
Abstract
Background
Gastric intestinal metaplasia (GIM) is an essential precancerous lesion. Although the reversal of GIM is challenging, it potentially brings a state-to-art strategy for gastric cancer therapeutics (GC). The lack of the appropriate in vitro model limits studies of GIM pathogenesis, which is the issue this work aims to address for further studies.
Method
The air-liquid interface (ALI) model was adopted for the long-term culture of GIM cells in the present work. This study conducted Immunofluorescence (IF), quantitative real-time polymerase chain reaction (qRT-PCR), transcriptomic sequencing, and mucoproteomic sequencing (MS) techniques to identify the pathways for differential expressed genes (DEGs) enrichment among different groups, furthermore, to verify novel biomarkers of GIM cells.
Result
Our study suggests that GIM-ALI model is analog to the innate GIM cells, which thus can be used for mucus collection and drug screening. We found genes MUC17, CDA, TRIM15, TBX3, FLVCR2, ONECUT2, ACY3, NMUR2, and MAL2 were highly expressed in GIM cells, while GLDN, SLC5A5, MAL, and MALAT1 showed down-regulated, which can be used as potential biomarkers for GIM cells. In parallel, these genes that highly expressed in GIM samples were mainly involved in cancer-related pathways, such as the MAPK signal pathway and oxidative phosphorylation signal pathway.
Conclusion
The ALI model is validated for the first time for the in vitro study of GIM. GIM-ALI model is a novel in vitro model that can mimic the tissue micro-environment in GIM patients and further provide an avenue for studying the characteristics of GIM mucus. Our study identified new markers of GIM as well as pathways associated with GIM, which provides outstanding insight for exploring GIM pathogenesis and potentially other related conditions.
Similar content being viewed by others


Introduction
Gastric intestinal metaplasia (GIM) is a pathological type of intestinal mucosa-like morphological and structural alternations in the gastric mucosa, and represents a risk factor for intestinal-type gastric cancer (GC) [1, 2]. Approximately 0.25% and 10% of GIM patients progress to GC annually in Western Europe and East Asia, respectively [3, 4]. GIM has been recognized as a precancerous lesion and its reverse can be significant for GC curing. GIM can be led by chronic inflammation in stomach, mainly caused by Helicobacter pylori infection, followed by autoimmune diseases [5]. However, its pathogenesis remains unclear and it is challenging to study GIM without an appropriate in vitro model. Thus, confirming an appropriate in vitro model that is analog to the tissues from GIM patients is essential to GIM and GC studies and treatments.
Clonal results from human gastric specimens suggest that GIM glands are clonal and can form large clonal plaques through the division of the glands. This process is known as “regional carcinogenesis”, indicating GIM originates from gastric mucosal stem cells [6]. The organoids model and the air-liquid interface (ALI) model are two main stem cell models [7], notably, organoids are miniature cell clusters formed by cells in a three-dimensional (3D) space in vitro, which can self-proliferate and differentiate into different cell types [8]. A necessary condition for a successful in vitro model for human disease is the persistent proliferation of certain cells [9]. It has been reported that Wnt and R-spondin administrated in vitro can maintain the stemness of stem cells and promote their continuous proliferation [10, 11]. The previous studies have shown that human gastric mucosal epithelial stem cells can be expanded indefinitely in 3D stromal cultures [12, 13]. The unique advantage of 3D organoids lies in their promising ability to faithfully recapitulate the intricate structure, functional properties, and genetic characteristics of the original tissue in vivo. This feature enables them to serve as a highly realistic and representative model, allowing for the accurate simulation of the complex dynamics observed in GIM tissue [14]. However, one limitation of 3D organoids is their tendency to form compact structures, which can hinder the penetration of drugs or microorganisms into the innermost cells [15]. Besides, the traditional 3D organoid model has certain restrictions when it comes to collecting mucus. The air-liquid interface (ALI) model addresses these limitations, which makes it ideal for culturing mucus-producing stem cells, in this case, GIM [16].
Due to the complexity of stem cell culture and the substantial differences in the composition of the medium required by various cells [17], researchers have only induced GIM-related phenotypes by overexpressing CDX2 in 3D organoid models at present. This induction method, however, has certain limitations as it predominantly leads to a reduction in the expression of gastric epithelium-related genes and an increase in the expression of intestinal epithelial-related genes [18]. At present, only human primary tracheal [19] and gastric epithelial cells have been successfully cultured on the ALI model [20], no successful cultivated GIM stem cells have been set on the ALI model.
In this study, human GIM cells were first cultured using the ALI model with the presence of Wnt and R-spondin to maintain the stemness of the GIM stem cells. We aim to construct an effective in vitro model of human GIM cells for further studies and applications, set up new ideas and strategies for the target, pathogenesis and related interventions of GIM, and provide an important reference for the early detection and treatment of GIM.
Materials and methods
The selection and exclusion criteria for GIM patients
Three participants with recognized GIM features in any part of the stomach, encompassing focal and extensive, complete and incomplete manifestations, were included in this investigation. The patients diagnosed as opportunistic by GIM through symptomatic services or population-based screening were included. GIM was defined by the histological identification of glands exhibiting phenotypic characteristics of the intestine. Three participants were included irrespective of documented H. pylori status, as infection may not be detectable once metaplasia occurred. Individuals with familial syndromes, such as autosomal dominant diffuse gastric cancer, were excluded [21]. Detailed patient information can be seen in Supplementary Table 1.
Source of cells
This study was approved by the Medical Ethics Committee of the Fifth Affiliated Hospital of Zhengzhou University with the approval number of K201811 (Supplementary File 1). Written consent was obtained from the patients with endoscopic diagnosis of GIM before starting this study. One piece of GIM tissue and another piece of surrounding normal gastric tissue were taken from three patients with biopsy forceps. The GIM and normal gastric tissues were equally divided into two sections, one was placed in 4% paraformaldehyde (PFA) for fixation and the other was used for cell culture.
Separation of cells
Collagen type II was thawed, and the shaker incubator was set as 37℃ for 35 min. The tissue obtained from the gastroscopy was immersed in medium containing 10% fetal bovine serum, and washed twice with cold (4℃) PBS. Then 2 mL of PBS (+ 40 µL penicillin/streptomycin + 8 µL gentamycin/amphotericin B) was added, and the sample and PBS solution were placed in a centrifuge tube. Next, they were kept at room temperature (18–25℃) for 30 min. PBS was discarded, 3 mL of collagenase type II solution was added to the tube, and the centrifuge tube was shaken at 37 °C for 30–40 min. The solution was fully mixed with a pipette, 3–5 mL of the precooled medium above was added, and the mixture was centrifuged at 1200 rpm under 4℃ for 5 min. The supernatant was discarded and 1 mL of cell basic medium was added. The cells containing the cell basic medium were gently aspirated five times with a 1 mL syringe (if there was still agglomeration, a 40 μm cell sieve was applied to remove the tissue block, and the cells were collected by reverse filtration and placed in another 1 mL centrifuge tube). Cells were counted with a common cell counter.
The medium composition of the GIM-ALI model
The basal medium widely utilized for culturing mammalian cells is Advanced DMEM/F-12 (ADF) medium [22]. To maintain cell stemness, Wnt3a, R-spondin1, and A83-01 were employed [23, 24]. Tissue culture buffer was provided by 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [25]. Stem cell development was facilitated by Glutamax, nicotinamide, and human epidermal growth factor (EGF) [26,27,28]. Stem cells were maintained by B27 and human noggin [29, 30], while N2 promoted their survival [31]. Human fibroblast growth factor (FGF)-10 and gastrin were capable of promoting stem cell proliferation [32, 33]. The ALI medium composition required for culturing GIM stem cells consists of these components in the following proportions (Table 1).
Generation of GIM mucosoid cultures
The inserts (Millipore) were put into 24 wells in advance and the pre-cooled collagen gel (Thermo Fischer) compound solution diluted with ddH2O was added. 200 µL ALI medium mixed with 200,000-250,000 primary cells was added to each insert, and 500 µL ALI medium combined with 1 µL Y-27,632 factor (Sigma) was added beneath each insert after the solution solidified. The 24-well plate was placed in a constant temperature incubator with 5% CO2, 95% humidity and 37℃ for cultivation. The day as the cells were cultured on the insert was defined to be day zero of cell culture. On the third day, the ALI medium above the insert was removed to start the ALI culture. The medium beneath the insert was subsequently changed twice a week while the mucus above the insert was collected (Supplementary video 1).
Immunofluorescence
4 µM paraffin-embedded sections were placed in an oven at 65 °C for 2 h and then placed in a xylene I dewaxing tank for 15 min, xylene II dewaxing tank for 15 min, anhydrous alcohol I dewaxing tank for 10 min, anhydrous alcohol II dewaxing tank for 10 min, 95% alcohol dewaxing tank for 5 min, 85% alcohol dewaxing tank for 5 min and 75% alcohol dewaxing tank for 5 min. Next, the sections were washed 3 times with 1×PBS solution for 5 min and then immersed in antigen retrieval solution composed of 12 mL antigen retrieval solution and 588 mL H2O for 5 min at 85℃ and 20 min at 65℃ successively, all at room temperature (18–25℃) and washed three times with 1×PBS solution for 5 min. GIM cells were circled with an immunohistochemical pen and 50–100 µL of blocking solution was added to each circle for 1–2 h at room temperature (18–25℃). The primary antibody diluted with the blocking solution was added to the circles and left to stand overnight at 4 °C. The slices were balanced at room temperature (18–25℃) for 1 h the next day and washed 3 times with 1×PBST solution for 5 min. The secondary antibody diluted with the blocking solution was added to the circles at room temperature (18–25℃) for 2 h and the slices were washed 5 times for 5 min with 1×PBST solution. Slices were mounted with the mounting medium containing 4’,6-diamidino-2-phenylindole (DAPI). The images were observed and collected under a fluorescence microscope.
Quantitative real-time PCR (qRT‐PCR)
The entire process of RNA extraction was carried out on ice. The inserts were washed with cold PBS twice, with 200 µL Trizol added to each wash. The cell suspension was aspirated and collected in a 1.5 mL sterile enzyme-free tube after waiting for 20 min. 400 µL of chloroform was added, the tube was shaken vigorously for 15 s, and left at room temperature (18–25℃) for 5 min. The samples were centrifuged at 4℃ and 12,000 rpm for 15 min. The upper aqueous phase was removed and transferred to a new 1.5 mL sterile enzyme-free tube. Isopropanol equal to the volume of upper aqueous phase was added, slowly shaken 15 times and centrifuged at 4℃, 12,000–13,000 rpm for 10 min. The supernatant was discarded and 1 mL of 75% alcohol was added to the tube, gently shaken, and centrifuged for 5 min. The supernatant was discarded, the remaining pellet was dried in air for 5 min, and dissolved 10 µL of diethyl pyrocarbonate (DEPC) water. The absorbance at A260/A280 was detected by Nando drop ND200, and the RIN value of RNA was determined by Agilent Bioanalyzer 4150. The Reverse Transcription Kit (Roche Diagnostics, Indianapolis, IN) was used to generate complementary DNA. qRT-PCR was performed on the Step One Plus Real-Time PCR System (Applied Biosystems, FosterCity, CA) using the SYBR-Green PCR kit (Roche Diagnostics, Indianapolis, I-N). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a reference gene. All primers are shown in Supplementary Tables 2, and the 2−ΔΔCt method [34] used made for qRT-PCR analysis.
Transcriptomic sequencing
The qualified RNA was prepared according to the instructions of the AB clonal transcriptomic analysis Lib Perp Kit to prepare the PE library. Library quality was assessed using an Agilent Bioanalyzer 4150 and the Illumina Novaseq 6000/MGISEQ-T7 sequencing platform was applied for sequencing. HISAT2 software (http://daehwankimlab.github.io/hisat2/) was employed to compare the clean reads obtained by processing the Perl script [35] with the reference genome [36] to obtain mapped reads, and further FPKM values for each gene were calculated in Feature Counts (http://subread.sourceforge.net/).
Proteomic sequencing
The GIM and normal mucus were extracted and quantitatively analyzed. The samples were separated by the HPLC liquid phase system Easynlc and analyzed using a Q-Exactive mass spectrometer (AGC, Automatic gain control). The target was set as 1e6, maximum IT as 50 ms and dynamic exclusion time as 60 s. The raw data files were analyzed and processed with the mass spectrometer.
Differentially expressed genes (DEGs) analysis
The processed transcriptomics data were imported into R software 4. 1. 3 [37], and differential analysis was performed by the “DESeq2” package [38]. The screening threshold for DEGs was |log2FC|>1 [39] and P value < 0.05 [40]. The “pheatmap” package [41] and “ggplot2” package [42] were applied to draw heat maps and bar graphs.
Differentially expressed proteins (DEPs) analysis
Quantitative analysis with MaxQuant was utilized to ascertain protein abundance while removing anti-library and contaminant proteins [43]. Proteins lacking any recorded features (those with zero counts across all samples) were excluded [44]. The dataset underwent logarithmic transformation and was then subjected to differential expression analysis employing “Limma” package. Differentially expressed proteins (DEPs) were defined based on a significance threshold of P value < 0.05 and a fold change (FC) greater than 1 [45]. The relative protein abundance in gastric tissues was depicted through the creation of heatmaps and bar graphs using ggplot2 in R software [46].
GO and KEGG enrichment analysis and PPI network
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were performed for the up- and down-regulated DEGs. The “clusterProfiler”, “richplot”, and “ggplot2” packages were used for analysis [47, 48]. The GO terms comprised 3 parts: Biological Process (BP), Cellular Component (CC), and Molecular Function (MF). The KEGG database included the systematic analysis, annotation, and visualization of gene functions [49]. The protein-protein interaction (PPI) network of DEGs was constructed by the STRING online website [50]. The PPI network was analyzed by Cytoscape software (version 3.9.0) [51].
GEO data processing
The GIM datasets GSE60427, GSE60662, GSE106656, and GSE78523 were retrieved from the Gene Expression Omnibus (GEO) database website (www.ncbi.nlm.nih.gov/geo) [52]. Specifically, GSE60427 comprised 7 normal cases and 8 GIM samples; GSE60662 contained 4 normal samples and 4 GIM samples; GSE106656 featured 7 GIM samples; and GSE78523 encompassed 15 normal samples and 16 GIM samples. These datasets consisted of various platform files, namely GPL17077, GPL13497, GPL6244, and GPL18990, housing clinical data of IM patients. ComBat, a classical Bayesian-based analysis method leveraging known batch information, was employed for batch correction on high-throughput data. The “Combat” algorithm within the R package “SVA” (version 3.29.1) was utilized to mitigate batch effects among different GSE datasets [53]. The data normalization was conducted post-batch effect removal, wherein the average value was considered as the expression value for a gene when multiple probes corresponded to it [54]. Finally, Principal Components Analysis (PCA) was employed to validate the results post-removal. To enhance the credibility of the analysis and verify the results, the GIM samples in GSE60427, GSE60662, and GSE78523 were designated as the train group, while those in GSE106656 were defined as the test group.
Cluster analysis of essential genes
The hub genes identified from the intersection of the DEGs obtained from the GEO datasets and the DEGs gained from the experimental groups were visualized by Venn diagrams [55]. The “ConsensusClusterPlus” package [56] was used to process cluster analysis on the samples from the GEO datasets. More details were shown in the heatmap.
Statistics
The statistical analysis was conducted using SPSS 23.0 (SPSS, Inc., Chicago, IL, USA), R software (version 4.2.1), and GraphPad Prism 8.0 software (GraphPad, Inc., La Jolla, CA, USA) [57, 58]. Each experiment was repeated 3 times on 3 samples to ensure robustness and reproducibility. For the identification of differential genes between the two groups of samples, The Wilcox test was utilized for statistical comparison. This non-parametric test is suitable for analyzing data that do not meet the assumptions of normality. It helps determine if there are significant differences between groups [59]. The hypergeometric distribution, a discrete distribution suitable for describing the number of sampling instances from a limited total sample pool, where n samples are drawn each time without replacement until a specified type of sample is obtained, was employed. The hypergeometric test was applied to conduct GO and KEGG enrichment analysis on differentially expressed genes (DEGs) [60]. For comparison of the means of two groups of independent samples, the t test was utilized [61]. Numeric expressions of P values were provided for each analysis, with statistical significance defined as P < 0.05 (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001) [62]. To control for type I errors (false positives) resulting from multiple comparisons, the P values of genes were corrected using various methods, including Bonferroni [63], Benjamini-Hochberg [64], Holm, Hochberg [65], Hommel [66], and BY [64]. These correction methods adjust the significance threshold to account for the increased chance of obtaining false positives when conducting multiple statistical tests simultaneously. By applying these correction methods, the study aimed to mitigate bias and enhance the reliability of the results.
The raw data, scripts and supplementary materials have been uploaded to https://www.jianguoyun.com/p/DSA9JDwQw5fKDBiEtc4FIAA.
Results
Verification of GIM tissue and cells
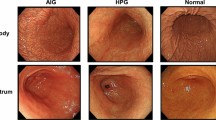
Prominent goblet cells were visible in the GIM tissue after HE staining (Fig. 1A-C). We found that GIM tissue expressed a large amount of MUC2 and a small amount of MUC5AC by immunohistochemistry and immunofluorescence, while normal gastric tissue specifically expressed MUC5AC (Figs. 1D-I and 2A-F; Supplementary Fig. 1), which agreed with their molecular characteristics.

Identification of the GIM organization. (A-C) HE staining of 3 GIM samples. The nucleus is stained blue, the cytoplasm is stained red and Red arrows indicate goblet cells. (C-F) Immunohistochemistry of 3 GIM samples. Nuclei are stained blue and MUC2 is stained brown. (G-I) Immunohistochemistry of 3 GIM samples. Nuclei are stained blue and MUC5AC is stained brown

Immunofluorescence of GIM and normal gastric samples. (A-C) Expression of MUC5AC and MUC2 in GIM samples. (D-F) The expression of MUC5AC and MUC2 in normal gastric tissue samples. The nucleus is stained blue, MUC5AC is stained green, and MUC2 is stained red
We found that GIM and gastric epithelial cells developed into a mature columnar epithelial morphology while GIM and normal samples separately specifically expressed MUC2 and MUC5AC based on immunofluorescence using the ALI model, which is consistent with in vivo findings (Fig. 3A&B; more details in Supplementary Fig. 2). The expression of intestinal markers (MUC2, CDX1, and CDX2), epithelial stemness markers (CD44, LGR5, and CTNNB), epithelial markers (KRT18, KRT19, and CDH1), and gastric gland cell type markers (PGC, MUC6, MUC5AC, CHGA, and ATP4B) kept relatively stable with the cells’ passage (Fig. 3C-F). Moreover, the importance of each component in the ALI medium was determined by qRT-PCR (Supplementary Fig. 3). These results suggest that our ALI model can be used to study GIM.

GIM ALI model immunofluorescence and qRT-PCR identification. (A) The expression of MUC2 in GIM samples using the ALI model after 10 days of culture under added Wnt and R-spondin (+ W + R) conditions visualized by red fluorescence (B) The expression of MUC2 in normal samples using the ALI model after 10 days of culture under added Wnt and R-spondin (+ W + R) conditions seen by red fluorescence. (C-F) Expression of intestinal markers, epithelial stemness markers, epithelial markers and gastric gland cell type markers in GIM samples remain relatively stable with cell passage
The differentially expressed genes (DEGs) functions between GIM and normal samples
To reveal the functions and relationships of DEGs, we conducted enrichment analysis and PPI network analysis. Compared with normal samples, the upregulated DEGs in GIM samples were mainly related to cell junction (Fig. 4A). In contrast, the down-regulated DEGs were primarily enriched in the negative regulation of Wnt signaling pathway by GO enrichment analysis (Fig. 4B). Moreover, the upregulated DEGs were relative to leukocyte transendothelial migration, proteoglycans in cancer (Fig. 4C), while down-regulated DEGs were abundant in the pentose-glucuronic acid conversion pathway and the PPAR lipid synthesis pathway through the KEGG pathway enrichment analysis (Fig. 4D). Eight nodes adjacent to SEMA5A and SPON2 were identified (Supplementary Fig. 4A). The top eight DEGs that were most connected were applied to construct a protein interaction network map (Supplementary Fig. 4B). Multiple pathways enriched in upregulated genes in the GIM samples were associated with GC progression, and these results suggested that GIM may affect GC occurrence through these genes.

The enrichment and PPI network of DEGs between the GIM and normal group. (A-D) GO and KEGG pathway enrichment analysis of up-regulated and down-regulated DEGs in GIM samples. Red and purple circles represent up-regulated and down-regulated DEGs in GIM samples, respectively. The larger the circle, the more the DEGs are enriched; the darker the circle, the more significant the difference
Enrichment analysis of differentially abundant proteins in GIM and normal samples
The enrichment analysis was similarly conducted to learn about the functions of differentially abundant proteins between GIM and normal samples. We found that the differentially abundant proteins were mainly enriched in the interleukin-like epithelial-mesenchymal transition (EMT) inducer domain (Fig. 5A). They were primarily associated with oxidative phosphorylation-related pathways and PPAR-Lipid synthesis-related pathways in the KEGG pathway enrichment analysis (Fig. 5B), and also related to the response to mis-folded proteins, nucleoside diphosphatase activity and polarized growth sites by GO analysis (Fig. 5C). In addition, they were mainly located in the cytoplasm and nucleus (Fig. 5D). These results indicate that GIM may also influence the development of GC by these proteins.

Comparison of differentially abundant proteins between the GIM and normal group. (A&B) The domain and KEGG pathway enrichment of differentially abundant proteins between GIM and normal samples. The circle size represents the number of differentially abundant proteins enriched in each case: the larger the circle, the higher the number. The color gradually changes from green to red, indicating that the difference is more distinguished. (C) The GO enrichment of differentially abundant proteins between the two sample groups. The color gradually changing from yellow to red shows that the difference is becoming more conspicuous. (D) The cellular distribution of differentially abundant proteins between the two sample groups
Batch correction of GEO datasets and identification of critical genes of GIM
To better make use of the GIM and normal samples in the GEO datasets and screen for potential markers of GIM, batch effects were removed among GSE60427, GSE60662 and GSE78523 as GIM train datasets (Fig. 6A&B). The data visualized by Venn diagrams allowed us to determine the intersection of the GEO datasets DEGs and the experimental group DEGs. The essential genes, MUC17, CDA, TRIM15, TBX3, FLVCR2, ONECUT2, ACY3, and NMUR2, were upregulated in GIM samples compared with normal samples; the down-regulated essential genes were GLDN, REP15, SLC5A5, and MAL (Fig. 6C&D). The P value of these genes was processed multiple testing corrections in Supplementary Table 3. In addition, GSEA enrichment analysis (Supplementary Fig. 5) was conducted on the 13 genes exhibiting differential expression between GIM and normal samples. It was observed that genes demonstrating elevated expression levels in GIM samples were primarily associated with cellular-cytokine interactions and immune-related functionalities. The samples from the above datasets were clustered into “Cluster1” and “Cluster2” according to these critical genes (Fig. 6E). “Cluster1” and “Cluster2” were intersected with the GIM samples and normal samples in the GEO databases by using a Venn diagram (Fig. 6F). Only 5 samples cannot be accurately divided into GIM and normal samples. The error rate is 5/55 = 9%<10%. Furthermore, the heatmap results also showed that these critical genes could be used to differentiate GIM samples from normal ones (Supplementary Fig. 6), suggesting that these genes could serve as potential markers for GIM. The analysis of the GIM test datasets revealed that the expression levels of CDA, TBX3, FLVCR2, ONECUT2, ACY3, and NMUR2 were significantly elevated in the GIM samples compared with the normal ones. Conversely, the expression of GLDN, REP15, and MAL showed significantly reduced in GIM patients (Supplementary Fig. 7, P < 0.05). These findings validate prior research and underscore a high level of conformity in the expression patterns of GIM biomarkers.

Batch correction between GEO datasets and identification of GIM key genes. (A&B) Batch correction between GEO datasets of GIM and normal samples. (C&D) The intersection of the upregulated and downregulated DEGs in the GIM sample from GEO datasets and the experimental group. (E) Cluster analysis of samples among GEO datasets. (F) The intersection of two clusters with GIM and normal samples in the GEO database
Validation of crucial genes linked to GIM
Based on qRT-PCR, we found that compared with normal samples, the expression of MUC17, CDA, TRIM15, TBX3, FLVCR2, ONECUT2, ACY3, and NMUR2 was significantly (P<0.05) higher in GIM (Fig. 7A&B), while expression of GLDN, REP15, SLC5A5, and MAL was significantly lower (P < 0.05, Fig. 7).

qRT-PCR validation of key GIM genes. (A&B) The key upregulated genes in GIM. (C) The key down-regulated key genes in GIM. *: P<0.05; **: P<0.01; ***: P<0.001; ****: P<0.0001
Discussion
This work confirmed that the ALI model provides the possibility for constructing GIM cells in vitro. We cultured human GIM and gastric mucosal epithelial cells using the ALI model for the first time, detected MUC5AC expression in normal gastric epithelium samples and MUC2 expression in GIM samples by IF, and verified the long-term stability of the GIM-ALI model for expressing intestinal markers, epithelial stem markers, stem markers and gland cell type markers using qRT-PCR. Consistent with our results, MUC5AC is secreted by the pit cells of the gastric antrum and corpus [67], and MUC2 is a gut marker highly expressed in GIM [68]. Studies had shown that CDX1, CDX2, and MUC2 are specifically expressed in GIM [69]; markers of epithelial stem include CD44, LGR5 and CTNNB [70,71,72], and KRT18, KRT19 and CDH1 are markers of epithelial cells [73,74,75]. Gastric gland cell type markers include PGC, MUC5AC, MUC6, and ATP4B. PGC is considered the final product of gastric mucosa maturity and differentiation [76]. The marker of gastric pit cells is MUC5AC, and the marker of gastric pyloric cells is MUC6 [77]; CHGA protein secreted by gastric mucosal enterochromaffin-like (ECL) cells is an acidic protein [78]. The protein expressed by ATP4B is a proton pump that plays a crucial role in gastric acid secretion [79]. Thus, adopting these selected genes to verify the stability of the GIM-ALI model in this study provided a proof-of-concept, representing a reliable and useful strategy for the future.
Transcriptomic and MS analyses were performed on human GIM and normal samples. Oxidative phosphorylation, an important source of energy and metabolic precursors in GC cells, provides a potential target for GC therapy [80]. The highly expressed protein in GIM samples was primarily associated with oxidative phosphorylation-related pathways, which suggests that these pathways may be necessary for GIM to develop into GC. By combining the transcriptomic analysis results of the human GIM and normal samples in the GEO datasets with the results in the experimental group, potential markers of GIM were: MUC17, CDA, TRIM15, TBX3, FLVCR2, ONECUT2, ACY3, NMUR2, GLDN, REP15, SLC5A5, and MAL. After verification by qRT-PCR experiments, the differences were all statistically significant (P<0.05).
Among the potential markers of GIM found in this study, only ONECUT2 has been reported in human GIM samples, which can induce and trigger the expression of ACSL5 through epigenetic changes in GIM. ONECUT2 and ACSL5 may synergistically promote the transdifferentiation of gastric mucosal epithelial cells to GIM cells and the progression of GIM to GC [81]. In GC, the expression of TRIM15 and TBX3 are specific and independent factors for poor prognosis [82, 83]. The suppression of GC cells invasion and metastasis is accomplished through the inhibition of STAT3 phosphorylation by MAL [84]. This supports the idea that the upregulation of these genes, TRIM15, TBX3, and MAL, are potentially responsible for the occurrence of GIM and GC.
Currently, stimulation of GES-1 cells by bile acid induces the GIM phenotype, which contains certain limitations, as the model solely exhibits an elevation in GIM markers through in vitro intervention [85]. Furthermore, GIM features can be induced by overexpressing CDX2 [85] or knocking out ATP4a [86] in the gastric mucosa of mice, albeit this model, utilizing mice and employing gene-editing technology to induce the emergence of the GIM phenotype, differs in species from humans. Furthermore, human induced pluripotent stem cells (hiPSCs) were differentiated into gastric organoids, subsequently in which overexpressed CDX2 via the tet-on system, the expression of partly gut genes and previously reported genes associated with GIM were enhanced. Despite the utilization of hiPSCs, this model is also gene-edited as well as incapable of collecting mucus secreted by human GIM goblet cells [18]. None of the three models featuring GIM phenotypes in the current study can realistically portray the state of human GIM.
Moreover, the GIM-ALI model represents a significant advancement, allowing for the long-term, stable in vitro cultivation of GIM cells. This achievement fills a critical void in the realm of in vitro GIM cell models, paving the way for enhanced biomarker discovery and more in-depth investigations into the mechanisms underlying GIM. Notably, this model offers a unique avenue for the collection of mucus secretions above the inserts, a distinguishing feature of GIM cells [87], thereby facilitating a deeper exploration of the role of mucus in GIM pathogenesis. In parallel, the GIM-ALI model presents novel strategies for investigating the impacts of diverse substances, encompassing drugs, metabolites, and bacteria co-cultivated with GIM cells treated under the insert, as well as for finding out the interactions of GIM cells with other cellular entities. The GIM-ALI model offers a novel framework, facilitating a deeper exploration of the pathogenic factors associated with the onset of GIM and elucidating how cytokines secreted by other cells influence the initiation and progression of GIM [16]. These attributes render the GIM-ALI model to simulate the real situation of GIM tissues in vivo. This provides a valuable tool for unraveling the mechanisms by which these agents either hinder or promote GIM progression.
While the GIM-ALI model presents certain advantages over conventional GIM in vitro models and in vivo animal models, it still harbors limitations. Primarily, the model solely comprises the GIM epithelial layer, thus lacking engagement with the vascular, immune, and nervous systems [88]. Moreover, the GIM-ALI models are relatively high costs attributable to the necessity for reagents and growth factors/inhibitors, alongside their reliance on extracellular matrices (e.g., matrix) and prolonged culture periods [89]. In addition, a significant amount of time is required for GIM-ALI model cultivation and the present GIM-ALI model fails to faithfully replicate the metabolism of its parent organ. To mitigate certain constraints, efforts are being made by bioengineers to develop precisely defined culture media and extracellular matrix systems [90]. Notably, a gastric epithelial ALI model has been developed by the German laboratory, and the identical GIM-ALI model has been established using our methodology for drug screening purposes (data in process).
Briefly, the development of the GIM-ALI model is a significant advancement, offering a novel in vitro platform for GIM studies. This model’s ability to mimic in vivo conditions and facilitate mucus secretion studies is particularly noteworthy. In addition, the identification of novel potential markers of GIM and the innovative GIM-ALI model represent significant contributions to the field, offering a new scene for research and therapeutic intervention. Our findings have significant implications for understanding GIM pathogenesis and its progression to GC, offering potential biomarkers for early detection and targets for therapeutic intervention [91]. In the future, our research aims to leverage the GIM-ALI model for crucial applications, including drug screening and the development of innovative therapeutic interventions. We also plan to delve into genetic and epigenetic studies to unravel the intricate mechanisms underlying GIM progression. Moreover, we envision adapting this model to individual patient cells to explore its potential in personalized medicine approaches. Furthermore, we are committed to expanding the utility of the GIM-ALI model in various aspects of gastrointestinal tract research. This includes investigating other precancerous lesions and conducting direct comparisons with in vivo observations to gain deeper insights. We are also dedicated to overcoming technical challenges such as batch effects and sample diversity that may arise during experimentation. To bolster the credibility and widespread adoption of the GIM-ALI model, we will actively collaborate with the broader research community. By engaging multiple research centers, we aim to assess and validate the model’s performance across different settings. This collaborative effort will enhance the reliability and generalizability of our findings.
Conclusion
GIM cells using the ALI model were successfully cultured for the first time, revealing 12 potential markers of GIM. This study fills the gap of lacking effective in vitro models of human GIM cells and provides insights into the GIM pathogenesis. It’s recommended that future studies explore the identified markers’ therapeutic potential, expand the GIM-ALI model’s applications, and strive for larger, more diverse study cohorts to validate and extend these findings.
Data availability
The data used in this study were from the patients with GIM and the GEO (https://www.ncbi.nlm.nih.gov/geo/) database. Our analyses’ protocols and raw figures or other information related to our study could be asked from the corresponding author on reasonable request.
Abbreviations
- CAG:
-
Chronic atrophic gastritis
- GIM:
-
Gastric intestinal metaplasia
- GC:
-
Gastric cancer
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- DEGs:
-
Differentially expressed genes
- DEPs:
-
Differentially expressed proteins
- PPI:
-
Protein-protein interaction
- GEO:
-
Gene Expression Omnibus
References
Shah SC, Gawron AJ, Li D. Surveillance of gastric intestinal metaplasia. Am J Gastroenterol. 2020;115(5):641–4.
Kinoshita H, Hayakawa Y, Koike K. Metaplasia in the stomach-precursor of gastric Cancer? Int J Mol Sci 2017; 18(10).
Song H, Ekheden IG, Zheng Z, Ericsson J, Nyren O, Ye W. Incidence of gastric cancer among patients with gastric precancerous lesions: observational cohort study in a low risk western population. BMJ. 2015;351:h3867.
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108.
Stairs DB, Kong J, Lynch JP. Cdx genes, inflammation, and the pathogenesis of intestinal metaplasia. Prog Mol Biol Transl Sci. 2010;96:231–70.
Hayakawa Y, Sethi N, Sepulveda AR, Bass AJ, Wang TC. Oesophageal adenocarcinoma and gastric cancer: should we mind the gap? Nat Rev Cancer. 2016;16(5):305–18.
Li X, Ootani A, Kuo C. An air-liquid Interface Culture System for 3D Organoid Culture of Diverse primary gastrointestinal tissues. Methods Mol Biol. 2016;1422:33–40.
Barbachano A, Fernandez-Barral A, Bustamante-Madrid P, Prieto I, Rodriguez-Salas N, Larriba MJ et al. Organoids and Colorectal Cancer. Cancers (Basel) 2021; 13(11).
Wang Y, Zhang H, Wu S, Wan W, Kang X, Gao B, et al. Substrate stiffness regulates the proliferation and Apoptosis of Periodontal Ligament Cells through integrin-linked kinase ILK. ACS Biomater Sci Eng. 2023;9(2):662–70.
Park SE, Georgescu A, Huh D. Organoids-on-a-chip. Science. 2019;364(6444):960–5.
Fennema E, Rivron N, Rouwkema J, van Blitterswijk C, de Boer J. Spheroid culture as a tool for creating 3D complex tissues. Trends Biotechnol. 2013;31(2):108–15.
Garreta E, Kamm RD, Chuva de Sousa Lopes SM, Lancaster MA, Weiss R, Trepat X, et al. Rethinking organoid technology through bioengineering. Nat Mater. 2021;20(2):145–55.
Bartfeld S, Bayram T, van de Wetering M, Huch M, Begthel H, Kujala P, et al. In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology. 2015;148(1):126–e136126.
Dutta D, Heo I, Clevers H. Disease modeling in stem cell-derived 3D Organoid systems. Trends Mol Med. 2017;23(5):393–410.
Wang H, Brown PC, Chow ECY, Ewart L, Ferguson SS, Fitzpatrick S, et al. 3D cell culture models: drug pharmacokinetics, safety assessment, and regulatory consideration. Clin Transl Sci. 2021;14(5):1659–80.
Boccellato F, Woelffling S, Imai-Matsushima A, Sanchez G, Goosmann C, Schmid M, et al. Polarised epithelial monolayers of the gastric mucosa reveal insights into mucosal homeostasis and defence against infection. Gut. 2019;68(3):400–13.
Jariyal H, Gupta C, Bhat VS, Wagh JR, Srivastava A. Advancements in Cancer Stem Cell isolation and characterization. Stem Cell Rev Rep. 2019;15(6):755–73.
Koide T, Koyanagi-Aoi M, Uehara K, Kakeji Y, Aoi T. CDX2-induced intestinal metaplasia in human gastric organoids derived from induced pluripotent stem cells. iScience. 2022;25(5):104314.
Cao X, Coyle JP, Xiong R, Wang Y, Heflich RH, Ren B, et al. Invited review: human air-liquid-interface organotypic airway tissue models derived from primary tracheobronchial epithelial cells-overview and perspectives. Vitro Cell Dev Biol Anim. 2021;57(2):104–32.
Ootani A, Toda S, Fujimoto K, Sugihara H. An air-liquid interface promotes the differentiation of gastric surface mucous cells (GSM06) in culture. Biochem Biophys Res Commun. 2000;271(3):741–6.
O’Connor A, McNamara D, O’Morain CA. Surveillance of gastric intestinal metaplasia for the prevention of gastric cancer. Cochrane Database Syst Rev 2013; (9):CD009322.
Wen H, Li F, Bukhari I, Mi Y, Guo C, Liu B et al. Comprehensive Analysis of Colorectal Cancer Immunity and Identification of Immune-Related Prognostic Targets. Dis Markers 2022; 2022:7932655.
Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T, et al. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003;423(6938):448–52.
Aguirre-Vazquez A, Salazar-Olivo LA, Flores-Ponce X, Arriaga-Guerrero AL, Garza-Rodriguez D, Camacho-Moll ME et al. 5-Aza-2’-Deoxycytidine and Valproic Acid in Combination with CHIR99021 and A83-01 Induce Pluripotency Genes Expression in Human Adult Somatic Cells. Molecules 2021; 26(7).
Luo J, Hein C, Pierson JF, Mucklich F. Erratum: early-stage corrosion, ion release, and the antibacterial effect of copper and cuprous oxide in physiological buffers: phosphate-buffered saline vs Na-4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [Biointerphases 14, 061004 (2019)]. Biointerphases. 2021;16(1):018601.
Chen PR, Redel BK, Spate LD, Ji T, Salazar SR, Prather RS. Glutamine supplementation enhances development of in vitro-produced porcine embryos and increases leucine consumption from the medium. Biol Reprod. 2018;99(5):938–48.
Yuan YG, Mesalam A, Song SH, Lee KL, Xu L, Joo MD, et al. Effect of nicotinamide supplementation in in vitro fertilization medium on bovine embryo development. Mol Reprod Dev. 2020;87(10):1070–81.
Martin KL, Barlow DH, Sargent IL. Heparin-binding epidermal growth factor significantly improves human blastocyst development and hatching in serum-free medium. Hum Reprod. 1998;13(6):1645–52.
Yoon SJ, Son HY, Shim JK, Moon JH, Kim EH, Chang JH, et al. Co-expression of cancer driver genes: IDH-wildtype glioblastoma-derived tumorspheres. J Transl Med. 2020;18(1):482.
Drost J, van Jaarsveld RH, Ponsioen B, Zimberlin C, van Boxtel R, Buijs A, et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature. 2015;521(7550):43–7.
Castillo-Diaz L, de la Cuetara-Bernal K, Garcia-Varona AY. [Marrow stromal cells cultured in N2-supplemented medium: implications on the generation of neural cells]. Rev Neurol. 2005;40(2):69–73.
Ma Y, Kakudo N, Morimoto N, Lai F, Taketani S, Kusumoto K. Fibroblast growth factor-2 stimulates proliferation of human adipose-derived stem cells via src activation. Stem Cell Res Ther. 2019;10(1):350.
Cheng CY, Zhou Z, Stone M, Lu B, Flesken-Nikitin A, Nanus DM, et al. Membrane metalloendopeptidase suppresses prostate carcinogenesis by attenuating effects of gastrin-releasing peptide on stem/progenitor cells. Oncogenesis. 2020;9(3):38.
Masihzadeh S, Amin M, Farshadzadeh Z. In vitro and in vivo antibiofilm activity of the synthetic antimicrobial peptide WLBU2 against multiple drug resistant Pseudomonas aeruginosa strains. BMC Microbiol. 2023;23(1):131.
Pires JG, da Silva GF, Weyssow T, Conforte AJ, Pagnoncelli D, da Silva FAB, et al. Galaxy and MEAN Stack to create a user-friendly workflow for the rational optimization of Cancer Chemotherapy. Front Genet. 2021;12:624259.
Wang D, Eraslan B, Wieland T, Hallstrom B, Hopf T, Zolg DP, et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol Syst Biol. 2019;15(2):e8503.
Liu XS, Zhou LM, Yuan LL, Gao Y, Kui XY, Liu XY, et al. NPM1 is a prognostic biomarker involved in Immune Infiltration of Lung Adenocarcinoma and Associated with m6A modification and Glycolysis. Front Immunol. 2021;12:724741.
Diboun I, Wernisch L, Orengo CA, Koltzenburg M. Microarray analysis after RNA amplification can detect pronounced differences in gene expression using limma. BMC Genomics. 2006;7:252.
Wang HB, Huang R, Yang K, Xu M, Fan D, Liu MX, et al. Identification of differentially expressed genes and preliminary validations in cardiac pathological remodeling induced by transverse aortic constriction. Int J Mol Med. 2019;44(4):1447–61.
Gobin E, Bagwell K, Wagner J, Mysona D, Sandirasegarane S, Smith N, et al. A pan-cancer perspective of matrix metalloproteases (MMP) gene expression profile and their diagnostic/prognostic potential. BMC Cancer. 2019;19(1):581.
Cheng Q, Wang L. LncRNA XIST serves as a ceRNA to regulate the expression of ASF1A, BRWD1M, and PFKFB2 in kidney transplant acute kidney injury via sponging hsa-mir-212-3p and hsa-miR-122-5p. Cell Cycle. 2020;19(3):290–9.
Gustavsson EK, Zhang D, Reynolds RH, Garcia-Ruiz S, Ryten M. ggtranscript: an R package for the visualization and interpretation of transcript isoforms using ggplot2. Bioinformatics 2022.
Duan S, Zhang M, Zeng H, Song J, Zhang M, Gao S, et al. Integrated proteomics and phosphoproteomics profiling reveals the cardioprotective mechanism of bioactive compounds derived from Salvia Miltiorrhiza Burge. Phytomedicine. 2023;117:154897.
Yang Y, Ma T, Zhang J, Tang Y, Tang M, Zou C, et al. An integrated multi-omics analysis of identifies distinct molecular characteristics in pulmonary infections of Pseudomonas aeruginosa. PLoS Pathog. 2023;19(8):e1011570.
Siegel PM, Barta BA, Orlean L, Steenbuck ID, Cosenza-Contreras M, Wengenmayer T, et al. The serum proteome of VA-ECMO patients changes over time and allows differentiation of survivors and non-survivors: an observational study. J Transl Med. 2023;21(1):319.
Jarousek R, Mikulova A, Dadova P, Taus P, Kurucova T, Plevova K, et al. Single-cell RNA sequencing analysis of T helper cell differentiation and heterogeneity. Biochim Biophys Acta Mol Cell Res. 2022;1869(10):119321.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7.
Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32(Database issue):D258–261.
Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto Encyclopedia of genes and genomes. Nucleic Acids Res. 1999;27(1):29–34.
Dai F, Chen G, Wang Y, Zhang L, Long Y, Yuan M, et al. Identification of candidate biomarkers correlated with the diagnosis and prognosis of cervical cancer via integrated bioinformatics analysis. Onco Targets Ther. 2019;12:4517–32.
Reimand J, Isserlin R, Voisin V, Kucera M, Tannus-Lopes C, Rostamianfar A, et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat Protoc. 2019;14(2):482–517.
Ma L, Hernandez MO, Zhao Y, Mehta M, Tran B, Kelly M, et al. Tumor Cell Biodiversity drives Microenvironmental Reprogramming in Liver Cancer. Cancer Cell. 2019;36(4):418–e430416.
Wu Z, Li W, Zhu H, Li X, Zhou Y, Chen Q, et al. Identification of cuproptosis-related subtypes and the development of a prognostic model in glioma. Front Genet. 2023;14:1124439.
Zhang Q, Liu J, Yao D, Shi JX, Liu YJ, Wei YG, et al. Comprehensive Analysis to identify Rh Family C glycoprotein (RHCG) as the Causative Gene for Psoriasis and search for Alternative Treatment modalities. Drug Des Devel Ther. 2023;17:2593–611.
Liu J, Zhou S, Li S, Jiang Y, Wan Y, Ma X, et al. Eleven genes associated with progression and prognosis of endometrial cancer (EC) identified by comprehensive bioinformatics analysis. Cancer Cell Int. 2019;19:136.
Qiu C, Shi W, Wu H, Zou S, Li J, Wang D, et al. Identification of Molecular subtypes and a prognostic signature based on inflammation-related genes in Colon Adenocarcinoma. Front Immunol. 2021;12:769685.
Amponsah-Tabi S, Djokoto R, Opoku S, Senu E, Boakye DK, Azanu WK, et al. Knowledge, attitude and acceptability of COVID-19 vaccine among residents in rural communities in Ghana: a multi-regional study. BMC Infect Dis. 2023;23(1):60.
Yue T, Chen S, Zhu J, Guo S, Huang Z, Wang P, et al. The aging-related risk signature in colorectal cancer. Aging. 2021;13(5):7330–49.
Li G, Wu XJ, Kong XQ, Wang L, Jin X. Cytochrome c oxidase subunit VIIb as a potential target in familial hypercholesterolemia by bioinformatical analysis. Eur Rev Med Pharmacol Sci. 2015;19(21):4139–45.
Wang W, Xing H, Huang C, Pan H, Li D. Identification of pancreatic cancer type related factors by Weighted Gene Co-expression Network Analysis. Med Oncol. 2020;37(4):33.
Mishra P, Singh U, Pandey CM, Mishra P, Pandey G. Application of student’s t-test, analysis of variance, and covariance. Ann Card Anaesth. 2019;22(4):407–11.
Martin A, Castells J, Allibert V, Emerit A, Zolotoff C, Cardot-Ruffino V, et al. Hypothalamic-pituitary-adrenal axis activation and glucocorticoid-responsive gene expression in skeletal muscle and liver of apc mice. J Cachexia Sarcopenia Muscle. 2022;13(3):1686–703.
Sedgwick P. Multiple hypothesis testing and Bonferroni’s correction. BMJ. 2014;349:g6284.
Shuken SR, McNerney MW. Costs and benefits of Popular P-Value correction methods in three models of quantitative omic experiments. Anal Chem. 2023;95(5):2732–40.
Zheng B, Keen KJ, Fritzler MJ, Ryerson CJ, Wilcox P, Whalen BA, et al. Circulating cytokine levels in systemic sclerosis related interstitial lung disease and idiopathic pulmonary fibrosis. Sci Rep. 2023;13(1):6647.
Walker TM, Miotto P, Koser CU, Fowler PW, Knaggs J, Iqbal Z, et al. The 2021 WHO catalogue of Mycobacterium tuberculosis complex mutations associated with drug resistance: a genotypic analysis. Lancet Microbe. 2022;3(4):e265–73.
Battista S, Ambrosio MR, Limarzi F, Gallo G, Saragoni L. Molecular Alterations in Gastric Preneoplastic Lesions and Early Gastric Cancer. Int J Mol Sci 2021; 22(13).
Yu JH, Zheng JB, Qi J, Yang K, Wu YH, Wang K, et al. Bile acids promote gastric intestinal metaplasia by upregulating CDX2 and MUC2 expression via the FXR/NF-kappaB signalling pathway. Int J Oncol. 2019;54(3):879–92.
Almeida R, Silva E, Santos-Silva F, Silberg DG, Wang J, De Bolos C, et al. Expression of intestine-specific transcription factors, CDX1 and CDX2, in intestinal metaplasia and gastric carcinomas. J Pathol. 2003;199(1):36–40.
Zhang H, Brown RL, Wei Y, Zhao P, Liu S, Liu X, et al. CD44 splice isoform switching determines breast cancer stem cell state. Genes Dev. 2019;33(3–4):166–79.
Fatehullah A, Terakado Y, Sagiraju S, Tan TL, Sheng T, Tan SH, et al. A tumour-resident Lgr5(+) stem-cell-like pool drives the establishment and progression of advanced gastric cancers. Nat Cell Biol. 2021;23(12):1299–313.
Dormeyer W, van Hoof D, Braam SR, Heck AJ, Mummery CL, Krijgsveld J. Plasma membrane proteomics of human embryonic stem cells and human embryonal carcinoma cells. J Proteome Res. 2008;7(7):2936–51.
Chen B, Xu X, Lin DD, Chen X, Xu YT, Liu X, et al. KRT18 modulates alternative splicing of genes involved in proliferation and apoptosis processes in both gastric Cancer cells and clinical samples. Front Genet. 2021;12:635429.
Gras D, Petit A, Charriot J, Knabe L, Alagha K, Gamez AS, et al. Epithelial ciliated beating cells essential for ex vivo ALI culture growth. BMC Pulm Med. 2017;17(1):80.
Corso G, Figueiredo J, De Angelis SP, Corso F, Girardi A, Pereira J, et al. E-cadherin deregulation in breast cancer. J Cell Mol Med. 2020;24(11):5930–6.
Ding HX, Wu YF, Xu Q, Yuan Y. Identification of PGC-related ncRNAs and their relationship with the clinicopathological features of gastric Cancer. J Cancer. 2021;12(14):4389–98.
Furukawa R, Oyama T, Takahashi A, Yorimitsu N, Shimoda T, Ota H, et al. [A case of gastric-type adenoma with chief cell and parietal cell differentiation surrounded by complete intestinal metaplasia]. Nihon Shokakibyo Gakkai Zasshi. 2017;114(3):450–5.
Engelstoft MS, Lund ML, Grunddal KV, Egerod KL, Osborne-Lawrence S, Poulsen SS, et al. Research Resource: a chromogranin a reporter for serotonin and histamine secreting Enteroendocrine cells. Mol Endocrinol. 2015;29(11):1658–71.
Lahner E, Marzinotto I, Lampasona V, Dottori L, Bazzigaluppi E, Brigatti C, et al. Autoantibodies toward ATP4A and ATP4B subunits of gastric Proton pump H+,K+-ATPase are Reliable Serological pre-endoscopic markers of Corpus Atrophic Gastritis. Clin Transl Gastroenterol. 2020;11(10):e00240.
Janku F, Beom SH, Moon YW, Kim TW, Shin YG, Yim DS, et al. First-in-human study of IM156, a novel potent biguanide oxidative phosphorylation (OXPHOS) inhibitor, in patients with advanced solid tumors. Invest New Drugs. 2022;40(5):1001–10.
Seo EH, Kim HJ, Kim JH, Lim B, Park JL, Kim SY, et al. ONECUT2 upregulation is associated with CpG hypomethylation at promoter-proximal DNA in gastric cancer and triggers ACSL5. Int J Cancer. 2020;146(12):3354–68.
Zhou W, Chen H, Ruan Y, Zeng X, Liu F. High expression of TRIM15 is Associated with Tumor Invasion and predicts poor prognosis in patients with gastric Cancer. J Invest Surg. 2021;34(8):853–61.
Lee YJ, Park JH, Oh SM. TOPK promotes epithelial-mesenchymal transition and invasion of breast cancer cells through upregulation of TBX3 in TGF-beta1/Smad signaling. Biochem Biophys Res Commun. 2020;522(1):270–7.
Geng Z, Li J, Li S, Wang Y, Zhang L, Hu Q, et al. MAL protein suppresses the metastasis and invasion of GC cells by interfering with the phosphorylation of STAT3. J Transl Med. 2022;20(1):50.
Jin D, Huang K, Xu M, Hua H, Ye F, Yan J, et al. Deoxycholic acid induces gastric intestinal metaplasia by activating STAT3 signaling and disturbing gastric bile acids metabolism and microbiota. Gut Microbes. 2022;14(1):2120744.
Liu W, Yang LJ, Liu YL, Yuan DS, Zhao ZM, Wang Q et al. Dynamic characterization of intestinal metaplasia in the gastric corpus mucosa of Atp4a-deficient mice. Biosci Rep 2020; 40(2).
Chlumska A, Mukensnabl P, Marecek P, Zamecnik M. Intestinal metaplasia of the stomach and esophagus: an immunohistochemical study of 60 cases including comparison with normal and inflamed intestinal mucosa. Cesk Patol. 2014;50(3):141–8.
Xu H, Jiao Y, Qin S, Zhao W, Chu Q, Wu K. Organoid technology in disease modelling, drug development, personalized treatment and regeneration medicine. Exp Hematol Oncol. 2018;7:30.
Choi KG, Wu BC, Lee AH, Baquir B, Hancock REW. Utilizing Organoid and Air-Liquid Interface Models as a screening method in the development of new host defense peptides. Front Cell Infect Microbiol. 2020;10:228.
Yui S, Azzolin L, Maimets M, Pedersen MT, Fordham RP, Hansen SL, et al. YAP/TAZ-Dependent reprogramming of Colonic epithelium links ECM remodeling to tissue regeneration. Cell Stem Cell. 2018;22(1):35–e4937.
Huang KK, Ma H, Chong RHH, Uchihara T, Lian BSX, Zhu F, et al. Spatiotemporal genomic profiling of intestinal metaplasia reveals clonal dynamics of gastric cancer progression. Cancer Cell. 2023;41(12):2019–37. e2018.
Acknowledgements
We acknowledge the participants and investigators of this study.
Funding
This work was supported by Key projects of discipline construction in Zhengzhou University (No. XKZDJC202001); National Key Research and Development Program in China (No.2020YFC2006100); Zhengzhou Major Collaborative Innovation Project (No.18XTZX12003); European Research Council (ERC) Advanced Grant (No.885008-MADMICS); and for this study, Simeng Liu also received financial support from the Preferential Scientific Research Funds for Overseas Chinese Students in 2020.
Author information
Authors and Affiliations
Contributions
SML constructed the ALI model, conceptualized the study and prepared the first draft. HJW did most of the experiments and analyzed the data. FZL organized data. XDS revised the first draft. XX, YM, FB and TFM thoroughly revised and reviewed for logical, grammatical and other linguistic errors. FHL, YUH and HYX did a small number of experiments. PYZ concepted study, oversaw overall work, reviewed and revised the final draft. All authors have read and approved the submission of the final manuscript.
Corresponding authors
Ethics declarations
Ethical approval
The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. Gastric cancer tissue specimens were obtained from the Fifth Affiliated Hospital of Zhengzhou University. The ethics committee of the Fifth Affiliated Hospital of Zhengzhou University approved the study (Ethics number: K201811). All the related patients have signed informed consent.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, S., Wen, H., Li, F. et al. Revealing the pathogenesis of gastric intestinal metaplasia based on the mucosoid air-liquid interface. J Transl Med 22, 468 (2024). https://doi.org/10.1186/s12967-024-05276-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-024-05276-7