
Abstract
Background
Chronic recurrent multifocal osteomyelitis (CRMO) is a rare auto-inflammatory bone disorder that primarily affects young girls, with a mean age of 10 years at onset. Generally, it is a self-limited disease. However, recent data indicate that more than 50% of patients have a chronic persistent disease and about 20% a recurring course of this condition. Also, there are more cases reported with associated auto-inflammatory and autoimmune diseases. In this case report, we present a rare case of sporadic CRMO in which the patient eventually developed C-ANCA (cytoplasmic anti-neutrophil cytoplasmic antibodies)-associated renal vasculitis and hyperparathyroidism.
Case presentation
A 14 year old female patient was brought to the emergency department with a sudden onset of left leg pain and oedema. After physical evaluation and initial investigation, she was diagnosed with femoral and pelvic deep vein thrombosis. While searching for possible thrombosis causes, osteomyelitis of the left leg was identified. Additional CT and MRI scans hinted at the CRMO diagnosis. Due to the multifocal lesions of CRMO, endocrinological evaluation of calcium metabolism was done. The results showed signs of hyperparathyroidism with severe hypocalcaemia. Moreover, when kidney damage occurred and progressed, a kidney biopsy was performed, revealing a C-ANCA associated renal vasculitis. Treatment was started with cyclophosphamide and prednisolone according to the renal vasculitis management protocol. Severe metabolic disturbances and hyperparathyroidism were treated with alfacalcidol, calcium and magnesium supplements. Secondary glomerulonephritis (GN) associated hypertension was treated with ACE (angiotenzine converting enzyme) inhibitors. Anticoagulants were prescribed for deep vein thrombosis. After 1.5 years of treatment, the patient is free of complaints. All microelement and parathormone levels are within normal range. Kidney function is now normal. To date, there are no clinical or diagnostic signs of deep vein thrombosis.
Conclusions
This case report presents a complex immunodysregulatory disorder with both auto-inflammatory and autoimmune processes. We hypothesize that the long lasting active inflammation of CRMO may induce an autoimmune response and result in concomitant diseases like C-ANCA-associated vasculitis in our patient. Any potential specific pathogenic relationships between these two rare pathologies may need to be further studied. Furthermore, there is a lack of specific biomarkers for CRMO and more studies are necessary to identify CRMO’s characteristic patterns and how to best monitor disease progression.
Similar content being viewed by others


Background
Chronic recurrent multifocal osteomyelitis (CRMO) is a rare auto-inflammatory bone disorder that primarily affects young girls, with a mean age of 10 years at onset [1, 2]. The disease can cause multiple bone lesions, mostly affecting long bones of the lower extremities and vertebrae [3]. Generally, it is a self-limited disease; however, recent data show that more than 50% of patients have a chronic persistent and about 20% have a recurring course of this condition [1]. According to the Eurofever registry [4], there are only slightly more then 460 cases registered world-wide so far, with a prevalence of 1–9/million. [5]. As CRMO is the diagnosis of exclusion and can imitate other inflammatory bone conditions, it is thought to be underestimated [6]. Also, there are more cases emerging associated with other auto-inflammatory and autoimmune diseases, for example related to disorders with skin diseases [7, 8], peripheral arthritis [9,10,11], concomitant inflammatory bowel disease [12, 13], granulomatosis with polyangiitis (GPA, formerly known as Wegener granulomatosis) [14, 15] and others [16,17,18,19]. An association with several auto-inflammatory conditions can be detected in about one-third of CRMO patients [3]. Despite being well-known for about 50 years, CRMO still does not have approved treatment guidelines as the aetiology is unknown, and none of the therapies used currently have provided consistent outcomes. In this case report, we present a rare case of prolonged sporadic CRMO which eventually developed cytoplasmic anti-neutrophil cytoplasmic antibody (C-ANCA)-associated renal vasculitis and hyperparathyroidism.
Case presentation
A 14-year-old female patient presented to the emergency department with a sudden onset of left leg pain and oedema. She had a fever of 38 °C once a few days before. Physical evaluation revealed a swollen and painful left leg with reduced range of motion of the left hip and bumps palpated on the right tibia proximal metaphysis. No signs of arthritis, possible intestinal inflammation or skin changes were seen during physical evaluation. The patient’s blood pressure and urinary output were normal. After the emergency ultrasound exam revealed a diagnosis of femoral and pelvic deep vein thrombosis, she was admitted to the hospital. According to her parents, the patient had had no chronic diseases to date. They indicated that she had a foot fracture at the age of 8 years. There were no autoimmune or auto-inflammatory disorders in the patient’s family history.
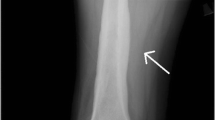
Initial investigations revealed high levels of inflammatory markers (CRP and ESR), and a severe microcytic anaemia as well as thrombocytosis, hypoalbuminemia and elevated fibrinogen concentration and D-dimer values (Table 1). Suspecting an infectious cause, a urinalysis was performed, and haematuria and proteinuria were detected. Both blood and urine cultures were sterile. X-rays of the legs were done (Fig. 1), showing sites of hyperostosis and sclerosis in the metaphysis of the right tibia along with a periosteal reaction, suggesting a possible osteomyelitis or oncological processes in the bones. As the diagnosis of acute osteomyelitis could not be disproved, broad-spectrum antibiotics were prescribed. However, the CRP level did not change significantly over the first few days, so other causes of acute thrombosis and inflammation were investigated. An abdominal ultrasound showed a giant, homogenous pelvic mass and hydronephrosis of the left kidney. Kidney function was quite abnormal as the creatinine level was 185 μmol/L and urinary protein excretion of 7 g over 24 h was detected. Within the next few days the patient’s kidney function deteriorated further, with a creatinine value increasing to 307 μmol/L with poor urine output and high blood pressure. The patient had no prior history of kidney problems.

X-ray of the legs.Hyperostosis on the left tibia metaphysis is indicated by a white arrow
Due to the findings of the abdominal ultrasound and leg X-ray, the child underwent a whole-body computed tomography (CT) scan. Multiple bone lesions were observed, the most severe of which were located in the left scapula, the fifth rib projection near the spine and a large deformity of the left pelvic bone close to the acetabulum (Fig. 2). The left iliopsoas muscle also appeared to be abnormal. Moreover, renal parenchymal thickening and oedema were found. Investigations for possible endocrine disorders were performed (Table 2). Signs of hyperparathyroidism were present together with hypocalcaemia, hypomagnesaemia and a low vitamin D3 concentration, as well as hypocalciuria in the 24-h urine test and hyperphosphaturia based on phosphate fractional excretion (Table 3).

Whole-body CT (pelvis region) showing destruction and sclerosis of the left hip bones
Bone and left kidney biopsies were performed. The bone biopsy from the affected site of the left pelvic bone demonstrated intertrabecular stromal fibrosis, several epithelioid granulomas with a central zone of necrosis and polymorphonuclear cells. Moreover, histological evaluation showed a few sites with plasma cell infiltration, including some cells positive for immunoglobulin G (IgG) and immunoglobulin G4 (IgG4). Histological examination of the surrounding connective tissue found mucoid oedema. The renal biopsy revealed an acute and active crescentic glomerulonephritis (GN) with ANCA-associated vasculitis (Fig. 3). Special staining was performed for the kidney sample, and no IgG subclasses were found.

Histological image of kidney tissue. PAS-stained sample, 400× magnification. The crescentic glomeruli, stromal fibrosis, normal mesangial region are indicated by arrows
Due to the results of left kidney biopsy together with thrombosis of the deep veins, additional screening for autoimmune diseases was carried out. The child tested positive for antinuclear antibodies (ANA 1:100), antibodies against centromere protein B (anti-CENP B), antibodies against proliferating cell nuclear antigens (anti-PCNA) and C-ANCA (Table 4). High levels of ferritin were also found (448 μg/L), indicating the possibility of both autoimmune and thrombotic causes of anaemia. However, antiphospholipid antibodies were not detected. Concerning the bone biopsy results, serum IgG levels were tested and only IgG4 was slightly increased (Table 4).
Since multiple bone lesions were detected, the fracture history was reviewed. At the age of 8 years our patient had not only a foot fracture, but also complained of pain in the left elbow and back. It appeared that she was diagnosed with CRMO at that time, based on the results of a biopsy from the fracture site as well as a CT of the left foot (Fig. 4) and a whole-body radionuclide scan. The radionuclide scan showed multiple sites of CRMO lesions, with three in the spine, one in the left hand, one in the left scapula, one in the left pelvic bone and two in the right leg. Surgery for the broken foot was performed and treatment with antibiotics and nonsteroidal anti-inflammatory drugs (NSAIDs) was prescribed for a month. When the patient’s pain decreased over time, analgesics were discontinued due to the impression of a self-limiting disease course. For the next 6 years she was free from complaints except for painless unilateral right periorbital oedema appearing on and off for the last 2–3 years and observed without medical supervision. Due to this oedema and slight exophthalmos of the right eye during current episode, additional head and spine magnetic resonance imaging (MRI) was performed, and a new CRMO lesion in a right periorbital region was diagnosed (Fig. 5) together with deformation and lipoid degeneration of the seventh neck vertebrae (Fig. 6).

Three-dimentional reconstruction CT of the left foot. The destruction and sclerosis of third metatarsus bone diaphysis and distal metaphysis parts are indicated by white arrows

MRI of the patient‘s head. Measuring of hyperostosis of frontal bone on the right is shown by a white line

MRI of the patient’s neck. Destruction and lipoid degeneration in the seventh neck vertebrae is indicated by a white arrow
Treatment was started for the vasculitis with cyclophosphamide and prednisolone according to the renal-limited vasculitis management protocol. No pain reliever was necessary. Severe metabolic disturbances and hyperparathyroidism were treated with alfacalcidol, calcium and magnesium supplements. Kidney and ureter stents were placed for 1 month due to hydronephrosis. Secondary GN-associated hypertension was treated with ACE (angiotensin converting enzyme) inhibitors. Anticoagulants were prescribed for deep vein thrombosis for 6 months until full recanalisation of the left femoral vein had occurred.
After 1.5 years of treatment, the child is free of complaints. Her auto-inflammatory, metabolic and endocrinological conditions are being monitored by a multidisciplinary team of physicians. All microelements and parathormone levels are within the normal range. There are no signs of hypertension and the patient’s kidney function has recovered. To date, there are no clinical or diagnostic signs of deep vein thrombosis. The patient is still on maintenance treatment with azathioprine according to the kidney vasculitis treatment protocol and receiving microelements supplementation.
Discussion
Chronic recurrent multifocal osteomyelitis has been recognized as a disease entity for several decades. It is a multi-faceted disease. The diagnosis is based on clinical signs, radiological findings and bone biopsy results. There are several diagnostic criteria suggested for this condition [3, 20], the latest ones are by Roderick et al. [20]. According to Zhao et al., these guidelines should be followed in clinical practice and their application in typical cases can prevent or minimise the need for bone biopsy [21]. Still, there is no unified protocol to identify and characterise this disease [22]. Regarding our case, biopsy results were essential, as various bone lesions and deformities were found together with highly elevated inflammation markers. Furthermore, possible oncological conditions such as osteoblastoma, osteosarcoma and others had to be ruled out [23]. Bone biopsy revealed all stages of bone inflammation, with fibrosis detected together with the presence of plasma cells (representing adaptive immunity) [24] and neutrophils (responsible for innate immune activation) [24]. These findings, together with symptomatic C-ANCA-associated kidney vasculitis and deep vein thrombosis, further illustrated the immune system disturbances.
Currently, there are an increasing number of CRMO cases associated with other auto-inflammatory and autoimmune conditions. According to different data, we found two more cases of CRMO that eventually developed GPA [14, 15]. However, both cases demonstrated typical signs of respiratory tract involvement. Our case presented with isolated C-ANCA-associated kidney vasculitis without respiratory tract injury by otorhinolaryngological examination or lung CT scans. Our case fulfils three criteria out of six for GPA diagnosis proposed by EULAR/PRINTO/PRES [25]. But the question remains if granulomatous lesions in bones are an expression of a separate skeletal inflammatory disorder which eventually caused kidney vasculitis (as a complication of persistent chronic inflammation) or if this sporadic CRMO could be the musculoskeletal expression of GPA [26]. The recent studies of CRMO pathogenesis show a disturbed balance between pro- and anti-inflammatory cytokines such as interleukin 6 (IL-6), tumour necrosis factor alfa (TNFα) and interleukin 10 (IL-10) [24, 27]. Also, a reduced number of IL-10 producing cells in ANCA-associated vasculitis was shown in a study by Wilde et all. [28]. It may suggest a possible connection between these two conditions.
Moreover, it is reported that persistent inflammation and endothelial dysfunction associated with ANCA vasculitis increases the rate of thrombotic events and is linked to the active disease phase [29, 30]. Our patient‘s kidney biopsy results showed active vasculitis. Also, she had nephrotic proteinuria, which is known to increase the risk of thrombosis [31]. However, tests for protein C, protein S or antithrombin deficiencies, antiphospholipid antibodies and factor V Leiden mutation were negative in this case, indicating the possibility of other hypercoagulation mechanisms. Several hypotheses have also been proposed for activation of the coagulation cascade when well-known hypercoagulable risk factors remain undetected, including necrosis of endothelial cells and their circulation in the blood, wide-spread endothelial dysfunction, or thrombocyte activation [30].
Interestingly, together with CRMO bone lesions, there were a few cells positive for IgG and IgG4 identified in the bone biopsy, pointing to another rare pathology, IgG4-related disease. To date, there are only 25 paediatric case reports of IgG-related disease identified and they usually involve ocular and parenchymal organs with positive histological results for IgG4 cells [32]. Although the IgG4 levels in the blood were slightly elevated compared to the reference values (Table 4), no other symptoms or signs of organ injury were detected in our patient. Also, the kidney histological examination was negative for IgG cells. A few studies have shown that increased IgG4 levels in serum can be found in a wide spectrum of diseases, including vasculitis [33, 34]. This could be the reason in our case too.
Our patient had several severe metabolic deficiencies including hypocalcaemia, hypomagnesemia and hyperparathyroidism leading to osteoporosis. In relation to these findings, the possibility of other metabolic bone diseases (like Camurati-Engelman syndrome and benign hyperostosis-pachydermoperiostosis) was considered. However, the lack of particular skin lesions and MRI results strongly supported CRMO diagnosis not the other two diagnoses. Information about the relationship between CRMO and calcium metabolism is still lacking.
We also considered a few possible conditions that could have caused hyperparathyroidism in our case. First, we think that the hypocalcaemia may have developed due to the kidney damage and calcitriol production disturbances, which is well known in secondary hyperparathyroidism. This theory could be supported by the renal insufficiency and vitamin D3 hypovitaminosis that lasted for an unknown period. As our patient was free of complaints, her serum creatinine and vitamin D3 levels were not checked for several years. On the other hand, it is less likely that only renal insufficiency could have had influence on calcium metabolism as it was not severe on admission. Finally, as shown by Ata et al. [35], a high turnover rate of bone metabolism could be the most probable explanation for osteoporosis and hyperparathyroidism in this CRMO case. Closer follow-up of calcium metabolism might have provided an earlier hint of hyperparathyroidism and renal insufficiency due to the multiple bone lesions diagnosed after the first episode of CRMO 6 years ago.
Chronic recurrent multifocal osteomyelitis is a very versatile disease. Several studies have been done to identify the possible connection between immunological defects and gene mutations. Different data show that genetic metabolism errors are responsible for some of CRMO syndromes [7, 16], for example, the PSTPIP1 gene is associated with pyogenic arthritis, pyoderma gangrenosum and acne (PAPA) syndrome [7] as well as defects in LPIN2 result in Majeed syndrome [16]. A few research groups suggested a link between sporadic CRMO and gene errors along the 18q chromosome [36]. However, more studies are needed. There is no data showing a specific genetic defects in most of the sporadic cases, especially those associated with other conditions like GPA, dermatomyositis and other auto-immune diseases. Genetic testing for the PSTPIP1 gene mutation was performed for our patient and was negative.
Despite various attempts to understand CRMO pathogenesis and favour diagnostic processes, no auto-antibody production has been observed to date, thus supporting an auto-inflammatory pathogenic mechanism of this condition [24, 37, 38]. We did not observe high auto-antibody titres in our case too. However, several different auto-antibodies were found. Anti-PCNA antibodies are known to occur in Sjogren’s syndrome [39], but no clinical signs of ocular or oral symptoms were present for this patient [40]. Also, we detected the presence of anti-CENP B antibodies, which are usually seen in systemic sclerosis [41]. Even so, our patient had no Raynaud’s phenomenon, skin lesions or other signs of systemic sclerosis. Moreover, an inflammatory processes and not antibody production was predominant throughout the course of the disease.
Recent studies of CRMO have led to a better understanding of pathogenic mechanisms and contributed the treatment improvements as following. NSAID monotherapy is thought to influence osteoclast activation by inhibiting prostaglandins, and thus impacting bone remodelling [24]. Moreover, glucocorticoids were shown to be important in stopping the general inflammatory response, thus having a positive effect in some cases [2]. Considering recent studies about bone remodelling in CRMO [35] and previous reports about increased osteoclast activity in bone histology specimens [42, 43], bone resorption inhibitors may be an effective treatment option. Moreover, bisphosphonates are strong osteoclast activity inhibitors and they may reduce bone turnover. Furthermore, they have shown to be effective in recurrent cases and in cases unresponsive to first-line medication [44, 45]. In very severe cases, biological therapy, for instance IL-1 or TNF-α inhibitors, have had a beneficial effect and limited progress of the disease [24, 46].
Our patient did not feel any pain and physical disability close to the CRMO lesion sites. Moreover, no vertebral fractures were detected to date and no additional treatment for CRMO was prescribed. Only treatment for vasculitis with a continuous low dose of prednisone and high doses of calcium and alfacalcidol supplements for the osteoporotic changes were given.
According to clinical guidelines, pain relief of the affected area is thought to be one of the most important criteria for clinical improvement. Despite no specific treatment, our patient had no complaints for 6 years after the initial diagnosis. Still, the progression of CRMO was obvious with a new lesion present in the MRI and painless bone deformities as a complication of the chronic course of the disease. Interestingly, this phenomenon has been described before [47] showing that active CRMO can be found in MRI, both, in periods without complaints and after various treatment options, even more than 10 years after the initial bone injury. Regardless of this data, it is still unknown if these clinically undetectable and painless lesions have clinical relevance to the disease progression and how long the treatment should be administered, i.e. to a complete radiological remission or improvement of clinical and laboratory signs [47]. Having this in mind and seeking to monitor possible severe complications (like vertebral fractures in the absence of complaints) the whole-body MRI with short tau inversion recovery (STIR) images is currently preferred as it was shown to be of superior sensitivity for evaluating CRMO lesion activity [48]. Well-timed MRI could show bone oedema prior to the appearance of erosions and/or sclerosis [48, 49]. In our case, comparative bone scintigraphy was done in parallel to the one done 6 years ago. One new CRMO lesion site was found in the right orbital area. No negative progression was observed in the previously diagnosed lesions. The MRI of the right orbital area detected sclerotic changes without bone oedema, indicating a continuous inflammatory processes that had likely developed within months or even years when the patient was supposedly symptoms-free and in a benign course of the disease. Taking into account other cases in which the patients eventually developed C-ANCA-associated vasculitis, we suggest that patients with multiple CRMO lesions should be monitored closely regarding chronic inflammation despite no signs or symptoms of disease activity.
Recently, research on new biomarkers for CRMO diagnosis and monitoring CRMO progression has been done by Hofmann et al. It showed that some changes in interleukin levels (IL-6, IL-12, eotaxin and others) were significantly altered during the CRMO disease course, meaning they could potentially be used as CRMO activity markers [37]. In our opinion, these biomarkers would be especially useful in unusual cases with atypical clinical courses, when some monitoring tools, for instance the PedsCNO score [50], are difficult to apply objectively.
Conclusions
This case report presents a rare case of a complex immunodysregulatory disorder with both auto-inflammatory and autoimmune elements. We hypothesize that the long lasting active inflammation of CRMO may induce an autoimmune response, resulting in a concomitant disease such as the C-ANCA-associated vasculitis in our patient. Any potential specific pathogenic relationships between these two rare diseases may need to be further studied. Furthermore, there is still a lack of specific biomarkers for CRMO and more studies are necessary to identify CRMO’s characteristic patterns and how to best monitor disease progression.
Abbreviations
- ACF:
-
Angiotenzine converting factor
- ANA:
-
Antinuclear antibodies
- ANCA:
-
Anti-neutrophil cytoplasmic antibodies
- anti-CENP B:
-
Antibodies against centromere protein B
- anti-PCNA:
-
Antibodies against proliferating cell nuclear antigens
- C-ANCA:
-
Cytoplasmic anti-neutrophil cytoplasmic antibodies
- CRMO:
-
Chronic recurrent multifocal osteomyelitis
- CRP:
-
C reactive protein
- CT:
-
Computed tomography
- ESR:
-
Erythrocyte sedimentation rate
- EULAR:
-
European League Against Rheumatism
- GN:
-
Glomerulonephritis
- GPA:
-
Granulomatosis with polyangiitis
- IgG:
-
Immunoglobulin G
- IgG4:
-
Immunoglobulin G4
- IL-10:
-
Interleukin 10
- IL-12:
-
Interleukin 12
- IL-6:
-
Interleukin 6
- MRI:
-
Magnetic resonance investigation
- NSAIDs:
-
Nonsteroidal anti-inflammatory drugs
- PAPA:
-
Pyogenic arthritis, pyoderma gangrenosum and acne syndrome
- PedsCNO score:
-
Paediatric chronic nonbacterial osteomyelitis score
- PRES:
-
Paediatric Rheumatology European Society
- PRINTO:
-
Paediatric Rheumatology INternational Trials Organization
- STIR:
-
Short tau inversion recovery
- TNFα:
-
Tumor necrosis factor alfa
References
Kaiser D, Bolt I, Hofer M, Relly C, Berthet G, Bolz D, Saurenmann T. Chronic nonbacterial osteomyelitis in children: a retrospective multicenter study. Pediatr Rheumatol Online J. 2015;13:25.
Wipff J, Costantino F, Lemelle I, Pajot C, Duquesne A, Lorrot M, et al. A Large National Cohort of French Patients With Chronic Recurrent Multifocal Osteitis. Arthritis and Rheumatology. 2015;67(4):1128–37.
Jansson A, Renner ED, Ramser J. Classification of non-bacterial osteitis: retrospective study of clinical, immunological and genetic aspects in 89 patients. Rheumatology (Oxford). 2007;46:154–60.
PRINTO. Eurofever registry. PRINTO web site. PRINTO. [Cited: June 12, 2017.] https://www.printo.it/eurofever/eurofever_registry.asp.
Orphanet. Orphanet Rare Disease Web site. [Cited: June 12, 2017] http://www.orpha.net.
Schnabel A, Range U, Hahn G, Siepmann T, Berner R, Hedrich CM. Unexpectedly high incidences of chronic non-bacterial as compared to bacterial osteomyelitis in children. Rheumatol Int. 2016;36(12):1737–45.
Wang D, Hoing S, Patterson HC, Ahmad UM, Rathinam VA, Rajewsky K, Fitzgerald KA, Golenbock DT. Inflammation in mice ectopically expressing human Pyogenic Arthritis, Pyoderma Gangrenosum, and Acne (PAPA) Syndrome-associated PSTPIP1 A230T mutant proteins. J Biol Chem. 2013;288(7):4594–601.
Tlougan BE, Podjasek JO, O’Haver J, Cordova KB, Nguyen XH, Tee R, Pinckard-Hansen KC, Hansen RC. Chronic recurrent multifocal osteomyelitis (CRMO) and synovitis, acne, pustulosis, hyperostosis, and osteitis (SAPHO) syndrome with associated neutrophilic dermatoses: a report of seven cases and review of the literature. Pediatr Dermatol. 2009;26(5):497–505.
Walsh P, Manners PJ, Vercoe J, Burgner D, Murray KJ. Chronic recurrent multifocal osteomyelitis in children: nine years’ experience at a statewide tertiary paediatric rheumatology referral centre. Rheumatology (Oxford). 2015;54(9):1688–91.
Tsitsami E, Dermentzoglou V, Moschovi M, Chrousos GP. Chronic recurrent multifocal osteomyelitis with concomitant features of juvenile idiopathic arthritis. Case Rep Rheumatol. 2011;2011:210795. https://doi.org/10.1155/2011/210795.
Zhao Y., Foster Shannon K., Murdock Todd J., Schlesinger M., Wallace Carol A. A rare case of chronic recurrent multifocal osteomyelitis with undifferentiated juvenile idiopathic arthritis, uveitis, and psoriasis. Case Rep Clin Med. 2016 5. DOI: https://doi.org/10.4236/crcm.2016.57041.
Girschick HJ, Zimmer C, Klaus G, Darge K, Dick A, Morbach H. Chronic recurrent multifocal osteomyelitis: what is it and how should it be treated? Nat Clin Pract Rheumatol. 2007;3(12):733–8.
Omidi CJ, Siegfried EC. Chronic recurrent multifocal osteomyelitis preceding pyoderma gangrenosum and occult ulcerative colitis in a pediatric patient. Pediatr Dermatol. 1998;15(6):435–8.
Pelkonen P, Ryoppy S, Jaaskelainen J, Rapola J, Repo H, Kaitila I. Chronic osteomyelitislike disease with negative bacterial cultures. Am J Dis Child. 1988;142(11):1167–73.
Brogan KS, Eleftheriou D, Biassoni L, Sebire N, Brogan PA. Multifocal noninfectious osteitis as a presentation of pediatric granulomatosis with polyangiitis (Wegener’s). Arthritis Rheumatol. 2015;67(10):2748.
Ferguson PJ, Chen S, Tayeh MK, Ochoa L, Leal SM, Pelet A, Munnich A, Lyonnet S, Majeed HA, El-Shanti H. Homozygous mutations in LPIN2 are responsible for the syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia (Majeed syndrome). J Med Genet. 2005;42(7):551–7.
Dagan O, Barak Y, Metzker A. Pyoderma gangrenosum and sterile multifocal osteomyelitis preceding the appearance of Takayasu arteritis. Pediatr Dermatol. 1995;12(1):39–42.
Daniela SA, Dent PB, Ferguson PJ, Laxer RM. Chronic Non-bacterial Osteomyelitis in a Child with Previous Juvenile Dermatomyositis. J Rheumatol. 2013;40(3):339–40.
Vettiyil G, Punnen A, Kumar S. An Unusual Association of Chronic Recurrent Multifocal Osteomyelitis, Pyoderma Gangrenosum, and Takayasu Arteritis. J Rheumatol. 2017;44(1):127–8.
Roderick MR, Shah R, Rogers V, Finn A, Ramanan AV. Chronic recurrent multifocal osteomyelitis (CRMO) - advancing the diagnosis. Pediatric Rheumatology. 2016;14(1):47.
Y Zhao, RM Laxer, PJ Ferguson. Treatment Advances in Chronic Non-Bacterial Osteomyelitis and Other Autoinflammatory Bone Conditions. Curr Treat Opt Rheumatol. 2017;3(1)17–32.
Ferrari J, Pilkington C. Chronic recurrent multifocal osteomyelitis: the prevalence of lower-limb and foot involvement. J Am Podiatr Med Assoc. 2014;104(6):583–7.
Costa-Reis P, Sullivan KE. Chronic Recurrent Multifocal Osteomyelitis. J Clin Immunol. 2013;33:1043–56.
Hedrich C. M., Hofmann S.R., Pablik J., Morbach H., Girschick, H. J. Autoinflammatory bone disorders with special focus on chronic recurrent multifocal osteomyelitis (CRMO). Pediatr Rheumatol Online J. 2013 11.
Ozen S, Pistorio A, Iusan SM, Bakkaloglu A, Herlin T, Brik R, Buoncompagni A, Lazar C, Bilge I, Uziel Y, Rigante D, Cantarini L, Hilario MO, Silva CA, Alegria MA, Norambuena X, Belot A, Berkun Y, Ibanez Estrella A, Olivieri AN, Alpigiani MG, Rumba I, Sztainbok F. EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis, and childhood Takayasu arteritis: Ankara 2008. Part II: final classification criteria. Ann Rheum Dis. 2010;69(5):798–806.
Pangoux C. Joint and bone manifestations in GPA. Can Rheumatol Assoc J. 2011;21(2):25–7.
Hofmann SR, Schwarz T, Möller JC, Morbach H, Schnabel A, Rösen-Wolff A, et al. Chronic non-bacterial osteomyelitis is associated with impaired Sp1 signaling, reduced IL10 promoter phosphorylation, and reduced myeloid IL-10 expression. Clin Immunol. 2011;141(3):317–27.
Wilde B, Thewissen M, Damoiseaux J, et al. Regulatory B cells in ANCA-associated vasculitis. Annals of the Rheumatic Diseases. 2013;72(8):1416–9.
Von Scheven E, Lu TT, Emery HM, Elder ME, Wara DW. Thrombosis and pediatric Wegener’s granulomatosis: Acquired and genetic risk factors for hypercoagulability. Arthr Rheum. 2003;49(6):862–5.
Tomasson G, Monach PA, Merkel PA. Thromboembolic disease in vasculitis. Curr Opin Rheumatol. 2009;21(1):41–6.
Singhal R, et al. Thromboembolic complications in the nephrotic syndrome: Pathophysiology and clinical management. Throm Res. 2006;118(3):397–407.
Karim F, Loeffen J, Bramer W, Westenberg L, Verdijk R, van Hagen M, et al. IgG4-related disease: a systematic review of this unrecognized disease in pediatrics. Pediatric Rheumatology. 2016;14:18.
Ryu JH, Horie R, Sekiguchi H, Peikert T, Yi ES. Spectrum of Disorders Associated with Elevated Serum IgG4 Levels Encountered in Clinical Practice. Int J Rheumatol. 2012;2012:232960. https://doi.org/10.1155/2012/232960.
Ebbo M, Grados A, Bernit E, et al. Pathologies Associated with Serum IgG4 Elevation. Int J Rheumatol. 2012;2012:602809. https://doi.org/10.1155/2012/602809.
Ata Y, Inaba Y, Choe H, Kobayashi N, Machida J, Nakamura N, et al. Bone metabolism and inflammatory characteristics in 14 cases of chronic nonbacterial osteomyelitis. Pediatr Rheumatol Online J. 2017;15(1):56.
Golla A, Jansson A, Ramser J, Hellebrand H, Zahn R, Meitinger T, et al. Chronic recurrent multifocal osteomyelitis (CRMO): evidence for a susceptibility gene located on chromosome 18q21.3-18q22. Eur J Hum Genet. 2002;10(3):217–21.
Hofmann SR, Kubasch AS, Range U, Laass MW, Morbach H, Girschick HJ, Hedrich CM. Serum biomarkers for the diagnosis and monitoring of chronic recurrent multifocal osteomyelitis (CRMO). Rheumatol Int. 2016;36(6):769–79.
Hofmann SR, Morbach H, Schwarz T, Rösen-Wolff A, Girschick HJ, Hedrich CM. Attenuated TLR4/MAPK signaling in monocytes from patients with CRMO resultsi n impaired IL-10 expression. Clin Immunol. 2012;145:69–76.
Mahler M, Silverman ED, Fritzler MJ. Novel diagnostic and clinical aspects of anti-PCNA antibodies detected by novel detection methods. Lupus. 2010;19(13):1527–33.
Beckman KA, Luchs J, Milner MS. Making the diagnosis of Sjögren’s syndrome in patients with dry eye. Clin Ophthalmol. 2015;2016(10):43–53.
Koenig M, Joyal F, Fritzler MJ, Roussin A, Abrahamowicz M, Boire G, Goulet J-R, Rich É, Grodzicky T, Raymond Y, Senécal J-L. Autoantibodies and microvascular damage are independent predictive factors for the progression of Raynaud’s phenomenon to systemic sclerosis: A twenty-year prospective study of 586 patients, with validation of proposed criteria for early systemic sclerosi. Arthritis Rheum. 2008;58:3902–12.
Bjorksten B, Boquist L. Histopathological aspects of chronic recurrent multifocal osteomyelitis. J Bone Joint Surg. 1980;62(3):376–80.
Girschick HJ, Raab P, Surbaum S, Trusen A, Kirschner S, Schneider P, et al. Chronic non-bacterial osteomyelitis in children. Ann Rheum Dis. 2005;64(2):279–85.
Pastore S, Ferrara G, Monasta L, Meini A, Cattalini M, Martino S, Alessio M, La Torre F, Teruzzi B, Gerloni V, Breda L, Taddio A, Lepore L. Chronic nonbacterial osteomyelitis may be associated with renal disease and bisphosphonates are a good option for the majority of patients. Acta Paediatr. 2016;105(7):328–33.
Roderick M, Shah R, Finn A, Ramanan AV. Efficacy of pamidronate therapy in children with chronic non-bacterial osteitis: disease activity assessment by whole body magnetic resonance imaging. Rheumatology (Oxford). 2014;53(11):1973–6.
Eleftheriou D, Gerschman T, Sebire N, Woo P, Pilkington CA, Brogan PA. Biologic therapy in refractory chronic non-bacterial osteomyelitis of childhood. Rheumatology. 2010;49(8):1505–12.
Voit AM, Arnoldi AP, Douis H, Bleisteiner F, Jansson MK, Reiser MF, et al. Whole-body magnetic resonance imaging in chronic recurrent multifocal osteomyelitis: clinical longterm assessment may underestimate activity. J Rheumatol. 2015;42:1455–62.
Fritz J, Tzaribatchev N, Claussen CD, Carrino JA, Horger MS. Chronic recurrent multifocal osteomyelitis: comparison of whole-body MR imaging with radiography and correlation with clinical and laboratory data. Radiology. 2009;252(3):842–51.
Roderick M, Shah R, Finn A, Ramanan AV. Efficacy of pamidronate therapy in children with chronic non-bacterial osteitis: disease activity assessment by whole body magnetic resonance imagin. Rheumatology (Oxford). 2014;53:1973–6.
Beck C, Morbach H, Beer M, Stenzel M, Tappe D, Gattenlöhner S, et al. Chronic nonbacterial osteomyelitis in childhood: prospective follow-up during the first year of anti-inflammatory treatment. Arthritis Res Ther. 2010;12(2):R74.
Acknowledgements
We are grateful for the assistance of Professor Arvydas Laurinavicius from Vilnius University Hospital and the team at the Lithuanian National Center of Pathology for histological evaluation of the kidney biopsies.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.
Author information
Authors and Affiliations
Contributions
AS was a major contributor in writing the manuscript. RS analysed and interpreted the patient‘s data regarding rheumatological diseases. JK analysed and interpreted the patient‘s data regarding oncological diseases and performed the bone biopsy. EJ analysed and interpreted the patient‘s data regarding endocrinological diseases. RU analysed and interpreted the patient‘s radiological scans. LJ designed and coordinated the writing of the manuscript, assisted in data interpretation and drafting of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Authors’ information
AS is a paediatric rheumatology resident at The Lithuanian University of Health Sciences (LUHS) and the Hospital of Lithuanian University of Health Sciences Kauno Klinikos (HLUHS KK).
RS is an Associated Professor at the Paediatric Clinic of LUHS, and is Head of the Department of Paediatric Rheumatology and Cardiology at the HLUHS KK.
JK is a paediatric heamatology doctor at the Department of Paediatric Oncology at the HLUHS KK.
EJ is a lecturer at the Endocrinology Clinic of the LUHS and Head of the Department of Paediatric Endocrinology at the HLUHS KK.
RU is a lecturer at the Radiology Clinic of the LUHS and a radiologist in the Radiology Department at the HLUHS KK.
LJ is a lecturer at the Paediatric Clinic of the LUHS, post-doc at the Department of Physiology and Pharmacology of the LUHS, and a paediatric pulmonology doctor in the Paediatric department at the HLUHS KK.
Ethics approval and consent to participate
The consent to use anonymic patient data for the case report was obtained from the parent of the patient.
Consent for publication
Written informed consent for publication of their clinical details and clinical images was obtained from the parent of the patient. A copy of the consent form is available for review by the Editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Snipaitiene, A., Sileikiene, R., Klimaite, J. et al. Unusual case of chronic recurrent multifocal osteomyelitis. Pediatr Rheumatol 16, 49 (2018). https://doi.org/10.1186/s12969-018-0267-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12969-018-0267-4