
Abstract
Objective
Blau syndrome (BS), a rare, autosomal-dominant autoinflammatory syndrome, is characterized by a clinical triad of granulomatous recurrent uveitis, dermatitis, and symmetric arthritis and associated with mutations of the nucleotide-binding oligomerization domain containing 2 (NOD2) gene. Aim of this study was to assess the efficacy of tofacitinib in Chinese paediatric patients with BS.
Methods
Tofacitinib was regularly administered to three BS patients (Patient 1, Patient 2, and Patient 3) at different dosages: 1.7 mg/day (0.11 mg/kg), 2.5 mg/day (0.12 mg/kg), and 2.5 mg/day (0.33 mg/kg). The clinical manifestations of the patients, magnetic resonance imaging results, serological diagnoses, therapeutic measures and outcomes of treatments are described in this report.
Results
The clinical characteristics and serological diagnoses of all BS patients were greatly improved after the administration of tofacitinib treatment. All patients reached clinical remission of polyarthritis and improvements in the erythrocyte sedimentation rate (ESR) and levels of C-reactive protein (CRP) and inflammatory cytokines.
Conclusion
Tofacitinib, a Janus kinase (JAK) inhibitor, is a promising agent for BS patients who have unsatisfactory responses to corticosteroids, traditional disease-modifying antirheumatic drugs, and biological agents.
Similar content being viewed by others

Background
Blau syndrome (BS) and early-onset sarcoidosis (EOS) are rare monogenic autoinflammatory diseases resulting from mutations of the nucleotide-binding oligomerization domain containing 2 (NOD2)/caspase recruitment domain family member 15 (CARD15) gene on chromosome 16 [1, 2]. BS is characterized by the syndrome of arthritis, uveitis, skin rash and granulomatous inflammation [3, 4]. The NOD2 gene is located on chromosome 16q12 and is highly expressed in antigen-presenting cells, such as monocytes and macrophages, and in intestinal Paneth cells [5]. CARD15 is expressed as a cytoplasmic monomer and constituted with 1040 amino acids. CARD15 has two isoforms, a full-length product (isoform 1) and isoform 2, which share an alternative initiation site corresponding to amino acid position 28. Both isoforms can activation of nuclear factor-κB (NF-κB) is a key step in the upregulation of many genes involved in inflammatory cascades [6]. It is critical to control the eye and joint involvements to improve the prognosis of BS. To date, treatments consist of non-steroid anti-inflammatory drugs, corticosteroids, methotrexate and azathioprine. Recently, biologic agents such as tocilizumab, interleukin (IL)-1 blockers and tumour necrosis factor (TNF)-α inhibitors have demonstrated effective in some cases [7,8,9]. In this study, we collected clinical data and laboratory studies from patients with BS. A missense heterozygous mutation in the NOD2 gene was revealed in each patient and their pedigrees. Furthermore, Janus kinase (JAK)-signal transducer and activator of transcription (JAK-STAT) are constitutively activated in sarcoidosis [10]. BS and EOS are paediatric granulomatous autoinflammatory syndromes that belong to a group of monogenic autoinflammatory syndromes [11,12,13]. JAK-STAT activation has been demonstrated to lead to an increase of levels of cytokines, such as interferon-γ (IFN-γ) and IL-6, that are produced by T cells and macrophages and play important roles in the pathogenesis of BS/EOS. Therefore, It appeared reasonable that inhibiting the JAK-STAT pathway could be an effective treatment strategy for BS/EOS. The treatment of BS/EOS in child patients is challenging. It also appeared reasonable that biologic treatment for BS may have diverse results due to varying genotypes and phenotypes of BS [10, 11].
In this study, we describe three Chinese patients at the Guangzhou Women and Children Medical Center. All mutations were located on exon 4 of the NOD2 gene; Patient 1 and Patient 2 (R334Q) had recurrent BS mutations [1]; however, a novel, unreported mutation in NOD2, p.M513K, but not p.M513T or p.M513R [2, 14], was found in Patient 3 by whole-exome sequencing. All of the patients were followed-up for more than half a year. Complete medical records and detailed clinical parameters were collected for further analysis. We assessed the therapeutic effect of treatment by measuring the white blood cell count (WBC); C-reactive protein (CRP) level; erythrocyte sedimentation rate (ESR); and the levels of TNF-α, IL-1β, and IL-6. This research was approved by the Ethics Committee of Guangzhou Women and Children Medical Center and performed according to the Declaration of Helsinki. Informed consent was obtained from all participants. Whole-exome sequencing was performed at the Center for Genetic Testing, Makino Beijing Gene Technology Co. Ltd., China.
Case presentation
Case report, Patient 1: The patient, A 16 month-old male child was brought to our clinic for arthritis. The onset of the arthritis was at age 8 months and included the knee, ankle, hand, foot and fingers. The patient presented with tender and swollen joints (knee, ankle, elbow, wrist, metacarpophalangeal joints, toe), and knee deformities caused by arthralgia. No fever, rash, or uveitis had been documented (Table 1 and Fig. 1A). Nuclear magnetic resonance (NMR) imaging showed synovitis, joint effusion and inflammatory oedema of the bone and periarticular soft tissue (Fig. 1A). Initially, at an age 16 months, he was diagnosed with systemic juvenile idiopathic arthritis. He was treated with ibuprofen, methotrexate (MTX), and prednisone; yet the symptoms persisted. Lab tests were again abnormal including WBC, ESR and CRP (Fig. 2A). Moreover, At 18 months of age, a genetic analysis revealed heterozygous missense mutation (R334Q) in the NOD2 region of the NOD2/CARD15 gene (Table 1 and Fig. 1D), He was diagnosed to have Blau syndrome at that point. He was begun on tocilizumab for 1 month without improvement. He then received a 2 year course of methotrexate and etanercept but still continued to have swollen and painful joints. His ESR varied between 4 and 26 mm/hour, his WBC 8.4–10.4 × 109 (ninth power)/liter, and his CRP level from 0.5–7.8 mg/dL (Fig. 2A). Plasma cytokines remained elevated despite this biologic and DMARD treatment (Fig. 2B). Etanercept treatment was stopped, and tofacitinib was orally administered starting on July 2018 with a final dosage of 1.7 mg/day. After 1 year of tofacitinib, his arthritis was in clinical remission and his lab tests were in the normal range.

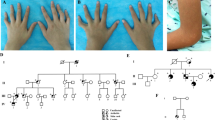
Clinical features of the patients. A Patient 1 had multiple swollen joints ( )including joints in both hands, knees, ankles and feet. Nuclear magnetic resonance (NMR) imaging of the wrist, knee and ankle joints of Patient 1 were obtained before tofacitinib treatment in December 2015. Synovitis (
)including joints in both hands, knees, ankles and feet. Nuclear magnetic resonance (NMR) imaging of the wrist, knee and ankle joints of Patient 1 were obtained before tofacitinib treatment in December 2015. Synovitis ( ), joint effusion and inflammatory oedema of the bone (
), joint effusion and inflammatory oedema of the bone ( ) and periarticular soft tissue were observed in the ankle and knee joints. B Features of paediatric granulomatous arthritis of Patient 2. Symmetric arthritis affecting the wrists, metacarpophalangeal and proximal interphalangeal joints, and knee in June 2018. NMR imaging of the knee and wrist joints of Patient 2 was carried out before tofacitinib treatment. Synovitis, joint effusion, and inflammatory oedema of the bone and periarticular soft tissue were observed in the wrist and knee joints. C Features of paediatric granulomatous arthritis of Patient 3. Symmetric arthritis affecting the wrists, ankle and knee. Nuclear magnetic resonance imaging (NMR) of the knee and ankle of Patient 3 in July 2019. Synovitis, joint effusion, inflammatory oedema of bone and periarticular soft tissue were observed in the wrist and knee joints. D Identification of the NOD2 R334Q (1001G>A) mutation in Patient 1, Patient 2 and their mother
) and periarticular soft tissue were observed in the ankle and knee joints. B Features of paediatric granulomatous arthritis of Patient 2. Symmetric arthritis affecting the wrists, metacarpophalangeal and proximal interphalangeal joints, and knee in June 2018. NMR imaging of the knee and wrist joints of Patient 2 was carried out before tofacitinib treatment. Synovitis, joint effusion, and inflammatory oedema of the bone and periarticular soft tissue were observed in the wrist and knee joints. C Features of paediatric granulomatous arthritis of Patient 3. Symmetric arthritis affecting the wrists, ankle and knee. Nuclear magnetic resonance imaging (NMR) of the knee and ankle of Patient 3 in July 2019. Synovitis, joint effusion, inflammatory oedema of bone and periarticular soft tissue were observed in the wrist and knee joints. D Identification of the NOD2 R334Q (1001G>A) mutation in Patient 1, Patient 2 and their mother

The laboratory results for the 3 patients. A Laboratory results for the patients, including WBC, CRP level and ESR. B Plasma levels of the proinflammatory cytokines IL-2, IL-4, IL-6, IL-10, TNF-α and IFN-γ in Patient 1 and Patient 2. Plasma levels of the proinflammatory cytokines IL-2, IL-4, IL-6, IL-10, TNF-α, IFN-γ and IL-1ß in Patient 3
Case report, Patient 2: The brother of patient 1 presented to the clinic at age 6 years and 10 months. He had developed chronic arthritis at age 6 months involving his ankles, knees, elbows, toes, and metacarpophalangeal joints. Like his brother, he had no history of rash, fevers, or uveitis (Table 1). He had been diagnosed to have systemic JIA at another hospital at age 5 years. At that time, he was started on ibuprofen and etanercept but did not improve. When he was seen in this hospital at almost 7 years of age, radiographs of his ankles, knee, and fingers showed joint damage (Fig. 1B). His ESR ranged from 16 to 32 mm/hour, WBC 7.5–9.3 × 109 (ninth power)/liter and CRP levels of 1.8–6.2 mg/dL (Fig. 2B). Genetic analysis appeared warranted and revealed a heterozygous missence mutation (R334D) in the NOD-2 region of the CARD15/NOD-2 gene (Table 1, Fig. 1D) (8). He was then diagnosed to have Blau syndrome. After the diagnosis of Blau syndrome, he was initially treated with methotrexate and tocilizumab with little response. He was then switched to subcutaneous methotrexate and etanercept. He improved for 1–2 months but then relapsed with abnormal labs (ESR 10–40 mm/hour, WBC 5.7–11.7 × 109 (ninth power) and CRP of 0.4–12.8 mg/dL). At the age of 9.3-years-old, after stopping etanercept, oral administration of tofacitinib was started with a final dosage 2.5 mg/day in July 2018. The tofacitinib therapy appeared to relieve his joint pain and swelling and was associated with a major decrease in his ESR and cytokine levels (Fig. 2A, B).
Case report, Patient-3. A 5 month-old infant developed arthritis in his ankles and wrists and a rash. No fever or uveitis problems were obvious (Table 1, Fig. 1C). His ESR was 17–55 mm/hour, his WBC 12.9–68.6 × 109 ninth power/L, and CRP 2.05–47.7 mg/dL (Fig. 2A). The levels of cytokines (IL-1B, IL-2, IL-6, IFN-γ, and TNF-α) were increased as well (Fig. 2B). NMR imaging of his knees and ankles in 2019 revealed synovitis, joint effusions, and inflammatory oedema of the bone and periarticular soft tissue (Fig. 1C). At that time, he was diagnosed with systemic onset juvenile idiopathic arthritis and begun on ibuprofen and methotrexate; however, no improvement followed. Genetic analysis at 21 months of age revealed a novel heterozygous missense mutation (p.M513K) in the NOD2 region of the NOD2/CARD15 gene (Table 1). Sequence data showed that the parents did not carry this mutation. He was diagnosed with BS based on the mutation and a compatible clinical scenario. He was started on tofacitinib 2.5 mg per day. The treatment appeared to improve his joint pain and swelling greatly over the next 4 months and his ESR, WBC, and CRP also decreased during that 4 month period (Fig. 2B).
Discussion
Blau syndrome is a rare systemic granulomatous disorder associated with NOD-2 mutations. NOD-2 mutations have been associated with other chronic inflammatory illnesses such as Crohn’s disease and EOS. NOD-2 is needed for induction of NFkB. It’s leucine-rich repeats, though, are apparently dispensable [5, 15,16,17]. The activation of NF-κB induced by NOD2 may rely on induced proximity of downstream effectors, including the RIP-like interacting CLARP kinase (RICK) and inhibitor of NF-κB (IκB)-kinase (IKK) complex, and promote caspase activation, which leads to secretion of the proinflammatory cytokines IL-1, IL-6, and TNF-α [18, 19]. BS and EOS are caused by mutations in the NOD2 gene, which encodes the cytosolic NOD2 protein; this protein is a pivotal molecule in the regulation in immunity that is primarily expressed in antigen-presenting cells [11, 17, 20,21,22,23]. Studies have shown that JAK-STAT signalling is constitutively activated in sarcoidosis [10, 24,25,26], In sarcoidosis, dysregulated signalling of cytokines, such as IFN-γ and IL-6, which help to develop granuloma formation, occurs via the JAK-STAT pathway (Fig. 3) [10]. A JAK inhibitor has produced rapid clinical improvements in RA patients who had previously failed other disease-modifying antirheumatic drug therapies or TNF antagonists [10, 27]. JAK inhibitors disrupt γc-chain cytokine signalling in CD4+ Th cells and block IL-6 signalling, which rapidly suppresses collagen-induced arthritis, the expression of inflammatory cytokines, and STAT1-dependent gene expression and interferes with the differentiation of Th1 and Th2 cells and the generation of inflammatory cytokines, such as IL-1β, IL-6 and IL-23, by Th17 cells [28].

Possible model of the activation of JAK and secretion of proinflammatory chemokines induced by the NOD2 mutation and their interference by tofacitinib
The mRNA levels of proinflammatory cytokines (such as IL-6, TNF-α, and IL-1β) have been reported to be increased in BS [29]. Accordingly, biological agents such as IL-1 and IL-6 inhibitors and TNF inhibitors have been used to treat BS [6]. Anti-TNF agents, such as infliximab, etanercept, adalimumab, and thalidomide, have shown promising results in several studies [6, 7, 9, 29] and are currently the most commonly used biologic therapies for BS in China. In three patients, we found that a single dose of the JAK inhibitor tofacitinib suppressed TNF and IL-6 production along with the production of other inflammatory cytokines. Substantial improvements in clinical symptoms and laboratory parameters in the tofacitinib-treated patients were observed. The symptoms of Patient 1 and Patient 2, who were treated with the TNF-α inhibitor and tocilizumab, have not yet improved. Replacement TNF-α inhibitor treatment resulted in relapse within the first year, and the joints began to swell again. It is speculated that TNF-α may not be the only cytokine that plays an important role in the pathogenesis of BS. In these 3 patients, TNF-alpha inhibitor therapy failed. This failure may suggest that TNF-alpha may not be the most important or the only important cytokine for BS. The reports and our results suggest that it is best to try a non-TNF inhibitor such as abatacept or tocilizumab if the initial TNF-alpha biologic does not help [30,31,32]. This report suggests that tofacitimib therapy may be a good option. There may be several reasons for this usefulness of tofacitimib treatment in BS but certainly one explanation might be that this drug inhibits not just TNF-alpha but appears to inhibit IL-1, IL-6, and IFN-gamma cytokines. Our 3 patients had no side effects in their short treatments of 1 year.
Conclusions
This case series reports the successful treatment of Blau syndrome in 3 young children using tofacitinib. Arthritis, uveitis, and rash improved on this drug after treatment failures of methotrexate, tocilizumab, and etanercept. No side effects were seen. This drug merits a multicenter study for Blau’s syndrome.
Availability of data and materials
Not applicable.
Abbreviations
- BS:
-
Blau syndrome
- CARD15:
-
caspase recruitment domain family member 15
- CRP:
-
C-reactive protein
- EOS:
-
early-onset sarcoidosis
- ESR:
-
erythrocyte sedimentation rate
- NF-ne:
-
nuclear factor-κB
- NOD2:
-
nucleotide-binding oligomerization domain containing 2
- RICK:
-
RIP-like interacting CLARP kinase
- TNF:
-
tumour necrosis factor
- WBC:
-
white blood cell count
- IKK:
-
inhibitor of NF-κB (IκB)-kinase
- IL:
-
interleukin
- IFN-γ:
-
interferon-γ
- JAK:
-
Janus kinase
- JAK-STAT:
-
Janus kinase-signal transducer and activator of transcription
References
Miceli-Richard C, Lesage S, Rybojad M, Prieur AM, Manouvrier-Hanu S, Hafner R, et al. CARD15 mutations in Blau syndrome. Nat Genet. 2001;29(1):19–20. https://doi.org/10.1038/ng720.
Kanazawa N, Okafuji I, Kambe N, Nishikomori R, Nakata-Hizume M, Nagai S, et al. Early-onset sarcoidosis and CARD15 mutations with constitutive nuclear factor-kappaB activation: common genetic etiology with Blau syndrome. Blood. 2005;105(3):1195–7. https://doi.org/10.1182/blood-2004-07-2972.
Blau EB. Familial granulomatous arthritis, iritis, and rash. J Pediatr. 1985;107(5):689–93. https://doi.org/10.1016/S0022-3476(85)80394-2.
Rose CD, Pans S, Casteels I, Anton J, Bader-Meunier B, Brissaud P, et al. Blau syndrome: cross-sectional data from a multicentre study of clinical, radiological and functional outcomes. Rheumatology (Oxford). 2015;54(6):1008–16. https://doi.org/10.1093/rheumatology/keu437.
Inohara N, Nunez G. NODs: intracellular proteins involved in inflammation and apoptosis. Nat Rev Immunol. 2003;3(5):371–82. https://doi.org/10.1038/nri1086.
Rose CD, Martin TM. Caspase recruitment domain 15 mutations and rheumatic diseases. Curr Opin Rheumatol. 2005;17(5):579–85. https://doi.org/10.1097/01.bor.0000169362.61443.52.
Yasui K, Yashiro M, Tsuge M, Manki A, Takemoto K, Yamamoto M, et al. Thalidomide dramatically improves the symptoms of early-onset sarcoidosis/Blau syndrome: its possible action and mechanism. Arthritis Rheum. 2010;62(1):250–7. https://doi.org/10.1002/art.25035.
Sfriso P, Caso F, Tognon S, Galozzi P, Gava A, Punzi L. Blau syndrome, clinical and genetic aspects. Autoimmun Rev. 2012;12(1):44–51. https://doi.org/10.1016/j.autrev.2012.07.028.
Chen J, Luo Y, Zhao M, Wu D, Yang Y, Zhang W, et al. Effective treatment of TNFalpha inhibitors in Chinese patients with Blau syndrome. Arthritis Res Ther. 2019;21(1):236. https://doi.org/10.1186/s13075-019-2017-5.
Damsky W, Young BD, Sloan B, Miller EJ, Obando JA, King B. Treatment of multiorgan sarcoidosis with tofacitinib. ACR Open Rheumatol. 2020;2(2):106–9. https://doi.org/10.1002/acr2.11112.
Caso F, Costa L, Rigante D, Vitale A, Cimaz R, Lucherini OM, et al. Caveats and truths in genetic, clinical, autoimmune and autoinflammatory issues in Blau syndrome and early onset sarcoidosis. Autoimmun Rev. 2014;13(12):1220–9. https://doi.org/10.1016/j.autrev.2014.08.010.
Caso F, Rigante D, Vitale A, Lucherini OM, Costa L, Atteno M, et al. Monogenic autoinflammatory syndromes: state of the art on genetic, clinical, and therapeutic issues. Int J Rheumatol. 2013;2013:513782.
Caso F, Cantarini L, Lucherini OM, Sfriso P, Fioretti M, Costa L, et al. Working the endless puzzle of hereditary autoinflammatory disorders. Mod Rheumatol. 2014;24(3):381–9. https://doi.org/10.3109/14397595.2013.843755.
Jimenez-Martinez MC, Cruz F, Groman-Lupa S, Zenteno JC. Immunophenotyping in peripheral blood mononuclear cells, aqueous humour and vitreous in a Blau syndrome patient caused by a novel NOD2 mutation. Int J Immunogenet. 2011;38(3):233–42. https://doi.org/10.1111/j.1744-313X.2011.00998.x.
Bertin J, Nir WJ, Fischer CM, Tayber OV, Errada PR, Grant JR, et al. Human CARD4 protein is a novel CED-4/Apaf-1 cell death family member that activates NF-kappaB. J Biol Chem. 1999;274(19):12955–8. https://doi.org/10.1074/jbc.274.19.12955.
Inohara N, Koseki T, Del Peso L, Hu Y, Yee C, Chen S, et al. Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-kappaB. J Biol Chem. 1999;274(21):14560–7. https://doi.org/10.1074/jbc.274.21.14560.
Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem. 2001;276(7):4812–8. https://doi.org/10.1074/jbc.M008072200.
Inohara N, Ogura Y, Chen FF, Muto A, Nunez G. Human Nod1 confers responsiveness to bacterial lipopolysaccharides. J Biol Chem. 2001;276(4):2551–4. https://doi.org/10.1074/jbc.M009728200.
Girardin SE, Tournebize R, Mavris M, Page AL, Li X, Stark GR, et al. CARD4/Nod1 mediates NF-kappaB and JNK activation by invasive Shigella flexneri. EMBO Rep. 2001;2(8):736–42. https://doi.org/10.1093/embo-reports/kve155.
Begue B, Dumant C, Bambou JC, Beaulieu JF, Chamaillard M, Hugot JP, et al. Microbial induction of CARD15 expression in intestinal epithelial cells via toll-like receptor 5 triggers an antibacterial response loop. J Cell Physiol. 2006;209(2):241–52. https://doi.org/10.1002/jcp.20739.
Lecat A, Piette J, Legrand-Poels S. The protein Nod2: an innate receptor more complex than previously assumed. Biochem Pharmacol. 2010;80(12):2021–31. https://doi.org/10.1016/j.bcp.2010.07.016.
Barnich N, Aguirre JE, Reinecker HC, Xavier R, Podolsky DK. Membrane recruitment of NOD2 in intestinal epithelial cells is essential for nuclear factor-{kappa} B activation in muramyl dipeptide recognition. J Cell Biol. 2005;170(1):21–6. https://doi.org/10.1083/jcb.200502153.
Chen G, Shaw MH, Kim YG, Nunez G. NOD-like receptors: role in innate immunity and inflammatory disease. Annu Rev Pathol. 2009;4(1):365–98. https://doi.org/10.1146/annurev.pathol.4.110807.092239.
Zhou T, Zhang W, Sweiss NJ, Chen ES, Moller DR, Knox KS, et al. Peripheral blood gene expression as a novel genomic biomarker in complicated sarcoidosis. PLoS One. 2012;7(9):e44818. https://doi.org/10.1371/journal.pone.0044818.
Damsky W, Thakral D, Emeagwali N, Galan A, King B. Tofacitinib treatment and molecular analysis of cutaneous sarcoidosis. N Engl J Med. 2018;379(26):2540–6. https://doi.org/10.1056/NEJMoa1805958.
Damsky W, Thakral D, McGeary MK, Leventhal J, Galan A, King B. Janus kinase inhibition induces disease remission in cutaneous sarcoidosis and granuloma annulare. J Am Acad Dermatol. 2020;82(3):612–21. https://doi.org/10.1016/j.jaad.2019.05.098.
Onuora S. Tofacitinib alleviates pain in RA, PsA and AS. Nat Rev Rheumatol. 2020;16:186.
Ghoreschi K, Jesson MI, Li X, Lee JL, Ghosh S, Alsup JW, et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J Immunol. 2011;186(7):4234–43. https://doi.org/10.4049/jimmunol.1003668.
Arostegui JI, Arnal C, Merino R, Modesto C, Antonia Carballo M, Moreno P, et al. NOD2 gene-associated pediatric granulomatous arthritis: clinical diversity, novel and recurrent mutations, and evidence of clinical improvement with interleukin-1 blockade in a Spanish cohort. Arthritis Rheum. 2007;56(11):3805–13. https://doi.org/10.1002/art.22966.
Minden K, Niewerth M, Zink A, Seipelt E, Foeldvari I, Girschick H, et al. Long-term outcome of patients with JIA treated with etanercept, results of the biologic register JuMBO. Rheumatology. 2012;51:1407–15.
Schmeling H, Minden K, Foeldvari I, Ganser G, Hospach T, Horneff G. Efficacy and safety of adalimumab as the first and second biologic agent in juvenile idiopathic arthritis: the German biologics JIA registry. Arthritis Rheum. 2014;66(9):2580–9. https://doi.org/10.1002/art.38741.
Ringold S, Angeles-Han ST, Beukelman T, Lovell D, Cuello CA, Becker ML, et al. 2019 American College of Rheumatology/Arthritis Foundation guideline for the treatment of juvenile idiopathic arthritis: therapeutic approaches for non-systemic polyarthritis, sacroiliitis, and enthesitis. Arthritis Rheum. 2019;71(6):846–63. https://doi.org/10.1002/art.40884.
Acknowledgements
We would like to thank all of the patients and their families for supporting this publication. The authors acknowledge support from the Department of Immunology and Rheumatology, Guangzhou Women and Children’s Medical Center.
Funding
The report was supported by the Internal Funding of Guangzhou Institute of Pediatrics, Guanzhou Women and Children’s Medical Center Post-doctoral Fellow Research Project (No. 3001098); the National Natural Science Foundation of China, General Research Project (No. 81873869); and the Natural Science Foundation of Guangdong Province (No. 2020A1515110193).
Author information
Authors and Affiliations
Contributions
All authors made substantial contributions to the study design, analysis and interpretation of data, manuscript preparation and editing for important intellectual content of this article. Study conception and design: Huasong Zeng and Song Zhang. Acquisition of data: Song Zhang and Xiaolan Mo. Manuscript preparation: Song Zhang and Zhe Cai. All authors have agreed to the submission of this manuscript to Pediatric Rheumatology.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethical Committees of the Guangzhou Women and Children’s Medical Center, Guangzhou, China. Written informed consent for participation in this study was obtained from the parents of the patients.
Consent for publication
Written informed consent was obtained from the parents of all participants.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, S., Cai, Z., Mo, X. et al. Tofacitinib effectiveness in Blau syndrome: a case series of Chinese paediatric patients. Pediatr Rheumatol 19, 160 (2021). https://doi.org/10.1186/s12969-021-00634-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12969-021-00634-x