantly reduced in the Aβ1-42 group and recovered to control levels following IFNβ1a treatment. This result further supports the anti-inflammatory properties of IFNβ1a.
Although the numerous mechanisms underlying the IFNβ1a anti-inflammatory effects have been clearly defined within the peripheral immune system, its role in the central nervous system function has been little explored. Indeed, the only data available in this field, in addition to already mentioned anti-inflammatory effect of IFNβ1a in patients with multiple sclerosis [17, 18, 64], derived from studies on experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. Indeed, IFNβ1a treatment prevents and reduces the progression of the experimental CNS demyelination [42, 65, 66] by inhibiting pro-inflammatory cytokines (IL-6, IL-1β, TNF-α, IFN-γ), astrocytes activation, and inducible nitric oxide synthase expression [20]. However, adverse effect of type I interferon (IFN-I) has been reported that in the aging brain chronically elevated IFN-I activity contributes to the pathology of various human CNS diseases and in animal models, including aging microglial phenotype, neurodegeneration, and microgliosis [48].
The present and above-listed data showed beneficial effects of IFNβ1a in a model of AD pathology, but it is still unclear how it exerts this effect within the CNS, since its passage from the bloodstream to the brain parenchyma is significantly restricted by the blood–brain and blood–cerebrospinal fluid barriers [67,68,69,70]. In this context, the effects of IFNβ1a in the brain have been associated with possible modulation of brain inflammatory events at capillaries level [41] or with a decreased permeability of the blood–brain barrier to inflammatory cell entry into to the brain [71], thus reducing CNS inflammatory response. Several studies have demonstrated anti-inflammatory effect of IFNβ1a outside the CNS [17, 18, 64]. Indeed, IFNβ1a regulates several immunological functions, including decrease in T cell activation, induction of cytokine shifts in favor of an anti-inflammatory effect, prevention of T cell adhesion, and extravasation across the blood–brain barrier, as well as induction of T-regulatory cells, all occurring within the peripheral immunological organs [12,13,14,15,16,17,18,19,20]. Recently, several pathways for the transport of cytokines from systemic circulation into the brain have been reviewed [70], but the mechanisms by which IFNβ1a treatment may affect pro-inflammatory response induced by Aβ1-42 peptide injection in the hippocampus need further investigations. Anyway, in the present work, it is possible to exclude a role of glucocorticoid in mediating anti-inflammatory effects, since we did not observe any significant change in cortisol levels among the experimental groups.
Another particular hypothesis that has received considerable interest in the pathogenesis and progression of AD is the oxidative damage in the brain [72, 73]. Therefore, strategies aimed to reduce oxidative stress in AD have been proposed, also supported by reports of beneficial effects in AD of various antioxidants treatments [74, 75]. The present study indicated potential neuroprotective effects of IFNβ1a possibly mediated also by its ability to reduce ROS and lipids peroxidation and to increase SOD1 protein levels, although we did not find a parallel increase in SOD activity. A recent report actually showed that treatment with IFNβ1a inhibits oxidative stress in an animal model of multiple sclerosis, but at present, there are no reports of similar findings in AD or AD models [20].
Conclusions
Based on the hypothesis that Aβ1-42-induced inflammation plays an important role in AD pathogenesis, using a rat model of AD, we provided evidence that IFNβ1a treatment may be a viable strategy to inhibit pro-inflammatory cytokines and oxidative stress. Therefore, IFNβ1a treatment, used for decades to contain inflammatory-mediated diseases of the brain, could be effective in AD patients, contributing to smolder the progression of this devastating disease.
Abbreviations
- AD:
-
Alzheimer disease
- DCF:
-
Dichlorofluorescein
- DCFH-DA:
-
Dichlorofluorescein diacetate
- ELISA:
-
Enzyme-linked immunosorbent assay
- IFNβ1a:
-
Interferon-β1a
- MDA:
-
Malondialdehyde
- NF-kB:
-
Transcription factor nuclear factor-kB
- PLSD:
-
Fisher’s Protected Least Significant Difference
- pNF-kB:
-
Phosphorylated NF-kB
- ROS:
-
Reactive oxygen species
- TBA:
-
Thiobarbituric acid
- TGF-β1:
-
Transforming growth factor-beta1
- Aβ1-42 :
-
Amyloid β Protein Fragment 1-42
References
Heneka MT, Carson MJ, El KJ, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14:388–405. https://doi.org/10.1016/S1474-4422(15)70016-5.
Rubio-Perez JM, Morillas-Ruiz JM. A review: inflammatory process in Alzheimer’s disease, role of cytokines. ScientificWorldJournal. 2012;2012:756357. https://doi.org/10.1100/2012/756357.
Domingues C, da Cruz E Silva OAB, Henriques AG. Impact of cytokines and chemokines on Alzheimer’s disease neuropathological hallmarks. Curr Alzheimer Res. 2017;14:870–82. https://doi.org/10.2174/1567205014666170317113606.
Rojo LE, Fernandez JA, Maccioni AA, Jimenez JM, Maccioni RB. Neuroinflammation: implications for the pathogenesis and molecular diagnosis of Alzheimer’s disease. Arch Med Res. 2008;39:1–16. https://doi.org/10.1016/j.arcmed.2007.10.001.
McGeer PL, McGeer EG. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: implications for therapy. Acta Neuropathol. 2013;126:479–97. https://doi.org/10.1007/s00401-013-1177-7.
Fernandez PL, Britton GB, Rao KS. Potential immunotargets for Alzheimer’s disease treatment strategies. J Alzheimers Dis. 2013;33:297–312. https://doi.org/10.3233/JAD-2012-121222.
Hensley K. Neuroinflammation in Alzheimer’s disease: mechanisms, pathologic consequences, and potential for therapeutic manipulation. J Alzheimers Dis. 2010;21:1–14. https://doi.org/10.3233/JAD-2010-1414.
Etminan M, Gill S, Samii A. Effect of non-steroidal anti-inflammatory drugs on risk of Alzheimer’s disease: systematic review and meta-analysis of observational studies. BMJ. 2003;327:128. https://doi.org/10.1136/bmj.327.7407.128.
Shadfar S, Hwang CJ, Lim MS, Choi DY, Hong JT. Involvement of inflammation in Alzheimer’s disease pathogenesis and therapeutic potential of anti-inflammatory agents. Arch Pharm Res. 2015;38:2106–19. https://doi.org/10.1007/s12272-015-0648-x.
Balducci C, Forloni G. Novel targets in Alzheimer’s disease: a special focus on microglia. Pharmacol Res. 2018;130:402–13. https://doi.org/10.1016/j.phrs.2018.01.017.
de Weerd NA, Nguyen T. The interferons and their receptors--distribution and regulation. Immunol Cell Biol. 2012;90:483–91. https://doi.org/10.1038/icb.2012.9.
Kawanokuchi J, Mizuno T, Kato H, Mitsuma N, Suzumura A. Effects of interferon-beta on microglial functions as inflammatory and antigen presenting cells in the central nervous system. Neuropharmacology. 2004;46:734–42. https://doi.org/10.1016/j.neuropharm.2003.11.007.
Okada K, Kuroda E, Yoshida Y, Yamashita U, Suzumura A, Tsuji S. Effects of interferon-beta on the cytokine production of astrocytes. J Neuroimmunol. 2005;159:48–54. https://doi.org/10.1016/j.jneuroim.2004.09.013.
Vermersch P, Czlonkowska A, Grimaldi LM, Confavreux C, Comi G, Kappos L, Olsson TP, Benamor M, Bauer D, Truffinet P, et al. Teriflunomide versus subcutaneous interferon beta-1a in patients with relapsing multiple sclerosis: a randomised, controlled phase 3 trial. Mult Scler. 2014;20:705–16. https://doi.org/10.1177/1352458513507821.
Patti F, Morra VB, Amato MP, Trojano M, Bastianello S, Tola MR, Cottone S, Plant A, Picconi O. Subcutaneous interferon beta-1a may protect against cognitive impairment in patients with relapsing-remitting multiple sclerosis: 5-year follow-up of the COGIMUS study. PLoS One. 2013;8:e74111. https://doi.org/10.1371/journal.pone.0074111.
Bastianello S, Romani A, Viselner G, Tibaldi EC, Giugni E, Altieri M, Cecconi P, Mendozzi L, Farina M, Mariani D, et al. Chronic cerebrospinal venous insufficiency in multiple sclerosis: clinical correlates from a multicentre study. BMC Neurol. 2011;11:132. https://doi.org/10.1186/1471-2377-11-132.
Graber JJ, Ford D, Zhan M, Francis G, Panitch H, Dhib-Jalbut S. Cytokine changes during interferon-beta therapy in multiple sclerosis: correlations with interferon dose and MRI response. J Neuroimmunol. 2007;185:168–74. https://doi.org/10.1016/j.jneuroim.2007.01.011.
Liu Y, Teige I, Ericsson I, Navikas V, Issazadeh-Navikas S. Suppression of EAE by oral tolerance is independent of endogenous IFN-beta whereas treatment with recombinant IFN-beta ameliorates EAE. Immunol Cell Biol. 2010;88:468–76. https://doi.org/10.1038/icb.2009.111.
Cheng W, Zhao Q, Xi Y, Li C, Xu Y, Wang L, Niu X, Wang Z, Chen G. IFN-beta inhibits T cells accumulation in the central nervous system by reducing the expression and activity of chemokines in experimental autoimmune encephalomyelitis. Mol Immunol. 2015;64:152–62. https://doi.org/10.1016/j.molimm.2014.11.012.
Lubina-Dabrowska N, Stepien A, Sulkowski G, Dabrowska-Bouta B, Langfort J, Chalimoniuk M. Effects of IFN-beta1a and IFN-beta1b treatment on the expression of cytokines, inducible NOS (NOS type II), and myelin proteins in animal model of multiple sclerosis. Arch Immunol Ther Exp. 2017;65:325–38. https://doi.org/10.1007/s00005-017-0458-6.
Patti F, Amato MP, Bastianello S, Caniatti L, Di ME, Ferrazza P, Goretti B, Gallo P, Morra VB, Lo FS, et al. Effects of immunomodulatory treatment with subcutaneous interferon beta-1a on cognitive decline in mildly disabled patients with relapsing-remitting multiple sclerosis. Mult Scler. 2010;16:68–77. https://doi.org/10.1177/1352458509350309.
Mokhber N, Azarpazhooh A, Orouji E, Rao SM, Khorram B, Sahraian MA, Foroghipoor M, Gharavi MM, Kakhi S, Nikkhah K, Azarpazhooh MR. Cognitive dysfunction in patients with multiple sclerosis treated with different types of interferon beta: a randomized clinical trial. J Neurol Sci. 2014;342:16–20. https://doi.org/10.1016/j.jns.2014.01.038.
Calabrese M, Bernardi V, Atzori M, Mattisi I, Favaretto A, Rinaldi F, Perini P, Gallo P. Effect of disease-modifying drugs on cortical lesions and atrophy in relapsing-remitting multiple sclerosis. Mult Scler. 2012;18:418–24. https://doi.org/10.1177/1352458510394702.
Grimaldi LM, Zappala G, Iemolo F, Castellano AE, Ruggieri S, Bruno G, Paolillo A. A pilot study on the use of interferon beta-1a in early Alzheimer’s disease subjects. J Neuroinflammation. 2014;11:30. https://doi.org/10.1186/1742-2094-11-30.
Jantaratnotai N, Ryu JK, Schwab C, McGeer PL, McLarnon JG. Comparison of vascular perturbations in an Abeta-injected animal model and in AD brain. Int J Alzheimers Dis. 2011;2011:918280. https://doi.org/10.4061/2011/918280.
McLarnon JG, Ryu JK. Relevance of abeta1-42 intrahippocampal injection as an animal model of inflamed Alzheimer’s disease brain. Curr Alzheimer Res. 2008;5:475–80.
Mazure CM, Swendsen J. Sex differences in Alzheimer’s disease and other dementias. Lancet Neurol. 2016;15:451–2. https://doi.org/10.1016/S1474-4422(16)00067-3.
Mielke MM, Vemuri P, Rocca WA. Clinical epidemiology of Alzheimer’s disease: assessing sex and gender differences. Clin Epidemiol. 2014;6:37–48. https://doi.org/10.2147/CLEP.S37929.
Carrotta R, Di CM, Manno M, Montana G, Picone P, Romancino D, San Biagio PL. Toxicity of recombinant beta-amyloid prefibrillar oligomers on the morphogenesis of the sea urchin Paracentrotus lividus. FASEB J. 2006;20:1916–7. https://doi.org/10.1096/fj.06-5716fje.
Mudo G, Salin T, Condorelli DF, Jiang XH, Dell’Albani P, Timmusk T, Metsis M, Funakoshi H, Belluardo N. Seizures increasetrkC mRNA expression in the dentate gyrus of rat hippocampus. Role of glutamate receptor activation. J Mol Neurosci. 1995;6:11–22. https://doi.org/10.1007/BF02736755.
Di Liberto V, Borroto-Escuela DO, Frinchi M, Verdi V, Fuxe K, Belluardo N, Mudo G. Existence of muscarinic acetylcholine receptor (mAChR) and fibroblast growth factor receptor (FGFR) heteroreceptor complexes and their enhancement of neurite outgrowth in neural hippocampal cultures. Biochim Biophys Acta. 2017;1861:235–45. https://doi.org/10.1016/j.bbagen.2016.10.026.
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–75.
Frinchi M, Scaduto P, Cappello F, Belluardo N, Mudo G: Heat shock protein (Hsp) regulation by muscarinic acetylcholine receptor (mAChR) activation in the rat hippocampus. J Cell Physiol. 2018;233:6107–16. https://doi.org/10.1002/jcp.26454
Zizzo MG, Frinchi M, Nuzzo D, Jinnah HA, Mudo G, Condorelli DF, Caciagli F, Ciccarelli R, Di IP, Mule F, et al. Altered gastrointestinal motility in an animal model of Lesch-Nyhan disease. Auton Neurosci. 2018;210:55–64. https://doi.org/10.1016/j.autneu.2017.12.007.
Di Liberto V, Frinchi M, Verdi V, Vitale A, Plescia F, Cannizzaro C, Massenti MF, Belluardo N, Mudo G. Anxiolytic effects of muscarinic acetylcholine receptors agonist oxotremorine in chronically stressed rats and related changes in BDNF and FGF2 levels in the hippocampus and prefrontal cortex. Psychopharmacology (Berl). 2017;234:559–73. https://doi.org/10.1007/s00213-016-4498-0.
Picone P, Carrotta R, Montana G, Nobile MR, San Biagio PL, Di Carlo M. Abeta oligomers and fibrillar aggregates induce different apoptotic pathways in LAN5 neuroblastoma cell cultures. Biophys J. 2009;96:4200–11. https://doi.org/10.1016/j.bpj.2008.11.056.
Yang T, Li S, Xu H, Walsh DM, Selkoe DJ. Large soluble oligomers of amyloid beta-protein from Alzheimer brain are far less neuroactive than the smaller oligomers to which they dissociate. J Neurosci. 2017;37:152–63. https://doi.org/10.1523/JNEUROSCI.1698-16.2016.
Tong JQ, Zhang J, Hao M, Yang J, Han YF, Liu XJ, Shi H, Wu MN, Liu QS, Qi JS. Leptin attenuates the detrimental effects of beta-amyloid on spatial memory and hippocampal later-phase long term potentiation in rats. Horm Behav. 2015;73:125–30. https://doi.org/10.1016/j.yhbeh.2015.06.013.
Ghasemi R, Zarifkar A, Rastegar K, Maghsoudi N, Moosavi M. Repeated intra-hippocampal injection of beta-amyloid 25-35 induces a reproducible impairment of learning and memory: considering caspase-3 and MAPKs activity. Eur J Pharmacol. 2014;726:33–40. https://doi.org/10.1016/j.ejphar.2013.11.034.
Maier CM, Yu F, Nishi T, Lathrop SJ, Chan PH. Interferon-beta fails to protect in a model of transient focal stroke. Stroke. 2006;37:1116–9. https://doi.org/10.1161/01.STR.0000208214.46093.d5.
Floris S, Ruuls SR, Wierinckx A, van der Pol SM, Dopp E, van der Meide PH, Dijkstra CD, De Vries HE. Interferon-beta directly influences monocyte infiltration into the central nervous system. J Neuroimmunol. 2002;127:69–79.
van der Meide PH, de Labie MC, Ruuls SR, Groenestein RJ, Botman CA, Olsson T, Dijkstra CD. Discontinuation of treatment with IFN-beta leads to exacerbation of experimental autoimmune encephalomyelitis in Lewis rats. Rapid reversal of the antiproliferative activity of IFN-beta and excessive expansion of autoreactive T cells as disease promoting mechanisms. J Neuroimmunol. 1998;84:14–23.
Gong S, Miao YL, Jiao GZ, Sun MJ, Li H, Lin J, Luo MJ, Tan JH. Dynamics and correlation of serum cortisol and corticosterone under different physiological or stressful conditions in mice. PLoS One. 2015;10:e0117503. https://doi.org/10.1371/journal.pone.0117503.
Meunier J, Ieni J, Maurice T. The anti-amnesic and neuroprotective effects of donepezil against amyloid beta25-35 peptide-induced toxicity in mice involve an interaction with the sigma1 receptor. Br J Pharmacol. 2006;149:998–1012. https://doi.org/10.1038/sj.bjp.0706927.
Stone LA, Frank JA, Albert PS, Bash C, Smith ME, Maloni H, McFarland HF. The effect of interferon-beta on blood-brain barrier disruptions demonstrated by contrast-enhanced magnetic resonance imaging in relapsing-remitting multiple sclerosis. Ann Neurol. 1995;37:611–9. https://doi.org/10.1002/ana.410370511.
Sundaram JR, Poore CP, Sulaimee NHB, Pareek T, Cheong WF, Wenk MR, Pant HC, Frautschy SA, Low CM, Kesavapany S. Curcumin ameliorates neuroinflammation, neurodegeneration, and memory deficits in p25 transgenic mouse model that bears hallmarks of Alzheimer’s disease. J Alzheimers Dis. 2017;60:1429–42. https://doi.org/10.3233/JAD-170093.
Garcez ML, Mina F, Bellettini-Santos T, Carneiro FG, Luz AP, Schiavo GL, Andrighetti MS, Scheid MG, Bolfe RP, Budni J. Minocycline reduces inflammatory parameters in the brain structures and serum and reverses memory impairment caused by the administration of amyloid beta (1-42) in mice. Prog Neuro-Psychopharmacol Biol Psychiatry. 2017;77:23–31. https://doi.org/10.1016/j.pnpbp.2017.03.010.
Deczkowska A, Matcovitch-Natan O, Tsitsou-Kampeli A, Ben-Hamo S, Dvir-Szternfeld R, Spinrad A, Singer O, David E, Winter DR, Smith LK, et al. Mef2C restrains microglial inflammatory response and is lost in brain ageing in an IFN-I-dependent manner. Nat Commun. 2017;8:717. https://doi.org/10.1038/s41467-017-00769-0.
Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, Berkutzki T, Barnett-Itzhaki Z, Bezalel D, Wyss-Coray T, et al. Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science. 2014;346:89–93. https://doi.org/10.1126/science.1252945.
Guerriero F, Sgarlata C, Francis M, Maurizi N, Faragli A, Perna S, Rondanelli M, Rollone M, Ricevuti G. Neuroinflammation, immune system and Alzheimer disease: searching for the missing link. Aging Clin Exp Res. 2017;29:821–31. https://doi.org/10.1007/s40520-016-0637-z.
Agostinho P, Cunha RA, Oliveira C. Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer’s disease. Curr Pharm Des. 2010;16:2766–78.
Meraz-Rios MA, Toral-Rios D, Franco-Bocanegra D, Villeda-Hernandez J, Campos-Pena V. Inflammatory process in Alzheimer’s disease. Front Integr Neurosci. 2013;7:59. https://doi.org/10.3389/fnint.2013.00059.
Morales I, Guzman-Martinez L, Cerda-Troncoso C, Farias GA, Maccioni RB. Neuroinflammation in the pathogenesis of Alzheimer’s disease. A rational framework for the search of novel therapeutic approaches. Front Cell Neurosci. 2014;8:112. https://doi.org/10.3389/fncel.2014.00112.
Dzamba D, Harantova L, Butenko O, Anderova M. Glial cells - the key elements of Alzheimer s disease. Curr Alzheimer Res. 2016;13:894–911.
Meda L, Cassatella MA, Szendrei GI, Otvos L Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. 1995;374:647–50. https://doi.org/10.1038/374647a0.
Xia Z, Wang F, Zhou S, Zhang R, Wang F, Huang JH, Wu E, Zhang Y, Hu Y. Catalpol protects synaptic proteins from beta-amyloid induced neuron injury and improves cognitive functions in aged rats. Oncotarget. 2017;8:69303–15. https://doi.org/10.18632/oncotarget.17951.
Vlad SC, Miller DR, Kowall NW, Felson DT. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology. 2008;70:1672–7. https://doi.org/10.1212/01.wnl.0000311269.57716.63.
Batarseh YS, Duong QV, Mousa YM, Al Rihani SB, Elfakhri K, Kaddoumi A. Amyloid-beta and astrocytes interplay in amyloid-beta related disorders. Int J Mol Sci. 2016;17:338. https://doi.org/10.3390/ijms17030338.
Gonzalez-Reyes RE, Nava-Mesa MO, Vargas-Sanchez K, Ariza-Salamanca D, Mora-Munoz L. Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front Mol Neurosci. 2017;10:427. https://doi.org/10.3389/fnmol.2017.00427.
Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–90.
Wang WY, Tan MS, Yu JT, Tan L. Role of pro-inflammatory cytokines released from microglia in Alzheimer's disease. Ann Transl Med. 2015;3:136. https://doi.org/10.3978/j.issn.2305-5839.2015.03.49.
Dugan LL, Ali SS, Shekhtman G, Roberts AJ, Lucero J, Quick KL, Behrens MM. IL-6 mediated degeneration of forebrain GABAergic interneurons and cognitive impairment in aged mice through activation of neuronal NADPH oxidase. PLoS One. 2009;4:e5518. https://doi.org/10.1371/journal.pone.0005518.
Erta M, Quintana A, Hidalgo J. Interleukin-6, a major cytokine in the central nervous system. Int J Biol Sci. 2012;8:1254–66. https://doi.org/10.7150/ijbs.4679.
Rio J, Montalban X. Interferon-beta 1b in the treatment of multiple sclerosis. Expert Opin Pharmacother. 2005;6:2877–86. https://doi.org/10.1517/14656566.6.16.2877.
Ruuls SR, de Labie MC, Weber KS, Botman CA, Groenestein RJ, Dijkstra CD, Olsson T, van der Meide PH. The length of treatment determines whether IFN-beta prevents or aggravates experimental autoimmune encephalomyelitis in Lewis rats. J Immunol. 1996;157:5721–31.
Yu M, Nishiyama A, Trapp BD, Tuohy VK. Interferon-beta inhibits progression of relapsing-remitting experimental autoimmune encephalomyelitis. J Neuroimmunol. 1996;64:91–100.
Thorne RG, Hanson LR, Ross TM, Tung D, Frey WH 2nd. Delivery of interferon-beta to the monkey nervous system following intranasal administration. Neuroscience. 2008;152:785–97. https://doi.org/10.1016/j.neuroscience.2008.01.013.
Greig NH, Fredericks WR, Holloway HW, Soncrant TT, Rapoport SI. Delivery of human interferon-alpha to brain by transient osmotic blood-brain barrier modification in the rat. J Pharmacol Exp Ther. 1988;245:581–6.
Pan W, Banks WA, Kastin AJ. Permeability of the blood-brain and blood-spinal cord barriers to interferons. J Neuroimmunol. 1997;76:105–11.
Capuron L, Miller AH. Immune system to brain signaling: neuropsychopharmacological implications. Pharmacol Ther. 2011;130:226–38. https://doi.org/10.1016/j.pharmthera.2011.01.014.
Weinstock-Guttman B, Ransohoff RM, Kinkel RP, Rudick RA. The interferons: biological effects, mechanisms of action, and use in multiple sclerosis. Ann Neurol. 1995;37:7–15. https://doi.org/10.1002/ana.410370105.
Sultana R, Butterfield DA. Oxidative modification of brain proteins in Alzheimer’s disease: perspective on future studies based on results of redox proteomics studies. J Alzheimers Dis. 2013;33(Suppl 1):S243–51. https://doi.org/10.3233/JAD-2012-129018.
Zhao Y, Zhao B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxidative Med Cell Longev. 2013;2013:316523. https://doi.org/10.1155/2013/316523.
Tonnies E, Trushina E. Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J Alzheimers Dis. 2017;57:1105–21. https://doi.org/10.3233/JAD-161088.
Wojtunik-Kulesza KA, Oniszczuk A, Oniszczuk T, Waksmundzka-Hajnos M. The influence of common free radicals and antioxidants on development of Alzheimer’s disease. Biomed Pharmacother. 2016;78:39–49. https://doi.org/10.1016/j.biopha.2015.12.024.
Acknowledgements
Not applicable.
Funding
This study was funded by Department of Biomedicine and Clinical Neurosciences (BIONEC) of the University of Palermo.
Availability of data and materials
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
GM, MF, and PS performed the experiments in vivo and analysis of immunostaining data. GC and LC performed the experiments in vivo and animal care. CC and FP planned and performed behavioral testing. MFM performed detection of cortisol levels. DN and MDC prepared Aβ1-42 oligomers aggregates and performed analysis of cytokines and oxidative stress. MR provided the IFNβ1a and planned the treatment. GM and MF performed the statistical analysis and figures. NB, LMG, and GM conceived and planned the work and contributed in writing the text. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The experiments were carried out in accordance with the National Institute of Health Guidelines for the Care and Use of Mammals in Neuroscience and Behavioral Research (The National Academics Press, WA, USA), with the rules and principles of the European Communities Council Directive 2010/63/EU revising Directive 86/609/EEC, in accordance with the national D.L. March 4, 2014, no. 26, and were approved by the local Animal Care Committee (OPBA) of University of Palermo, Italy and Ministry of Health, Italy. The consent to participate is not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional information
Giuseppa Mudò and Monica Frinchi are co-first authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Mudò, G., Frinchi, M., Nuzzo, D. et al. Anti-inflammatory and cognitive effects of interferon-β1a (IFNβ1a) in a rat model of Alzheimer’s disease. J Neuroinflammation 16, 44 (2019). https://doi.org/10.1186/s12974-019-1417-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-019-1417-4