
Abstract
Background
TAS-102 (Lonsurf®) is an oral fluoropyrimidine consisting of a combination of trifluridine (a thymidine analog) and tipiracil (a thymidine phosphorylation inhibitor). The drug is effective in metastatic colorectal cancer (mCRC) patients refractory to fluorouracil, irinotecan and oxaliplatin. This study is a real-world analysis, investigating the interplay of genotype/phenotype in relation to TAS-102 sensitivity.
Methods
Forty-seven consecutive mCRC patients were treated with TAS-102 at the National Cancer Institute of Naples from March 2019 to March 2021, at a dosage of 35 mg/m2, twice a day, in cycles of 28 days (from day 1 to 5 and from day 8 to 12). Clinical-pathological parameters were described. Activity was evaluated with RECIST criteria (v1.1) and toxicity with NCI-CTC (v5.0). Survival was depicted through the Kaplan-Meyer curves. Genetic features of patients were evaluated with Next Generation Sequencing (NGS) through the Illumina NovaSeq 6000 platform and TruSigt™Oncology 500 kit.
Results
Median age of patients was 65 years (range: 46–77). Forty-one patients had 2 or more metastatic sites and 38 patients underwent to more than 2 previous lines of therapies. ECOG (Eastern Cooperative Oncology Group) Performance Status (PS) was 2 in 19 patients. The median number of TAS-102 cycles was 4 (range: 2–12). The most frequent toxic event was neutropenia (G3/G4 in 16 patients). There were no severe (> 3) non-haematological toxicities or treatment-related deaths. Twenty-six patients experienced progressive disease (PD), 21 stable disease (SD). Three patients with long-lasting disease control (DC: complete, partial responses or stable disease) shared an FGFR4 (p.Gly388Arg) mutation. Patients experiencing DC had more frequently a low tumour growth rate (P = 0.0306) and an FGFR4 p.G388R variant (P < 0.0001). The FGFR4 Arg388 genotype was associated with better survival (median: 6.4 months) compared to the Gly388 genotype (median: 4 months); the HR was 0.25 (95% CI 0.12- 0.51; P = 0.0001 at Log-Rank test).
Conclusions
This phenotype/genotype investigation suggests that the FGFR4 p.G388R variant may serve as a new marker for identifying patients who are responsive to TAS-102. A mechanistic hypothesis is proposed to interpret these findings.
Graphical Abstract

Similar content being viewed by others

Background
Fluoropyrimidines (FPDs) represent a milestone in the treatment of colorectal cancer (CRC) both in the adjuvant and metastatic settings. They inhibit the thymidylate synthetase (TS), a fundamental enzyme in the synthesis pathway of pyrimidine nucleotides [1]. 5-flurouracil (5-FU) and capecitabine (pro-drug of 5-FU) belong to this category and constitute an essential component of chemotherapy regimens, in combination with irinotecan and/or oxaliplatin, currently used in metastatic CRC (mCRC) in association with new biologic drugs (bevacizumab, aflibercept, panitumumab, cetuximab) [2]. TAS-102 (Lonsurf®) is a combination of two drugs: trifluridine (a thymidine analogue) and tipiracil (a thymidine phosphorylation inhibitor) [3]. The triphosphate form of trifluridine (TAS-102) is incorporated into the DNA determining its anti-tumor effects while tipiracil prevents the degradation of TAS-102 by maintaining adequate plasma levels. It is administered at a dosage of 35 mg/m2, twice a day, in cycles of 28 days, from day 1 to 5 and from day 8 to 12 [4, 5]. In previous studies, the drug has been shown to be effective in mCRC patients refractory to fluorouracil, irinotecan and oxaliplatin [6, 7] determining an increase median patients’ survival of approximately 2 months (with a significant reduction of death risk by 32%) versus placebo and an acceptable toxicity profile. The benefit was evident and significant in all patients’ subgroups (ECOG PS 0 vs 1, KRAS mutated vs not mutated, right vs left colon, young vs elderly, Asian vs Caucasian, poly-metastatic vs oligo-metastatic disease, etc.). The most frequent toxic events were mainly haematological (neutropenia) while non-haematological toxicities were not significantly higher than placebo. Due to its good risk/benefit ratio, TAS-102 is increasingly used in the treatment of mCRC after failure of previous therapies (including 5-FU, oxaliplatin and irinotecan).
Recently, genetic assessments have provided a useful tool to optimize treatment of mCRC patients including analysis of genetic variants of: 1. DPD (dihydropyrimidine dehydrogenase) and UGT1 (uridine diphosphate glucuronosyl-transferase), causing reduced/abrogated metabolism and risk of extremely severe toxicities from FDPs [8] and irinotecan [9], respectively, 2. RAS (RAt Sarcoma viral oncogene homolog) and BRAF (v-raf murine sarcoma viral oncogene homolog B), determining a constitutive activation of EGFR-dependent proliferative pathway of MAP kinases and consequent unresponsiveness to anti-EGFR blocking agents (panitumumab and cetuximab) [10], 3. micro-satellites status (MSS), short and repetitive predominantly non-coding DNA sequences which can accumulate alterations (becoming unstable) [11], 4. mismatch repair (MMR) genes (including MSH2, MLH1, MSH6, PMS2) [12]. In the last two cases, the tumours are more prone to mutate and accumulate neo-antigens and, consequently, to respond to immunotherapy (immune check-point inhibitors) [13].
In this study, we present a real-world, consecutive, two-year analysis of TAS-102 administration, investigating the genomic profiles of patients who benefited clinically. Our aim is to generate hypotheses regarding potential biomarkers and molecular factors associated with TAS-102 responsiveness.
Methods
Patients, treatments and follow-up
Patients were enrolled at the SSD-Innovative Abdominal Metastasis Therapies of the Department of Abdominal Oncology of the National Cancer Institute of Naples from March 2019 to March 2021. We reported the following clinical-pathological parameters: age, gender, histological characteristics, tumor site, PS (Performance Status) according to ECOG (Eastern Cooperative Oncology Group), disease sites, previous treatments.
The first- and second-line treatments were carried out in accordance with the ESMO guidelines [14]. No BRAF mutated patients of this cohort received “on-label” encorafenib for reasons of timing approval of the Italian regulatory authorities and clinical appropriateness. All patients had cytologically and/or histologically confirmed CRC. TNM staging (according to AJCC) was performed with total-body Computerized Tomography (CT) before the start of treatments (regardless of the line). Considering the real-word nature of this cohort, CT scan monitoring was not centralized, and it was performed every three months (± 14 days) or anticipated in case of clinically suspected progression of disease. Other examinations (ultrasound, MRI, PET, RX, etc.) were carried out at the discretion of the reference oncologist and in relation to specific clinical questions. TAS-102 was administered per os at a dose of 35 mg/m2 twice daily on days 1–5 and days 8–12 in a 28-day cycle. Response to treatments was assessed according to RECIST v1.1 criteria [15]. Disease Control (DC) rate was defined as the percentage of patients who have achieved complete response (CR), partial response (PR) and stable disease (SD). Duration of DC was the time elapsing from the date of TAS-102 start to the date of the first progressive disease (PD) documentation. Treatment toxicity has been reported in this article according to CTC v5.0 criteria (www.eortc.be/services/doc/ctc). According to the internal policies of our Institute, the institutional review board approval was not required for the retrospective analysis of this clinical cohort. All patients signed a written informed consent before treatment administration and molecular assessments.
NGS assessment and genetic data reporting
Gene mutations were assessed on formalin-fixed paraffin-embedded (FFPE) of metastatic tumor tissues if available (biopsies or tumour resections). DNA was extracted from three 10 µm FFPE sections by means of the MGF03-Genomic DNA FFPE One-Step Kit, according to the manufacturer’s protocol (MagCore Diatech). DNA quality was evaluated in triplicate with the FFPE QC Kit according to the manufacturer’s protocol (Illumina, San Diego, USA). Libraries were prepared thorough the TruSigt TMOncology 500 kit. Sequencing was performed on an Illumina NovaSeq 6000 (San Diego, USA) platform. The assay detects small nucleotide variants (SNVs), indels, splice variants and immunotherapy biomarkers in 523 cancer-relevant genes. An Illumina TruSigth Oncology 500 bioinformatics pipeline was applied to analyse the sequencing results, as previously reported [16, 17]. The prioritization of variants was done according to a four-tiered structure, adopting the joint consensus recommendation by AMP/ACMG [18]. Missense genetic variants were reported through the p. notation (p. followed by the new amino acid which replaces the wild-type amino acid) and Venn-Diagrams were applied in order to depict intersections among them (shared and private mutations) in different patients.
Tumor Mutational Burden (TMB) was estimated by counting all coding alterations within the targeted genomic region [19]. A robust and precise model was utilized for predicting the MSI phenotype, based on TMB dichotomization into low/intermediate (0–19 mutations/mb) and high (≥ 20 mutations/mb) categories, achieving both positive and negative predictive values of approximately 99% [19, 20].
Tumour growth rate assessment
The tumour growth rate (TGR) was calculated with the algorithm 100x(STLprogression-STLpreceding)/STLpreceding/T) were STL is the sum of target lesions (STLprogression: at progression; STLpreceding: immediately preceding STLprogression) and T is the time elapsed between these evaluations. TGRs was classified in low TGR and high TGR as previously reported [21]. Briefly, low TGR was < 0.33%/day and no new metastatic sites, high TGR was ≥ 0.33%/day or emergence of new metastatic sites.
Setting of LDH values
The cut-off value to discriminate between high vs low LDH (Lactic DeHydrogenase) serum levels was chosen as previously reported [22]. It was settled at 1.2 × ULN (Upper Limit Normal) U/L: 270 U/L.
Statistical analysis
Data were extracted from an internal electronic database prospectively updated. To avoid negative prognostic interferences, patients with PS ECOG > 2, age > 80 years, and an oncologist-estimated life expectancy < 3 months were excluded. Survival was intended as the time elapsed from the first administration of TAS-102 until death or the last available follow-up and was depicted with Kaplan-Meyer curves. The statistical significance of time-to-outcome divergences according to selected prognostic factors was studied with the Log-Rank test (P < 0.05 was considered statistically significant). The reported Hazard Ratios (HRs) are the estimate of the survival probability, and they can be interpreted as the instantaneous relative risk of death, at any time, for an individual. 95% confidence intervals (CI) of HR are also reported. Analyses of time-to-outcome are predominantly descriptive and no attempts to other statistical inferences have been made. Associations between FGFR4 p.G388R, TGR, basal CEA, NLR, LDH and response to TAS-102 were evaluated by χ2 test. P < 0.05 was considered statistically significant. Statistical analysis was performed using the MedCalc® 20.011 and Excel software.
Results
Clinico-pathological characteristics of clinical cohort
Forty-seven consecutive patients treated with trifluridine/tipiracil for metastatic CRC were enrolled. Clinico-pathological characteristics are reported in Table 1. The median age was 65 years (range: 46–77). Twenty-seven were male, 20 female. Four patients had a mucinous histology. The site of the primary tumor was right colon in 25, left in 22. Nineteen patients started TAS-102 therapy with PS ECOG 2. Six patients had single organ involvement (5, liver; 1, peritoneum), 41 had lesions in multiple sites (liver, lungs and lymph nodes, 13; liver and lungs, 9; liver and peritoneum, 8; liver, lymph nodes and bones, 4; lungs, liver, lymph nodes and peritoneum, 4; lungs and peritoneum, 2; peritoneum, 1). KRAS oncogene was mutated (mutKRAS) in 13 patients (27%) and wild-type (wtKRAS) in 34. BRAF p.V600E mutation was present in 4 patients (9%). Thirty-eight patients received more than two previous treatment lines. All patients were refractory to oxaliplatin- and irinotecan-based schedules. Twenty-eight patients were treated with regorafenib before TAS-102. Sixteen patients experienced objective responses (complete or partial according to RECIST v1.1) to previous first-line therapy (Table 2).
Toxicity and response
The median number of administered cycles (1 cycle = 28 days) was 4 (range: 2–12). The most frequent toxic event attributable to the administration of TAS-102 was neutropenia (G3/G4 in 16 patients). There were no severe (> 3) non-haematological toxicities or treatment-related deaths (Table 3). Nine patients started therapy at reduced doses (− 5 mg/m2/dose). A dose adjustment (reduction of at least -5 mg/m2/dose) was applied in 29 patients (Table 4) during treatment course. A radiologic re-evaluation of the disease was carried out in 33 patients. Fifteen patients did not undergo to instrumental restaging because of a clear clinical evidence of progressive disease and rapid deterioration of general conditions. The outcome of these patients is formally indicated as PD. No responses were recorded according to RECIST v1.1 criteria. Eleven patients showed instrumental PD, 21 had SD (DC rate: 44.7%). Figure 1 shows representative CT restagings in patients experiencing long-lasting DC.

Representative computed tomography (CT) restaging images depict FA, CC, and NA patients with long-lasting disease control (FA, CC, and NA represent patients' initials). Baseline CT scans at TAS-102 treatment initiation are outlined in green frames, while CT scans at the last available restaging (third for FA and CC patients, second for NA patients) are framed in blue. Measurable lesions in the peritoneum (FA: 58 mm vs 59 mm), abdominal lymph nodes (CC: 22 mm vs 0 mm; 25 mm vs 23 mm), and lungs (NA: 10 mm vs 8 mm, 6 mm and 8 mm vs 0 mm and 0 mm) are delineated by their longest diameters, with measurements in millimeters reported within the images
Genetic sharing in tumours of patients experiencing long-lasting DC to TAS-102 therapy
Since the genetic characteristics of tumors can be linked to specific clinical behaviors, including responses to treatments and the extent of time to outcomes, we selected three patients who experienced the longest disease control durations with TAS-102 (10, 12, and 14 months of disease control). We then correlated their genetic landscape, analyzed through NGS. The results are depicted in Venn Diagrams in Fig. 2. Interestingly, these patients shared only one genetic variant, FGFR4 p.G388R.

Venn diagram illustrating the coding genetic variant shared among patients exhibiting long-lasting disease control with TAS-102 (FA, CC, and NA represent patients' initials). The sole shared genetic variation (highlighted in red) is FGFR4 p.G388R. DNA extraction was performed from formalin-fixed and paraffin-embedded (FFPE) primary colorectal cancer tissue specimens. Libraries were prepared using the TruSigtTMOncology 500 kit, which assesses 523 cancer-relevant genes, and sequenced on an Illumina NovaSeq 6000 platform. Sequences were aligned to the human reference genome GRCh37 using the Burrows–Wheeler Aligner tool (for further details, refer to the Methods section)
Time-to-outcome
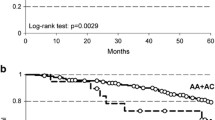
OS was considered the best time-to-outcome descriptor since CT scans were not centralized and 15 patients did not undergo instrumental restaging. The median survival of the entire cohort from TAS-102 start was 4.8 months; at last follow-up, there were 42 events (deaths). Since FGFR4 p.G388R was the only genetic variant shared by long-lasting DC patients, its prognostic impact on survival of the entire clinical cohort of metastatic CRC patients treated with TAS-102 was studied (Fig. 3). All primary tumours were genetically characterized. Fifteen patients had the FGFR4 p.G388R variant (31.9%). Median survival of patients with the FGFR4 Arg388 genotype (events: 10/15) was 6.4 months compared to 4.0 months of patients with the Gly388 genotype (events: 32/32). The HR was 0.25 (95% CI 0.12- 0.51; P = 0.0001 at Log-Rank test). No attempts were done to perform multi-variate analysis.

Kaplan–Meier survival curves depict patient outcomes from the initiation of TAS-102 therapy until death, stratified by the presence or absence of the rs351855 FGFR4 polymorphism. Time is represented on the x-axis, while survival probability is depicted on the y-axis. The number of patients at risk is reported below the figure, with data categorized based on FGFR4 polymorphism status
Association between FGFR4 variants, TGR, basal CEA, NLR, LDH and DC
Very few background data about genetic, clinical and/or biochemical factors predicting response to TAS-102 are available, among these, TGR, CEA, NLR and LDH at treatment start, seem the most promising. Assessments of potential factors predicting DC to TAS-102 were dichotomized: FGFR4 p.G388R (not present vs present), TGR (low vs high), basal CEA (low vs high), NLR (low vs high), LDH (low vs high). Applied cut-offs for TGR, CEA, NLR, and LDH were 0.33%/day, 200 ng/ml, 5, and 1.2xULN U/L, respectively (Table 5). Interestingly, patients experiencing DC had more frequently an FGFR4 p.G388R variant (P < 0.0001), and a low TGR (P = 0.0306). There were no significant associations between basal CEA, NLR, LDH values and DC to TAS-102, even if higher CEA, NLR and LDH levels were more frequent in patients whose disease was not controlled by treatment.
Discussion
In this article, we present the genetic evaluation of a cohort of clinical patients undergoing treatment with TAS-102 for extensive metastatic disease, with only 6 patients exhibiting single-organ involvement. These patients were heavily pre-treated, with 38 out of 47 having undergone 3 or 4 prior lines of therapy, and displaying poor performance status (ECOG PS 2 in 19 out of 47). Hence, TAS-102 was administered in a typical real-world clinical setting. Our findings indicate that TAS-102 is well tolerated when managed through tailored approaches, including early dose reductions, and is associated with a disease control rate of 44.7%. Given these observations, we opted to investigate any genetic characteristics that might influence response to treatment and prognosis. Indeed, gathering clinical data from routine practice offers a scientific opportunity to validate efficacy in unselected populations and to gain translational insights into specific subgroups.
Durations of responses in all patients ranged from 2 to 14 months (median: 4.5 months). Interestingly, the best outcome from TAS-102 emerged in 3 patients (NA, FA, CC) whose metastatic disease was controlled for 10, 12 and 14 months, respectively. In two of these patients, a volumetric reduction of the disease was found even if failing to meet the criteria for an objective response. Consequently, we conducted a comprehensive genetic assessment of primary tumor tissue. To the best of our knowledge, this represents the initial description of human genomic landscape characterization specifically aimed at exploring determinants of clinical benefit from TAS-102. A prevalent genetic variant, FGFR4 p.Gly388Arg, was identified. Therefore, we focused our attention to generate hypotheses on genotype/phenotype association on FGFR4 p.Gly388Arg.
Interestingly, it has been demonstrated that FGFR4 p.Gly388Arg determines the exposure of a cytoplasmic signal transducer and activator of transcription 3 (STAT3) binding site. This concurs to increase the recruitment of STAT3 enhancing tyrosine phosphorylation increasing cancer progression [23]. Interestingly, FGFR4 p.Gly388Arg was found to be correlated with worse outcomes in breast and lung cancer [24, 25], and very recently, in silico analysis in a Mexican population with colorectal cancer showed an association between rs351855 and the disease [26]. Moreover, it has been shown that trifluridine, can modulate through complex and still unknown mechanisms the phosphorylation status of ERK/AKT/STAT3 pathway [27, 28]. Taken together, our previous observations could unveil the existence of composite and complex relationships between genetic alterations in biologic pathways of FGFR4 and the effect of TAS-102. Most importantly, FGFR4 p.G388R variant was found in patients whose disease was long-term controlled by TAS-102 and it associates with DC and prognosis. The FGFR4 pathway has been reported as a putative targetable regulator of drug resistance in CRC. In fact, silencing of FGFR4 is able to reduce CRC cell viability and it has synergistic activity with 5-fluorouracil (5-FU) and oxaliplatin chemotherapy in CRC chemotherapy-refractory cell lines by decreasing the activity of STAT3 transcription factor [29]. FGFR4 p.G388R (rs351855) has been reported to prompt progression and affect prognosis of several cancers including CRC [30, 31]. Interestingly, this amino acid change (G > A) shows oncogenic properties by inducing tumour growth and migration of malignant cells [32]. Furthermore, this mutation in FGFR4 has been observed alongside other gene mutations in tumor calcinosis, a rare, obscure, and debilitating disorder of phosphate metabolism characterized by the formation of hard masses in soft tissues [33]. Recently, there have been reports of a small intestine tumor and a history of surgery for lung squamous cell carcinoma. The patient subsequently developed jejunal squamous cell carcinoma, gastric adenocarcinoma, and rectal adenocarcinoma concurrently. The intestinal cancer exhibited the same rare mutations observed in the lung cancer tissue, including FGFR4 p.G388R, suggesting a metastatic origin of the intestinal tumor from the lung [34]. The oncogenic role of the FGFR4 p.G388R variant is further supported by a recent genetic study in a large population. Peng et al. evaluated 13,793 cancer patients and 16,179 controls and found that the G388R polymorphism is associated with increased susceptibility to cancer in the homozygous state. Stratified analysis by cancer type and ethnicity revealed similar findings for prostate cancer, breast cancer, and individuals of Asian descent [35].
In the present study, we found that FGFR4 p.G388R is a predictive marker of response to TAS-102 in mCRC patients. This strongly suggests, in conjunction with the aforementioned previous observations, that this genetic characteristic could be a driver of tumor promotion influenced by TAS-102 administration. It is also noteworthy that FGFR4 represents an attractive new therapeutic target in cancer treatment, and several small kinase inhibitors are in preclinical and clinical development for the treatment of various cancers, including mCRC [36]. Therefore, based on these findings, potential approaches involving the design of combinations between TAS-102 and FGFR4 inhibitors could be considered for FGFR4-mutated CRCs. Clinical and biochemical factors including TGR, CEA, NLR and LDH values, have been recently explored [37]. Only TGR displayed an association with TAS-102-related DC. However, although easy to assess, none of these factors can be used to precisely predict the clinical benefit from TAS-102.
Our study has some limitations. The sample size and the retrospective nature do not allow us to make definitive conclusions and to exclude any biases in patients’ selection. However, the monocentric and consecutive characteristics of the clinical cohort and the short range of patients’ accrual could limit the detrimental effects of both factors and strength its hypothesis-generating power.
Conclusions
Our data generate the hypothesis that FGFR4 assessment could represent a tool to rationally select patients for TAS-102 treatment and for shifting this therapy to an earlier phase of treatment. Larger studies are being planned by our group to confirm these interesting findings.
Availability of data and materials
The genetic data supporting the findings of this study are available upon reasonable request, which can be sent to giovanni.savarese@centroames.it. Examples of TSO500 reports are available at https://zenodo.org/records/10968790.
Abbreviations
- FDPs:
-
Fluoropyrimidines
- CRC:
-
Colorectal cancer
- TS:
-
Thymidylate synthetase
- 5-FU:
-
5-Flurouracil (5-FU)
- mCRC:
-
Metastatic CRC
- TAS-102:
-
Trifluridine/tipiracil
- ECOG PS:
-
Eastern Cooperative Oncology Group Performance Status
- UGT1:
-
Uridine diphosphate glucuronosyl-transferase
- RAS:
-
RAt Sarcoma viral oncogene homolog
- BRAF:
-
V-raf murine sarcoma viral oncogene homolog B
- EGFR:
-
Epithelial Growth Factor Receptor
- MAP:
-
Mitogen Activated Protein
- MSS:
-
Micro-satellites status
- MMR:
-
Mismatch repair
- ESMO:
-
European Society of Medical Oncology
- CT:
-
Computerized Tomography
- MRI:
-
Magnetic Resonance Imaging
- PET:
-
Positron Emission Tomography
- RX:
-
Radiography
- RECIST:
-
Recist response evaluation criteria in solid tumors
- DC:
-
Disease control
- CR:
-
Complete response
- PR:
-
Partial response
- SD:
-
Stable disease
- PD:
-
Progressive disease
- CTC:
-
Common toxicity criteria
- NGS:
-
Next generation sequencing
- FFPE:
-
Formalin-fixed paraffin-embedded
- SNVs:
-
Small nucleotide variants
- AMP:
-
Association for Molecular Pathology
- ACMG:
-
American College of Medical Genetics
- TMB:
-
Tumor Mutational Burden
- TGR:
-
Tumour growth rate
- NLR:
-
Neutrophil-to-lymphocyte ratio
- STL:
-
Sum of target lesions
- LDH:
-
Lactic dehydrogenase
- ULN:
-
Upper limit normal
- HR:
-
Hazard ratio
- CI:
-
Confidence interval
- FGFR4:
-
Fibroblast Growth Factor Receptor 4
- CEA:
-
Carcinoembryonic antigen
- Mut:
-
Mutant
- STAT 3:
-
Signal transducer and activator of transcription 3
- ERK:
-
Extracellular signal-regulated kinase
- AKT:
-
Proetin kinase B
References
Vodenkova S, Buchler T, Cervena K, Veskrnova V, Vodicka P, Vymetalkova V. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: past, present and future. Pharmacol Ther. 2020;206: 107447. https://doi.org/10.1016/j.pharmthera.2019.107447.
Nappi A, Berretta M, Romano C, et al. Metastatic colorectal cancer: role of target therapies and future perspectives. Curr Cancer Drug Targets. 2018;18:421–9.
Uboha N, Hochster HS. TAS-102: a novel antimetabolite for the 21st century. Future Oncol. 2016;12:153–63. https://doi.org/10.2217/fon.15.276.
Doi T, Ohtsu A, Yoshino T, et al. Phase I study of TAS-102 treatment in Japanese patients with advanced solid tumours. Br J Cancer. 2012;107:429–34.
Patel MR, Bendell JC, Mayer RJ, et al. A phase I dose-escalation study of TAS-102 in patients (pts) with refractory metastatic colorectal cancer (mCRC). J Clin Oncol. 2012;30:3631–3631.
Yoshino T, Mizunuma N, Yamazaki K, et al. TAS-102 monotherapy for pretreated metastatic colorectal cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2012;13:993–1001.
Mayer RJ, Van Cutsem E, Falcone A, et al. RECOURSE Study Group. Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N Engl J Med. 2015;372:1909–19. https://doi.org/10.1056/NEJMoa1414325.
Palles C, Fotheringham S, Chegwidden L, Lucas M, Kerr R, Mozolowski G, Rosmarin D, Taylor JC, Tomlinson I, Kerr D. An evaluation of the diagnostic accuracy of a panel of variants in DPYD and a single variant in ENOSF1 for predicting common capecitabine related toxicities. Cancers. 2021;13(7):1497. https://doi.org/10.3390/cancers13071497.
Chen S, Yueh MF, Bigo C, Barbier O, Wang K, Karin M, Nguyen N, Tukey RH. Intestinal glucuronidation protects against chemotherapy-induced toxicity by irinotecan (CPT-11). Proc Natl Acad Sci USA. 2013;110(47):19143–8. https://doi.org/10.1073/pnas.1319123110.
Bellio H, Fumet JD, Ghiringhelli F. Targeting BRAF and RAS in colorectal cancer. Cancers. 2021;13(9):2201. https://doi.org/10.3390/cancers13092201.PMID:34063682;PMCID:PMC8124706.
Heinimann K. Toward a molecular classification of colorectal cancer: the role of microsatellite instability status. Front Oncol. 2013;31(3):272. https://doi.org/10.3389/fonc.2013.00272.
Quaas A, Rehkaemper J, Rueschoff J, Pamuk A, Zander T, Hillmer A, Siemanowski J, Wittig J, Buettner R, Plum P, Popp F, Gebauer F, Bruns CJ, Loeser H, Alakus H, Schoemig-Markiefka B. Occurrence of high microsatellite-instability/mismatch repair deficiency in nearly 2000 human adenocarcinomas of the gastrointestinal tract, pancreas, and bile ducts: a study from a large german comprehensive cancer center. Front Oncol. 2021;22(11): 569475. https://doi.org/10.3389/fonc.2021.569475.
Morse MA, Hochster H, Benson A. Perspectives on treatment of metastatic colorectal cancer with immune checkpoint inhibitor therapy. Oncologist. 2020;25(1):33–45. https://doi.org/10.1634/theoncologist.2019-0176.
Van Cutsem E, Cervantes A, Adam R, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. 2016;27:1386–422.
Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228–47. https://doi.org/10.1016/j.ejca.2008.10.026.
Ottaiano A, Caraglia M, Di Mauro A, Botti G, Lombardi A, Galon J, Luce A, D’Amore L, Perri F, Santorsola M, Hermitte F, Savarese G, Tatangelo F, Granata V, Izzo F, Belli A, Scala S, Delrio P, Circelli L, Nasti G. Evolution of mutational landscape and tumor immune-microenvironment in liver oligo-metastatic colorectal cancer. Cancers. 2020;12(10):3073. https://doi.org/10.3390/cancers12103073.
Ottaiano A, Circelli L, Lombardi A, Scala S, Martucci N, Galon J, Buonanno M, Scognamiglio G, Botti G, Hermitte F, Savarese G, D’Amore L, Tatangelo F, Di Mauro A, Liguori G, Trotta AM, Napolitano M, Capozzi M, Tafuto S, Perri F, La Rocca A, Caraglia M, Nasti G. Genetic trajectory and immune microenvironment of lung-specific oligometastatic colorectal cancer. Cell Death Dis. 2020;11(4):275. https://doi.org/10.1038/s41419-020-2480-6.
Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, Tsimberidou AM, Vnencak-Jones CL, Wolff DJ, Younes A, Nikiforova MN. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the association for molecular pathology, american society of clinical oncology, and college of American pathologists. J Mol Diagn. 2017;19(1):4–23. https://doi.org/10.1016/j.jmoldx.2016.10.002.
Buchhalter I, Rempel E, Endris V, Allgäuer M, Neumann O, Volckmar AL, Kirchner M, Leichsenring J, Lier A, von Winterfeld M, Penzel R, Christopoulos P, Thomas M, Fröhling S, Schirmacher P, Budczies J, Stenzinger A. Size matters: dissecting key parameters for panel-based tumor mutational burden analysis. Int J Cancer. 2019;144(4):848–58. https://doi.org/10.1002/ijc.31878.
Cortes-Ciriano I, Lee S, Park WY, Kim TM, Park PJ. A molecular portrait of microsatellite instability across multiple cancers. Nat Commun. 2017;8:15180. https://doi.org/10.1038/ncomms15180.
Goodman AM, Sokol ES, Frampton GM, Lippman SM, Kurzrock R. Microsatellite-stable tumors with high mutational burden benefit from immunotherapy. Cancer Immunol Res. 2019;7(10):1570–3. https://doi.org/10.1158/2326-6066.CIR-19-0149.
Masuishi T, Taniguchi H, Kawakami T, Kawamoto Y, Kadowaki S, Onozawa Y, Muranaka T, Tajika M, Yasui H, Nakatsumi H, Yuki S, Muro K, Omae K, Komatsu Y, Yamazaki K. Impact of tumour growth rate during preceding treatment on tumour response to regorafenib or trifluridine/tipiracil in refractory metastatic colorectal cancer. ESMO Open. 2019;4(6):e000584. https://doi.org/10.1136/esmoopen-2019-000584.
Scartozzi M, Giampieri R, Maccaroni E, Del Prete M, Faloppi L, Bianconi M, Galizia E, Loretelli C, Belvederesi L, Bittoni A, Cascinu S. Pre-treatment lactate dehydrogenase levels as predictor of efficacy of first-line bevacizumab-based therapy in metastatic colorectal cancer patients. Br J Cancer. 2012;106(5):799–804. https://doi.org/10.1038/bjc.2012.17.
Wei W, You Z, Sun S, Wang Y, Zhang X, Pang D, Jiang Y. Prognostic implications of fibroblast growth factor receptor 4 polymorphisms in primary breast cancer. Mol Carcinog. 2018;57(8):988–96. https://doi.org/10.1002/mc.22819.
Quintanal-Villalonga Á, Carranza-Carranza A, Meléndez R, Ferrer I, Molina-Pinelo S, Paz-Ares L. Prognostic Role of the FGFR4-388Arg variant in lung squamous-cell carcinoma patients with lymph node involvement. Clin Lung Cancer. 2017;18(6):667-674.e1. https://doi.org/10.1016/j.cllc.2017.05.008.
Carrillo-Dávila IA, Garibaldi-Ríos AF, Figuera LE, Gómez-Meda BC, Zúñiga-González GM, Puebla-Pérez AM, García-Verdín PM, Castro-García PB, Gutiérrez-Hurtado IA, Torres-Mendoza BM, Gallegos-Arreola MP. Association of the rs1966265 and rs351855 FGFR4 variants with colorectal cancer in a Mexican population and their analysis in silico. Biomedicines. 2024;12(3):602. https://doi.org/10.3390/biomedicines12030602.
Ulaganathan VK, Sperl B, Rapp UR, Ullrich A. Germline variant FGFR4 p.G388R exposes a membrane-proximal STAT3 binding site. Nature. 2015;528(7583):570–4. https://doi.org/10.1038/nature16449.
Baba Y, Tamura T, Satoh Y, Gotou M, Sawada H, Ebara S, Shibuya K, Soeda J, Nakamura K. Panitumumab interaction with TAS-102 leads to combinational anticancer effects via blocking of EGFR-mediated tumor response to trifluridine. Mol Oncol. 2017;11(8):1065–77. https://doi.org/10.1002/1878-0261.12074.
Bijnsdorp IV, Kruyt FA, Fukushima M, Smid K, Gokoel S, Peters GJ. Molecular mechanism underlying the synergistic interaction between trifluorothymidine and the epidermal growth factor receptor inhibitor erlotinib in human colorectal cancer cell lines. Cancer Sci. 2010;101(2):440–7. https://doi.org/10.1111/j.1349-7006.2009.01375.x.
Turkington RC, Longley DB, Allen WL, Stevenson L, McLaughlin K, Dunne PD, Blayney JK, Salto-Tellez M, Van Schaeybroeck S, Johnston PG. Fibroblast growth factor receptor 4 (FGFR4): a targetable regulator of drug resistance in colorectal cancer. Cell Death Dis. 2014;5(2): e1046. https://doi.org/10.1038/cddis.2014.10.
Cho SH, Hong CS, Kim HN, Shin MH, Kim KR, Shim HJ, Hwang JE, Bae WK, Chung IJ. FGFR4 Arg388 is correlated with poor survival in resected colon cancer promoting epithelial to mesenchymal transition. Cancer Res Treat. 2017;49(3):766–77. https://doi.org/10.4143/crt.2016.457.
Bange J, Prechtl D, Cheburkin Y, Specht K, Harbeck N, Schmitt M, Knyazeva T, Muller S, Gartner S, Sures I, et al. Cancer progression and tumor cell motility are associated with the FGFR4 Arg(388) allele. Cancer Res. 2002;62:840–7.
Leow MKS, Ang J, Bi X, Koh ET, McFarlane C. Alterations in SAMD9, AHSG, FRG2C, and FGFR4 genes in a case of late-onset massive tumoral calcinosis. AACE Clin Case Rep. 2023;9(5):153–7. https://doi.org/10.1016/j.aace.2023.05.004.
Zhu F, Lin J, Liao H, Wang Y, Pan J. Gene profiling of metastatic small intestinal squamous cell carcinoma after lung squamous cell carcinoma surgery: a case report. Transl Cancer Res. 2022;11(4):967–72. https://doi.org/10.21037/tcr-22-481.
Peng T, Sun Y, Lv Z, Zhang Z, Su Q, Wu H, Zhang W, Yuan W, Zuo L, Shi L, Zhang LF, Zhou X, Mi Y. Effects of FGFR4 G388R, V10I polymorphisms on the likelihood of cancer. Sci Rep. 2021;11(1):1373. https://doi.org/10.1038/s41598-020-80146-y.
Levine KM, Ding K, Chen L, Oesterreich S. FGFR4: a promising therapeutic target for breast cancer and other solid tumors. Pharmacol Ther. 2020;214: 107590. https://doi.org/10.1016/j.pharmthera.2020.107590.
Stavraka C, Pouptsis A, Synowiec A, Angelis V, Satterthwaite L, Khan S, Chauhan M, Holden C, Young S, Karampera C, Martinou M, Mills-Baldock T, Baxter M, Barry A, Eccles B, Iveson T, Shiu KK, Hill M, Abdel-Raouf S, Graham JS, Thomas A, Ross PJ. Trifluridine/tipiracil in metastatic colorectal cancer: a UK multicenter real-world analysis on efficacy, safety, predictive and prognostic factors. Clin Colorectal Cancer. 2021;20(4):342–9. https://doi.org/10.1016/j.clcc.2021.09.009.
Acknowledgements
We thank Daniela Capobianco for technical editing and writing assistance. We would like to express our gratitude to the TRIAL scientific association (CF: 92088670622) for their invaluable and unwavering collaboration on this work.
Funding
We acknowledge the “Lega Italiana per la Lotta contro I Tumori (LILT)‐sezione di Napoli” for collaboration and financial support. We also greatly thank Mrs. Antonietta Nacca (private citizen) for free and unconditional financial support. The current study was financially supported by the “Ministero delle Imprese e del Made in Italy” project number F/310034/01-03/X56, “Epi-MET - Funzionalizzazione delle aberrazioni (epi)genomiche nei tumori metastatici”.
Author information
Authors and Affiliations
Contributions
Conceptualization, A.O., Michele Caraglia, G.N.; methodology, A.O., G.S., A.C., Michele Caraglia, N.P.; software, A.O., M.S., S.Z.; validation, M.I., G.S.; formal analysis, R.S., A.O., G.N., A.C., Marco Cascella; investigations, genetic assessments, M.I., Marika Casillo, N.P., F.C., G.S.; investigations, radiologic assessments, C.P.; resources, A.O. and G.N.; data curation, F.P., Michele Caraglia, F.C., F.S., G.N.; writing-original draft preparation, A.O., Michele Caraglia, F.P.; writing-review and editing, G.N., M.S.; supervision, R.S. and A.O. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
In accordance with local and national laws, ethics approval and consent to participate were exempted due to the retrospective/observational and non-interventional nature of the work. However, all patients involved in this study provided written informed consent before undergoing treatment administration and molecular assessments.
Consent for publication
Not applicable.
Competing interests
Roberto Sirica, Giovanni Savarese, Monica Ianniello, and Marika Casillo are employed by AMES, Centro Polidiagnostico Strumentale srl, 80013 Naples, Italy. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ottaiano, A., Santorsola, M., Ianniello, M. et al. Predictive significance of FGFR4 p.G388R polymorphism in metastatic colorectal cancer patients receiving trifluridine/tipiracil (TAS-102) treatment. J Transl Med 22, 379 (2024). https://doi.org/10.1186/s12967-024-05184-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-024-05184-w