
Abstract
Background
Chemokine (C-C motif) receptor 8 (CCR8) is a chemokine receptor selectively expressed on tumor-infiltrating regulatory T cells (Tregs). Strong immunosuppression mediated by CCR8+ Tregs observed in breast and lung malignancies suggest for their functional significance in cancer therapy. To date, detailed characterization of tumor-infiltrating CCR8+ Tregs cells in colorectal cancer (CRC) is limited.
Methods
To study the presence and functional involvement of CCR8+ Tregs in CRC, we analyzed the proportions of CCR8-expressing T cells in different T cell subsets in tumor and adjacent normal tissues and peripheral blood mononuclear cells (PBMCs) from CRC patients by Flow cytometry. Also, we compared the distribution of CCR8+ T cells in malignant tissues and peripheral lymphoid organs from a subcutaneous CRC murine model. Bioinformatic analysis was performed to address the significance of CCR8 expression levels in CRC prognosis, immune regulatory gene expression profiles and potential molecular mechanisms associated with CCR8+ Tregs in CRC tumors. Further, we administrated an anti-CCR8 monoclonal antibody to CT26 tumor-bearing mice and examined the antitumor activity of CCR8-targeted therapy both in vivo and in an ex vivo confirmative model.
Results
Here, we showed that Tregs was predominantly presented in the tumors of CRC patients (13.4 ± 5.8, p < 0.0001) and the CRC subcutaneous murine model (35.0 ± 2.6, p < 0.0001). CCR8 was found to be preferentially expressed on these tumor-infiltrating Tregs (CRC patients: 63.6 ± 16.0, p < 0.0001; CRC murine model: 65.3 ± 9.5, p < 0.0001), which correlated with poor survival. We found that majority of the CCR8+ Tregs expressed activation markers and exhibited strong suppressive functions. Treatment with anti-CCR8 antibody hampered the growth of subcutaneous CRC tumor through effectively restoring the anti-tumor immunity of CD4+ conventional T cells (CD4+ Tconvs) and CD8+ T cells, which was confirmed in the ex vivo examinations.
Conclusions
Collectively, these findings illustrate the importance of CCR8+ Tregs for an immunosuppressive microenvironment in CRC tumors by functional inhibition of CD4+ Tconvs and CD8+ T cells, and suggest for the applicable value of CCR8-targeted therapy for CRC.
Similar content being viewed by others

Background
Colorectal cancer (CRC) has become the second leading cause of cancer death, with estimated global incidence of 1.93 to 3.2 million by 2040 [1, 2]. Despite the benefits of screening in reducing the morbidity and mortality of CRC patients, about 50% CRC patients have exhibited the spread of cancer at the time of diagnosis [3, 4]. The 5-year survival rate for individuals diagnosed with metastatic colorectal cancer (mCRC) is less than 15% [5]. Conventional treatments for CRC include surgery and adjuvant chemotherapy. Fluoropyrimidines and their combination with oxaliplatin, tyrosine-kinase inhibitors, and antibodies to VEGF and EGFR have been shown to partially increase the therapeutic outlook [6]. However, these treatments become largely ineffective with the occurrence of metastasis [6]. Therefore, the development of new treatment strategies to improve CRC patient survival is an ever growing need in the field of medical oncology [7,8,9,10,11,12].
In recent years, immune checkpoint therapy has remarkably improved the overall survival of CRC patients [13,14,15]. These treatments restore the anti-tumor activity of cytotoxic T cells and relieve tumor-mediated immunosuppression [16]. Currently, the complete response rate to immunotherapy is still not sufficient for CRC. Enriched presence and proficient infiltration of immunosuppressive cells in the tumors, such as Tregs, is one possible reason for the limited responses to immunotherapy in these CRC patients.
Tregs are a subset of CD4+ T cells that normally functions in maintaining self-tolerance and immune homeostasis. Accumulating evidences have revealed that the contribution of tumor-infiltrating Tregs to the immunosuppressive tumor microenvironment (TME) can be achieved through competitive interaction of Treg-expressed CD25 to IL-2 for inhibiting effector T cell activation and function, enhanced release of immunosuppressive molecules, such as IL-10, TGF-β, and IL-35, and direct functional inhibition of antigen-presenting cells (APCs) [17,18,19,20]. High infiltration of Tregs is often associated with worse prognosis in multiple types of cancers, including malignancies in the lung [21, 22], breast [23, 24], liver [25, 26], ovary [18, 27] and colorectal [28]. Therefore, approaches through depletion or functional inhibition of Tregs have drawn recent attentions in expectation to restore anti-tumor cellular immunity.
Recent attempts have been made targeting surface molecules on Tregs, including CTLA4, GITR, CCR4, PD-1, OX-40, and LAG3 and CD25 [17]. The lack of Treg-specificity of these molecules limits the anti-tumor immune response and reduced the efficacy of these targeted approaches [29]. Therefore, the development of more selective strategies to target tumor-infiltrating Tregs is needed. Chemokine (C-C motif) receptor 8 (CCR8) is a G protein-coupled receptor and a member of the chemokine receptor subfamily [30]. Elevated expression of CCR8 has been associated with poor immunotherapeutic outcomes in breast cancer, non-small-cell lung cancer (NSCLC) and bladder cancer patients [31]. Recent studies report that CCR8 is selectively up-regulated on tumor-infiltrating Tregs [23, 31,32,33,34,35,36], which is in conjunction with effective anti-tumor immune response by targeted approaches against CCR8 [29, 32, 34]. Despite these updated findings, the functional characteristics of CCR8+ Tregs in CRC patients have not been clearly elucidated, and the effect of CCR8+ Tregs on effector T cells remains to be further investigated.
In the present study, we examined the features of CCR8+ Tregs and their functional profiles in CRC patients and CRC subcutaneous murine model. We demonstrated that the proportion of Tregs in CD4+ T cells was significantly elevated in CRC tumors, and CCR8 was predominantly enriched on these tumor-infiltrating Tregs. High proportion of CCR8+ Treg cells in CRC tumors were correlated with poor patient survival. We found that majority of CCR8+ Treg cells exhibited activation markers with high functional immune suppression. CCR8-targeting antibodies effectively delayed CT26 subcutaneous tumor growth, by proficiently reducing the proportion of tumor-infiltrating Tregs in CD4+ T cells. Specifically, we demonstrated a marked reverse of the exhausted state of CD4+ Tconvs and CD8+ T cells by targeting CCR8, both in vivo and ex vivo. Collectively, these findings highlight the potential applicable value of targeted therapy against CCR8+ Treg for CRC treatment.
Materials and methods
Colorectal carcinoma patients
9 colorectal carcinoma tissues, 12 peripheral blood samples and 4 normal autologous colonic tissues of CRC patients during primary surgical treatment were collected from department of gastrointestinal surgery, the First Affiliated Hospital of Chongqing Medical University. No patient received neoadjuvant chemo- and/or radiotherapy. This project was approved by the ethics committee of Chongqing Medical University (2020 − 557). Informed consent was obtained from all individual donors before this research.
Cell line
Murine tumor cell lines CT26. WT cells (CRL-2638) were cultured in RPMI 1640 medium (Gibco, USA) supplemented with 10% (v/v) fetal bovine serum (ExCell, China), 2 mM L-glutamine, 100 µg/ml streptomycin and 100 units/ml penicillin at 37℃ in a 5% CO2 incubator.
Mice and tumour models
All animal experiments described in this study were reviewed and approved by the Institutional Animal Care and Use Committee of Chongqing Medical University (No. IACUC-CQMU-2024-0014). Six-week-old female BALB/c mice were supplied by the Animal Center of Chongqing Medical University (Chongqing, China) and raised under specific pathogen-free (SPF) standard conditions. 5 × 105 CT26 cells in 100 µl Dulbecco’s Phosphate Buffered Saline (PBS) were injected subcutaneously into the right shaved flanks of syngeneic BALB/c mice. CT26-bearing mice were subcutaneously inoculated with 4 µg purified rat IgG2b isotype antibody (rat IgG2b, k, Clone RTK4530; BioLegend) or purified anti-CCR8 antibody (rat IgG2b, k, clone SA214G2; BioLegend) on day 5, day 6, day 8, day 9, day 12, day 14 and day 15. Tumor volume (mm3) was measured using caliper and calculated according to the formula: V = (length × width2)/2. To analyze tumor-infiltrating T cells upon anti-CCR8 antibody treatment, tumors from all groups were harvested 20 days after tumor implantation.
Human tissue digestion and PBMCs isolation for flow cytometry
To prepare single-cell suspensions of human colorectal carcinoma, normal autologous colonic tissues, necrotic tissue and fat were first mechanically separated. Samples were cut into 1–9 mm2 fragments and digested for 30 min at 37℃ under agitation (RPMI 1640 medium supplemented with 1.5 mg/ml collagenase type II (Sigma-Aldrich, USA), 10 µg/mL DNase I (Solarbio, China), 2% fetal bovine serum). Digestion was stopped with RPMI 1640 medium containing 10% fetal bovine serum. Cell suspensions were filtered through a 70 μm cell strainer and resuspended in PBS containing 2% fetal bovine serum. Human whole blood was diluted with an equal volume of PBS with 2% fetal bovine serum and mixed gently. Peripheral blood mononuclear cells (PBMCs) separation was performed directly in a SepMate™ PBMCs Isolation Tubes (STEMCELL, Canada) with pre added Lymphoprep™ (STEMCELL, Canada). The diluted blood samples were centrifuged at 1200×g for 10 min at room temperature. PBMCs were washed twice using PBS with 2% fetal bovine serum.
Mouse tissue digestion and PBMCs isolation for flow cytometry
To obtain mouse tumor-infiltrating cells, separated tumor tissues were cut into 1–9 mm2 fragments and digested for 30 min at 37℃ under agitation (RPMI 1640 medium supplemented with 0.5 mg/ml collagenase type IV (Sigma-Aldrich, USA), 10 µg/ml DNase I (Solarbio, China), 2% fetal bovine serum). Digestion was stopped with RPMI 1640 medium containing 10% fetal bovine serum. Cell suspensions were filtered through a 70 μm cell strainer and resuspended in PBS containing 2% fetal bovine serum. PBMCs were isolated from mice peripheral blood and red blood cells lysed using lysis buffer (Biosharp, China). Cells were washed twice using PBS and resuspended in PBS containing 2% fetal bovine serum. Murine spleen and lymph nodes was first extirpated and mechanically squashed on nylon filter. The obtained cell suspensions were isolated by density gradient centrifugation with lymphocyte separation medium (DAKEWEI, China) and resuspended in PBS containing 2% fetal bovine serum. Single cell suspensions were used for flow cytometry.
Analysis of single-cell RNA-seq data for GO and KEGG
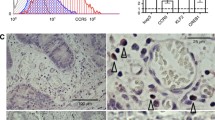
Single-cell RNA-seq data of human CRC tumor-infiltrating T cells were obtained from CRC_GSE108989 [37]. According to the expression density curve of CCR8 in Tregs, cells were divided into CCR8+ Tregs (1163 cells) and CCR8− Tregs (560 cells) with a threshold of 0.8. The FC values of all genes in the CCR8+ Tregs and CCR8− Tregs were calculated. The wilcoxon rank sum test was then used to calculate the significance of gene differences (p-value) and BH correction (fdr value). Differential genes were extracted according to fdr < 0.01 and log2 (FC) > 0.3, including 52 up-regulated genes (red) and 3 down-regulated genes (blue). The collated core targets were imported into Metascape (https://meta-scape.org/gp/index.html#/main/step1) for Gene ontology (GO) enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, and the results were visualized using bioinformatics.
Analysis of CCR8 antibody treatment on single-cell suspensions derived from ex vivo tumors
Three CT26 tumors were harvested 20 days after tumor implantation. The single cell suspension were prepared as above, then further isolated by density gradient centrifugation with lymphocyte separation medium (DAKEWEI, China) to remove most of the tumor cells. Cells were pelleted and then resuspended in RPMI supplemented with 10% fetal bovine serum and filtered again using a 70 μm cell strainer, then into 24-well plates and cultured for 72 h in the presence or absence of anti-CCR8 antibody (5 µg/ml) on plates coated with anti-CD3 antibody (2 µg/ml, BioLegend, USA) in the presence of anti-CD28 antibody (1 µg/ml, BioLegend, USA) and IL-2 (200 U/ml, Peprotech, USA) in flow cytometry assays as described below.
Flow cytometry analysis
For human samples, the following fluorochrome-conjugated antibodies were used: BV510-anti-CD3 (clone SK7, BioLegend), PerCP-Cy5.5-anti-CD4 (clone RPA-T4, BioLegend), FITC-anti-CD8 (clone HIT8a, BioLegend), BV650-anti-CD25 (clone BC96, BioLegend), APC-anti-CD127 (clone A019D5, BioLegend), BV785-anti-CD127 (clone A019D5, BioLegend), PE-anti-CCR8 (clone L263G8, BioLegend), APC-anti-CD134 (clone Ber-ACT35 (ACT35), BioLegend), APC-anti-CD137 (clone 4B4-1, BioLegend), BV605-anti-CD39 (clone A1, BioLegend), BV605-anti-PD-1 (clone EH12.1, BioLegend), APC-anti-TIM-3 (clone F38-2E2, BioLegend). The following fluorochrome-conjugated antibodies and reagents were used for flow cytometry of murine samples: BV510-anti-CD3 (clone 145-2C11, BioLegend), PerCP-Cy5.5-anti-CD4 (clone RM4-5, BioLegend), FITC-anti-CD8 (clone 53 − 6.7, BioLegend), BV785-anti-CD25 (clone PC61, BioLegend), BV650-anti-CD127 (clone A7R34, BioLegend), PE-anti-CCR8 (clone SA214G2, BioLegend), BV421-anti-CD134 (clone OX-86, BioLegend), APC-anti-CD137 (clone 17B5, BioLegend), APC-anti-CD39 (clone DUha59, BioLegend), APC-anti-PD-1 (clone RMP1-30, BioLegend), BV421-anti-TIM-3 (clone PMT3-23, BioLegend). Cells were stained by commercial antibodies at room temperature for 20 min. Then, the cells were washed by PBS and stained for 15 min using LIVE/DEAD viability dye (Thermo Fisher Scientific). Data were acquired using the FACSCelesta cytometer and analyzed by FlowJo software.
Statistical analysis
GraphPad Prism 8.0 (GraphPad Software, San Diego, CA, USA) was used for statistical analysis. Data are shown as the mean ± standard deviation (mean ± SD). Differences between groups were determined using unpaired t-test. When comparing more than two groups, one-way ANOVA or two-way ANOVA was used. p values < 0.05 were considered statistically significant (* p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001).
Results
CCR8 is selectively expressed on tumor-infiltrating Tregs
We first analyzed the proportion of different T cell subsets in the primary tumors of CRC patients (as shown in Table 1) by flow cytometry. The results showed that a significantly higher percentage of Tregs (CD4+CD25+CD127−) was presented in tumor-infiltrating T cell (TIL) population than in the para-neoplastic tissues (Para) or the PBMCs (Fig. 1A, B). This was in line with a markedly increased ratio of Tregs to CD8+ T cells in the TILs relative to those in the other two compartments (Fig. 1C). Next, we determined CCR8 expression in different T cell subsets by flow cytometry analysis. The result showed that the proportion of CCR8-expressing Tregs was markedly higher on tumor-infiltrating Tregs than that on Tregs from the PBMCs (Fig. 1D). These findings suggested that CCR8+ Tregs were predominantly enriched within the tumors of CRC patients.

CCR8 is selectively expressed on tumor-infiltrating Tregs in CRC patients. A-B Representative flow cytometry plots for Tregs (CD4+ CD25+ CD127-) staining of CD4+ T cells and the proportion of Tregs in CD4+ T cells from the peripheral blood mononuclear cells (PBMCs, n = 12), para-neoplastic tissues (Para, n = 4) and tumour-infiltrating T cells (TIL, n = 9) in CRC patients. C Ratio of Tregs to CD8+ T cells in PBMCs, Para and TIL. D Representative flow cytometry plots of CCR8 expression on CD8+ T, CD4+ Tconv (CD4+ CD25-) and Tregs within PBMCs (n = 12) and TIL (n = 9). The error bars represent SD, and B-Cp-values were calculated via unpaired t-test. Dp-value of the right graph was calculated using one-way ANOVA. *** p < 0.001, **** p < 0.0001. ns, no significant
This phenomenon was confirmed in a CRC murine model with subcutaneous CT26 tumors established in wild-type BALB/c mice. Similar to what was observed from CRC patient samples, the highest percentage of Tregs in CD4+ T cells was found in the tumor-infiltrating T cell population in CT26 tumor-bearing mice, compared with T cells obtained from the spleen, lymph nodes (LNs) and PBMCs (Fig. 2A). We also investigated CCR8 expression on Tregs in CT26 tumor-bearing mice. The result of flow cytometric analysis showed that the proportion of CCR8-expressing Tregs was significantly higher in the tumor-infiltrating Treg population, relative to those from the spleen, LNs and PBMCs (Fig. 2B). In addition, we showed that CCR8 expression was absent in CT26 tumor cells (Additional file 1: Figure S1). These data suggested that CCR8 was selectively expressed on tumor-infiltrating Tregs in both CRC patients and the subcutaneous tumors of CRC.

CCR8 is selectively expressed on tumor-infiltrating Tregs in CRC murine model with subcutaneous CT26 tumors. Spleen (Spl), PBMCs, lymph nodes (LNs) and TIL (n = 4 / group) from CT26 tumor-bearing mice were harvested 20 days after tumor implantation. A Representative flow cytometry plots for Tregs (CD4+ CD25+ CD127-) staining of CD4+ T cells and the proportion of Tregs in CD4+ T cells from the Spl, PBMCs, LNs and TIL in CT26 tumor-bearing mice. B Representative flow cytometry plots of CCR8 expression on Tregs within Spl, PBMCs, LNs and TIL in CT26 tumor-bearing mice. The error bars represent SD, and A-Bp-values were calculated using one-way ANOVA. * p < 0.05, **** p < 0.0001. ns, no significant
CRC tumor-infiltrating CCR8+ tregs exhibit strong immunosuppressive functions
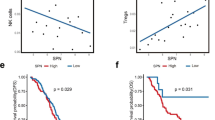
To confirm the significance of CCR8 expression in CRC prognosis, we first analyzed CRC patient data obtained from The Cancer Genome Atlas (TCGA). The results showed that the overall survival of CRC patients with high level CCR8 expression was significantly reduced than that of the CCR8 low-level group (Fig. 3A). Next, we analyzed immune regulatory gene expression profile associated with CCR8+ Tregs in CRC tumors using the CRC_GSE108989 dataset. Genes functionally correlated with immune suppression, such as CTLA4, IL2RA (CD25), TIGIT and ENTPD1 (CD39), and T cell activation signature genes, such as TNFRSF4 (CD134) and TNFRSF9 (CD137), were all expressed at higher levels in CCR8+ Tregs compared with their CCR8− counterparts (Fig. 3B, C). These data indicated that tumor-infiltrating CCR8+ Tregs might be functionally activated and preferentially contribute to an immune suppressive microenvironment.
To elucidate possible molecular mechanisms associated with CCR8+ Tregs, we analyzed the differentially expressed gene (DEG) profile. GO enrichment results showed that the DEGs between CCR8+ and CCR8− Tregs were predominantly enriched in cytokine-mediated signaling pathway, regulation of leukocyte activation, regulation of I-kappaB kinase/NF-kappaB signaling and inflammatory response (Fig. 3D). KEGG pathways enrichment analysis revealed that cytokine-cytokine receptor interaction, viral carcinogenesis, TNF signaling pathway and Glycolysis/Gluconeogenesis exhibited high level distinguishment between these groups with diverse CCR8 expressions (Fig. 3E).

CRC tumor-infiltrating CCR8+ Tregs exhibit strong immunosuppressive function by bioinformatics analysis. A Overall survival curves showing the significant differences of CRC patients stratified based on the median CCR8 mRNA expression in the cohorts from The Cancer Genome Atlas (TCGA). B Volcano plot of genes significantly differentially expressed between CCR8- Tregs and CCR8+ Tregs in the CRC TME were obtained from the CRC_GSE108989 dataset [37]. Up-regulated genes were indicated in red and down-regulated genes were indicated in blue. C Heatmap of Treg signature genes expressed by CCR8- Tregs and CCR8+ Tregs in B. D-E GO functional enrichment analysis (D) and KEGG pathway enrichment analysis (E) based on differential expressed genes between CCR8- Tregs and CCR8+ Tregs
Up-regulated expression of Treg signature markers in intratumoral CCR8+Tregs
We utilized tumor samples collected from CRC patients to verify the results of bioinformatics analysis shown above. CRC tumor-infiltrating Tregs were divided into CCR8+ Tregs and CCR8− Tregs in human CRC tumor by flow cytometry. We assessed the expressions of Treg-related activation and suppression functional molecules and found significantly increased levels of CD134, CD137, CD39 and PD-1 in CCR8+ Tregs compared with CCR8− Tregs (Fig. 4A). Similarly, tumor-infiltrating CCR8+ Tregs in CT26 tumor-bearing mice demonstrated marked elevation in CD137, CD39, PD-1 and TIM-3 expressions relative to CCR8− Tregs, while no significant changes were observed for CD134 levels (Fig. 4B). These data demonstrated that tumor-infiltrating CCR8+ Tregs exhibited up-regulation of Treg signature molecules, suggesting for enhanced immune suppressive functions of these Tregs in CRC tumors.

Up-regulated expression of Treg signature markers in intratumoral CCR8+ Tregs measured by flow cytometry. A Frequency of CD134, CD137, CD39, PD-1 and TIM-3 expression on tumor-infiltrating CCR8- Tregs (blue) and CCR8+ Tregs (red) subsets from CRC patients (n = 3), respectively. B Frequency of CD134, CD137, CD39, PD-1 and TIM-3 expression on tumor-infiltrating CCR8- Tregs (blue) and CCR8+ Tregs (red) from CT26 tumor-bearing mice (n = 7), respectively. The error bars represent SD, and A-B p-values were calculated using unpaired t-test. * p < 0.05, ** p < 0.01, *** p < 0.001. ns, no significant
CCR8-targeted therapy reduces Tregs immunosuppression and enhances T cell antitumor immunity
To determine the potential antitumor activity of CCR8-targeted therapy, we treated CT26 tumor-bearing mice with an anti-CCR8 antibody. We found that the anti-CCR8 antibody treatment markedly delayed tumor growth than the isotype IgG2b control group after the initial three doses, and further treatment with the anti-CCR8 antibody completely eradicated the tumor by 18 days (Fig. 5A). Given the effective antibody-induced obliteration of tumors, the T cell subsets in LNs of the CT26 tumor-bearing mice were analyzed by flow cytometric analysis. The results showed that the anti-CCR8 antibody not only significantly reduced the frequency of Tregs in LNs, but also decreased the ratio of Treg/CD8+ T cells within LNs (Fig. 5B, C). Examination of functional markers in Tregs in LNs showed that CD39, PD-1 and TIM-3 were expressed at low levels in the anti-CCR8 antibody treated group than the IgG2b isotype control group (Fig. 5D). Moreover, we assessed exhaustion markers in CD4+ Tconv and CD8+ T cells in LNs. The results showed that both CD4+ Tconv cells and CD8+ T cells of the anti-CCR8 therapy group exhibited significantly lower levels of PD-1, TIM-3 and LAG-3 than those of the control group (Fig. 5E, F).

Targeting CCR8 reduces Tregs immunosuppression and enhances T cell antitumor immunity in vivo. A Growth curves of CT26-bearing mice treated with anti-CCR8 antibody and isotype IgG2b control (n = 5 / group). B Frequency of Tregs in CD4+ T cells of LNs in CT26 tumor-bearing mice after anti-CCR8 antibody treatment (n = 5). C Ratio of Tregs to CD8+ T of LNs in CT26 tumor-bearing mice after anti-CCR8 antibody treatment (n = 5). D Frequency of CD39, PD-1 and TIM-3 expression on Tregs of LNs in CT26 tumor-bearing mice after anti-CCR8 antibody treatment (n = 4–5), respectively. E Frequency of PD-1, TIM-3 and LAG-3 expression on CD4+ Tconvs of LNs in CT26 tumor-bearing mice after anti-CCR8 antibody treatment (n = 4–5), respectively. F Frequency of PD-1, TIM-3 and LAG-3 expression on CD8+ T cells of LNs in CT26 tumor-bearing mice after anti-CCR8 antibody treatment (n = 3–5), respectively. The error bars represent SD, and A p-values were calculated using two-way ANOVA. B-F p-values were calculated using unpaired test. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. ns, no significant
To further confirm the effect of anti-CCR8 therapy on Tregs immunosuppression, we prepared single cell suspension of the CT26 subcutaneous tumors established in BALB/c mice and subjected these cell suspensions to the anti-CCR8 antibody treatment ex vivo. Flow cytometric analysis results showed that the proportion of Tregs was significantly decreased after treatment with CCR8 antibody for 3 days, in comparison with the control group (Fig. 6A). This was accompanied by a markedly reduced ratio of Treg/CD8+ T cells (Fig. 6B). The proportions of Tregs with surface expressions of PD-1, TIM-3 and LAG-3 were significantly reduced by the treatment of the anti-CCR8 antibody, indicative of decreased immune suppressive functions of Tregs (Fig. 6C). Meanwhile, we observed that the proportions of PD-1+, TIM3+ and LAG-3+ CD4+ Tconv (Fig. 6D) and CD8+ T cells (Fig. 6E) were also robustly reduced after treatment with the CCR8 antibody, suggesting an effective reversal of cellular immunity. Collectively, the anti-CCR8 antibody treatment could selectively deplete the CCR8+ Treg population with profound immune suppressive functions, and enhance the antitumor immunity of effector T cells by antagonizing T cell exhaustion.

Targeting CCR8 reduces Tregs immunosuppression and enhances T cell antitumor immunity ex vivo. Single cell suspension of the CT26 subcutaneous tumors established in BALB/c mice were harvested from day 20 implanted CT26 nontreated tumors and cultured for 72 h in the presence or absence of anti-CCR8 antibody on plates coated with anti-CD3 antibody in the presence of anti-CD28 antibody and IL-2. A Frequency of Tregs in CD4+ T cells in ex vivo CT26-bearing tumor after anti-CCR8 antibody treatment (n = 4). B Ratio of Treg to CD8+ T in ex vivo CT26-bearing tumor after anti-CCR8 antibody treatment (n = 4). C Frequency of PD-1, TIM-3 and LAG-3 expression on Tregs in ex vivo CT26-bearing tumor after anti-CCR8 antibody treatment (n = 4). D Frequency of PD-1, TIM-3 and LAG-3 expression on CD4+ Tconvs in ex vivo CT26-bearing tumor after anti-CCR8 antibody treatment (n = 4). E Frequency of PD-1, TIM-3 and LAG-3 expression on CD8+ T cells in ex vivo CT26-bearing tumor after anti-CCR8 antibody treatment (n = 3). The error bars represent SD, and A-E p-values were calculated using unpaired t-test. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. ns, no significant
Discussion
Tumor-infiltrating Tregs is one of the major obstacles for driving effective tumor-specific immune responses, and Treg-targeted therapy is expected to improve the anti-tumor immunity. In the present study, we found that the proportion of Tregs in CD4+ T cell populations were remarkably higher in the tumors of CRC patients and a subcutaneous CRC murine model, relative to that in PBMCs, in the spleen or in tumor-draining LNs. Moreover, we showed that CCR8 expression was preferentially enriched in tumor-infiltrating Tregs rather than Tregs from PBMCs in CRC patient samples. Similar result was found in the CT26 subcutaneous tumor-baring immune competent BALB/c mice. Notably, we found that CCR8 was expressed at comparatively low levels in the CT26 tumor cells, or in Tregs from the PBMCs, LNs or the spleen. Further, the tumor-infiltrating CCR8+ Tregs were shown to be correlated with an activated functional state and exhibited strong immune suppressive features. Targeting approach against these CCR8+ Tregs by a CCR8 specific antibody demonstrated effective abolishment of Tregs functions and a re-establishment of the anti-tumor activities of effector T cells, both in vivo and in the CRC tumor single cell suspension ex vivo examinations. The complete obliteration of subcutaneous tumors with continued CCR8-antagonizing antibody treatment highlighted a potential applicable basis for the CCR8-targeted strategies in CRC clinical therapeutic development.
The observed elevation in the proportion of tumor-infiltrating Tregs suggests for poor anti-cancer immune activities in CRC patients as well as a less desired immunotherapy outcome in these patients. The selective expression of CCR8 on infiltrating-Tregs of CRC tumor provided a unique role of this surface receptor as an indicator of Treg percentages in CRC. Indeed, analysis of CRC patient data obtained from TCGA showed a shorter overall survival in CRC patients with high CCR8 expression, with similar prognostic tendencies in breast, lung and bladder cancer patients [23, 29, 31]. Additional analysis revealed the functional significance of CCR8+ Tregs, as the expression of Treg-related molecules and the immune inhibitory effect were markedly higher with this cell population than the CCR8− Tregs. These findings, together with the observed low level of CCR8 expression on effector T cells, imply for a practical therapeutic window for CCR8-targeted therapy.
To date, focused study utilizing single-cell RNA-seq data from human tumor samples and mice samples to address the features of CCR8+ Tregs is rare [29, 31,32,33,34]. In the present study, we took advantage of available single-cell RNA-seq data [37] and systematically analyzed the immune regulatory gene expression profile associated with CCR8+ Tregs in CRC tumors. We revealed a significance of CCR8+ Treg in CRC disease progression, which shared functional similarity to those observed in other types of cancer and yet had not been reported in gastrointestinal malignancies from this perspective. The essential role of CCR8+ Treg in CRC tumor regulation was further validated with patient tumor samples and in a CRC subcutaneous murine model. In addition, we explored potential signaling activities associated with CCR8+ Tregs in CRC patient tumors, and uncovered a series of molecular explanations with comparatively higher association to the immune suppressive functions of these cells rather than their CCR8− counterparts. For example, GO analysis and KEGG analysis both identified the enrichment of cytokine-mediated signaling activities in CRC tumor infiltrating CCR8+ Tregs. The activation of this pathway has been shown to foster the differentiation and enhance the immunosuppressive capabilities of Tregs, both of which are essential in facilitating the immune suppressive secretome and antagonizing the tumor cell killing of cytotoxic TILs [38]. This could serve as a potential functional explanation of the sharpened tumor inhibition observed in the CRC murine model by CCR8-targeting, although further validation with broader CRC cell lines choices and mechanism characterization and confirmation are definitely needed.
Other than inhibiting the cytotoxic execution of effector T cells, Tregs can hamper the functions of a variety of immune cells in the tumor microenvironment [17, 39]. We observed enhanced expression of CTLA4 and TIGIT in CCR8+ Tregs than in CCR8− Tregs in our study, which encouraged the speculation of a preferential functional suppression of APCs by the CCR8+ Tregs. Also, we found elevated expression of ENTPD1 (CD39) in CCR8+ Tregs relative to the CCR8− Tregs, which suggested for a possible reduction in effector T cell activation and function through CD39/CD73-mediated metabolite restrain [40, 41]. Similarly, the enhanced IL2RA (CD25) levels in CCR8+ Tregs might be account for increased intracellular signal transduction in response to IL-2, highlighting a possible feed-forward loop for the enrichment of CCR8+ Tregs in the TME, which further the immune escape of CRC tumors. In depth analysis is needed to determine vital mechanisms unique to CCR8+ Tregs in the suppression of effector T cell activation and functions.
Utilizing a commercially available CCR8 antibody, we demonstrated complete tumor growth regression in the CT26 subcutaneous model. In absence of tumors, we moved to an examination of T cell subsets in lymph nodes, the main sites of immune response, which was confirmed by an additional analysis with tumor single-cell suspension. The abolishment of Tregs functions and the re-establishment of anti-tumor functions of effector T cells confirmed successful disengagement in the Treg-modulated immunosuppressive network. Given the lack of CCR8 expression in CT26 cells, the robust tumor growth inhibition was largely due to the cytotoxicity of effector T cells that experienced a gain-of-function subsequent to targeting of CCR8+ Tregs. The observed decrease in the expression of apoptosis indicators (PD-1, TIM-3 and LAG-3) in the effector T cell populations indicated a reduce of cellular apoptosis and an effective reversal of cellular immunity. This is of particular importance for improving the immunotherapeutic efficacies clinically applied for CRC treatment. On one hand, as the CCR8+ Tregs represented a group of Tregs with advanced immunosuppression, the depletion of these cells might provide fundamental increase in effector T cell functions both in the lymph nodes and tumors. On the other hand, as we observed a close correlation in the expression of CCR8 and PD-1 on CCR8+ Treg cell surface, the alternate targeting of CCR8+ Tregs might be beneficial for overcoming the reported activation of PD-1+ Treg cells induced by anti-PD-1 therapy [42, 43], thus pausing the Immune Checkpoint Blockade (ICB)-related hyperprogression of malignancies.
Immunotherapy is a currently wide-applied therapeutic approach with characterized clinical benefits to many cancers. Targeting immune checkpoint proteins, like CTLA4, PD-1 and its ligand (PD-L1) has been shown to restore and enhance the anti-tumor function of effector T cells in a fraction of CRC patients [44]. But the improvement of ICB therapeutic effectiveness still stands as a central focus of medical research of CRC [45]. Given the identified unique expression of CCR8 in CRC tumors, CCR8-targeted therapy can weaken the immune-suppresive function of CCR8+ Treg and inhibit tumor growth with minimum off-target risks. The combination efficacy of ICB and CCR8-targeted therapy and the corresponding molecular profiles need to be further addressed.
Conclusions
Our study demonstrated that CCR8 was selectively expressed on CRC tumor-infiltrating Tregs. These CCR8+ Tregs were predominantly activated and exhibited strong immune suppressive features. CCR8-targeted therapy could induce complete tumor growth regression, suggesting for a possible duo role of CCR8 in CRC prognosis and immunotherapies. Together, these findings evidence a basis for the development of CCR8-targeted approaches for the treatment of CRC.
Data availability
Data generated and used and/or analyzed during are available from the corresponding author on reasonable request.
Abbreviations
- CCR8:
-
Chemokine (C-C motif) receptor 8
- Tregs:
-
Regulatory T cells
- CRC:
-
Colorectal cancer
- Tconvs :
-
Conventional T cells
- TME:
-
Tumor microenvironment
- APCs:
-
Antigen-presenting cells
- NSCLC:
-
Non-small-cell lung cancer
- TIL:
-
Tumor-infiltrating T cell
- Para:
-
Para-neoplastic tissues
- PBMCs:
-
Peripheral blood mononuclear cells
- LNs:
-
Lymph nodes
- TCGA:
-
The Cancer Genome Atlas
- DEG:
-
Differentially expressed gene
- GO:
-
Gene ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- SPF:
-
Specific pathogen-free
- PBS:
-
Dulbecco’s Phosphate Buffered Saline
References
Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, Jemal A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74:229–63.
Xi Y, Xu P. Global colorectal cancer burden in 2020 and projections to 2040. Transl Oncol. 2021;14:101174.
Edwards BK, Ward E, Kohler BA, Eheman C, Zauber AG, Anderson RN, Jemal A, Schymura MJ, Lansdorp-Vogelaar I, Seeff LC, et al. Annual report to the nation on the status of cancer, 1975–2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer. 2010;116:544–73.
Sargent D, Sobrero A, Grothey A, O’Connell MJ, Buyse M, Andre T, Zheng Y, Green E, Labianca R, O’Callaghan C, et al. Evidence for cure by adjuvant therapy in colon cancer: observations based on individual patient data from 20,898 patients on 18 randomized trials. J Clin Oncol. 2009;27:872–7.
Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC, Cercek A, Smith RA, Jemal A. Colorectal cancer statistics, 2020. CA Cancer J Clin. 2020;70:145–64.
Martinez-Perez J, Torrado C, Dominguez-Cejudo MA, Valladares-Ayerbes M. Targeted treatment against Cancer Stem cells in Colorectal Cancer. Int J Mol Sci. 2024;25.
Ali I, Wani WA, Haque A, Saleem K. Glutamic acid and its derivatives: candidates for rational design of anticancer drugs. Future Med Chem. 2013;5:961–78.
Ali I, Wani WA, Khan A, Haque A, Ahmad A, Saleem K, Manzoor N. Synthesis and synergistic antifungal activities of a pyrazoline based ligand and its copper(II) and nickel(II) complexes with conventional antifungals. Microb Pathog. 2012;53:66–73.
Ali I, Wani WA, Saleem K, Wesselinova D, Syntheses. DNA binding and anticancer profiles of L-glutamic acid ligand and its copper(II) and ruthenium(III) complexes. Med Chem. 2013;9:11–21.
Ali I, Alsehli M, Scotti L, Tullius Scotti M, Tsai ST, Yu RS, Hsieh MF, Chen JC. Progress in polymeric nano-medicines for theranostic cancer treatment. Polym (Basel). 2020;12.
Ali I, Aboul-Enein HY, Ghanem A. Enantioselective toxicity and carcinogenesis. Curr Pharm Anal. 2005;1:109–25.
Ali I, Wani WA, Saleem K, Hsieh MF. Anticancer metallodrugs of glutamic acid sulphonamides: in silico, DNA binding, hemolysis and anticancer studies. RSC Adv. 2014;4:29629–41.
Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, Desai J, Hill A, Axelson M, Moss RA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18:1182–91.
Andre T, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, Morse MA, Van Cutsem E, McDermott R, Hill A, et al. Nivolumab plus low-dose ipilimumab in previously treated patients with microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer: 4-year follow-up from CheckMate 142. Ann Oncol. 2022;33:1052–60.
Lenz HJ, Van Cutsem E, Luisa Limon M, Wong KYM, Hendlisz A, Aglietta M, Garcia-Alfonso P, Neyns B, Luppi G, Cardin DB, et al. First-line nivolumab plus low-dose ipilimumab for microsatellite Instability-High/Mismatch repair-deficient metastatic colorectal Cancer: the phase II CheckMate 142 study. J Clin Oncol. 2022;40:161–70.
Kanani A, Veen T, Soreide K. Neoadjuvant immunotherapy in primary and metastatic colorectal cancer. Br J Surg. 2021;108:1417–25.
Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27:109–18.
Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–9.
Olguín JE, Medina-Andrade I, Rodríguez T, Rodríguez-Sosa M, Terrazas LI. Relevance of Regulatory T Cells during colorectal Cancer Development. Cancers. 2020;12.
Nishikawa H, Koyama S. Mechanisms of regulatory T cell infiltration in tumors: implications for innovative immune precision therapies. J Immunother Cancer. 2021;9.
Tao H, Mimura Y, Aoe K, Kobayashi S, Yamamoto H, Matsuda E, Okabe K, Matsumoto T, Sugi K, Ueoka H. Prognostic potential of FOXP3 expression in non-small cell lung cancer cells combined with tumor-infiltrating regulatory T cells. Lung Cancer. 2012;75:95–101.
Soo RA, Chen Z, Yan Teng RS, Tan HL, Iacopetta B, Tai BC, Soong R. Prognostic significance of immune cells in non-small cell lung cancer: meta-analysis. Oncotarget. 2018;9:24801–20.
Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV, Chudakov DM, Rudensky AY. Regulatory T cells exhibit distinct features in human breast Cancer. Immunity. 2016;45:1122–34.
Liu S, Foulkes WD, Leung S, Gao D, Lau S, Kos Z, Nielsen TO. Prognostic significance of FOXP3 + tumor-infiltrating lymphocytes in breast cancer depends on estrogen receptor and human epidermal growth factor receptor-2 expression status and concurrent cytotoxic T-cell infiltration. Breast Cancer Res. 2014;16:432.
Sun L, Xu G, Liao W, Yang H, Xu H, Du S, Zhao H, Lu X, Sang X, Mao Y. Clinicopathologic and prognostic significance of regulatory T cells in patients with hepatocellular carcinoma: a meta-analysis. Oncotarget. 2017;8:39658–72.
Sasaki A, Tanaka F, Mimori K, Inoue H, Kai S, Shibata K, Ohta M, Kitano S, Mori M. Prognostic value of tumor-infiltrating FOXP3 + regulatory T cells in patients with hepatocellular carcinoma. Eur J Surg Oncol. 2008;34:173–9.
Wolf D, Wolf AM, Rumpold H, Fiegl H, Zeimet AG, Muller-Holzner E, Deibl M, Gastl G, Gunsilius E, Marth C. The expression of the regulatory T cell-specific forkhead box transcription factor FoxP3 is associated with poor prognosis in ovarian cancer. Clin Cancer Res. 2005;11:8326–31.
Ladoire S, Martin F, Ghiringhelli F. Prognostic role of FOXP3 + regulatory T cells infiltrating human carcinomas: the paradox of colorectal cancer. Cancer Immunol Immunotherapy. 2011;60:909–18.
Haruna M, Ueyama A, Yamamoto Y, Hirata M, Goto K, Yoshida H, Higuchi N, Yoshida T, Kidani Y, Nakamura Y, et al. The impact of CCR8 + regulatory T cells on cytotoxic T cell function in human lung cancer. Sci Rep. 2022;12:5377.
Islam SA, Ling MF, Leung J, Shreffler WG, Luster AD. Identification of human CCR8 as a CCL18 receptor. J Exp Med. 2013;210:1889–98.
Wang T, Zhou Q, Zeng H, Zhang H, Liu Z, Shao J, Wang Z, Xiong Y, Wang J, Bai Q, et al. CCR8 blockade primes anti-tumor immunity through intratumoral regulatory T cells destabilization in muscle-invasive bladder cancer. Cancer Immunol Immunother. 2020;69:1855–67.
Villarreal DO, L’Huillier A, Armington S, Mottershead C, Filippova EV, Coder BD, Petit RG, Princiotta MF. Targeting CCR8 induces protective antitumor immunity and enhances Vaccine-Induced responses in Colon cancer. Cancer Res. 2018;78:5340–8.
Van Damme H, Dombrecht B, Kiss M, Roose H, Allen E, Van Overmeire E, Kancheva D, Martens L, Murgaski A, Bardet PMR et al. Therapeutic depletion of CCR8(+) tumor-infiltrating regulatory T cells elicits antitumor immunity and synergizes with anti-PD-1 therapy. J Immunother Cancer. 2021;9.
Kidani Y, Nogami W, Yasumizu Y, Kawashima A, Tanaka A, Sonoda Y, Tona Y, Nashiki K, Matsumoto R, Hagiwara M et al. CCR8-targeted specific depletion of clonally expanded Treg cells in tumor tissues evokes potent tumor immunity with long-lasting memory. Proc Natl Acad Sci U S A. 2022;119.
Campbell JR, McDonald BR, Mesko PB, Siemers NO, Singh PB, Selby M, Sproul TW, Korman AJ, Vlach LM, Houser J, et al. Fc-Optimized Anti-CCR8 antibody depletes Regulatory T cells in human tumor models. Cancer Res. 2021;81:2983–94.
De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, Bonnal RJP, Provasi E, Sarnicola ML, Panzeri I, et al. Transcriptional Landscape of Human Tissue Lymphocytes Unveils Uniqueness of Tumor-Infiltrating T Regulatory Cells. Immunity. 2016;45:1135–47.
Zhang L, Yu X, Zheng LT, Zhang YY, Li YS, Fang Q, Gao RR, Kang BX, Zhang QM, Huang JY, et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature. 2018;564:268–.
Zong Y, Deng K, Chong WP. Regulation of Treg cells by cytokine signaling and co-stimulatory molecules. Front Immunol. 2024;15:1387975.
Olguin JE, Medina-Andrade I, Rodriguez T, Rodriguez-Sosa M, Terrazas LI. Relevance of Regulatory T Cells during colorectal Cancer Development. Cancers (Basel). 2020;12.
Kumar V. Adenosine as an endogenous immunoregulator in cancer pathogenesis: where to go? Purinergic Signalling. 2013;9:145–65.
Antonioli L, Pacher P, Vizi ES, Haskó G. CD39 and CD73 in immunity and inflammation. Trends Mol Med. 2013;19:355–67.
Kamada T, Togashi Y, Tay C, Ha D, Sasaki A, Nakamura Y, Sato E, Fukuoka S, Tada Y, Tanaka A, et al. PD-1(+) regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc Natl Acad Sci U S A. 2019;116:9999–10008.
Kumagai S, Togashi Y, Kamada T, Sugiyama E, Nishinakamura H, Takeuchi Y, Vitaly K, Itahashi K, Maeda Y, Matsui S, et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat Immunol. 2020;21:1346–58.
Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018;8:1069–86.
Ou QL, Chang YL, Liu JH, Yan HX, Chen LZ, Guo DY, Zhang SF. Mapping the intellectual structure and landscape of colorectal cancer immunotherapy: a bibliometric analysis. Hum Vaccin Immunother. 2024;20:2323861.
Funding
This work was supported by the China Postdoctoral Science Foundation (Grant No. 2019M663444), Science and Technology Research Program of Chongqing Municipal Education Commission (Grant No. KJQN202300440).
Author information
Authors and Affiliations
Contributions
A.J. and C.H. conceived and designed the experiments; Y.L. and Z.W. collected CRC patient samples; Q.C., C.H., M.S., X.H., S.M., L.L., Y.W., T.L and Y.W. conducted experiments; Q.C., C.H., M.Y. and S.L. performed data analysis and incorporation; C.H., Q.C., W.W. and A.J. wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The studies involving human participants were reviewed and approved by the Ethical Committee of the First Affiliated Hospital of Chongqing Medical University (No. 2020 − 557). All animal experiments described in this study were reviewed and approved by the Institutional Animal Care and Use Committee of Chongqing Medical University (No. IACUC-CQMU-2024-0014).
Consent for publication
The authors consent for publication.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.

Supplementary Material 1
: Figure S1. CCR8 expression was absent in CT26 tumor cells
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it.The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

{kind=link}
Cite this article
Chen, Q., Shen, M., Yan, M. et al. Targeting tumor-infiltrating CCR8+ regulatory T cells induces antitumor immunity through functional restoration of CD4+ Tconvs and CD8+ T cells in colorectal cancer. J Transl Med 22, 709 (2024). https://doi.org/10.1186/s12967-024-05518-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-024-05518-8