
Abstract
Background
Zinc-α2-glycoprotein (ZAG) is a 42-kDa protein reported as an anti-inflammatory adipocytokine. Evidences from clinical and experimental studies revealed that brain inflammation plays important roles in epileptogenesis and seizure. Interestingly, closely relationship between ZAG and many important inflammatory mediators has been proven. Our previous study identified ZAG in neurons and found that ZAG is decreased in epilepsy and interacts with TGFβ and ERK. This study aimed to investigate the role of ZAG in seizure and explore its effect on seizure-related neuroinflammation.
Methods
We overexpressed AZGP1 in the hippocampus of rats via adeno-associated virus vector injection and observed their seizure behavior and EEG after pentylenetetrazol (PTZ) kindling. The level of typical inflammation mediators including TNFα, IL-6, TGFβ, ERK, and ERK phosphorylation were determined.
Results
The overexpression of AZGP1 reduced the seizure severity, prolonged the latency of kindling, and alleviated epileptiform discharges in EEG changes induced by PTZ. Overexpression of AZGP1 also suppressed the expression of TNFα, IL-6, TGFβ, and ERK phosphorylaton in PTZ-kindled rats.
Conclusions
ZAG may inhibit TGFβ-mediated ERK phosphorylation and inhibit neuroinflammation mediated by TNFα and IL-6, suggesting ZAG may suppress seizure via inhibiting neuroinflammation. ZAG may be a potential and novel therapeutic target for epilepsy.
Similar content being viewed by others

Background
Zinc-α2-glycoprotein (ZAG) is a 42-kDa, soluble, secretory protein encoded by the AZGP1 gene located on chromosome 7q22.1 [1, 2]. The structure and amino acid sequence of ZAG are highly homologous to proteins in the major histocompatibility complex class I (MHC-I) family, which has important function in immunity [3]. In our previous study, we found that both ZAG protein and AZGP1 mRNA levels were significantly decreased in brain tissues of refractory TLE patients and pentylenetetrazol (PTZ)-kindled rats [4], but the role of ZAG in epilepsy and seizure is still unclear.
ZAG is known to be involved in many molecular pathways linked to epilepsy and seizure, and it can regulate many epilepsy- or siezure-related molecules, although its role has never been studied in epilepsy and/or seizure. AZGP1 overexpression can inhibit the mammalian target of rapamycin (mTOR) pathway activity [5], and ZAG can inhibit transforming growth factor-β (TGFβ)-mediated vimentin expression and extracellular regulated protein kinase (ERK) phosphorylation [6]. Meanwhile, mTOR, TGFβ, ERK, and phosphorylated ERK (pERK) were known to play important roles in epilepsy and seizure. The mTOR pathway regulates various cellular processes involved in growth, metabolism, structure, and cell–cell interactions of neurons and glia [7]. Inhibiting mTOR by rapamycin can suppress seizure, delay seizure development, or prevent epileptogenesis [7]. Increase of TGFβ in neurons was proven to be involved in epileptogenesis via regulating dendrite growth and synaptogenesis [8, 9]. The level of pERK in refractory epilepsy patients is significantly higher than that in controls [10], indicating an increased ERK activity in epilepsy. Inhibiting sodium glucose co transporter 2 (SGLT2) has been proven to increase the level of ZAG via activating PPARγ [11], while activating PPARγ was confirmed to increase AZGP1 mRNA [12]. Interestingly, both SGLT2 and PPARγ are known to participate in epileptogenesis, especially PPARγ was considered as a promising therapeutic target of epilepsy [13,14,15]. ZAG can also increase the level of mitochondrial uncoupling proteins (UCP) [16], and PPARγ-upregulated mitochondrial UCP2 expression can ameliorate neuronal death in the hippocampus following status epilepticus [17]. Interactions between ZAG and these epilepsy- or seizure-related molecules or pathways suggest a potential role of ZAG in epilepsy and/or seizure.
Evidences from clinical and experimental studies revealed that brain inflammation plays important roles in epileptogenesis [18]. ZAG was reported as one of the anti-inflammatory adipocytokines including adiponectin, omentin, SFRP5, vaspin, and interleukin-10 (IL-10) [19]. ZAG was reported to block transforming growth factor-β (TGFβ)-mediated extracellular regulated protein kinase (ERK) phosphorylation [7]. While TGFβ plays a significant role in inflammation, inhibition of TGFβ receptor 1 or TGFβ1 diminished TGFβ1-induced inflammation [20]. ERK pathway also plays an important role in the inflammatory response [21,22,23,24]. We have verified the interaction between ZAG and TGFβ or pERK previously [4]. Therefore, it is possible that ZAG may participate in the pathogenesis and pathophysiology of epilepsy via regulating TGFβ-mediated ERK phosphorylation. In addition, increase of various inflammation-related cytokines has been observed in epilepsy [25,26,27]. Pro-inflammatory cytokines, such as tumor-necrosis factor (TNFα) and interleukin-6 (IL-6), were increased in epilepsy patients and epilepsy models [28,29,30,31,32]. Interestingly, TNFα has been identified to reduce ZAG production [33], while the interaction between ZAG and IL-6 has not been found.
The role of ZAG in seizure and seizure-related inflammation has not yet been discussed. In this study, we injected adeno-associated virus (AAV) that overexpress AZGP1 to the hippocampus of rats and investigated the effect of AZGP1 overexpression on PTZ kindling-induced seizures in rats by behavior tests and scalp electroencephalogram. To further explore the molecular mechanism by which ZAG affects seizure and seizure-related inflammation, the effect of AZGP1 overexpression on pERK, total ERK (tERK), TGFβ, TNFα, and IL-6 were also measured in the hippocampus of PTZ-kindled rats and controls.
Methods
Experimental animals
Adult, male, 200–300 g, specific-pathogen-free Sprague–Dawley rats (Experimental Animal Center of Chongqing Medical University) were raised in a temperature and humidity-controlled room (temperature 27 °C, humidity 55–65%) with a 12-h light/12-h dark cycle (lights on 6:00 and off 18:00), and they were allowed free access to food and water. All rats were housed for 1 week before the experiment.
Adeno-associated virus (AAV) vector construction and stereotaxic injections
A DNA sequence that amplifies AZGP1 expression was incorporated into the adeno-associated virus (AAV) vector (pHBAAV-CMV-ZsGreen) containing green fluorescent protein (GFP) sequence and was named AAV–AZGP1. The same AAV vector containing only GFP was used as a control and named as AAV–GFP. Both vectors were manufactured by Hanbio Biotechnology (Shanghai, China). The final titer was 1.35 × 1012 vector genomes/ml for AAV–AZGP1 and 1.5 × 1012 vector genomes/ml for AAV–GFP.
To verify the successful transfection of AAV, 60 rats were randomly divided into 4 groups and treated as following (n = 15 for each group): (1) controls: rats received stereotaxic injection of saline; (2) GFP group: rats received AAV–GFP injection; (3) AZGP1-3W group: rats received AAV–AZGP1 injection and recovered for 3 weeks; (4) AZGP1-9W group: rats received AAV–AZGP1 injection and recovered for 9 weeks. After injection, GFP fluorescence was observed in the hippocampus of rats from GFP group, AZGP1-3W group and AZGP1-9W group (n = 5 for each group) as described below. Quantitative real-time polymerase chain reaction (qrt-PCR) (n = 5 for each group) and western blot (n = 5 for each group) were also performed in controls, GFP group, AZGP1-3W group, and AZGP1-9W group as described below.
When performing stereotaxic injection, rats were anesthetized by intraperitoneal injections of pentobarbital (60 mg/kg) and then placed in a stereotaxia frame (RWD Life Science, Shenzhen, China). After disinfection, skin of the dorsal surface of rat skull was cut apart, and two parallel holes were created using dental drill in the skull. The stereotactic coordinates of bilateral hippocampus region were 3.0 mm posterior to bregma, 2.0 mm lateral to median line of the skull, and 2.8 mm deep beneath the skull. 4 μL AAV–AZGP1 or AAV–GFP was injected into the hippocampus through a glass pipette at a speed of 0.2 μL/min. To prevent backflow of vectors, the pipette was kept in the hippocampus for an additional 5 min and then retracted steadily and slowly. For the controls, an equal volume of saline was injected in the same way. In most cases, the surgical procedure last about 45 min for each rat, and most rats recovered from anesthesia 1 h after surgery ending, subsequent injection of pentobarbital was not necessary in our experiment. Rarely, rats had respiratory failure after surgery was rescued using atropine (1 mg/kg, intraperitoneally).
PTZ kindling
After the AAV transfection and AZGP1 expression was verified successful, another 45 rats were randomly divided into three groups (n = 15 for each group): controls, AZGP1 + PTZ group and GFP + PTZ group. Rats in AZGP1 + PTZ group and GFP + PTZ group recovered for 3 weeks after AAV injection and then received intraperitoneal injection of PTZ (35 mg/kg, Sigma-Aldrich, St. Louis, USA) for 28 days daily. After each injection of PTZ, all rats were observed for 30 min in plastic cages to assess and record the seizure severity according to a modified Racine scale as follows [34]: grade 0, no response; grade 1, facial myoclonus; grade 2, head nodding; grade 3, forelimb clonus; grade 4, rearing and severe forelimb clonus; grade 5, rearing, falling, and severe forelimb clonus. Controls (n = 15) received equal amount of saline instead of AAV and PTZ. Rats that exhibited stage 4 or 5 seizures on 3 consecutive days were considered to be fully kindled. Latency was defined as the days between the first PTZ injection and fully kindling. PTZ injection was conducted between 13:00 and 16:00 in order to minimize possible complicating effects on the behavior of the animals’ circadian rhythms.
Scalp EEG recording
Scalp EEG was performed at the last time of PTZ injection. Two unipole scalp electrodes were placed on bilateral temporal skin of rats. After the place of electrodes, rats were allowed to move freely in plastic cages. EEG baseline was recorded for approximately 5 min before the injection of PTZ or saline, then EEG was recorded for at least 30 min using the nicolet vEGG system (natus, USA) and analyzed using nicoleton bms 5000 (natus, USA). The parameters of EEG are set as follows: filtering 30 Hz, paper speed 30 mm/s, and sensitivity 70 μV/mm. All remaining rats that received PTZ injection for 28 days were sacrificed for further research.
Tissue processing
Rats were deeply anesthetized by intraperitoneal injection of pentobarbital (60 mg/kg). For western blot and quantitative real-time polymerase chain reaction (qrt-PCR), rat brains were removed and stored at − 80 °C. For GFP fluorescence observation, the rat brains were removed after perfusion with saline and 4% paraformaldehyde in phosphate-buffered saline (PBS) by cardiac puncture via the left ventricle. For comparing level of ZAG protein and AZGP1 mRNA 3 and 9 weeks after AAV injection, some rat brain tissues were stored for 6 weeks at − 80 °C before western blot and qrt-PCR examination. The rest rat brain tissues were examined in 2 weeks after removal.
qrt-PCR
Total RNA was extracted from the brain tissue using RNAiso plus (Takara, Dalian, China) and was reverse transcribed into complementary deoxyribonucleic acid (cDNA) with the Applied Biosystems Veriti-Well Thermal Cycler (Thermo, Wilmington, USA) using the PrimeScript RT reagent Kit with genome DNA Eraser (Takara, Dalian, China) following the manufacturer’s instructions. Briefly, 2 μg total RNA was mixed with 4 μl Reverse Transcriptase and 0.5 μg oligo (dT) primer and incubated at 37 °C for 15 min, and the reaction was then terminated at 85 °C for 5 s. Each qrt-PCR reaction contained 2 μl cDNA, 0.8 μl forward primer, 0.8 μl reverse primer, 6.4 μl DEPC water, and 10 μl SYBR Premix ExTaq II (Takara, Dalian, China). The PCR protocol consisted of an initial denaturation step at 95 °C for 30 s, followed by 40 cycles of amplification at 95 °C for 5 s and at 60 °C for 34 s, and then terminated at 95 °C for 15 s. Melting curve analyses were also performed (65.0 to 95.0 °C, 0.5 °C increments for 5 s). The relative gene expression levels in the hippocampus of PTZ-kindled rats were calculated using the 2−ΔΔCt method [35]. The primer sequences for rat were as follows: AZGP1: forward 5′-TTCAAGCCACCGCATTTCTC-3′, reverse 5′-TCCTTCTCCCAGTCCTCCATTC-3′. GAPDH: forward 5′-ACGGTCAGGTCATCACTATCG-3′, reverse 5′-GGCATAGAGGTCTTTACGGATG-3′.
Observation of fluorescence of GFP
The fixed brain tissues were successively immersed in 20 and 30% sucrose solution for 48 h (24 h per solution) and sliced into 10-μm-thick frozen sections. Finally, the sections were mounted using 80% glycerol. Images were collected using laser scanning confocal microscopy (Nikon 1R, Japan).
Western blot
Brain tissue was homogenized in RIPA lysis buffer (Beyotime, Haimen, China) containing proteinase inhibitor mixture and phosphorylase inhibitor mixture and centrifuged at 12000 rpm, 4 °C for 25 min. The protein concentrations in the supernatant were determined using a BCA Protein Assay Kit (Beyotime, Haimen, China). The extracted total proteins were mixed with 5× sodium dodecylsulfate (SDS) loading buffer and boiled for 5 min. Equal amounts of total protein (80 μg/lane) were separated by SDS-polyacrylamide gel electrophoresis (PAGE) and then transferred onto polyvinylidene fluoride (PVDF) membranes (Immobilon, Merck Millipore, Darmstadt, Germany). The membranes were blocked with 5% BSA at room temperature for 2 h and then incubated with the anti-ZAG antibody (1:400, Santa Cruz, USA), anti-TNFα antibody (1:500, Bosterbio, USA), anti-IL6 antibody (1:500, Bosterbio, USA), anti-pERK antibody (1:2000, Cell Signaling Technology, Danvers, USA), anti-tERK antibody (1:2000, Proteintech, Wuhan, China), anti-TGFβ antibody (1:1000, Proteintech, Wuhan, China), or anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody (1:3000, Proteintech, Wuhan, China) at 4 °C overnight. After washed with tris-buffered saline with Tween-20 (TBST), the membranes were incubated with the horseradish peroxidase-conjugated rabbit anti-mouse antibody (1:3000, Abcam, Cambridge, UK) or mouse anti-rabbit antibody (1:3000, Abcam, Cambridge, UK) at room temperature for 1 h and washed again. Immunoreactivity was visualized using chemiluminescence substrate kit (Beyotime, Haimen, China) and quantified by densitometric scanning with the Fusion-FX7 system (Vilber Lourmat, Collégien, France). The mean optic density (OD) was normalized by GAPDH.
Statistical analysis
The results were expressed as mean ± standard deviation (SD). SPSS 20.0 (IBM, Armonk, USA) and GraphPad prism 6.01 (GraphPad software, La Jolla, USA) were used for data analysis and graph drawing. Mann–Whitney U test was used to compare the differences of Racine’s scores between AAV–AZGP1 group and AAV–GFP group. Student’s t test was used to compare the differences of latency between AAV–AZGP1 group and AAV–GFP group. One-way ANOVA with Bonferroni or Dunnett’s T3 post hoc analysis was used to compare the level of proteins between the three groups. p < 0.05 (two tailed) was regarded statistically significant.
Results
The transfection of AAV and the overexpression of AZGP1
To confirm the efficiency and stability of AAV-induced AZGP1 expression, we detected GFP distribution and measured the level of AZGP1 mRNA and ZAG protein in rat hippocampus 3 weeks and 9 weeks after AAV injection. The GFP-positive cells in the CA3 region of hippocampus were visualized (Fig. 1a). The level of AZGP1 mRNA (Fig. 1b) and ZAG protein (Fig. 1c, d) was significantly increased on 3 and 9 weeks in AZGP1 group compared to GFP group. There was no difference of AZGP1 mRNA (Fig. 1b) and ZAG protein (Fig. 1c, d) levels between 3 and 9 weeks in AZGP1 group, suggesting a steady expression of AZGP1. In addition, no significant difference of AZGP1 mRNA (Fig. 1b) and ZAG protein (Fig. 1c, d) level was found between GFP group and controls, suggesting no effect of AAV vectors on AZGP1 mRNA and ZAG protein expression.

Expression of GFP and AZGP1 after injection of AAV vectors. a Fluorescent images showing GFP expression in the hippocampus of rats 3 and 9 weeks after AAV injection. The scale bar = 500 μm. b qrt-PCR showed increased AZGP1 mRNA level in the hippocampus of rats in AAV–AZGP1 group compared to AAV–GFP group 3 and 9 weeks after AAV injection. c Western blots showed increased ZAG protein level in the hippocampus of rats in AAV–AZGP1 group compared to AAV–GFP group 3 and 9 weeks after AAV injection. Optic density was normalized by GAPDH. n = 5 for each group, *p < 0.05
Overexpression of AZGP1 suppresses seizures in PTZ-treated rats
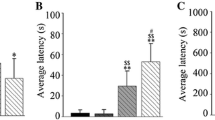
ZAG protein level in rats of AZGP1 + PTZ group (OD 1.496 ± 0.086) was significantly higher than in the GFP + PTZ group (OD 0.778 ± 0.080) and controls (OD 1.104 ± 0.074) (n = 5, p = 0.0001, degrees of freedom (df) = 12, one-way ANOVA) (Fig. 2a). Rats in AZGP1 + PTZ group had significantly milder seizure severity (Fig. 2b) before being fully kindled and longer latency (Fig. 2c) compared to rats in GFP + PTZ group (latency: AZGP1 + PTZ 16.07 ± 2.786, n = 14 vs. GFP + PTZ 20.00 ± 2.530, n = 11, p = 0.0014, df = 23, Student’s t test). In addition, scalp EEG results showed significant decrease in the frequency and amplitude of seizure spike wave in AZGP1 + PTZ groups compared to GFP + PTZ group (Fig. 3).

Overexpression of AZGP1 suppressed seizure in PTZ-treated rats. a ZAG protein level in rats of AZGP1 + PTZ group was significantly increased compared to GFP + PTZ group and controls. b Rats in AZGP1 + PTZ group had significantly alleviated seizure severity during 12th to 23rd day of PTZ injection compared to GFP + PTZ group, while after 24th day of PTZ injection, there is no difference in seizure severity. c Overexpression of AZGP1 significantly prolonged the latency of PTZ kindling. n = 15 for each group, *p < 0.05

Scalp electroencephalogram (EEG) changes in AZGP1 + PTZ group and GFP + PTZ group after PTZ kindling. a Typical EEG record of seizure. b Representative EEG record among the three groups of rats. Rats in AZGP1 + PTZ group had significantly decreased frequency and amplitude of epileptiform spike waves in EEG compared to GFP + PTZ group
Overexpression of AZGP1 decreased the level of TGFβ and pERK in PTZ-kindled rats without affecting total ERK level
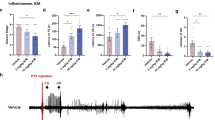
To explore the possibility that ZAG affects seizure via TGFβ-mediated ERK signaling pathway, we measured the level of pERK, total ERK, and TGFβ in the hippocampus of rats in the three groups using western blot. The level of pERK (Fig. 4a, c), pERK/total ERK ratio (Fig. 4c), and TGFβ (Fig. 4d, e) was significantly increased in both AZGP1 + PTZ and GFP + PTZ groups compared to controls, while rats in AZGP1 + PTZ group had decreased pERK (Fig. 4a, c), pERK/total ERK ratio (Fig. 4c), and TGFβ (Fig. 4d, e) compared to rats in GFP + PTZ group (pERK: control 0.674 ± 0.045, GFP + PTZ 1.344 ± 0.071, AZGP1 + PTZ 0.787 ± 0.070, p = 0.0001, df = 12, one-way ANOVA); (pERK/total ERK ratio: control 0.410 ± 0.026, GFP + PTZ 0.840 ± 0.073, AZGP1 + PTZ 0.482 ± 0.040, p = 0.0001, df = 12, one-way ANOVA); (TGFβ: control 0.490 ± 0.021, GFP + PTZ 0.862 ± 0.070, AZGP1 + PTZ 0.660 ± 0.072, p = 0.001, df = 12, one-way ANOVA), (n = 5 for each group). In addition, there was no significant difference in total ERK level between the three groups (Fig. 4b) (control 1.642 ± 0.123, GFP + PTZ 1.614 ± 0.183, AZGP1 + PTZ 1.636 ± 0.133, p = 1.000, df = 12, one-way ANOVA).

Overexpression of AZGP1 decreased the level of TGFβ and pERK in PTZ-kindled rats without affecting total ERK level. a, b Representative western blot bands of p-ERK and t-ERK in hippocampal tissues of rats. c Western blot showed GFP + PTZ group had significantly increased level of p-ERK and p-ERK/t-ERK ratio in hippocampal tissues compared to controls, and rats in AZGP1 + PTZ group had significantly decreased the level of pERK and pERK/tERK ratio compared to GFP + PTZ group. The level of tERK in hippocampal tissues is of no significant difference among the three groups. d Representative western blot bands of TGFβ in hippocampal tissues of rats. e Western blot showed increased level of TGFβ in the hippocampal tissues of rats in GFP + PTZ group compared to controls, and rats in AZGP1 + PTZ group had significantly decreased the level of TGFβ compared to GFP + PTZ group. Optic density was normalized by GAPDH, n = 5 for each group, *p < 0.05
Overexpression of AZGP1 suppressed the increase of TNFα and IL-6 in PTZ-kindled rats
To assess the inflammatory state of the hippocampus, two typical pro-inflammatory cytokines, TNFα and IL-6, were measured using western blot in the hippocampus of AZGP1 + PTZ and GFP + PTZ group rats and controls. Hippocampal TNFα (Fig. 5a) level was increased in AZGP1 + PTZ and GFP + PTZ groups compared to controls, while in AZGP1 + PTZ group, TNFα (Fig. 5a) was significantly decreased compared to GFP + PTZ group (TNFα: control 0.415 ± 0.055, GFP + PTZ 0.722 ± 0.032, AZGP1 + PTZ 0.554 ± 0.029, p = 0.001, df = 12, one-way ANOVA). Hippocampal IL6 level was significantly decreased in AZGP1 + PTZ group compared to GFP + PTZ group (IL6: AZGP1 + PTZ 0.472 ± 0.068, GFP + PTZ 0.692 ± 0.081, p = 0.001, df = 12, one-way ANOVA) while there is no statistical significance between AZGP1 + PTZ group and control (IL6: AZGP1 + PTZ 0.472 ± 0.068, control 0.413 ± 0.058, p = 0.609, df = 12, one-way ANOVA) (Fig. 5b).

Overexpression of AZGP1 suppressed the increase of TNFα and IL-6 in PTZ-kindled rats. a Representative western blot bands and expression level of TNFα in the hippocampal tissues of rats. Western blot showed increased level of TNFα in the hippocampal tissues of rats in GFP + PTZ group compared to controls, and rats in AZGP1 + PTZ group had significantly decreased the level of TNFα compared to GFP + PTZ group. b Representative western blot bands and expression level of IL-6 in the hippocampus of rats. Western blot showed increased level of IL-6 in the hippocampal tissues of rats in GFP + PTZ group compared to controls, and rats in AZGP1 + PTZ group had significantly decreased the level of IL-6 compared to GFP + PTZ group. Optic density normalized by GAPDH, n = 5 for each group, *p < 0.05
Discussion
In our previous study, we had found that the level of AZGP1 mRNA and ZAG protein was significantly decreased in the neocortex of refractory epilepsy patients as well as in the hippocampus and neocortex of PTZ-kindled rats compared to controls [4]. However, the specific role of ZAG in epilepsy and/or seizure remains unclear.
In this study, behavior tests showed that overexpression of AZGP1 in the hippocampus prolonged the latency of PTZ kindling and alleviated the seizure severity in PTZ-treated rats. Similarly, in scalp EEG recording, decreased frequency and amplitude of spike wave were also identified in AAV–AZGP1 group. These results indicate a protective role of ZAG against seizure. Interestingly, we found that the seizure severity score of AAV–AZGP1 group was significantly lower than AAV–GFP group during 12–23 days of PTZ treatment, while after the 24th day of PTZ treatment, the seizure severity was of no difference between the two groups, indicating that overexpression of ZAG can delay but not fully prevent the kindling. Moreover, on the 28th day of PTZ treatment, although the seizure severity of rats in both AAV–AZGP1 group and AAV–GFP group were similar, the scalp EEG recording demonstrated decreased frequency and amplitude of spike waves in AAV–AZGP1 group compared to AAV–GFP group. This result indicates that although rats in the two groups had similar seizure severity in behavior, their epileptiform discharge in brain is different. It is possible that overexpression of ZAG may have protective effect on seizure even if the rats are fully kindled. Although ZAG only delayed PTZ kindling in this study, it also alleviated the epileptiform discharges after the rats were fully kindled. It is possible that ZAG may prevent seizure in the early stage of kindling and has protective effect in the late stage of kindling, indicating a possible role of ZAG in different stage of epileptogenesis or seizure. This is the first study on the role of ZAG in seizure; further study is needed to explore its effect on seizure and mechanism.
ZAG was reported as an anti-inflammatory adipocytokine [24]. Clinical and experimental evidences have revealed that brain inflammation plays an important role in epileptogenesis [23]. The most extensively studied prototypical inflammatory cytokines in the central nervous system are TNFα and IL-6 [36,37,38]. The enhanced production of pro-inflammatory cytokines, including TNFα and IL-6, were known to play pro-epileptic roles in the brain [39]. The dynamic modulation of inflammatory processes has potential to be a novel therapeutic strategy for pharmacologic treatment to control seizures, delay disease progression, or retard epileptogenesis [40]. Moreover, overexpression of cytokines such as TNFα or IL-6 results in age-dependent increase of seizure susceptibility and spontaneous seizures [41, 42]. In our study, overexpression of AZGP1 attenuated the increase of TNFα and IL6 induced by PTZ kindling. Interestingly, TNFα has been identified to reduce ZAG production [33]. This is suggesting that there may be a circuit or feedback regulation mechanism between ZAG and TNFα. Therefore, ZAG may prevent seizure and protect the brain via alleviating neuroinflammation. This is the first time that the relationship between ZAG and IL-6 was identified.
We had previously verified the interaction between ZAG and TGFβ or p-ERK in the hippocampus of rats [4]. In this study, we found that overexpression of ZAG could decrease the level of TGFβ and ERK phosphorylation. Human recombinant ZAG was found to specifically block TGFβ-mediated ERK phosphorylation [6]. And TGFβ and p-ERK/ERK ratio were upregulated in patients with refractory epilepsy [8, 10]. TGFβ can promote epileptogenesis via upregulating IL-6 [43] and inhibiting TGFβ by losartan can suppress epileptogenesis [44]. ERK activation induced by its phosphorylation is known to cause seizure by activating N-methy-d-aspartate (NMDA) receptors [45, 46]. In addition, TGFβ and ERK are known as inflammatory mediators [20,21,22,23,24]. Thus, it is possible that ZAG may prevent seizure via inhibiting TGFβ-mediated ERK signaling pathway and alleviate inflammation induced by seizure. As the effect of ZAG on TGFβ and ERK has not been investigated in epilepsy and/or seizure before, further study is needed to clarify the existence of ZAG–TGFβ–ERK pathway and its specific role in seizure and epilepsy.
Many studies showed that various inflammatory cytokines are associated with seizure susceptibility [47,48,49,50]. Overexpressing IL-6 or TNFα can decrease seizure threshold and exacerbate seizure-induced neuronal loss [51, 52]. Inflammatory signaling is also known to worsen the loss of GABAergic neurons in the hippocampus and thus resulted in an increased susceptibility for seizure [53]. TNFα have also been associated with the regulation of seizure duration in amygdala-kindled rats [54]. In our study, overexpression of AZGP1 in the hippocampus prolonged the latency of PTZ kindling and attenuated PTZ kindling-induced increase of TNFα and IL-6. This result suggests that overexpression of AZGP1 may decrease seizure susceptibility in PTZ-treated rats, and this effect of overexpression of AZGP1 may possibly be attributed to its inhibition of neuroinflammation. This is the first study relating ZAG to neuroinflammation in seizure; further study is needed to explore the relationship between ZAG and neuroinflammation in seizure. This study is a preliminary discuss on the role of ZAG in seizure, but more specific mechanism, such as ZAG–TGFβ–ERK pathway and the effect of ZAG on NMDA/AMPA/GABA receptors, still needs to be further researched. Furthermore, researches on the role of ZAG deficiency in epilepsy and/or seizure are also needed.
Conclusion
Our study found that overexpression of AZGP1 delayed PTZ kindling, alleviated seizure and epileptiform discharges, inhibited TGFβ-mediated ERK phosphorylation, and decreased TNFα and IL-6 in PTZ-treated rats. Our study indicated that ZAG may suppress seizure via inhibiting neuroinflammation. As an anti-inflammatory cytokine, ZAG may be a novel target for research and clinical treatment of seizure and possibly for epilepsy.
Abbreviations
- AAV:
-
Adeno-associated virus
- cDNA:
-
Complementary deoxyribonucleic acid
- EEG:
-
Electroencephalography
- ERK:
-
Extracellular regulated protein kinase
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- GFP:
-
Green fluorescent protein
- IL-10:
-
Interleukin-10
- IL-6:
-
Interleukin-6
- MHC-I:
-
Major histocompatibility complex class I
- mTOR:
-
Mammalian target of rapamycin
- NMDA:
-
N-methy-d-aspartate
- OD:
-
Optic density
- PAGE:
-
Polyacrylamide gel electrophoresis
- PBS:
-
Paraformaldehyde in phosphate-buffered saline
- pERK:
-
Phosphorylated extracellular regulated protein kinase
- PPARγ:
-
Peroxisome proliferator-activated receptor gamma
- PTZ:
-
Pentylenetetrazol
- PVDF:
-
Polyvinylidene fluoride
- qrt-PCR:
-
Quantitative real-time polymerase chain reaction
- SD:
-
Standard deviation
- SDS:
-
Sodium dodecylsulfate
- SGLT2:
-
Sodium–glucose cotransporter 2
- TBST:
-
Tris-buffered saline with Tween-20
- TGFβ:
-
Transforming growth factor-β
- TNFα:
-
Tumor-necrosis factor
- UCP:
-
Uncoupling proteins
- ZAG:
-
Zinc-α2-glycoprotein
References
Burgi W, Schmid K. Preparation and properties of Zn-alpha 2-glycoprotein of normal human plasma. J Biol Chem. 1961;236:1066–74.
Tada T, Ohkubo I, Niwa M, Sasaki M, Tateyama H, Eimoto T. Immunohistochemical localization of Zn-alpha 2-glycoprotein in normal human tissues. J Histochem Cytochem. 1991; https://doi.org/10.1177/39.9.1918940.
Hassan MI, Waheed A, Yadav S, Singh TP, Ahmad F. Zinc alpha 2-glycoprotein: a multidisciplinary protein. Mol Cancer Res. 2008; https://doi.org/10.1158/1541-7786.MCR-07-2195.
Liu Y, Wang T, Liu X, Wei X, Xu T, Yin M, Ding X, Mo L, Chen L. Neuronal zinc-α2-glycoprotein is decreased in temporal lobe epilepsy in patients and rats. Neuroscience. 2017; https://doi.org/10.1016/j.neuroscience.2017.05.043.
Chang L, Tian X, Lu Y, Jia M, Wu P, Huang P. Alpha-2-glycoprotein 1(AZGP1) regulates biological behaviors of LoVo cells by down-regulating mTOR signaling pathway and endogenous fatty acid synthesis. PLoS One. 2014;9(6):e99254.
Kong B, Michalski CW, Hong X, Valkovskaya N, Rieder S, Abiatari I, Streit S, Erkan M, Esposito I, Friess H, Kleeff J. AZGP1 is a tumor suppressor in pancreatic cancer inducing mesenchymal-to-epithelial transdifferentiation by inhibiting TGF-β-mediated ERK signaling. Oncogene. 2010; https://doi.org/10.1038/onc.2010.258.
Ostendorf AP, Wong M. mTOR inhibition in epilepsy: rationale and clinical perspectives. CNS Drugs. 2015;29(2):91–9.
Cacheaux LP, Ivens S, David Y, Lakhter AJ, Bar-Klein G, Shapira M, Heinemann U, Friedman A, Kaufer D. Transcriptome profiling reveals TGF-beta signaling involvement in epileptogenesis. J Neurosci. 2009; https://doi.org/10.1523/JNEUROSCI.0430-09.2009.
Yu CY, Gui W, He HY, Wang XS, Zuo J, Huang L, Zhou N, Wang K, Wang Y. Neuronal and astroglial TGFβ-Smad3 signaling pathways differentially regulate dendrite growth and synaptogenesis. NeuroMolecular Med. 2014;16(2):457–72.
Xi ZQ, Wang XF, He RQ, Li MW, Liu XZ, Wang LY, Zhu X, Xiao F, Sun JJ, Li JM, Gong Y, Guan LF. Extracellular signal-regulated protein kinase in human intractable epilepsy. Eur J Neurol. 2007; https://doi.org/10.1111/j.1468-1331.2007.01777.x.
Liao X, Wang X, Li H, Li L, Zhang G, Yang M, Yuan L, Liu H, Yang G, Gao L. Sodium-glucose cotransporter 2 (SGLT2) inhibitor increases circulating zinc-α2-glycoprotein levels in patients with type 2 diabetes. Sci Rep. 2016; https://doi.org/10.1038/srep32887.
Bao Y, Bing C, Hunter L, Jenkins JR, Wabitsch M, Trayhurn P. Zinc-alpha2-glycoprotein, a lipid mobilizing factor, is expressed and secreted by human (SGBS) adipocytes. FEBS Lett. 2005; https://doi.org/10.1016/j.febslet.2004.11.042.
Melo IS, Santos YM, Costa MA, Pacheco AL, Silva NK, Cardoso-Sousa L, Pereira UP, Goulart LR, Garcia-Cairasco N, Duzzioni M, Gitaí DL, Tilelli CQ, Sabino-Silva R, Castro OW. Inhibition of sodium glucose cotransporters following status epilepticus induced by intrahippocampal pilocarpine affects neurodegeneration process in hippocampus. Epilepsy Behav. 2016;61:258–68.
Auvin S. Fatty acid oxidation and epilepsy. Epilepsy Res. 2012;100(3):224–8.
Taha AY, Burnham WM, Auvin S. Polyunsaturated fatty acids and epilepsy. Epilepsia. 2010;51(8):1348–58.
Sanders PM, Tisdale MJ. Effect of zinc-alpha2-glycoprotein (ZAG) on expression of uncoupling proteins in skeletal muscle and adipose tissue. Cancer Lett. 2004;212(1):71–81.
Chuang YC, Lin TK, Huang HY, Chang WN, Liou CW, Chen SD, Chang AY, Chan SH. Peroxisome proliferator-activated receptors γ/mitochondrial uncoupling protein 2 signaling protects against seizure-induced neuronal cell death in the hippocampus following experimental status epilepticus. J Neuroinflammation. 2012;9:184.
Vezzani A, Friedman A, Dingledine RJ. The role of inflammation in epileptogenesis. Neuropharmacology. 2013; https://doi.org/10.1016/j.neuropharm.2012.04.004.
Leal Vde O, Mafra D. Adipokines in obesity. Clin Chim Acta. 2013; https://doi.org/10.1016/j.cca.2013.02.003.
Patel RK, Prasad N, Kuwar R, Haldar D, Abdul-Muneer PM. Transforming growthfactor-beta1 signaling regulates neuroinflammation and apoptosis in mild traumatic brain injury. Brain Behav Immun. 2017; https://doi.org/10.1016/j.bbi.2017.04.012.
Xu G, Feng L, Song P, Xu F, Li A, Wang Y, Shen Y, Wu X, Luo Q, Wu X, Sun Y, Wu X, Xu Q. Isomeranzin suppresses inflammation by inhibiting M1 macrophage polarization through the NF-κB and ERK pathway. Int Immunopharmacol. 2016; https://doi.org/10.1016/j.intimp.2016.05.027.
Huang Y, Zeng J, Chen G, Xie X, Guo W, Tian W. Periodontitis contributes to adipose tissue inflammation through the NF-<kappa>B, JNK and ERK pathways to promote insulin resistance in a rat model. Microbes Infect. 2016; https://doi.org/10.1016/j.micinf.2016.08.002.
Li C, Yan Y, Shi Q, Kong Y, Gao L, Bao H, Li Y. Recuperating lung decoction attenuates inflammation and oxidation in cigarette smoke-induced COPD in rats via activation of ERK and Nrf2 pathways. Cell Biochem Funct. 2017; https://doi.org/10.1002/cbf.3273.
Zeng KW, Yu Q, Liao LX, Song FJ, Lv HN, Jiang Y, Tu PF. Anti-neuroinflammatory effect of MC13, a novel coumarin compound from condiment Murraya, through inhibiting lipopolysaccharide-induced TRAF6-TAK1-NF-κB, P38/ERK MAPKS and Jak2-Stat1/Stat3 pathways. J Cell Biochem. 2015; https://doi.org/10.1002/jcb.25084.
Turrin NP, Rivest S. Innate immune reaction in response to seizures: implications for the neuropathology associated with epilepsy. Neurobiol Dis. 2004;16(2):321–34.
Oprica M, Eriksson C, Schultzberg M. Inflammatory mechanisms associated with brain damage induced by kainic acid with special reference to the interleukin-1 system. J Cell Mol Med. 2003;7(2):127–40.
Vezzani A, Conti N, De Luigi A, Ravizza T, Moneta D, Marchesi F, et al. Interleukin-l beta immunoreactivity and microglia are enhanced in the rat hippocampus by focal kainate application: functional evidence for enhancement of electrographic seizures. J Neurosci. 1999;19(12):5054–65.
Tombini M, Squitti R, Cacciapaglia F, Ventriglia M, Assenza G, Benvenga A, Pellegrino G, Campana C, Assenza F, Siotto M, Pacifici L, Afeltra A, Rossini PM. Inflammation and iron metabolism in adult patients with epilepsy: does a link exist? Epilepsy Res. 2013; https://doi.org/10.1016/j.eplepsyres.2013.09.010.
Lehtimäki KA, Keränen T, Palmio J, Mäkinen R, Hurme M, Honkaniemi J, Peltola J. Increased plasma levels of cytokines after seizures in localization-related epilepsy. Acta Neurol Scand. 2007; https://doi.org/10.1111/j.1600-0404.2007.00882.x.
Peltola J, Palmio J, Korhonen L, Suhonen J, Miettinen A, Hurme M, et al. Interleukin-6 and interleukin-1 receptor antagonist in cerebrospinal fluid from patients with recent tonic-clonic seizures. Epilepsy Res. 2000;41(3):205–11.
Virta M, Hurme M, Helminen M. Increased plasma levels of pro- and anti-inflammatory cytokines in patients with febrile seizures. Epilepsia. 2002;43(8):920–3.
Bauer S, Cepok S, Todorova-Rudolph A, Nowak M, Köller M, Lorenz R, Oertel WH, Rosenow F, Hemmer B, Hamer HM. Etiology and site of temporal lobe epilepsy influence postictal cytokine release. Epilepsy Res. 2009; https://doi.org/10.1016/j.eplepsyres.2009.05.009.
Mracek T, Gao D, Tzanavari T, Bao Y, Xiao X, Stocker C, Trayhurn P, Bing C. Downregulation of zinc-α2-glycoprotein in adipose tissue and liver of obese ob/ob mice and by tumour necrosis factor-alpha in adipocytes. J Endocrinol. 2010; https://doi.org/10.1677/JOE-09-0299.
Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32(3):281–94.
Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–8.
Bartfai T, Sanchez-Alavez M, Andell-Jonsson S, Schultzberg M, Vezzani A, Danielsson E, Conti B. Interleukin-1 system in CNS stress: seizures, fever, and neurotrauma. Ann N Y Acad Sci. 2007; https://doi.org/10.1196/annals.1391.022.
Allan SM, Rothwell NJ. Cytokines and acute neurodegeneration. Nat Rev Neurosci. 2001; https://doi.org/10.1038/35094583.
Bartfai T, Schultzberg M. Cytokines in neuronal cell types. Neurochem Int. 1993;22(5):435–44.
Ho YH, Lin YT, Wu CW, Chao YM, Chang AY, Chan JY. Peripheral inflammation increases seizure susceptibility via the induction of neuroinflammation and oxidative stress in the hippocampus. J Biomed Sci. 2015; https://doi.org/10.1186/s12929-015-0157-8.
Yang T, Zhou D, Stefan H. Why mesial temporal lobe epilepsy with hippocampal sclerosis is progressive: uncontrolled inflammation drives disease progression? J Neurol Sci. 2010; https://doi.org/10.1016/j.jns.2010.06.002.
Akassoglou K, Probert L, Kontogeorgos G, Kollias G. Astrocyte-specific but not neuron-specific transmembrane TNF triggers inflammation and degeneration in the central nervous system of transgenic mice. J Immunol. 1997;158(1):438–45.
Stalder AK, Carson MJ, Pagenstecher A, Asensio VC, Kincaid C, Benedict M, Powell HC, Masliah E, Campbell IL. Late-onset chronic inflammatory encephalopathy in immune-competent and severe combined immune-deficient (SCID) mice with astrocyte-targeted expression of tumor necrosis factor. Am J Pathol. 1998; https://doi.org/10.1016/S0002-9440(10)65620-9.
Levy N, Milikovsky DZ, Baranauskas G, Vinogradov E, David Y, Ketzef M, Abutbul S, Weissberg I, Kamintsky L, Fleidervish I, Friedman A, Monsonego A. Differential TGF-β signaling in glial subsets underlies IL-6-mediated epileptogenesis in mice. J Immunol. 2015; https://doi.org/10.4049/jimmunol.1401446.
Bar-Klein G, Cacheaux LP, Kamintsky L, Prager O, Weissberg I, Schoknecht K, Cheng P, Kim SY, Wood L, Heinemann U, Kaufer D, Friedman A. Losartan prevents acquired epilepsy via TGF-β signaling suppression. Ann Neurol. 2014; https://doi.org/10.1002/ana.24147.
Nateri AS, Raivich G, Gebhardt C, Da Costa C, Naumann H, Vreugdenhil M, Makwana M, Brandner S, Adams RH, Jefferys JG, Kann O, Behrens AERK. Activation causes epilepsy by stimulating NMDA receptor activity. EMBO J. 2007;26(23):4891–901.
Itoh T, Kaibuchi K, Masuda T, Yamamoto T, Matsuura Y, Maeda A, Shimizu K, Takai Y. A protein factor for ras p21-dependent activation of mitogen-activated protein (MAP) kinase through MAP kinase kinase. Proc Natl Acad Sci U S A. 1993;90(3):975–9.
Vezzani A. Anti-inflammatory drugs in epilepsy: does it impact epileptogenesis? Expert Opin Drug Saf. 2015;14(4):583–92.
Vezzani A, Friedman A. Brain inflammation as a biomarker in epilepsy. Biomark Med. 2011;5(5):607–14.
Frigerio F, Flynn C, Han Y, Lyman K, Lugo JN, Ravizza T, Ghestem A, Pitsch J, Becker A, Anderson AE, Vezzani A, Chetkovich D, Bernard C. Neuroinflammation alters integrative properties of rat hippocampal pyramidal cells. Mol Neurobiol. https://doi.org/10.1007/s12035-018-0915-1.
Rao RS, Prakash A, Medhi B. Role of different cytokines and seizure susceptibility: a new dimension towards epilepsy research. Indian J Exp Biol. 2009;47(8):625–34.
Cunningham AJ, Murray CA, O’Neill LA, Lynch MA, O’Connor JJ. Interleukin-1 beta (IL-1 beta) and tumour necrosis factor (TNF) inhibit long-term potentiation in the rat dentate gyrus in vitro. Neurosci Lett. 1996;203(1):17–20.
Probert L, Akassoglou K, Pasparakis M, Kontogeorgos G, Kollias G. Spontaneous inflammatory demyelinating disease in transgenic mice showing central nervous system-specific expression of tumor necrosis factor alpha. Proc Natl Acad Sci U S A. 1995;92(24):11294–8.
Samland H, Huitron-Resendiz S, Masliah E, Criado J, Henriksen SJ, Campbell IL. Profound increase in sensitivity to glutamatergic—but not cholinergic agonist-induced seizures in transgenic mice with astrocyte production of IL-6. J Neurosci Res. 2003;73(2):176–87.
Shandra AA, Godlevsky LS, Vastyanov RS, Oleinik AA, Konovalenko VL, Rapoport EN, et al. The role of TNF-alpha in amygdala kindled rats. Neurosci Res. 2002;42(2):147–53.
Acknowledgements
Not applicable.
Funding
This work is funded by the National Natural Science Foundation of China (81771391) and the Chongqing Municipal Public Health Bureau, Chongqing People’s Municipal Government (2015ZDXM011). This study is also supported by the Program for Innovative Research Team of Chongqing Kuanren Hospital.
Availability of data and materials
Data of this study could be accessed freely under reasonable request.
Author information
Authors and Affiliations
Contributions
LC, XT, and XL designed the research. YL, TW, YW, TX, XY, XW, XD, LM, and MY performed the research. YL and TW analyzed the data. YL and XL wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
This study protocol was approved by the Ethics Committee of The Second Affiliated Hospital of Chongqing Medical University (2017-009). Animal experiment was performed in accordance with the guidelines of Chongqing Medical University and Guide for the care and use of laboratory animals (National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals, 2011). All efforts were made to minimize animal suffering and to reduce the number of animals used.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Liu, Y., Wang, T., Liu, X. et al. Overexpression of zinc-α2-glycoprotein suppressed seizures and seizure-related neuroflammation in pentylenetetrazol-kindled rats. J Neuroinflammation 15, 92 (2018). https://doi.org/10.1186/s12974-018-1132-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-018-1132-6