
Abstract
Background
Familial chilblain lupus (FCL) is a rare, chronic form of cutaneous lupus erythematosus, which is characterized by painful bluish-red inflammatory cutaneous lesions in acral locations. Mutations in TREX1, SAMHD1 and STING have been described in FCL patients. Less than 10 TREX1 mutation positive FCL families have been described in the literature.
Case presentation
Genetic study was performed in a large, nonconsanguineous Chinese family with 13 members over 4 generations affected by chilblain lupus. Whole exome sequencing was performed for the index patient. Significant variant detection was subsequently validated by resequencing using Sanger sequencing in the index patient and other family members. A novel pathogenic mutation TREX1 p.Asp18His was iditified in the index patient. The mutation was present in affected individuals and was absent in non-affected individuals in the familiy.
Conclusions
We present a four-generation Chinese family with FCL caused by a novel heterozygous mutation TREX1 p.Asp18His, which had been reported in a patient with Aicardi–Goutie’res syndrome. This is the first reported Chinese family with FCL based on mutation in TREX1.
Similar content being viewed by others


Background
Chilblain lupus erythematosus (CHLE) is a rare, chronic form of cutaneous lupus erythematosus, characterized by painful bluish-red inflammatory cutaneous lesions in acral locations such as fingers, toes, nose, cheeks, and ears, and tend to ulcerate [1]. Cutaneous lesions are precipitated by cold and wet exposure and usually improve during summer. Sporadic CHLE usually affects middle-aged females, whilst familial chilblain lupus (FCL) manifests in early childhood, which was first described by Lee-Kirsch MA. et al. [2] in 2006. FCL is a monogenic form of cutaneous lupus erythematosus, and mostly inherited in an autosomal-dominant trait. Mutations in Three Prime Repair Exonuclease 1 (TREX1) [2,3,4,5,6,7,8,9,10,11], SAMHD1 [12] and STING [13] have been described in FCL patients. Less than 10 TREX1 mutation positive FCL families have been described in the literature [2,3,4,5,6,7,8,9,10,11]. Here, we report a novel TREX1 mutation in a Chinese FCL family by whole exome sequencing. This is the first reported Chinese family with FCL based on mutation in TREX1.
Case presentation
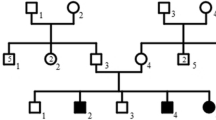
In this study, we describe a large, nonconsanguineous Chinese family with 13 members over 4 generations affected by chilblain lupus (Fig. 1, Table 1). All affected individuals showed painful bluish-red papular, or nodular lesions, or even ulcerations of the skin in acral locations including fingers, toes, ears, and nose since early childhood, which became significantly worse in the winter months (Fig. 2). Patients II-3, III-3, III-4, III-5 and III-7 showed great improvements of cutaneous lupus lesions as they grew older, having a few skin lesions only in cold weather now. The condition of Patient III-6 and Patient II-5 did not improve as they aged, having severe skin lesions especially in cold weather now. Patient II-5 even had destruction of the distal interphalangeal joints because of the ulcerations. Except for arthritis in patients III-3, III-6, IV-1, IV-2, there was no history of associated disease of any internal organ (including the central nervous system), immune deficiency, or malignancy in this family.

Pedigree of the family with dominant Familial chilblain lupus erythematosus. The arrow shows the index patient (IV-1). Black squares and circles indicate affected males and females; open squares and circles indicate unaffected males and females

Chilblain lesions on skin of the patients. Skin features observed in the affected families(II-5, III-6, III-7, IV-1). Previous ulcerations have led to a loss of the distal interphalangeal joints in patient II-5
More data was available from three affected individuals who had been hospitalized.
Patient IV-1
The index patient (IV-1), a 3.8-year-old boy, was born at 39 weeks after an uncomplicated pregnancy to unrelated parents. His birth weight was 3100 g and no congenital infections were documented. He has developed chilblains on his fingers, toes and ears since the first winter when he was 6 months old. He also reported recurrent arthritis of the knees and hip joints since 2 years old. Physical examination was normal except for skin findings such as crusty wounds, hyperemic ulcers on acral surfaces and swelling of the knees. Except for mildly anemia, laboratory test findings were unremarkable, including liver and kidney function tests, urinalysis, erythrocyte sedimentation rate, C reactive protein, rheumatic factor, anticardiolipin antibodies, complement levels, as well as antibodies of extractable nuclear antigens, double-stranded DNA, and cyclic citrullinated peptide. There was no evidence for hypergammaglobulinemia, cold agglutinins, viral or bacterial infection. His cranial CT scan was normal, brain MRI disclosed an abnormal signal in bilateral occipital white matter, suggesting the possibility of poor myelination. MRI of right knee revealed synovitis with effusion. No abnormalities were found in ophthalmological examination. Skin biopsy was not performed.
Patient IV-2
Patient IV-2 is the sister of the index patient, who was 1.6 years old. She has got chilblains on her fingers and toes since her first winter just like what her brother has. She got the swelling of right knee at 1.2 years old, regressing a few days later. Physical examination showed painful bluish-red inflammatory cutaneous lesions in fingers and toes. Laboratory investigations were unremarkable. Her cranial CT scan was normal. MRI of right knee indicated arthritis.
Patient III-6
Patient III-6 was an aunt of the index patient, who was in her 20s. She has reported chilblains on her fingers, toes and knees, and arthralgia of the knees since early childhood. Laboratory investigations were unremarkable, except for slightly elevated of erythrocyte-sedimentation rate, serum IgA and IgG. Knees MRI revealed arthritis. Histologic examination of lesional skin from the knee showed lymphocytes, neutrophils and eosinophils dermal inflammatory infiltrate, and focal dermal interstitial edema with cystic degeneration(Fig. 3), which was consistent with lupus erythematosus.

Histology of lesional skin biopsy from Patient III-6
In order to identify the genetic etiology of the disease in this family, whole exome sequencing (WES) (Additional file 1) was performed for the index patient. Significant variant detected was subsequently validated by resequencing using Sanger sequencing in the index patient and other family members, including II-1, II-5, III-2, III-3, III-6, III-7, IV-2 and the index patient’s father. The participants in this study gave written informed consent. This study was approved by the ethical committee of The First Affilated Hospital of Xiamen University.
Acrroding to “Mayo Clinic Diagnostic Criteria” [1], all patients in this family can be diagnosed as FCL. WES revealed a heterozygous novel missense mutation c.52 G > C in TREX1 gene leading to a Aspartate to Histidine substitution (p.Asp18His) in the index patient, which was validated by Sanger sequencing (Fig. 4). The mutation was presented in affected individuals II-5, III-3, III-6, III-7, and IV-2. Patient III-3 was the index patient’s mother. The mutation was absent in non-affected individuals II-1, III-2, and the index patient’s father.

Sequence data of TREX1 gene in the index patient. The nucleotide exchange is marked by an arrow, with two peaks representing G and C at location 52
Discussions
TREX1 is a 314 amino acid protein encoded by gene TREX1, which is located on chromosome 3p21. It represents the major DNA-specific 3-prime-to-5-prime exonuclease activity measured in mammalian cells. It is anchored in the outer nuclear membrane that degrading short DNA metabolites derived from the nucleus leak into the cytosol [1, 4, 14]. In TREX1-deficient cells, self-DNA accumulates in the cytosol and leads to inappropriate activation of chronic type I interferons, which can break immune tolerance and promote autoimmunity or autoinflammatory diseases [4, 14].
Nine TREX1 mutation-positive FCL families have been described in the literature [2,3,4,5,6,7,8,9,10,11] (Table 2). Among the nine families, five of them had a mutation resulting in a Aspartate to Asparagine substitution at the acid residue 18 (p.Asp18Asn). It shows aspartate 18 is a hot-spot mutation. In contrast, in this study, we found a heterozygous mutation resulting in a Aspartate to Histidine substitution at the acid residue 18, which has not been reported in FCL patients before. Several lines of evidence indicated that the mutation TREX1 p.Asp18His was pathogenic. First, the acid residue 18 constitutes a highly conserved Mg 2+ − coordinating aspartate residue within the catalytic centre of the dimeric TREX1 enzyme [4], suggesting that it could affect enzymatic function. Second, the mutation was a rare variant not found in 1000 genome, ExAC, gnomAD. Third, the mutation was predicted to be a disease-causing mutation by several different computational prediction methods, including SIFT, Polyphen, Mutation Taster, and PROVEAN. Fourth, this mutation completely cosegregated with affected family members and was absent in non-affected family members. Finally, the clinical course of our patients was similar to those observed in previously reported FCL patients with TREX1 mutation [2,3,4,5,6,7,8,9,10,11] (Table 2).
In addition, the heterozygous TREX1 mutation (c.52G > C; p.Asp18His) has been reported in a patient with Aicardi-Goutières syndrome (AGS) [9]. AGS is a rare syndrome characterized by calcification, diffuse demyelination, and variable degree of brain atrophy caused by inherited defects in nucleic acid metabolism [15]. About 24% of AGS patients have mutations in TREX1 [16, 17]. And chilblain-like lesions are observed in 36.7% of TREX1 AGS patients [15]. Haaxma CA et al. [18] reported a de novo heterozygous p.Asp18Asn mutation in TREX1 in an AGS patient, which was the most frequent mutation in FCL patients. Abe J et al. [8] reported a case of AGS and FCL in a three-generation family with chilblains caused by the same heterozygous TREX1 p. Asp18Asn mutation. Apart from AGS and FCL, mutations in TREX1 are also responsible for systemic lupus erythematosus (SLE). SLE is a heterogeneous multisystem autoimmune disease, characterized by a variety of clinical manifestations and a wide profile of autoantibodies. An upregulation of type I interferon signaling has been reported in some SLE patients [19]. About 2% SLE patients have mutations in TREX1 [15]. Namjou et al. [20] reported a mutation TREX1 p.Arg114His in an SLE patient, which was the most frequently TREX1 mutation in AGS patients. There are some clinical, genetic, and basic science considerations that underline a possible overlap between AGS, FCL and SLE. But the exact molecular mechanisms and the different modes of inheritance remain to be clarified.
Apart from cutaneous lesions, signs of systemic involvement have been observed in FCL patients, including arthralgia, cerebral thrombosis and hematologic system involvement including apenia, leukopenia, thrombocytopenia, and some patients have elevated of antinuclear antibodies titer [2,3,4,5,6,7,8,9,10,11] (Table 2). Millard LG et al. [21] reported that up to 18% of affected sporadic CHLE individuals progressed to SLE after a long time of follow up, which was not found in FCL patients. The high prevalence of systemic clinical manifestations may suggest that TREX1-associated FCL may be a systemic disease with prominent cutaneous involvement.
The expression of the phenotype may vary among the members of an individual FCL family with TREX1 mutation. In the family described in this study, patients II-3, III-3, III-4, III-5 and III-7 had cold-induced infiltrates and ulcerations in childhood that declined in severity as they aged, whereas patient II-5 had destruction of the distal interphalangeal joints because of the ulcerations. Arthralgia was presented in patients IV-1, III-3, IV-2 and III-6, but not in other affected patients in our study. TREX1 mutation FCL patients may have variable penetrance, and the same mutation can cause an exclusive skin phenotype, or a neurological phenotype, or a hematologic system involvement, even in the same family [2,3,4,5,6,7,8,9,10,11] (Table 2). Gillian Rice et al. [7] reported one individual in an FCL family was unaffected on clinical examination but carried the same molecular changes observed in her affected siblings. Modifier genes and their epistatic interactions, epigenetic or environmental factors may also play a role in the result of incomplete penetrance, though more cases are needed for a better understanding about these effects.
Patient IV-1 and patient IV-2 have undergone treatment with JAK inhibitor tofacitinib for 2 months. Their symptoms of arthritis had a complete remission and their skin lesions also had a significantly improvement. Patient III-6 was considered to be treated with tofacitinib recently. The exact effect needs a longer follow up of patients IV-1, IV-2 and III-6, especially in winter. The other patients did not have any treatment.
In conclusion, we presented a four-generation Chinese family with FCL caused by a novel heterozygous mutation TREX1 p.Asp18His, which had been reported in a patient with AGS. This is the first reported Chinese family with FCL based on mutation in TREX1.
Availability of data and materials
The datasets supporting the results of this article are included within the article and its additional file.
Abbreviations
- CHLE:
-
Chilblain lupus erythematosus
- FCL:
-
Familial chilblain lupus
- WES:
-
Whole exome sequencing
- TREX1:
-
Three prime repair exonuclease 1
- AGS:
-
Aicardi-Goutières syndrome
- SLE:
-
Systemic lupus erythematosus
References
Hedrich CM, Fiebig B, Hauck FH, Sallmann S, Hahn G, Pfeiffer C, et al. Chilblain lupus erythematosus-a review of literature. Clin Rheumatol. 2008;27:1341.
Lee-Kirsch MA, Gong M, Schulz H, Rüschendorf F, Stein A, Pfeiffer C, et al. Familial chilblain lupus, a monogenic form of cutaneous lupus erythematosus, maps to chromosome 3p. Am J Hum Genet. 2006;79:731–7.
Kisla Ekinci RM, Balci S, Bisgin A, Altintas DU, Yilmaz M. A homozygote TREX1 mutation in two siblings with different phenotypes: chilblains and cerebral vasculitis. Eur J Med Genet. 2017;60:690–4.
Lee-Kirsch MA, Chowdhury D, Harvey S, Gong M, Senenko L, Engel K, et al. A mutation in TREX1 that impairs susceptibility to granzyme A-mediated cell death underlies familial chilblain lupus. J Mol Med (Berl). 2007;85:531–7.
Yamashiro K, Tanaka R, Li Y, Mikasa M, Hattori N. A TREX1 mutation causing cerebral vasculopathy in a patient with familial chilblain lupus. J Neurol. 2013;260:2653–5.
Günther C, Berndt N, Wolf C, Lee-Kirsch MA. Familial chilblain lupus due to a novel mutation in the exonuclease III domain of 3′ repair exonuclease 1 (TREX1). JAMA Dermatol. 2015;151:426–31.
Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, et al. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am J Hum Genet. 2007;80:811–5.
Abe J, Izawa K, Nishikomori R, Awaya T, Kawai T, Yasumi T, et al. Heterozygous TREX1 p.Asp18Asn mutation can cause variable neurological symptoms in a family with Aicardi-Goutieres syndrome/familial chilblain lupus. Rheumatology (Oxford). 2013;52:406–8.
Tüngler V, Silver RM, Walkenhorst H, Günther C, Lee-Kirsch MA. Inherited or de novo mutation affecting aspartate 18 of TREX1 results in either familial chilblain lupus or Aicardi-Goutières syndrome. Br J Dermatol. 2012;167:212–4.
Sugiura K, Takeichi T, Kono M, Ito Y, Ogawa Y, Muro Y, et al. Severe chilblain lupus is associated with heterozygous missense mutations of catalytic amino acids or their adjacent mutations in the exonuclease domains of 3′-repair exonuclease 1. J Invest Dermatol. 2012;132:2855–7.
Günther C, Hillebrand M, Brunk J, Lee-Kirsch MA. Systemic involvement in TREX1-associated familial chilblain lupus. J Am Acad Dermatol. 2013;69:e179–81.
Ravenscroft JC, Suri M, Rice GI, Szynkiewicz M, Crow YJ. Autosomal dominant inheritance of a heterozygous mutation in SAMHD1 causing familial chilblain lupus. Am J Med Genet A. 2011;155A:235–7.
König N, Fiehn C, Wolf C, Schuster M, Cura Costa E, Tüngler V, et al. Familial chilblain lupus due to a gain-of-function mutation in STING. Ann Rheum Dis. 2017;76:468–72.
Zimmermann N, Wolf C, Schwenke R, Lüth A, Schmidt F, Engel K, et al. Assessment of clinical response to Janus kinase inhibition in patients with familial chilblain lupus and TREX1 mutation. JAMA Dermatol. 2019;155:342–6.
Rice GI, Rodero MP, Crow YJ. Human disease phenotypes associated with mutations in TREX1. J Clin Immunol. 2015;35:235–43.
Abe J, Nakamura K, Nishikomori R, Kato M, Mitsuiki N, Izawa K, et al. A nationwide survey of Aicardi-Goutières syndrome patients identifies a strong association between dominant TREX1 mutations and chilblain lesions: Japanese cohort study. Rheumatology (Oxford). 2014;53:448–58.
Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, et al. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am J Hum Genet. 2007;81:713–25.
Haaxma CA, Crow YJ, van Steensel MA, Lammens MM, Rice GI, Verbeek MM, et al. A de novo p.Asp18Asn mutation in TREX1 in a patient withAicardi-Goutières syndrome. Am J Med Genet A. 2010;152A:2612–7.
Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197:711–23.
Namjou B, Kothari PH, Kelly JA, Glenn SB, Ojwang JO, Adler A, et al. Evaluation of the TREX1 gene in a large multi-ancestral lupus cohort. Genes Immun. 2011;12:270–9.
Millard LG, Rowell NR. Chilblain lupus erythematosus (Hutchinson). A clinical and laboratory study of 17 patients. Br J Dermatol. 1978;98:497–506.
Acknowledgements
We thank the patients and their families for their participation in this study. We thank the team of Qiyuan Li for their help in the experiment of whole exome sequencing and sanger sequencing.
Funding
No funding.
Author information
Authors and Affiliations
Contributions
Jihong Xiao initiated this study and collected the materials of all participants in this study. Qiyuan Li analyzed and interpreted the result of whole exome sequencing and sanger sequencing of the particiants. Cuili Yi took part in the analyzed of the finial result of the genetic suty and draft this manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the ethical committee of The First Affilated Hospital of Xiamen University. Oral informed assent and written informed consent was obtained from all participants.
Consent for publication
Publication consent was obtained from all participants.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yi, C., Li, Q. & Xiao, J. Familial chilblain lupus due to a novel mutation in TREX1 associated with Aicardi–Goutie’res syndrome. Pediatr Rheumatol 18, 32 (2020). https://doi.org/10.1186/s12969-020-00423-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12969-020-00423-y