
Abstract
Platelets not only participate in thrombosis and hemostasis but also interact with tumor cells and protect them from mechanical damage caused by hemodynamic shear stress and natural killer cell lysis, thereby promoting their colonization and metastasis to distant organs. Platelets can affect the tumor microenvironment via interactions between platelet-related factors and tumor cells. Metastasis is a key event in cancer-related death and is associated with platelet-related factors in lung, breast, and colorectal cancers. Although the factors that promote platelet expression vary slightly in terms of their type and mode of action, they all contribute to the overall process. Recognizing the correlation and mechanisms between these factors is crucial for studying the colonization of distant target organs and developing targeted therapies for these three types of tumors. This paper reviews studies on major platelet-related factors closely associated with metastasis in lung, breast, and colorectal cancers.
Similar content being viewed by others

Introduction
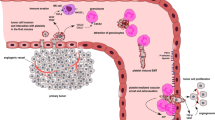
Platelets protect tumor cells from mechanical damage caused by natural killer cell lysis and hemodynamic shear stress, which can discharge cytokines, secondary mediators, and growth factors, thus increasing invasion, epithelial–mesenchymal transition (EMT), and extravasation of tumor cells and promoting angiogenesis and vascular remodeling [1,2,3,4,5,6]. In addition, platelets can induce resistance to anoikis by activating YAP1 signaling [7,8,9]. Anoikis is a form of apoptosis caused by the loss of extracellular matrix (ECM) adhesion or improper cell adhesion [10]. Anoikis is a barrier to tumor metastasis, and tumor progression and metastasis need to overcome anoikis [11]. Cancer cells can overcome anoikis by changing the integrin repertoire expression [12, 13], promoting EMT [14, 15], and regulating anoikis resistance caused by oxidative stress or hypoxia [16,17,18]. Cancer cells also act on platelets. Cancer cells can release platelet agonists (ADP, thrombin, and TXA2), induce platelet activation and factor release, and interact with platelets to promote tumor angiogenesis and metastasis [19,20,21,22,23,24]. Tumor cells from primary tumors invade the matrix and basement membrane, enter the blood flow, and form circulating complexes with leukocytes and platelets [25]. The interplay between platelets and circulating cancer cells produces tumor microemboli, which may block distant organs and promote the interplay of the endothelium [26]. The formation of tumor microemboli is deemed a crucial step in tumor metastasis and colonization during the intravascular phase. Metastasis is a key event in cancer-related deaths [27]. Tumor metastasis is closely associated with platelet-related factors. This strategy opens new prospective avenues for developing therapeutic strategies for lung, breast, and colorectal cancers.
Lung cancer and platelet-related factors
Lung cancer is the leading cause of cancer-related death worldwide [28]. According to the National Center for Health Statistics, approximately 127,750 individuals in the United States die annually from lung cancer [29]. Non-small cell lung cancer (NSCLC) accounts for approximately 80% of all lung cancers, and its incidence is increasing rapidly [30]. There are usually no obvious in the early stage of lung cancer, so it is necessary to find an appropriate screening method. MicroRNAs (miRNAs), long noncoding RNAs (lncRNAs) and exosomal lncRNAs can be regarded as noninvasive early biomarkers for lung cancer detection [31,32,33]. At present, many antitumor drugs, such as curcumin, cisplatin and doxorubicin, have been developed for the treatment of lung cancer [34,35,36]. In addition, treatment methods targeting the AMPK signaling pathway in lung cancer have been proposed [37]. However, it is crucial to clarify the mechanism of lung cancer for its diagnosis and treatment because platelets are related to the growth and metastasis of lung cancer [38].
Programmed death-ligand 1 (PD-L1)
PD-L1 is the principal ligand of programmed death 1 (PD-1) [39]. PD-1 (CD279) plays an important role in maintaining the tolerance of peripheral and central immune cells by binding to the ligands PD-L1 (B7-H1) and PD-L2 (B7-DC). Platelets from patients with NSCLC express PD-L1, which restrains CD4 + and CD8 + T cells [38, 40, 41]. Blood platelets often contact lung cancer cells both in vivo and in vitro. Platelets can also absorb PD-L1 from cancer cells in a manner dependent on fibronectin, GPIbα, and integrin α5β1 [38]. EGFR activation by EGF stimulation or mutation can decrease the expression of PD-L1 in cancer cells through the p-ERK1/2/p-c-Jun pathway, which plays an important role in the platelet-induced upregulation of PD-L1 in tumor cells [42]. The expression of PD-L1 can predict the response and overall survival (OS) rate in lung cancer patients [43]. PD-L1 is expressed in many malignant tumors and can be transferred from tumor cells to platelets in the form of integrins, thereby playing important roles in tumor immune escape [38, 43]. PD-L1 is related not only to tumor stage and metastasis but also to the prediction of immune checkpoint inhibition (ICI) [38].
P-selectin
Selectin is a key cell adhesion molecule that usually exists in platelet α particles. Generally, no or persistently low expression levels are observed in the resting state. When platelets are stimulated, P-selectin (CD62P) is promptly transferred to their surface through membrane fusion; thus, P-selectin is usually chosen as a marker of platelet activation [44, 45]. Sialyl-LewisX (sLeX) and its isomer sialyl-LewisA (sLeA) are the minimal recognition motifs for all selectins and are synthesized by α2,3-sialyltransferases, α1,3-fucosyltransferases IV or VII, N-acetyl glucosaminyltransferases, and β1,4-galactosyltranferases. The combination of selectins with carbohydrates normally requires a glycoprotein scaffold, and P-selectin glycoprotein ligand-1 (PSGL-1) is the most characteristic ligand assembled at the tips of microvilli on the surface of white blood cells [46,47,48]. PSGL-1 is expressed on different cell surfaces. PSGL-1 interacts with P-selectin to initiate platelet-mediated cell adhesion. Activated platelets and endothelial cells express P-selectin, which interacts with PSGL-1 to aggregate activated platelets on leukocytes, progressing to activated endothelial cells. P-selectin mediates the aggregation of activated platelets with cancer cells and the adhesion of cancer cells to activated endothelial cells [49]. Circulating tumor cells act on the normal endothelium in a leukocyte-like manner and adhere to the endothelium of the metastatic site via adhesion molecules [50] (Fig. 1). Activated platelets act on lung cancer cells via PSGL-1, leading to distant hematogenous metastasis of lung cancer cells [51].

Stimulated endothelial cells and activated platelets express P-selection (CD62P), which interacts with PSGL-1 for leukocyte rolling on stimulated endothelial cells. P-selectin mediates the heterotypic aggregation of activated platelets with cancer cells and adhesion of cancer cells to stimulated endothelial cells. Circulating tumor cells interact with the normal endothelium of the target organ in a leukocyte-like manner and attach to the endothelium of the future metastasis site by using the adhesion molecules of the leukocyte adhesion cascade
Integrins
Integrin is a cell-matrix adhesion molecule that not only provides mechanical engagement of the cell to the extracellular matrix but also transduces signals related to cancer and malignant tumors. Integrins have two primary functions: mechanically linking cells to the extracellular matrix (ECM) and initiating signal transduction pathways. In other words, they serve as both the physical connection between cells and the ECM and as initiators of signaling processes [52]. Integrins are large glycoproteins composed of a group of noncovalently related type I transmembrane α- and β-subunits [53, 54]. There are two integrin subgroups in platelets, β1 and β3, which can compose five human platelet integrins [55]. Two β3 integrins exist on platelets, namely, αIIbβ3 and αvβ3 [56, 57]. Integrins are primary regulators of cell adhesion, diffusion, and migration. Integrins play important roles in promoting oncogenic growth factor receptor (GFR) signaling and GFR-dependent cancer cell invasion, as well as in determining the colonization of metastatic sites and promoting the survival of circulating tumor cells [58].
Integrin αIIbβ3
The integrin αIIbβ3 is the main integrin on platelets and is also referred to as the glycoprotein GPIIb/IIIa (CD41/CD61) complex. This integrin is essential for normal platelet function. The integrin αIIbβ3 is also produced in lung cancer cells [59]. Notably, integrin αIIbβ3 can recognize RGD peptide-binding sequences on different adhesive proteins, such as fibrinogen and von Willebrand factor (VWF). The main function of integrin αIIbβ3 is to promote platelet aggregation through its binding with plasma fibrinogen. Its dimeric structure ensures the effective linkage of platelets [60]. Transmitting bidirectional signals is a key feature of integrin αIIbβ3. In the resting state, integrin αIIbβ3 is in an inactive conformation. However, the affinity of the extracellular domain for this ligand is low. Under agonist stimulation, the cytoplasmic tail of integrin αIIbβ3 can bind to intracellular proteins, especially talin or kinin. This combination leads to intracellular and transmembrane separation. The integrin αIIbβ3 complex undergoes a conformational change in its extracellular domain, transitioning from a low-affinity (inactive) state to a high-affinity (active) state for its ligand (fibrinogen) [61]. According to the literature, integrin αIIbβ3 exists in different tumor cells [62,63,64,65], promoting cancer cell adhesion and invasion [63,64,65,66].
Integrin αvβ3
Integrin αvβ3 is considered a recognized marker of breast, lung, and pancreatic cancers [67, 68]. Integrin αvβ3 can trigger nonanchored cell survival and tumor metastasis without ligand binding [69]. The expression of integrin αvβ3 is necessary for inducing the stem-like properties of lung cancer cells [67]. Integrin αvβ3 is usually not produced by epithelial cells and has been shown to be a remarkable regulator of tumor angiogenesis [70,71,72]. The fibrin-fibronectin complex induces the activation of integrin αvβ3, which triggers proinvasive EMT signaling and invasive protrusions in cancer cells [73, 74]. In tumor cells, integrin αvβ3 not only phosphorylates the adaptor protein p130 CRK-associated substrate (p130CAS) but also induces adhesion-dependent activation of steroid receptor coactivator (Src) and focal adhesion kinase (FAK). These signaling events lead to the survival, proliferation, and invasion of tumor cells in combination with the ECM [67, 68]. The inhibition of integrin αvβ3 binding to ECM ligands can directionally block endothelial cell-mediated tumor metastasis and angiogenesis. Moreover, integrin-blocking agents have become a potential strategy for targeted therapy [52]. In lung cancers, clusters of integrin αvβ3 emerge on the surface of suspended cells. This clustering is mediated by the interplay between galectin-3 and integrin αvβ3, which is irrelevant to its ligand binding domain [67]. Unligated integrin αvβ3 can drive tumor cells toward a stem-like state, whereas when connected to its ligands, it can contribute to ECM-driven cell invasion and proliferation [68] (Fig. 2). Compared with blocking tumor integrin αvβ3 alone, blocking both platelet integrin αIIbβ3 and tumor integrin αvβ3 simultaneously yields greater antiangiogenic and antitumor effects. These findings indicate that antagonists targeting both platelets and endothelial integrins may have clinical efficacy.

Integrin signaling generated by binding to extracellular matrix (ECM) ligands occurs. In the absence of ligand binding, αvβ3 integrin recruited kras and src to drive cell reprogramming events, which led to phenotypic changes, thus promoting stem dryness, metastasis and drug resistance. Fibrin-fibronection complex induces the activation of αvβ3 integrin, which triggers Survival, prolifera on, adhesion, migra? on, invasion in cancer cells
Autotaxin
Autotaxin (ATX) is a unique member of the nucleotide pyrophosphatase family. ATX induces lysophospholipase D (lysoPLD) activity, which catalyzes lysophosphatidic acid (LPA) production [9]. By producing LPA, ATX and/or lysoPLD can promote tumor progression by providing a favorable microenvironment for tumor cell invasion and angiogenesis [75]. ATX is a multidomain protein consisting of two somatomedin B (SMB1,2)-like domains, a catalytic phosphodiesterase (PDE) domain and a nuclease-like domain [76]. Compared with that in healthy lung tissue, ATX overexpression in lung cancer tissue is significantly related to poorly differentiated or undifferentiated cells [77, 78].
VEGF and bFGF
Angiogenesis is an extremely important process in the development and metastasis of tumors. Tumor cells attempt to obtain an independent blood supply through a series of processes, including the release of proangiogenic factors and binding to receptors on vascular endothelial cells [79,80,81]. The interaction between tumor cells and platelets leads to platelet activation. The major angiogenic factors released by the alpha granules of activated platelets include vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF) [82,83,84]. Both VEGF and bFGF are considered key regulators of angiogenesis [85, 86]. The VEGF family consists of seven secreted glycoproteins, namely, VEGF-A, -B, -C, -D, -E, -F, and placental growth factor (PlGF). VEGF-A is the most effective factor for angiogenesis [87,88,89]. In addition, VEGF can be secreted by multiple cell types, including endothelial cells, epithelial cells of the retina, macrophages, stromal cells, and malignant cells. The main receptors of VEGF include VEGFR-1, VEGFR-2, and VEGFR-3 [90, 91]. The binding of VEGF and VEGFR induces receptor dimerization, which leads to the activation of tyrosine kinases in cells, thus exerting their biological effects in cells [87]. In vitro, VEGF and bFGF can induce the proliferation, migration and differentiation of angioblasts [92, 93]. Angioblasts build the primary vascular plexus [94]. However, VEGF and bFGF regulate angiogenesis differentially. Angiogenesis can be driven by VEGF alone but not by bFGF [95]. NSCLC cells can secrete VEGF, increase the number of VEGF and VEGF receptors, and subsequently promote angiogenesis and metastasis [96,97,98]. However, bFGF- targeted therapy for lung cancer has limitations. Blocking bFGF can inhibit cell growth but promote cell invasion [99]. The overexpression of bFGF may indicate poor prognosis in patients with lung cancer [100].
Breast cancer and platelet-related factors
Breast cancer (BC) is the most common malignancy in women [101]. Currently, the 5-year overall survival rate of patients with BC without metastasis is > 80% [102]. However, 20–30% of patients with BC develop metastasis after primary tumor treatment [103]. Furthermore, metastasis is the primary cause of death in patients with BC [104], and platelet-related factors are associated with tumor metastasis.
PD-L1
PD-L1 expression was independently detected in circulating tumor cells and platelets from patients with metastatic BC [105]. The platelet PD-L1 expression level markedly differed between and within patients [105]. This heterogeneity aligns with the varying sensitivities of patients to immune checkpoint inhibition therapy [106, 107].
P-selectin
P-selectin exists on the surface of endothelial cells and platelets. PSGL-1 is the primary ligand of P-selectin and is responsible for leukocyte rolling on active endothelial cells [108]. In the resting state, P-selectin is expressed at a low level. In the activated state, most P-selectin molecules are transferred from in-granules to platelet membranes [109]. P-selectin initiates interactions between platelets and sialylated fucosylated mucins in circulating tumor cells [25, 110]. Furthermore, P-selectin participates in platelet signaling through protein kinase B (Akt), leading to the phosphorylation of the Src family kinases Fyn and Hck, as well as Erk. These processes appear to be prerequisites for platelet granule secretion and aggregation [111,112,113]. P-selectin interacts with intracellular talin-1 and subsequently activates integrin GP IIb/IIIa, resulting in the P-selectin-GP IIb/IIIa-talin complex and the accumulation of platelets in tumor tissues [114]. P-selectin can activate additional intracellular signaling pathways that are beneficial for the secretion and aggregation of α-granules and dense granules [109]. Low-molecular-weight heparin (LMWH) combines with P-selectin and simultaneously inhibits the plasma coagulation cascade. Therefore, it is a potentially valuable drug for cancer treatment [109].
Lysophosphatidic acid
Lysophosphatidic acid (LPA) is a bioactive lipid. It serves as a multifunctional lipid mediator that regulates cell growth, movement, and differentiation [115]. LPA induces several cellular activities, including adenylyl cyclase activation, Ca2+ mobilization, and mitogen-activated protein kinase stimulation [116]. There are six distinct G protein-coupled receptors: LPA1, LPA2, LPA3, LPA4, LPA5, and LPA6 [75, 116,117,118]. LPA is produced by aggregated platelets during tumor cell-induced platelet aggregation. It actss as a paracrine factor in tumor cells through the LPA1 receptor, thereby promoting the proliferation, migration, and secretion of proinflammatory factors [119].
Autotaxin
ATX can be stored in platelet α-particles. Platelet-derived lysophosphatidylcholine degrades LPA [9]. β3 integrin may bind ATX on the surface of cancer cells/platelets, providing a mechanism for the production of LPA near its receptor, thus enhancing the spread of cancer cells [119]. The interplay between circulating tumor cells and platelets induces platelet aggregation and LPA release. In the blood, LPA acts on tumor LPA1 to promote survival and invasion and may act on platelet LPA5 to promote platelet aggregation [119,120,121]. Moreover, LPA promotes the migration, invasion, and proliferation of BC cells in vivo [122].
Integrin αvβ3
Breast and lung cancers share similarities, as both express the integrin αvβ3. Furthermore, their mechanisms of action in cancer cells are similar. Integrin αvβ3 has different functions in tumors, such as promoting angiogenesis, cell proliferation, invasion, and metastasis in different cancers [67, 69]. The integrin αvβ3 ligand L1 cell adhesion molecule (L1-CAM) expressed on BC cells is necessary for BC metastasis to the lungs, where it allows tumor cells to combine and extravasate through the lung endothelium [123]. Specific integrins can dominate the localization and activity of matrix metalloproteases to promote invasive migration. For example, integrin αvβ3 controls matrix metalloproteinase 9 (MMP9) in MDA-MB-435 BC cells [124]. Integrin β3 and KRAS interact via galectin-3 propels to activate RALB. RALB subsequently activates TANK-binding kinase 1, which activates the NF-κB pathway, thus promoting cell survival [69]. Ligated integrins activate FAKs and other downstream signaling molecules, resulting in anchorage-dependent survival and proliferation [125]. However, unligated integrins can induce a form of death called integrin-mediated death (IMD) by activating the apoptosis pathway, thus negatively affecting the malignant characteristics of tumor cells [126].
VEGF and bFGF
Angiogenesis is an important process related to tumor development [79, 80], which regulated by proangiogenic factors (VEGF, bFGF, and PDGF) and the microenvironment (hypoxia) [127, 128]. In breast cancer cells, VEGFR1 mainly activates the MAPK/ERK1/2 and PI3K/AKT signaling pathways, leading to tumor growth and EMT and thus promoting tumor invasion and metastasis [129]. In addition to promoting angiogenesis, bFGF is involved in plasminogen activator synthesis, cell growth and differentiation, and tumor invasion [130, 131]. Some studies have shown that angiogenesis, tumor growth and metastasis of breast cancer cells can be inhibited by blocking VEGFR1 and VEGFR2 [132,133,134,135,136], and the expression of bFGF is related to a shorter survival time in patients with tumors [137]. Bevacizumab is an effective treatment for metastatic breast cancer targeting VEGF ligands [138]. The success of VEGF- targeted drugs has encouraged the research on targeted therapy for breast cancer, indicating that targeting VEGF is a potentially valuable treatment for breast cancer.
Colorectal cancer and platelet-related factors
Colorectal cancer (CRC) is a major cause of death worldwide [139]. CRC occurs mainly in the older population, with a median age > 60 years at diagnosis. However, population-based studies have reported that the incidence of CRC is greater in younger populations than in those aged > 50 years [140, 141]. Fecal occult blood tests and colonoscopy are the main screening methods for CRC; however, the invasive nature of colonoscopy limits its application [142]. Therefore, it is important to develop sensitive screening methods for colorectal cancer. Investigations on the expression of platelet-related factors have revealed their association with colorectal cancer cells [142]. These findings could lead to encouraging prospects for future colorectal cancer screening. Understanding the role of platelet-related factors in CRC may offer new insights for the diagnosis, treatment, and prognosis of CRC.
Platelet-derived growth factor (PDGF)
PDGF is a nonhomogeneous molecule that exists in its active form as a dimer through the formation of four chain proteins: PDGF-A, PDGF-B, PDGF-C, and PDGF-D. PDGF-A and PDGF-B can simultaneously form homodimers and heterodimers, whereas the PDGF-C and PDGF-D chains can only form homodimers. PDGF-AB is the most commonly detected form of PDGF-AB in the serum [143]. PDGFs have a series of biological functions and are induced by activating the tyrosine kinase receptors (TKRs) PDGFR-α and PDGFR-β [144]. PDGFR-αα combines with all growth factors, including PDGF-AA, PDGF-AB, PDGF-BB, and PDGF-CC but not PDGF-DD. PDGFR-αβ combines with all growth factors, namely, PDGF-AB, PDGF-BB, PDGF-CC, and PDGF-DD, but not PDGF-AA. PDGFR-β combines with PDGF-BB and PDGF-DD. However, its interaction with PDGF-DD has not been determined [145] (Fig. 3). PDGFR/PDGF signaling results from interactions between dimeric PDGF isoforms and PDGFRs. The specific binding of PDGF ligands to PDGFRs leads to dimerization of these receptors, thus enhancing their stability through interactions with the receptors [146]. Changes in PDGFR family signaling play important roles in CRC. CRC is associated with PDGFR overexpression in tumors and tumor-related stromal cells [145, 147]. Overexpression of PDGFRs in CRC is associated with invasion, angiogenesis, metastasis, low survival rates, and targeted therapies [142]. A high PDGF-AB blood concentration may be an important parameter for CRC recurrence [142]. In summary, PDGF-BB may be involved in the progression of CRC, maintaining angiogenesis by augmenting pericytes within tumors, which is related to disease severity [142]. PDGF-CC has similar mitogenic activity comparable to that of PDGF-AB and PDGF-BB and is also considered to be an important oncogene in the PDGF/PDGFR signaling pathway due to its affinity for both PDGFR-αα and PDGFR-αβ. PDGF-CC induces angiogenesis in vivo [148]. Peripheral blood PDGF-C levels may be useful for the early diagnosis of CRC. In different types of cancers, PDGF-DD is upregulated; however, its role in CRC has not yet been determined [142]. The interplay between platelets and tumor cells improves their survival rate in the blood and promotes tumor metastasis [149, 150].

PDGFR-αα binds to all growth factors except PDGF-DD, PDGFR-bb binds to PDGF-BB and PDGF-DD, and PDGFR-αβ binds to all proteins except PDGF-AA. PDGFR/PDGF system initiate a complex cascade of MAP-kinase signaling to activate genes involved in angiogenesis, proliferation, invasion and metastasis
Glycoprotein VI (GPVI)
GPVI is a receptor for collagen, laminin, and fibrin [151,152,153] and can regulate platelet functions such as adhesion, aggregation, and procoagulant activity. GPVI is a member of the Ig superfamily and has two Ig domains (D1 and D2), a stalk containing an O-glycosylation site, and a cytoplasmic tail for binding Src kinase and calmodulin [152, 154]. The signal transduction of GPVI relies on its association with the dimeric Fc receptor chain (FcRγ). GPVI activation results in the phosphorylation of two conserved tyrosine residues that are dependent on Src kinase, which binds to the tandem SH2 domain of Syk. Subsequently, a signaling cascade is initiated, leading to the activation of phospholipase Cγ2 [155]. Glycoprotein VI is thought to bind to galectin-3 (Gal-3) in tumor cells, inducing platelet activation and promoting metastasis in CRC cells [156].
Gal-3 is a member of the β-galactoside-binding lectin family that is located mainly in the cytoplasm [157]. Gal-3 is present in the nucleus and on the cell surface and can be secreted into the circulation [158, 159]. As the main GPVI ligand in tumor cells, Gal-3 induces platelet activation and promotes BC metastasis [160]. The interplay between Gal-3 and GPVI promotes platelet activation, degranulation, and tumor cell transendothelial migration [160].
Autotaxin
ATX is a unique member of the ectonucleotide pyrophosphatase/phosphodiesterase (ENPP) family, which has lysoPLD activity and can convert lysophosphatidylcholine (LPC) into LPA [161,162,163,164]. LPA interacts with G protein-coupled receptors on the cell membrane, which can activate downstream signaling molecules, such as Ras, Rho, PLC, and PI3K [165]. In the early stage of CRC, the expression of ATX is positively correlated with tumor angiogenesis [166]. The up-regulation of ATX is related to cancer invasion and metastasis [9, 167].
VEGF and bFGF
Related studies have shown that the density of blood vessels at the infiltrating edge of CRC tissue is significantly greater than that in other areas of the tumor [168], and a high density of blood vessels is related to CRC progression and metastasis [169]. Both VEGF and bFGF are considered key regulators of angiogenesis [85, 86]. VEGF-A is the main angiogenic factor in CRC and is related to poor prognosis [170]. VEGFR-1 is expressed in human CRC cells and participates in tumor progression and metastasis [171]. Inhibition of VEGF signaling can lead to a decrease in protein activity related to cell movement, which further reduces the invasion of CRC cells [172]. There is a self-regulating mechanism for angiogenesis in colon cancer. VEGF expression is up-regulated, while bFGF expression is down-regulated [173]. Increased angiogenesis is associated with poor prognosis in patients with CRC, and targeting angiogenesis is a good therapeutic option. In the future, more drugs targeting angiogenesis will be developed, and we need to further explore drugs with high efficacy and minimal adverse effects.
Conclusions and perspectives
As an important source of circulating angiogenesis-related factors, platelets can affect the tumor microenvironment through interactions with tumor cells. Different platelet-related factors have independent or overlapping effects on the proliferation and metastasis of tumors, and they cross-talk with each other to regulate tumor angiogenesis and vascular integrity. High PD-L1 expression is observed in NSCLC, BC, and CRC [174]. Additionally, P-selectin expression is strongly and positively correlated with PD-L1 expression [38]. In particular, lung and breast cancers highly express P-selectin, integrin αvβ3, VEGF, bFGF, and ATX. Moreover, breast cancer cells express high levels of LPA and ATX in addition to PD-L1, P-selectin, integrin αvβ3, VEGF, bFGF, and PDGF. Integrin αvβ3 promotes bone metastasis through strengthened breast cancer cell adhesion [69]. Colorectal and breast cancers express high levels of ATX, PDGF, VEGF, bFGF, and GPVI. Overexpression of PDGF-AA/BB in patients with stage 4 breast cancer is associated with a relatively shorter survival time [175].
The inhibition of tumor proliferation and metastasis has always been a focus of research. The interaction between platelet-related factors and tumors opens a new direction for research. In lung, breast, and colorectal cancers, we found that tumor cells interact with platelets and that different platelet-related factors have independent or overlapping effects on the proliferation and metastasis of tumors, as these factors can predict the degree of tumor progression, prognosis, and metastasis. Platelet-related factors are interconnected and engage in crosstalk, which introduces a novel concept for tumor treatment. Targeting coexpressed platelet-related factors independently expressed by certain tumor cells to block signaling pathways may inhibit tumor metastasis.
Therapeutic options targeting platelet-related factors are currently being investigated. PD-L1 is expressed in many different types of tumors and platelets in patients with metastatic BC [176]. Immune checkpoint inhibitors targeting PD-L1 and PD-L1 receptors have been verified for tumor treatment [177]. Medi4736 is an antagonist of PD-L1 that can inhibit the growth of human tumors [178]. Low-molecular-weight heparin (LMWH) and unfractionated heparin can bind to P-selectin and inhibit its function [110, 179, 180]. Targeting the activation and inhibition of integrin αIIbβ3 is a promising therapeutic strategy. Adapter protein (ADAP) promotes the activation of integrin αIIbβ3 [181, 182]. Some proteins are also believed to directly bind to the cytoplasmic tails of integrin αIIb or β3 to inhibit the activation of integrin αIIbβ3 [61]. Moreover, α-actin is valuable for keeping integrin αIIbβ3 inactive [183]. Therapeutic drugs targeting integrin αIIbβ3, such as the integrin αIIbβ3 antibody fragment abciximab, antagonists, and small molecule inhibitors, have been used in clinical settings [184]. Therapeutic drugs targeting the integrin αvβ3 molecule, such as cilengitide MRL-123, have been widely investigated for cancer and osteoporosis treatment [185]. Bevacizumab is an antiangiogenic agent, and the FDA approved bevacizumab for the treatment of advanced NSCLC, metastatic breast cancer (mBC) and metastatic colorectal cancer (mCRC) [138, 186, 187]. Inhibitors of Src, Syk, and Tec tyrosine kinases block platelet activation via CLEC-2 and GPVI. Phase II trials using human GPVI-blocking F(ab) ACT-017 have achieved encouraging results [155]. Glenzocimab targets platelet GPVI by binding to the D2 domain of GPVI, inducing steric hindrance and structural modifications, thus inhibiting the interaction between GPVI and its main ligands [188]. PD173074 is an FGFR inhibitor that blocks small cell lung carcinoma (SCLC) growth both in vitro and in vivo [189]. LPA receptors are expressed in the vasculature and brain, which has led to consideration of the toxicity of LPA inhibitors. LPA3 is restricted and abnormally expressed in many cancer lineages, making it a particularly attractive target [75]. Inhibitors against LPA and ATX monoclonal antibodies have been used in clinical trials for treating fibrosis but have not yet entered clinical trials for cancer treatment [190]. GLPG1690 is a new ATX inhibitor [191,192,193]. PDGFs play important roles in tumor occurrence and are upregulated in many different malignant tumors [194]. At present, numerous drug studies are underway with the aim of inhibiting cancer progression by targeting PDGF. For example, 6B3 is a high-affinity monoclonal antibody that can effectively neutralize PDGF-CC-induced PDGFR-α phosphorylation and activation [195]. MOR8457 is a PDGF antibody that can effectively bind to and neutralize PDGF-BB [196]. Compound P2 can effectively inhibit PDGF-BB-induced autophosphorylation of PDGFR-β with low toxicity [197] (Table 1). However, the interactions of platelet-related factors with tumors are complex and require further exploration. Understanding these new mechanisms and exploring novel approaches to treat tumors in the future are therefore warranted.
Data availability
Not applicable.
Abbreviations
- EMT:
-
epithelial–mesenchymal transition
- ECM:
-
extracellular matrix
- NSCLC:
-
non-small cell lung cancer
- miRNAs:
-
MicroRNAs
- lncRNAs:
-
long noncoding RNAs
- PD-L1:
-
programmed death-ligand 1
- LPA:
-
lysophosphatidic acid
- ATX:
-
autotaxin
- PDGF:
-
Platelet-derived growth factor
- GPVI:
-
Glycoprotein VI
- PD-1:
-
programmed death 1
- OS:
-
overall survival
- ICIs:
-
immune checkpoint inhibitors
- sLeX:
-
Sialyl-Lewis X
- sLeA:
-
sialyl-Lewis A
- PSGL-1:
-
P-selectin glycoprotein ligand-1
- GFR:
-
growth factor receptor
- vWF:
-
von Willebrand factor
- ADAP:
-
Adapter protein
- Src:
-
a steroid receptor coactivator
- FAK:
-
focal adhesion kinase
- lysoPLD:
-
lysophospholipase D
- PDE:
-
phosphodiesterase
- VEGF:
-
vascular endothelial growth factor
- bFGF:
-
basic fibroblast growth factor
- PlGF:
-
placental growth factor
- BC:
-
breast cancer
- Akt:
-
protein kinase B
- LMWH:
-
low molecular weight heparin
- PDE:
-
phosphodiesterase domain
- L1-CAM:
-
L1 cell adhesion molecule
- MMP9:
-
matrix metalloproteinase-9
- IMD:
-
integrin-mediated death
- ITAM:
-
immunoreceptor tyrosine-based activation motif
- ENPP:
-
ectonucleotide pyrophosphatase/phosphodiesterase
- LPC:
-
lysophosphatidylcholine
- Gal-3:
-
Galectin-3
- CRC:
-
colorectal cancer
- TKRs:
-
tyrosine kinase receptors
- mBC:
-
metastatic breast cancer
- mCRC:
-
metastatic colorectal cancer
- SCLC:
-
small cell lung carcinoma
References
Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011;20(5):576–90.
Malik AB. Pulmonary microembolism. Physiol Rev. 1983;63(3):1114–207.
Schumacher D, Strilic B, Sivaraj KK, Wettschureck N, Offermanns S. Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell. 2013;24(1):130–7.
Stegner D, Dütting S, Nieswandt B. Mechanistic explanation for platelet contribution to cancer metastasis. Thromb Res. 2014;133(Suppl 2):S149–57.
Strilic B, Offermanns S. Intravascular survival and extravasation of Tumor cells. Cancer Cell. 2017;32(3):282–93.
Wojtukiewicz MZ, Sierko E, Hempel D, Tucker SC, Honn KV. Platelets and cancer angiogenesis nexus. Cancer Metastasis Rev. 2017;36(2):249–62.
Haemmerle M, Taylor ML, Gutschner T, Pradeep S, Cho MS, Sheng J, et al. Platelets reduce anoikis and promote metastasis by activating YAP1 signaling. Nat Commun. 2017;8(1):310.
Ward Y, Lake R, Faraji F, Sperger J, Martin P, Gilliard C, et al. Platelets promote metastasis via binding tumor CD97 leading to bidirectional signaling that coordinates Transendothelial Migration. Cell Rep. 2018;23(3):808–22.
Leblanc R, Lee SC, David M, Bordet JC, Norman DD, Patil R, et al. Interaction of platelet-derived autotaxin with tumor integrin αVβ3 controls metastasis of breast cancer cells to bone. Blood. 2014;124(20):3141–50.
Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. 1994;124(4):619–26.
Simpson CD, Anyiwe K, Schimmer AD. Anoikis resistance and tumor metastasis. Cancer Lett. 2008;272(2):177–85.
Felding-Habermann B, Fransvea E, O’Toole TE, Manzuk L, Faha B, Hensler M. Involvement of tumor cell integrin alpha v beta 3 in hematogenous metastasis of human melanoma cells. Clin Exp Metastasis. 2002;19(5):427–36.
Gehlsen KR, Davis GE, Sriramarao P. Integrin expression in human melanoma cells with differing invasive and metastatic properties. Clin Exp Metastasis. 1992;10(2):111–20.
Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer. 2004;4(2):118–32.
Gheldof A, Berx G. Cadherins and epithelial-to-mesenchymal transition. Prog Mol Biol Transl Sci. 2013;116:317–36.
Giannoni E, Parri M, Chiarugi P. EMT and oxidative stress: a bidirectional interplay affecting tumor malignancy. Antioxid Redox Signal. 2012;16(11):1248–63.
Cosse JP, Sermeus A, Vannuvel K, Ninane N, Raes M, Michiels C. Differential effects of hypoxia on etoposide-induced apoptosis according to the cancer cell lines. Mol Cancer. 2007;6:61.
Sermeus A, Cosse JP, Crespin M, Mainfroid V, de Longueville F, Ninane N, et al. Hypoxia induces protection against etoposide-induced apoptosis: molecular profiling of changes in gene expression and transcription factor activity. Mol Cancer. 2008;7:27.
Heinmöller E, Weinel RJ, Heidtmann HH, Salge U, Seitz R, Schmitz I, et al. Studies on tumor-cell-induced platelet aggregation in human lung cancer cell lines. J Cancer Res Clin Oncol. 1996;122(12):735–44.
Asanuma K, Wakabayashi H, Okamoto T, Asanuma Y, Akita N, Yoshikawa T, et al. The thrombin inhibitor, argatroban, inhibits breast cancer metastasis to bone. Breast Cancer. 2013;20(3):241–6.
Plantureux L, Crescence L, Dignat-George F, Panicot-Dubois L, Dubois C. Effects of platelets on cancer progression. Thromb Res. 2018;164(Suppl 1):S40–7.
Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9(1):28–39.
de Leval X, Benoit V, Delarge J, Julémont F, Masereel B, Pirotte B, et al. Pharmacological evaluation of the novel thromboxane modulator BM-567 (II/II). Effects of BM-567 on osteogenic sarcoma-cell-induced platelet aggregation. Prostaglandins Leukot Essent Fat Acids. 2003;68(1):55–9.
Adams GN, Rosenfeldt L, Frederick M, Miller W, Waltz D, Kombrinck K, et al. Colon Cancer Growth and Dissemination relies upon Thrombin, stromal PAR-1, and Fibrinogen. Cancer Res. 2015;75(19):4235–43.
Varki NM, Varki A. Heparin inhibition of selectin-mediated interactions during the hematogenous phase of carcinoma metastasis: rationale for clinical studies in humans. Semin Thromb Hemost. 2002;28(1):53–66.
Karpatkin S, Pearlstein E. Role of platelets in tumor cell metastases. Ann Intern Med. 1981;95(5):636–41.
Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331(6024):1559–64.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30.
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72(1):7–33.
Wu F, Wang L, Zhou C. Lung cancer in China: current and prospect. Curr Opin Oncol. 2021;33(1):40–6.
Entezari M, Ghanbarirad M, Taheriazam A, Sadrkhanloo M, Zabolian A, Goharrizi M, et al. Long non-coding RNAs and exosomal lncRNAs: potential functions in lung cancer progression, drug resistance and tumor microenvironment remodeling. Biomed Pharmacother. 2022;150:112963.
Gao Z, Yuan H, Mao Y, Ding L, Effah CY, He S, et al. In situ detection of plasma exosomal microRNA for lung cancer diagnosis using duplex-specific nuclease and MoS(2) nanosheets. Analyst. 2021;146(6):1924–31.
Abadi AJ, Zarrabi A, Gholami MH, Mirzaei S, Hashemi F, Zabolian A et al. Small in size, but large in action: microRNAs as potential modulators of PTEN in breast and lung cancers. Biomolecules. 2021;11(2).
Ashrafizadeh M, Najafi M, Makvandi P, Zarrabi A, Farkhondeh T, Samarghandian S. Versatile role of curcumin and its derivatives in lung cancer therapy. J Cell Physiol. 2020;235(12):9241–68.
Jin Y, Wang Y, Liu X, Zhou J, Wang X, Feng H, et al. Synergistic combination chemotherapy of Lung Cancer: Cisplatin and Doxorubicin Conjugated Prodrug Loaded, glutathione and pH sensitive nanocarriers. Drug Des Devel Ther. 2020;14:5205–15.
Ashrafizadeh M, Zarrabi A, Hushmandi K, Hashemi F, Moghadam ER, Owrang M, et al. Lung cancer cells and their sensitivity/resistance to cisplatin chemotherapy: role of microRNAs and upstream mediators. Cell Signal. 2021;78:109871.
Ashrafizadeh M, Mirzaei S, Hushmandi K, Rahmanian V, Zabolian A, Raei M, et al. Therapeutic potential of AMPK signaling targeting in lung cancer: advances, challenges and future prospects. Life Sci. 2021;278:119649.
Hinterleitner C, Strähle J, Malenke E, Hinterleitner M, Henning M, Seehawer M, et al. Platelet PD-L1 reflects collective intratumoral PD-L1 expression and predicts immunotherapy response in non-small cell lung cancer. Nat Commun. 2021;12(1):7005.
Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. 2016;375(18):1767–78.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–64.
Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7:12632.
Guo Q, Malloy MW, Roweth HG, McAllister SS, Italiano JE, Battinelli EM. Platelets upregulate tumor cell programmed death ligand 1 in an epidermal growth factor receptor-dependent manner in vitro. Blood Adv. 2022;6(20):5668–75.
Kythreotou A, Siddique A, Mauri FA, Bower M, Pinato DJ. PD-L1. J Clin Pathol. 2018;71(3):189–94.
Massaguer A, Engel P, Tovar V, March S, Rigol M, Solanes N, et al. Characterization of platelet and soluble-porcine P-selectin (CD62P). Vet Immunol Immunopathol. 2003;96(3–4):169–81.
Ludwig RJ, Schön MP, Boehncke WH. P-selectin: a common therapeutic target for cardiovascular disorders, inflammation and tumour metastasis. Expert Opin Ther Targets. 2007;11(8):1103–17.
Rosen SD. Ligands for L-selectin: homing, inflammation, and beyond. Annu Rev Immunol. 2004;22:129–56.
Sperandio M, Gleissner CA, Ley K. Glycosylation in immune cell trafficking. Immunol Rev. 2009;230(1):97–113.
Kansas GS. Selectins and their ligands: current concepts and controversies. Blood. 1996;88(9):3259–87.
Chen M, Geng JG. P-selectin mediates adhesion of leukocytes, platelets, and cancer cells in inflammation, thrombosis, and cancer growth and metastasis. Arch Immunol Ther Exp (Warsz). 2006;54(2):75–84.
Heidemann F, Schildt A, Schmid K, Bruns OT, Riecken K, Jung C, et al. Selectins mediate small cell lung cancer systemic metastasis. PLoS ONE. 2014;9(4):e92327.
Gong L, Cai Y, Zhou X, Yang H. Activated platelets interact with lung cancer cells through P-selectin glycoprotein ligand-1. Pathol Oncol Res. 2012;18(4):989–96.
Aksorn N, Chanvorachote P. Integrin as a molecular target for anti-cancer approaches in Lung Cancer. Anticancer Res. 2019;39(2):541–8.
Caswell PT, Vadrevu S, Norman JC. Integrins: masters and slaves of endocytic transport. Nat Rev Mol Cell Biol. 2009;10(12):843–53.
Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10(1):9–22.
Staatz WD, Rajpara SM, Wayner EA, Carter WG, Santoro SA. The membrane glycoprotein Ia-IIa (VLA-2) complex mediates the Mg++-dependent adhesion of platelets to collagen. J Cell Biol. 1989;108(5):1917–24.
Ill CR, Engvall E, Ruoslahti E. Adhesion of platelets to laminin in the absence of activation. J Cell Biol. 1984;99(6):2140–5.
Sonnenberg A, Modderman PW, Hogervorst F. Laminin receptor on platelets is the integrin VLA-6. Nature. 1988;336(6198):487–9.
Hamidi H, Ivaska J. Every step of the way: integrins in cancer progression and metastasis. Nat Rev Cancer. 2018;18(9):533–48.
Lavergne M, Janus-Bell E, Schaff M, Gachet C, Mangin PH. Platelet integrins in Tumor Metastasis: do they represent a therapeutic target? Cancers (Basel). 2017;9(10).
Varga-Szabo D, Pleines I, Nieswandt B. Cell adhesion mechanisms in platelets. Arterioscler Thromb Vasc Biol. 2008;28(3):403–12.
Huang J, Li X, Shi X, Zhu M, Wang J, Huang S, et al. Platelet integrin αIIbβ3: signal transduction, regulation, and its therapeutic targeting. J Hematol Oncol. 2019;12(1):26.
Grossi IM, Hatfield JS, Fitzgerald LA, Newcombe M, Taylor JD, Honn KV. Role of tumor cell glycoproteins immunologically related to glycoproteins ib and IIb/IIIa in tumor cell-platelet and tumor cell-matrix interactions. Faseb j. 1988;2(8):2385–95.
Honn KV, Chen YQ, Timar J, Onoda JM, Hatfield JS, Fligiel SE, et al. Alpha IIb beta 3 integrin expression and function in subpopulations of murine tumors. Exp Cell Res. 1992;201(1):23–32.
Timar J, Trikha M, Szekeres K, Bazaz R, Honn K. Expression and function of the high affinity alphaIIbbeta3 integrin in murine melanoma cells. Clin Exp Metastasis. 1998;16(5):437–45.
Boukerche H, Berthier-Vergnes O, Tabone E, Doré JF, Leung LL, McGregor JL. Platelet-melanoma cell interaction is mediated by the glycoprotein IIb-IIIa complex. Blood. 1989;74(2):658–63.
Cooper J, Giancotti FG. Integrin signaling in Cancer: mechanotransduction, stemness, epithelial plasticity, and Therapeutic Resistance. Cancer Cell. 2019;35(3):347–67.
Seguin L, Kato S, Franovic A, Camargo MF, Lesperance J, Elliott KC, et al. An integrin β3-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat Cell Biol. 2014;16(5):457–68.
Seguin L, Desgrosellier JS, Weis SM, Cheresh DA. Integrins and cancer: regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol. 2015;25(4):234–40.
Desgrosellier JS, Barnes LA, Shields DJ, Huang M, Lau SK, Prévost N, et al. An integrin alpha(v)beta(3)-c-Src oncogenic unit promotes anchorage-independence and tumor progression. Nat Med. 2009;15(10):1163–9.
Stucci S, Tucci M, Passarelli A, Silvestris F. Avβ3 integrin: Pathogenetic role in osteotropic tumors. Crit Rev Oncol Hematol. 2015;96(1):183–93.
Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science. 1994;264(5158):569–71.
Drake CJ, Cheresh DA, Little CD. An antagonist of integrin alpha v beta 3 prevents maturation of blood vessels during embryonic neovascularization. J Cell Sci. 1995;108(Pt 7):2655–61.
Knowles LM, Gurski LA, Engel C, Gnarra JR, Maranchie JK, Pilch J. Integrin αvβ3 and fibronectin upregulate slug in cancer cells to promote clot invasion and metastasis. Cancer Res. 2013;73(20):6175–84.
Malik G, Knowles LM, Dhir R, Xu S, Yang S, Ruoslahti E, et al. Plasma fibronectin promotes lung metastasis by contributions to fibrin clots and tumor cell invasion. Cancer Res. 2010;70(11):4327–34.
Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. 2003;3(8):582–91.
Pamuklar Z, Federico L, Liu S, Umezu-Goto M, Dong A, Panchatcharam M, et al. Autotaxin/lysopholipase D and lysophosphatidic acid regulate murine hemostasis and thrombosis. J Biol Chem. 2009;284(11):7385–94.
Magkrioti C, Oikonomou N, Kaffe E, Mouratis MA, Xylourgidis N, Barbayianni I, et al. The Autotaxin-Lysophosphatidic Acid Axis promotes Lung Carcinogenesis. Cancer Res. 2018;78(13):3634–44.
Yang Y, Mou L, Liu N, Tsao MS. Autotaxin expression in non-small-cell lung cancer. Am J Respir Cell Mol Biol. 1999;21(2):216–22.
Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1(1):27–31.
Carmeliet P. Manipulating angiogenesis in medicine. J Intern Med. 2004;255(5):538–61.
Kuhn H, Hammerschmidt S, Wirtz H. Targeting tumorangiogenesis in lung cancer by suppression of VEGF and its receptor - results from clinical trials and novel experimental approaches. Curr Med Chem. 2007;14(30):3157–65.
Folkman J. Angiogenesis and proteins of the hemostatic system. J Thromb Haemost. 2003;1(8):1681–2.
Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6(4):389–95.
Cecerska-Heryć E, Heryć R, Wiśniewska M, Michalczyk A, Dołęgowska B. Regenerative potential of platelets in patients with chronic kidney disease. Int Urol Nephrol. 2019;51(10):1831–40.
Sato Y. The vasohibin family: novel regulators of angiogenesis. Vascul Pharmacol. 2012;56(5–6):262–6.
Wang S, Olson EN. AngiomiRs–key regulators of angiogenesis. Curr Opin Genet Dev. 2009;19(3):205–11.
Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9(6):669–76.
Houck KA, Ferrara N, Winer J, Cachianes G, Li B, Leung DW. The vascular endothelial growth factor family: identification of a fourth molecular species and characterization of alternative splicing of RNA. Mol Endocrinol. 1991;5(12):1806–14.
Suto K, Yamazaki Y, Morita T, Mizuno H. Crystal structures of novel vascular endothelial growth factors (VEGF) from snake venoms: insight into selective VEGF binding to kinase insert domain-containing receptor but not to fms-like tyrosine kinase-1. J Biol Chem. 2005;280(3):2126–31.
Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol. 2002;20(21):4368–80.
Karaman S, Paavonsalo S, Heinolainen K, Lackman MH, Ranta A, Hemanthakumar KA et al. Interplay of vascular endothelial growth factor receptors in organ-specific vessel maintenance. J Exp Med. 2022;219(3).
Kalka C, Masuda H, Takahashi T, Gordon R, Tepper O, Gravereaux E, et al. Vascular endothelial growth factor(165) gene transfer augments circulating endothelial progenitor cells in human subjects. Circ Res. 2000;86(12):1198–202.
Poole TJ, Finkelstein EB, Cox CM. The role of FGF and VEGF in angioblast induction and migration during vascular development. Dev Dyn. 2001;220(1):1–17.
Flamme I, Risau W. Induction of vasculogenesis and hematopoiesis in vitro. Development. 1992;116(2):435–9.
Kazemi S, Wenzel D, Kolossov E, Lenka N, Raible A, Sasse P, et al. Differential role of bFGF and VEGF for vasculogenesis. Cell Physiol Biochem. 2002;12(2–3):55–62.
Volm M, Koomägi R, Mattern J. Prognostic value of vascular endothelial growth factor and its receptor Flt-1 in squamous cell lung cancer. Int J Cancer. 1997;74(1):64–8.
Tanno S, Ohsaki Y, Nakanishi K, Toyoshima E, Kikuchi K. Human small cell lung cancer cells express functional VEGF receptors, VEGFR-2 and VEGFR-3. Lung Cancer. 2004;46(1):11–9.
Popper HH. Progression and metastasis of lung cancer. Cancer Metastasis Rev. 2016;35(1):75–91.
Wang Z, Xu H, Zhang J, Jin H, Wei P. Basic fibroblast growth factor blockade enhances lung cancer cell invasion by activating the AKT/MMP-2/VEGF pathway. Basic Clin Pharmacol Toxicol. 2020;126(1):43–50.
Hu M, Hu Y, He J, Li B. Prognostic Value of Basic Fibroblast Growth factor (bFGF) in Lung Cancer: a systematic review with Meta-analysis. PLoS ONE. 2016;11(1):e0147374.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34.
Allemani C, Matsuda T, Di Carlo V, Harewood R, Matz M, Nikšić M, et al. Global surveillance of trends in cancer survival 2000-14 (CONCORD-3): analysis of individual records for 37 513 025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet. 2018;391(10125):1023–75.
Comprehensive molecular portraits. Of human breast tumours. Nature. 2012;490(7418):61–70.
Felding-Habermann B, O’Toole TE, Smith JW, Fransvea E, Ruggeri ZM, Ginsberg MH, et al. Integrin activation controls metastasis in human breast cancer. Proc Natl Acad Sci U S A. 2001;98(4):1853–8.
Darga EP, Dolce EM, Fang F, Kidwell KM, Gersch CL, Kregel S, et al. PD-L1 expression on circulating tumor cells and platelets in patients with metastatic breast cancer. PLoS ONE. 2021;16(11):e0260124.
Davis AA, Patel VG. The role of PD-L1 expression as a predictive biomarker: an analysis of all US Food and Drug Administration (FDA) approvals of immune checkpoint inhibitors. J Immunother Cancer. 2019;7(1):278.
Doroshow DB, Sanmamed MF, Hastings K, Politi K, Rimm DL, Chen L, et al. Immunotherapy in Non-small Cell Lung Cancer: facts and hopes. Clin Cancer Res. 2019;25(15):4592–602.
Vandendries ER, Furie BC, Furie B. Role of P-selectin and PSGL-1 in coagulation and thrombosis. Thromb Haemost. 2004;92(3):459–66.
Schwarz S, Gockel LM, Naggi A, Barash U, Gobec M, Bendas G et al. Glycosaminoglycans as tools to decipher the platelet Tumor Cell Interaction: a focus on P-Selectin. Molecules. 2020;25(5).
Borsig L, Wong R, Feramisco J, Nadeau DR, Varki NM, Varki A. Heparin and cancer revisited: mechanistic connections involving platelets, P-selectin, carcinoma mucins, and tumor metastasis. Proc Natl Acad Sci U S A. 2001;98(6):3352–7.
Flevaris P, Li Z, Zhang G, Zheng Y, Liu J, Du X. Two distinct roles of mitogen-activated protein kinases in platelets and a novel Rac1-MAPK-dependent integrin outside-in retractile signaling pathway. Blood. 2009;113(4):893–901.
Li Z, Xi X, Du X. A mitogen-activated protein kinase-dependent signaling pathway in the activation of platelet integrin alpha IIbbeta3. J Biol Chem. 2001;276(45):42226–32.
Estevez B, Du X. New concepts and mechanisms of platelet activation signaling. Physiol (Bethesda). 2017;32(2):162–77.
Qi C, Wei B, Zhou W, Yang Y, Li B, Guo S, et al. P-selectin-mediated platelet adhesion promotes tumor growth. Oncotarget. 2015;6(9):6584–96.
Goetzl EJ, Lee H, Dolezalova H, Kalli KR, Conover CA, Hu YL, et al. Mechanisms of lysolipid phosphate effects on cellular survival and proliferation. Ann N Y Acad Sci. 2000;905:177–87.
Bandoh K, Aoki J, Hosono H, Kobayashi S, Kobayashi T, Murakami-Murofushi K, et al. Molecular cloning and characterization of a novel human G-protein-coupled receptor, EDG7, for lysophosphatidic acid. J Biol Chem. 1999;274(39):27776–85.
Noguchi K, Ishii S, Shimizu T. Identification of p2y9/GPR23 as a novel G protein-coupled receptor for lysophosphatidic acid, structurally distant from the Edg family. J Biol Chem. 2003;278(28):25600–6.
McIntyre TM, Pontsler AV, Silva AR, St Hilaire A, Xu Y, Hinshaw JC, et al. Identification of an intracellular receptor for lysophosphatidic acid (LPA): LPA is a transcellular PPARgamma agonist. Proc Natl Acad Sci U S A. 2003;100(1):131–6.
Leblanc R, Houssin A, Peyruchaud O. Platelets, autotaxin and lysophosphatidic acid signalling: win-win factors for cancer metastasis. Br J Pharmacol. 2018;175(15):3100–10.
Martin TJ. Manipulating the environment of cancer cells in bone: a novel therapeutic approach. J Clin Invest. 2002;110(10):1399–401.
de la Mata J, Uy HL, Guise TA, Story B, Boyce BF, Mundy GR, et al. Interleukin-6 enhances hypercalcemia and bone resorption mediated by parathyroid hormone-related protein in vivo. J Clin Invest. 1995;95(6):2846–52.
Boucharaba A, Serre CM, Grès S, Saulnier-Blache JS, Bordet JC, Guglielmi J, et al. Platelet-derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer. J Clin Invest. 2004;114(12):1714–25.
Zhang H, Wong CC, Wei H, Gilkes DM, Korangath P, Chaturvedi P, et al. HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene. 2012;31(14):1757–70.
Rolli M, Fransvea E, Pilch J, Saven A, Felding-Habermann B. Activated integrin alphavbeta3 cooperates with metalloproteinase MMP-9 in regulating migration of metastatic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(16):9482–7.
Reddig PJ, Juliano RL. Clinging to life: cell to matrix adhesion and cell survival. Cancer Metastasis Rev. 2005;24(3):425–39.
Stupack DG, Puente XS, Boutsaboualoy S, Storgard CM, Cheresh DA. Apoptosis of adherent cells by recruitment of caspase-8 to unligated integrins. J Cell Biol. 2001;155(3):459–70.
Kerbel R, Folkman J. Clinical translation of angiogenesis inhibitors. Nat Rev Cancer. 2002;2(10):727–39.
Rosen LS. Clinical experience with angiogenesis signaling inhibitors: focus on vascular endothelial growth factor (VEGF) blockers. Cancer Control. 2002;9(2 Suppl):36–44.
Ning Q, Liu C, Hou L, Meng M, Zhang X, Luo M, et al. Vascular endothelial growth factor receptor-1 activation promotes migration and invasion of breast cancer cells through epithelial-mesenchymal transition. PLoS ONE. 2013;8(6):e65217.
D’Amore PA. Modes of FGF release in vivo and in vitro. Cancer Metastasis Rev. 1990;9(3):227–38.
Yiangou C, Gomm JJ, Coope RC, Law M, Luqmani YA, Shousha S, et al. Fibroblast growth factor 2 in breast cancer: occurrence and prognostic significance. Br J Cancer. 1997;75(1):28–33.
Farzaneh Behelgardi M, Zahri S, Mashayekhi F, Mansouri K, Asghari SM. A peptide mimicking the binding sites of VEGF-A and VEGF-B inhibits VEGFR-1/-2 driven angiogenesis, tumor growth and metastasis. Sci Rep. 2018;8(1):17924.
Farzaneh Behelgardi M, Zahri S, Gholami Shahvir Z, Mashayekhi F, Mirzanejad L, Asghari SM. Targeting signaling pathways of VEGFR1 and VEGFR2 as a potential target in the treatment of breast cancer. Mol Biol Rep. 2020;47(3):2061–71.
Assareh E, Mehrnejad F, Mansouri K, Esmaeili Rastaghi AR, Naderi-Manesh H, Asghari SM. A cyclic peptide reproducing the α1 helix of VEGF-B binds to VEGFR-1 and VEGFR-2 and inhibits angiogenesis and tumor growth. Biochem J. 2019;476(4):645–63.
Sadremomtaz A, Kobarfard F, Mansouri K, Mirzanejad L, Asghari SM. Suppression of migratory and metastatic pathways via blocking VEGFR1 and VEGFR2. J Recept Signal Transduct Res. 2018;38(5–6):432–41.
Sadremomtaz A, Mansouri K, Alemzadeh G, Safa M, Rastaghi AE, Asghari SM. Dual blockade of VEGFR1 and VEGFR2 by a novel peptide abrogates VEGF-driven angiogenesis, tumor growth, and metastasis through PI3K/AKT and MAPK/ERK1/2 pathway. Biochim Biophys Acta Gen Subj. 2018;1862(12):2688–700.
Faridi A, Rudlowski C, Biesterfeld S, Schuh S, Rath W, Schröder W. Long-term follow-up and prognostic significance of angiogenic basic fibroblast growth factor (bFGF) expression in patients with breast cancer. Pathol Res Pract. 2002;198(1):1–5.
Cobleigh MA, Langmuir VK, Sledge GW, Miller KD, Haney L, Novotny WF, et al. A phase I/II dose-escalation trial of bevacizumab in previously treated metastatic breast cancer. Semin Oncol. 2003;30(5 Suppl 16):117–24.
Leslie A, Carey FA, Pratt NR, Steele RJ. The colorectal adenoma-carcinoma sequence. Br J Surg. 2002;89(7):845–60.
Chew MH, Koh PK, Ng KH, Eu KW. Improved survival in an Asian cohort of young colorectal cancer patients: an analysis of 523 patients from a single institution. Int J Colorectal Dis. 2009;24(9):1075–83.
O’Connell JB, Maggard MA, Liu JH, Etzioni DA, Livingston EH, Ko CY. Rates of colon and rectal cancers are increasing in young adults. Am Surg. 2003;69(10):866–72.
Manzat Saplacan RM, Balacescu L, Gherman C, Chira RI, Craiu A, Mircea PA, et al. The role of PDGFs and PDGFRs in Colorectal Cancer. Mediators Inflamm. 2017;2017:4708076.
Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999;79(4):1283–316.
Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22(10):1276–312.
Cao Y. Multifarious functions of PDGFs and PDGFRs in tumor growth and metastasis. Trends Mol Med. 2013;19(8):460–73.
Omura T, Heldin CH, Ostman A. Immunoglobulin-like domain 4-mediated receptor-receptor interactions contribute to platelet-derived growth factor-induced receptor dimerization. J Biol Chem. 1997;272(19):12676–82.
Appiah-Kubi K, Wang Y, Qian H, Wu M, Yao X, Wu Y, et al. Platelet-derived growth factor receptor/platelet-derived growth factor (PDGFR/PDGF) system is a prognostic and treatment response biomarker with multifarious therapeutic targets in cancers. Tumour Biol. 2016;37(8):10053–66.
Cao R, Bråkenhielm E, Li X, Pietras K, Widenfalk J, Ostman A, et al. Angiogenesis stimulated by PDGF-CC, a novel member in the PDGF family, involves activation of PDGFR-alphaalpha and -alphabeta receptors. Faseb j. 2002;16(12):1575–83.
Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nat Rev Cancer. 2011;11(2):123–34.
Labelle M, Hynes RO. The initial hours of metastasis: the importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov. 2012;2(12):1091–9.
Mammadova-Bach E, Ollivier V, Loyau S, Schaff M, Dumont B, Favier R, et al. Platelet glycoprotein VI binds to polymerized fibrin and promotes thrombin generation. Blood. 2015;126(5):683–91.
Nieswandt B, Watson SP. Platelet-collagen interaction: is GPVI the central receptor? Blood. 2003;102(2):449–61.
Clemetson JM, Polgar J, Magnenat E, Wells TN, Clemetson KJ. The platelet collagen receptor glycoprotein VI is a member of the immunoglobulin superfamily closely related to FcalphaR and the natural killer receptors. J Biol Chem. 1999;274(41):29019–24.
Moroi M, Jung SM. Platelet glycoprotein VI: its structure and function. Thromb Res. 2004;114(4):221–33.
Rayes J, Watson SP, Nieswandt B. Functional significance of the platelet immune receptors GPVI and CLEC-2. J Clin Invest. 2019;129(1):12–23.
Dovizio M, Maier TJ, Alberti S, Di Francesco L, Marcantoni E, Münch G, et al. Pharmacological inhibition of platelet-tumor cell cross-talk prevents platelet-induced overexpression of cyclooxygenase-2 in HT29 human colon carcinoma cells. Mol Pharmacol. 2013;84(1):25–40.
Barondes SH, Castronovo V, Cooper DN, Cummings RD, Drickamer K, Feizi T, et al. Galectins: a family of animal beta-galactoside-binding lectins. Cell. 1994;76(4):597–8.
Díaz-Alvarez L, Ortega E. The many roles of Galectin-3, a multifaceted molecule, in Innate Immune responses against pathogens. Mediators Inflamm. 2017;2017:9247574.
Sciacchitano S, Lavra L, Morgante A, Ulivieri A, Magi F, De Francesco GP et al. Galectin-3: one molecule for an alphabet of diseases, from a to Z. Int J Mol Sci. 2018;19(2).
Mammadova-Bach E, Gil-Pulido J, Sarukhanyan E, Burkard P, Shityakov S, Schonhart C, et al. Platelet glycoprotein VI promotes metastasis through interaction with cancer cell-derived galectin-3. Blood. 2020;135(14):1146–60.
Goding JW, Grobben B, Slegers H. Physiological and pathophysiological functions of the ecto-nucleotide pyrophosphatase/phosphodiesterase family. Biochim Biophys Acta. 2003;1638(1):1–19.
Tokumura A, Majima E, Kariya Y, Tominaga K, Kogure K, Yasuda K, et al. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J Biol Chem. 2002;277(42):39436–42.
Hausmann J, Perrakis A, Moolenaar WH. Structure-function relationships of autotaxin, a secreted lysophospholipase D. Adv Biol Regul. 2013;53(1):112–7.
Aoki J, Inoue A, Okudaira S. Two pathways for lysophosphatidic acid production. Biochim Biophys Acta. 2008;1781(9):513–8.
Choi JW, Herr DR, Noguchi K, Yung YC, Lee CW, Mutoh T, et al. LPA receptors: subtypes and biological actions. Annu Rev Pharmacol Toxicol. 2010;50:157–86.
Kazama S, Kitayama J, Aoki J, Mori K, Nagawa H. Immunohistochemical detection of autotaxin (ATX)/lysophospholipase D (lysoPLD) in submucosal invasive colorectal cancer. J Gastrointest Cancer. 2011;42(4):204–11.
David M, Wannecq E, Descotes F, Jansen S, Deux B, Ribeiro J, et al. Cancer cell expression of autotaxin controls bone metastasis formation in mouse through lysophosphatidic acid-dependent activation of osteoclasts. PLoS ONE. 2010;5(3):e9741.
Bolatai A, He Y, Wu N. Vascular endothelial growth factor and its receptors regulation in gestational diabetes mellitus and eclampsia. J Transl Med. 2022;20(1):400.
Takahashi Y, Tucker SL, Kitadai Y, Koura AN, Bucana CD, Cleary KR, et al. Vessel counts and expression of vascular endothelial growth factor as prognostic factors in node-negative colon cancer. Arch Surg. 1997;132(5):541–6.
Ellis LM, Takahashi Y, Liu W, Shaheen RM. Vascular endothelial growth factor in human colon cancer: biology and therapeutic implications. Oncologist. 2000;5(Suppl 1):11–5.
Fan F, Wey JS, McCarty MF, Belcheva A, Liu W, Bauer TW, et al. Expression and function of vascular endothelial growth factor receptor-1 on human colorectal cancer cells. Oncogene. 2005;24(16):2647–53.
Bhattacharya R, Fan F, Wang R, Ye X, Xia L, Boulbes D, et al. Intracrine VEGF signalling mediates colorectal cancer cell migration and invasion. Br J Cancer. 2017;117(6):848–55.
Zhao M, Yu Z, Li Z, Tang J, Lai X, Liu L. Expression of angiogenic growth factors VEGF, bFGF and ANG1 in colon cancer after bevacizumab treatment in vitro: a potential self-regulating mechanism. Oncol Rep. 2017;37(1):601–7.
Ohaegbulam KC, Assal A, Lazar-Molnar E, Yao Y, Zang X. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol Med. 2015;21(1):24–33.
Seymour L, Dajee D, Bezwoda WR. Tissue platelet derived-growth factor (PDGF) predicts for shortened survival and treatment failure in advanced breast cancer. Breast Cancer Res Treat. 1993;26(3):247–52.
Mazel M, Jacot W, Pantel K, Bartkowiak K, Topart D, Cayrefourcq L, et al. Frequent expression of PD-L1 on circulating breast cancer cells. Mol Oncol. 2015;9(9):1773–82.
Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019;19(3):133–50.
Stewart R, Morrow M, Hammond SA, Mulgrew K, Marcus D, Poon E, et al. Identification and characterization of MEDI4736, an antagonistic Anti-PD-L1 monoclonal antibody. Cancer Immunol Res. 2015;3(9):1052–62.
Koenig A, Norgard-Sumnicht K, Linhardt R, Varki A. Differential interactions of heparin and heparan sulfate glycosaminoglycans with the selectins. Implications for the use of unfractionated and low molecular weight heparins as therapeutic agents. J Clin Invest. 1998;101(4):877–89.
Stevenson JL, Choi SH, Varki A. Differential metastasis inhibition by clinically relevant levels of heparins–correlation with selectin inhibition, not antithrombotic activity. Clin Cancer Res. 2005;11(19 Pt 1):7003–11.
Kasirer-Friede A, Kang J, Kahner B, Ye F, Ginsberg MH, Shattil SJ. ADAP interactions with talin and kindlin promote platelet integrin αIIbβ3 activation and stable fibrinogen binding. Blood. 2014;123(20):3156–65.
Gao J, Huang M, Lai J, Mao K, Sun P, Cao Z, et al. Kindlin supports platelet integrin αIIbβ3 activation by interacting with paxillin. J Cell Sci. 2017;130(21):3764–75.
Tadokoro S, Nakazawa T, Kamae T, Kiyomizu K, Kashiwagi H, Honda S, et al. A potential role for α-actinin in inside-out αIIbβ3 signaling. Blood. 2011;117(1):250–8.
Giordano A, Musumeci G, D’Angelillo A, Rossini R, Zoccai GB, Messina S, et al. Effects of glycoprotein IIb/IIIa antagonists: anti platelet aggregation and Beyond. Curr Drug Metab. 2016;17(2):194–203.
Goodman SL, Picard M. Integrins as therapeutic targets. Trends Pharmacol Sci. 2012;33(7):405–12.
Das M, Wakelee H. Targeting VEGF in lung cancer. Expert Opin Ther Targets. 2012;16(4):395–406.
Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus Irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335–42.
Billiald P, Slater A, Welin M, Clark JC, Loyau S, Pugnière M, et al. Targeting platelet GPVI with glenzocimab: a novel mechanism for inhibition. Blood Adv. 2023;7(7):1258–68.
Pardo OE, Latigo J, Jeffery RE, Nye E, Poulsom R, Spencer-Dene B, et al. The fibroblast growth factor receptor inhibitor PD173074 blocks small cell lung cancer growth in vitro and in vivo. Cancer Res. 2009;69(22):8645–51.
Benesch MGK, Yang Z, Tang X, Meng G, Brindley DN. Lysophosphatidate Signaling: the Tumor Microenvironment’s New Nemesis. Trends Cancer. 2017;3(11):748–52.
Jia Y, Li Y, Xu XD, Tian Y, Shang H. Design and development of Autotaxin inhibitors. Pharmaceuticals (Basel). 2021;14(11).
Tang X, Wuest M, Benesch MGK, Dufour J, Zhao Y, Curtis JM, et al. Inhibition of Autotaxin with GLPG1690 increases the efficacy of Radiotherapy and Chemotherapy in a mouse model of breast Cancer. Mol Cancer Ther. 2020;19(1):63–74.
Maher TM, van der Aar EM, Van de Steen O, Allamassey L, Desrivot J, Dupont S, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of GLPG1690, a novel autotaxin inhibitor, to treat idiopathic pulmonary fibrosis (FLORA): a phase 2a randomised placebo-controlled trial. Lancet Respir Med. 2018;6(8):627–35.
Zou X, Tang XY, Qu ZY, Sun ZW, Ji CF, Li YJ, et al. Targeting the PDGF/PDGFR signaling pathway for cancer therapy: a review. Int J Biol Macromol. 2022;202:539–57.
Roswall P, Bocci M, Bartoschek M, Li H, Kristiansen G, Jansson S, et al. Microenvironmental control of breast cancer subtype elicited through paracrine platelet-derived growth factor-CC signaling. Nat Med. 2018;24(4):463–73.
Kuai J, Mosyak L, Brooks J, Cain M, Carven GJ, Ogawa S, et al. Characterization of binding mode of action of a blocking anti-platelet-derived growth factor (PDGF)-B monoclonal antibody, MOR8457, reveals conformational flexibility and avidity needed for PDGF-BB to bind PDGF receptor-β. Biochemistry. 2015;54(10):1918–29.
Zarei O, Sarri N, Dastmalchi S, Zokai F, Papadopoulos N, Lennartsson J, et al. Structure-based discovery of novel small molecule inhibitors of platelet-derived growth factor-B. Bioorg Chem. 2020;94:103374.
Acknowledgements
We thank Professor Haiyan Wang and Editage (www.editage.cn) for English language editing.
Funding
This study was supported by the Clinical Medicine + X Project of Qingdao University (Grant No. 2020018) and the Joint Fund for Innovation and Development of the Shandong Natural Science Foundation (Grant No. ZR2022LSW024).
Author information
Authors and Affiliations
Contributions
XJ and DJZ performed the scientific literature search; collected, arranged, and summarized the data; and wrote the manuscript. QHW was responsible for sorting and analyzing the data. YSX, ZZ, QLF and LJ helped substantively revised it. WHY provided ideas for the manuscript and helped modify it. All the authors have reviewed and agreed to the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xue, J., Deng, J., Qin, H. et al. The interaction of platelet-related factors with tumor cells promotes tumor metastasis. J Transl Med 22, 371 (2024). https://doi.org/10.1186/s12967-024-05126-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-024-05126-6