
Abstract
Background
Mevalonate kinase deficiency (MKD) is a rare autoinflammatory condition caused by biallelic loss-of-function (LOF) mutations in mevalonate kinase (MVK) gene encoding the enzyme mevalonate kinase. Patients with MKD display a variety of non-specific clinical manifestations, which can lead to diagnostic delay. We report the case of a child presenting with vasculitis that was found by genetic testing to be caused by MKD, and now add this autoinflammatory disease to the ever-expanding list of causes of monogenic vasculitides.
Case presentation
A 2-year-old male presented with an acute 7-day history of high-grade fever, abdominal pain, diarrhoea, rectal bleeding and extensive purpuric and necrotic lesions, predominantly affecting the lower limbs. He had been suffering from recurrent episodes of fever from early in infancy, associated with maculopapular/petechial rashes triggered by intercurrent infection, and after vaccines. Extensive infection screen was negative. Skin biopsy revealed small vessel vasculitis. Visceral digital subtraction arteriography was normal. With a diagnosis of severe idiopathic cutaneous vasculitis, he was treated with corticosteroids and mycophenolate mofetil. Despite that his acute phase reactants remained elevated, fever persisted and the vasculitic lesions progressed. Next-generation sequencing revealed compound heterozygous mutation in MVK c.928G > A (p.V310M) and c.1129G > A (p.V377I) while reduced mevalonate enzyme activity was confirmed suggesting a diagnosis of MKD as a cause of the severe vasculitis. Prompt targeted treatment with IL-1 blockade was initiated preventing escalation to more toxic vasculitis therapies and reducing unnecessary exposure to cytotoxic treatment.
Conclusions
Our report highlights the broad clinical phenotype of MKD that includes severe cutaneous vasculitis and emphasizes the need to consider early genetic screening for young children presenting with vasculitis to exclude a monogenic vasculitis which may be amenable to targeted treatment.
Similar content being viewed by others


Background
Mevalonate kinase deficiency (MKD) is an inherited autoinflammatory condition caused by biallelic loss-of-function (LOF) mutations in mevalonate kinase (MVK) encoding the enzyme mevalonate kinase [1,2,3]. MKD is now viewed as a phenotypic continuum based on the degree of enzyme deficiency, with mevalonic aciduria (MA) being the most severe phenotype and hyperimmunoglobulinemia D with periodic fever syndrome (HIDS) on the mild end of the spectrum [4]. HIDS is often characterized by lifelong recurrent autoinflammatory episodes, typically lasting anywhere from 3 to 7 days in duration [5]. These episodes are frequently accompanied by various cutaneous manifestations, arthritis, abdominal pain, vomiting, splenomegaly, and lymphadenopathy, with elevated markers of inflammation [1, 5]. Conversely, MA is the more severe phenotype with varying neurological abnormalities such as psychomotor retardation, ataxia, failure to thrive, cataracts, growth retardation and dysmorphic features [6]. The clinical heterogenicity of MKD is therefore well recognised. In addition, patients may often present with a variety of non-specific clinical manifestations mimicking other inflammatory or infectious disorders which can lead to diagnostic delay [7, 8]. We report the case of a child presenting with vasculitis that was found by genetic testing to be caused by MKD thus highlighting the expanding spectrum of phenotypes associated with MKD and now adding this autoinflammatory disease to the ever-expanding list of causes of monogenic vasculitides [9,10,11].
Case presentation
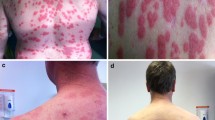
A 2-year-old male of non-consanguineous mixed White/Asian background was referred to us with an acute 7-day history of high-grade fever of up to 40 °C, abdominal pain, diarrhoea, rectal bleeding and extensive purpuric and necrotic lesions, predominantly affecting the lower limbs (Fig. 1A). There was no organomegaly or arthritis noted on clinical examination. There were no concerns about his development and his weight and height were along the 9th centile for his age. Extensive and exhaustive screening for infectious triggers was negative including the following: PCRs for meningococcus, adenovirus, cytomegalovirus, Ebstein-B-virus, mycoplasma, 16S (bacterial), 18S (fungal), Herpes Simplex Virus, parechovirus, parvovirus B19, enterovirus; stool viral screen and NPA respiratory viruses screen; mycoplasma antibodies; anti-streptococcal antibodies; blood cultures and urine cultures; quantiferon test; hepatitis A, B and C screening.

A. Palpable purpura mainly affecting the lower limbs in a 2-year-old boy with Mevalonate Kinase Deficiency (MKD). B. Skin biopsy of this patient showed vasculitis affecting superficial and deep dermal small vessels in addition to vessels in the subcutis. There is a predominant mononuclear perivascular infiltrate composed of lymphocytes and macrophages, with nuclear dust. (Haematoxylin and Eosin stain, original magnification × 200). C. Pedigree of a 2-year-old boy with mevalonate kinase deficiency presenting with vasculitis (black square). Genetic testing revealed compound heterozygosity of c.928G > A (p.V310M) and c.1129G > A (p.V377I) variant in MVK gene in this patient. D. Sanger sequencing electropherogram of MVK c.928G > A (p.V310M) and c.1129G > A (p.V377I) mutations in the patient (II-1) and their heterozygosity in the parents. The c.928G > A mutation was inherited from the mother (I-1), and the c.1129G > A variant was inherited from the father (I-2)
C-reactive protein was grossly elevated (253 mg/L, reference range, RR < 10); erythrocyte sedimentation rate was 41 mm/h (RR < 15); and serum amyloid A was 211 mg/L (RR < 10). Initial full blood count showed: HgB of 92 g/L (RR 105–135 g/L); white cell count of 12.16 × 10 9 /L, (RR 5–15 × 10 9 /L) with neutrophil count of 9.13 × 10 9 /L (RR 1.5–8.5 × 10 9 /L) and platelet count of 288 × 10 9 /L (RR 150–450 × 10 9 /L). Abnormal liver function tests included: alanine aminotransferase of 156 (RR 5–45) U/L and gamma glutamyltransferase of 69 (RR 6–19) U/L; ferritin was mildy elevated at 147 μg/L (RR of < 62 μg/L) while there was normal renal function (urea 4 mmol/L, RR 2.5–6 mmol/L and creatine 15 umol/L, RR < 31 umol/L) and normal clotting screen. Urinalysis showed no proteinuria or haematuria. Autoantibody testing was negative for ANA/ANCA/ENA; levels of C1Q, C3 and C4, immunoglobulins (IgG 6.3 G/L, RR of 3.5–15.70; IgA 1.3, RR of 0.30–1.3 G/L; IgM 0.48, RR of 0.4–2.2 G/L), nitroblue tetrazolium test, and lymphocyte subsets were normal. Chest x-ray was normal (no evidence of lung inflammation), and ultrasonography of abdomen was normal too (specifically no organomegaly and no bowel thickening or other intestinal pathology); echocardiography showed no serositis or coronary artery changes. Computed tomography imaging of his brain was normal, specifically with no evidence of intracerebral calcification. Magnetic resonance imaging and angiography of brain showed no intracerebral pathology or vasculopathy. Ophthalmology review showed no ocular inflammation. Skin biopsy revealed a patchy perivascular infiltrate comprising of lymphocytes, macrophages and neutrophils, nuclear dust and endothelial swelling, but no fibrinoid vasculitis or fibrin thrombi (Fig. 1B). Immunofluorescence was negative for IgG, IgA, IgM, C3 and C1Q. The histology was suggestive of small vessel leucocytoclastic vasculitis, cause undetermined. Visceral digital subtraction arteriography was normal.
With a diagnosis of severe idiopathic cutaneous vasculitis, he was treated with intravenous methylprednisolone (30 mg/kg/day × 3 days) followed by oral prednisolone 2 mg/kg/day, weaning over 8 weeks; and oral mycophenolate mofetil (MMF, 600 mg/m2 twice a day). Despite that, his SAA remained elevated at 561 mg/L with CRP of 18 mg/L, fever persisted and the vasculitic lesions progressed.
More detailed past medical history revealed that at aged 3 months old he had a perianal abscess requiring hospital admission and treatment with antibiotics. He also had been suffering from recurrent episodes of fever from early in infancy, associated with maculopapular/petechial rashes lasting 2–6 days every 2 weeks. These episodes were noted to be triggered by intercurrent infection, and after vaccines. In view of this, custom next-generation sequencing using a gene panel for autoinflammation was undertaken (https://www.ucl.ac.uk/amyloidosis/national-amyloidosis-centre/molecular-genetic-testing). This revealed compound heterozygous mutation in MVK (NM_000431.2): c.928G > A (p.V310M) and c.1129G > A (p.V377I) (Fig. 1C). Sanger sequencing confirmed the presence of both mutations in the proband and their heterozygosity in the parents (Fig. 1D). Mevalonate kinase enzyme activity measured at the Laboratory Genetics Metabolic Diseases (Amsterdam UMC; https://www.amc.nl), in patient derived lymphocytes was markedly reduced at 5 pmol/ (min.mg protein), reference values (mean ± SD) 213 ± 59 pmol/(min.mg protein), and to the activity of a healthy control sample analysed on the same day 192 pmol/(min.mg protein).
These results confirmed the diagnosis of MKD as the cause of the extensive cutaneous vasculitis. MMF was stopped, and he commenced anakinra (2 mg/kg/day, titrated up to 5 mg/kg/day) with normalisation of his acute phase reactants, and complete resolution of the vasculitis over 3–4 days. Breakthrough periodic fevers are managed with short courses of oral prednisolone (0.5–1 mg/kg/day). Canakinumab therapy was initiated 8 months later with excellent response (no febrile episodes, no cutaneous lesions and normalisation of acute phase response noted at 6 months since treatment initiation).
Discussion and conclusions
Patients with MKD display a variety of non-specific clinical manifestations, often mimicking other disorders [1, 7]. In particular, leucocytoclastic vasculitis (and other causes of cutaneous vasculitis) have many differential diagnoses in the young, and we now add to this list MKD as a cause of monogenic vasculitis which responds to IL-1 blockade. MKD may also be misdiagnosed as Periodic Fever, Aphthous Stomatitis, Pharyngitis, Adenitis (PFAPA), Behçet’s disease, or inflammatory bowel disease in view of painful oral ulcers, pseudo-pustular skin lesions, gut inflammation and arthritis, delaying targeted treatment with IL-1 blockade [7]. The prompt diagnosis of MKD in this case prevented further escalation of treatment with more toxic vasculitis therapies such as cyclophosphamide, thus reducing unnecessary exposure to cytotoxic treatment. Targeted therapy with IL-1 blockade in this case also facilitated glucocorticoid sparing, minimising toxicity.
It is now increasingly recognised that vasculitis may be a presenting or early-onset feature of an ever expanding list of monogenic autoinflammatory diseases, including: deficiency of adenosine deaminase type 2; STING-associated vasculitis of infancy; and other emerging genetic immunodysregulatory diseases; monogenic defects in complement; and miscellaneous even rarer genetic syndromes [9, 10, 12]. Skin involvement in MKD often manifests with erythematous macules, papules, urticarial lesions, and erythematous nodules [13], but lymphocytic vasculitis with or without immune complexes is also on occasions described mainly in adults with MKD [7]. Cases of severe cutaneous vasculitis such as seen in the case we report herein are however uncommon and may lead to misdiagnosis of MKD as another inflammatory or infectious disorder [11, 12]. For instance IgA vasculitis was considered in the differential diagnosis of the case presented here in view of the cutaneous and gastrointestinal manifestations. The recurrent nature of the fever and vasculitis attacks and persistent acute phase response prompted diagnostic suspicion of an autoinflammatory disorder and subsequently led to a prompt genetic and biochemical confirmation of MKD in this case with immediate therapeutic implications. The use of either anakinra or canakinumab have been found to be efficacious in controlling and preventing flares in most patients with MKD [14, 15] such as for the patient described herein. However, in some cases the disease may not be well controlled and for these patients haematopoetic stem cell transplantation may be a therapeutic option [16, 17]. An earlier diagnosis of MKD could have been considered prior to us seeing this patient given the medical history of early onset recurrent fevers associated with maculopapular rashes and some gastrointestinal symptoms, all features of MKD [7, 18].
In summary, our report highlights the broad clinical phenotype of MKD that includes severe cutaneous vasculitis and emphasizes the need to consider early genetic screening for young children presenting with vasculitis to exclude a monogenic vasculitis which may be amenable to targeted treatment.
Availability of data and materials
All data generated or analysed during this study are included in this article.
Abbreviations
- MKD:
-
Mevalonate kinase deficiency
- MVK:
-
Mevalonate kinase
- HIDS:
-
Hyperimmunoglobulinemia D with periodic fever syndrome
- PFAPA:
-
Periodic Fever, Aphthous Stomatitis, Pharyngitis, Adenitis
- CRP:
-
C-reactive protein
- SAA:
-
Serum amyloid A
- RR:
-
Reference range
- MMF:
-
mycophenolate mofetil
References
Ter Haar NM, Jeyaratnam J, Lachmann HJ, Simon A, Brogan PA, Doglio M, et al. The phenotype and genotype of mevalonate kinase deficiency: a series of 114 cases from the Eurofever registry. Arthritis Rheumatol Hoboken NJ. 2016;68(11):2795–805. https://doi.org/10.1002/art.39763.
Houten SM, Romeijn GJ, Koster J, Gray RGF, Darbyshire P, Smit GPA, et al. Identification and characterization of three novel missense mutations in mevalonate kinase cDNA causing Mevalonic aciduria, a disorder of isoprene biosynthesis. Hum Mol Genet. 1999 Aug 1;8(8):1523–8. https://doi.org/10.1093/hmg/8.8.1523.
Drenth JPH, Cuisset L, Grateau G, Vasseur C, van de Velde-Visser SD, de Jong JGN, et al. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. Nat Genet. 1999 Jun;22(2):178–81. https://doi.org/10.1038/9696.
Favier LA, Schulert GS. Mevalonate kinase deficiency: current perspectives. Appl Clin Genet. 2016 Jul 20;9:101–10. https://doi.org/10.2147/TACG.S93933.
van der Hilst JCH, Frenkel J. Hyperimmunoglobulin D syndrome in childhood. Curr Rheumatol Rep. 2010 Apr 1;12(2):101–7. https://doi.org/10.1007/s11926-010-0086-1.
Haas D, Hoffmann GF. Mevalonate kinase deficiencies: from mevalonic aciduria to hyperimmunoglobulinemia D syndrome. Orphanet J Rare Dis. 2006 Apr 26;1(1):13. https://doi.org/10.1186/1750-1172-1-13.
De Pieri C, Taddio A, Insalaco A, Barbi E, Lepore L, Ventura A, et al. Different presentations of mevalonate kinase deficiency: a case series. Clin Exp Rheumatol. 2015 Jun;33(3):437–42.
Correa ARE, Gupta N, Bagri N, Vignesh P, Alam S, Yamaguchi S. Mevalonate kinase deficiency as cause of periodic fever in two siblings. Indian Pediatr. 2020 Feb;57(2):180–1. https://doi.org/10.1007/s13312-020-1742-9.
Omoyinmi E, Mohamoud I, Gilmour K, Brogan PA, Eleftheriou D. Cutaneous Vasculitis and Digital Ischaemia Caused by Heterozygous Gain-of-Function Mutation in C3. Front Immunol [Internet]. 2018 [cited 2020 May 4];9. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6221951/
Nanthapisal S, Eleftheriou D, Gilmour K, Leone V, Ramnath R, Omoyinmi E, et al. Cutaneous Vasculitis and recurrent infection caused by deficiency in complement factor I. Front Immunol. 2018;9:735. https://doi.org/10.3389/fimmu.2018.00735.
Jain A, Misra DP, Sharma A, Wakhlu A, Agarwal V, Negi VS. Vasculitis and vasculitis-like manifestations in monogenic autoinflammatory syndromes. Rheumatol Int. 2018 Jan;38(1):13–24. https://doi.org/10.1007/s00296-017-3839-6.
Demir S, Sag E, Dedeoglu F, Ozen S. Vasculitis in Systemic Autoinflammatory Diseases. Front Pediatr [Internet]. 2018 [cited 2021 Apr 29];6. Available from: https://www.frontiersin.org/articles/10.3389/fped.2018.00377/full
Drenth JP, Boom BW, Toonstra J, Van der Meer JW. Cutaneous manifestations and histologic findings in the hyperimmunoglobulinemia D syndrome. International hyper IgD study group. Arch Dermatol. 1994;130(1):59–65. https://doi.org/10.1001/archderm.1994.01690010063008.
De Benedetti F, Gattorno M, Anton J, Ben-Chetrit E, Frenkel J, Hoffman HM, et al. Canakinumab for the treatment of autoinflammatory recurrent fever syndromes. N Engl J Med. 2018;378(20):1908–19. https://doi.org/10.1056/NEJMoa1706314.
Bodar EJ, Kuijk LM, Drenth JPH, van der Meer JWM, Simon A, Frenkel J. On-demand anakinra treatment is effective in mevalonate kinase deficiency. Ann Rheum Dis. 2011;70(12):2155–8. https://doi.org/10.1136/ard.2011.149922.
Neven B, Valayannopoulos V, Quartier P, Blanche S, Prieur A-M, Debré M, et al. Allogeneic Bone Marrow Transplantation in Mevalonic Aciduria [Internet]. https://doi.org/10.1056/NEJMoa070715. 2009 [cited 2021 Sep 14]. Available from: https://www.nejm.org/doi/10.1056/NEJMoa070715
Faraci M, Giardino S, Podestà M, Pierri F, Dell’Orso G, Beccaria A, et al. Haploidentical α/β T-cell and B-cell depleted stem cell transplantation in severe mevalonate kinase deficiency. Rheumatology [Internet]. 2021 Jan 7 [cited 2021 Sep 14];(keaa912). Available from: https://doi.org/10.1093/rheumatology/keaa912, 60, 10, 4850, 4854.
Zhang S. Natural history of mevalonate kinase deficiency: a literature review. Pediatr Rheumatol Online J. 2016;14(1):30. https://doi.org/10.1186/s12969-016-0091-7.
Acknowledgements
Not applicable.
Funding
Despina Eleftheriou is funded by Versus Arthritis (grants 21791 and 21593).
Author information
Authors and Affiliations
Contributions
EO drafted the manuscript. NS generated the histology data. EO and DR contributed to the genetic data acquisition and analysis. DE, PAB participated in clinical care and contributed to the critical revisions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All research at Great Ormond Street Hospital NHS Foundation Trust and UCL Great Ormond Street Institute of Child Health is made possible by the NIHR Great Ormond Street Hospital Biomedical Research Centre. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. Written informed consent was obtained from the parents of the patient.
Consent for publication
Parental consent for publication was obtained.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Omoyinmi, E., Rowczenio, D., Sebire, N. et al. Vasculitis in a patient with mevalonate kinase deficiency (MKD): a case report. Pediatr Rheumatol 19, 161 (2021). https://doi.org/10.1186/s12969-021-00645-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12969-021-00645-8