
Abstract
Machine learning (ML) methods are increasingly becoming crucial in genome-wide association studies for identifying key genetic variants or SNPs that statistical methods might overlook. Statistical methods predominantly identify SNPs with notable effect sizes by conducting association tests on individual genetic variants, one at a time, to determine their relationship with the target phenotype. These genetic variants are then used to create polygenic risk scores (PRSs), estimating an individual's genetic risk for complex diseases like cancer or cardiovascular disorders. Unlike traditional methods, ML algorithms can identify groups of low-risk genetic variants that improve prediction accuracy when combined in a mathematical model. However, the application of ML strategies requires addressing the feature selection challenge to prevent overfitting. Moreover, ensuring the ML model depends on a concise set of genomic variants enhances its clinical applicability, where testing is feasible for only a limited number of SNPs. In this study, we introduce a robust pipeline that applies ML algorithms in combination with feature selection (ML-FS algorithms), aimed at identifying the most significant genomic variants associated with the coronary artery disease (CAD) phenotype. The proposed computational approach was tested on individuals from the UK Biobank, differentiating between CAD and non-CAD individuals within this extensive cohort, and benchmarked against standard PRS-based methodologies like LDpred2 and Lassosum. Our strategy incorporates cross-validation to ensure a more robust evaluation of genomic variant-based prediction models. This method is commonly applied in machine learning strategies but has often been neglected in previous studies assessing the predictive performance of polygenic risk scores. Our results demonstrate that the ML-FS algorithm can identify panels with as few as 50 genetic markers that can achieve approximately 80% accuracy when used in combination with known risk factors. The modest increase in accuracy over PRS performances is noteworthy, especially considering that PRS models incorporate a substantially larger number of genetic variants. This extensive variant selection can pose practical challenges in clinical settings. Additionally, the proposed approach revealed novel CAD-genetic variant associations.
Similar content being viewed by others

Introduction
A major goal of genetics researchers is to identify group of interacting genetic loci that contribute to complex phenotypic traits and human diseases. This is often conducted through genome-wide association studies (GWAS), which are a research approach used to associate specific genetic variations—often single nucleotide polymorphisms (SNPs)—with particular diseases. GWAS involve scanning genomes from many individuals to find genetic markers that correlate with observable traits or diseases. The process starts with association tests that evaluate one SNP at a time across the genome to find variants with statistically significant associations with the target phenotype. These tests typically measure the frequency of each SNP in individuals with the phenotype (cases) versus control individuals without the phenotype (controls). SNPs that show a significant difference in frequency between cases and controls are considered to be associated with the phenotype. SNPs have the potential to enhance the differentiation of complex phenotypes where clinical data alone may be insufficient. They can help identify new disease subtypes or trajectories, offering utility beyond clinical settings [1]. For example, SNPs can act as accurate markers for distinguishing between plant species or varieties [2]. Once these associated SNPs are identified, they are used to construct polygenic risk scores (PRS). A PRS is a number that represents an individual's genetic predisposition to a disease, derived from the sum of effect sizes of SNPs associated with the disease. PRS has shown effectiveness in distinguishing complex phenotypes, such as coronary artery disease (CAD), by aggregating the effects of many genetic variants across the genome. For instance, studies like the CARDIoGRAMplusC4D consortium have demonstrated the utility of PRS in CAD risk prediction [3, 4]. However, a significant challenge with PRS is that it may rely on a large number of genomic variants. This creates a barrier to clinical translation, as testing for hundreds or thousands of SNPs is not always practical or cost-effective in clinical settings. Moreover, incorporating too many weak predictors may lead to biased results and challenges in replicating findings across different cohorts [5,6,7]. While prior research has largely concentrated on association testing and polygenic risk scores, the application of Machine Learning (ML) to discern significant genotype–phenotype correlations has been marginally explored. ML-based strategies approach can discover associations that do not necessarily meet statistical significance at the level of single genetic locus, yet still contributing to the combined predictive power at the level of variant panels. However, using ML algorithms with a large number of features, such as those generated by genome-wide association studies (GWASs), results in complex models that are slower to execute and, most importantly, prone to overfitting. One way to reduce overfitting is feature selection [8,9,10]. Feature selection aims to reduce data dimensionality, remove noisy and irrelevant data, and thus it can preserve the most useful variables from the dataset. Furthermore, feature selection helps identify a concise set of omics-based features for complex classification tasks [11,12,13], enhancing the development of cost-effective biomarker panels. In scenarios where two predictive models show similar performance but one uses significantly fewer features, the more compact biomarker model would be the preferred choice for clinicians due to its potential for cost-effective implementation. There are mainly three types of feature selection methods: filtering, embedded, and wrapper methods [14, 15]. Filter-based feature selection is the most used method for selecting relevant genotypes for GWAS, since it relies on statistical measures to score the association between a single genetic variant and the target variable (e.g., known phenotypic trait, disease status, etc.). Then, the computed scores (e.g., size effects, p-values, etc.) can be used to choose (or prioritize) the most relevant features or genetic loci. However, such a strategy does not try to remove redundant features, making it challenging to select genetic variants for accurate ML models.
In this study, we systematically evaluate three methods for feature selection in ML models: (1) using univariate test statistics with PLINK; (2) employing the Maximum Relevance Minimum Redundancy (mRMR) algorithm [16], which tries to identify predictive and uncorrelated features; (3) utilizing Random Forest (RF) feature importance scores to account for complex interactions. The goal of all tested methods is to identify a compact, effective combination of single-nucleotide polymorphisms (SNPs) for use in machine learning models that classify individuals with Coronary Artery Disease (CAD) in the UK Biobank, while discovering pertinent CAD variants. Each feature selection strategy is systematically combined with linear and non-linear ML-based classification algorithms: Logistic Regression (LR), Lasso (LA), Support Vector Machine (SVM) and Random Forests (RF). We assessed the quality of selected SNP combinations using the Area Under the Receiver Operating Characteristic Curve (AUC-ROC), a comprehensive performance measure for binary classifiers, considering both sensitivity and specificity to distinguish between CAD and non-CAD cases [17]. We also compared classification performance of feature selection (FS)-driven ML models with polygenic risk scores (PRSs) using three methods: P + T, LDPred2, and Lassosum, since they represent the primary approach for disease risk prediction [18, 19]. To evaluate the performance of ML- and PRS-based approaches, we employed tenfold cross-validation (10FCV), a method that enhances the accuracy of classification performance estimates and enables the assessment of feature selection stability. The stability refers to the robustness of the feature selection process, with respect to data sampling in cross validation studies [20]. Finally, functional annotation of frequently selected genetic variants from the FS-ML approaches was performed using FUMA [21].
Results
Machine learning-based identification of risk loci for CAD via feature selection
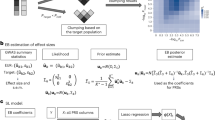
Here we describe a computational framework to perform machine learning and feature selection algorithms on large-scale genotyped data. Figure 1 provides a graphic overview of the main steps. The first step, depicted in Fig. 1A, aimed at gathering individuals for analyzing CAD phenotypes.

Computational framework comparing different feature selection strategies for the selection of risk loci panels for CAD. A–C The data collected from the UKB is subjected to preprocessing to extract the phenotype of interest (CAD vs. non-CAD) and generate high-quality genotype imputed data. D, E To identify the most relevant covariates for association tests, we performed an analysis of CAD-associated risk factors and conducted principal component analysis on the genotype data. F tenfold cross-validation was used to perform a fair comparison between ML-based methods and PRSs. G Genomic variants for predicting CAD were selected based on three feature selection strategies encompassing filter-based and embedded methods. H, I Three PRS methods were implemented and combined with Logistic Regression-based classifier for the classification. J Genomic variants selected through FS were systematically uses to train three different classification algorithms: Lasso, RF and SVM. K The Area Under the ROC Curve (AUC) statistics as the main accuracy metric. Moreover, we also recorded the frequency of each feature being selected across different training set and feature selection methods. L The most informative SNPs were further analyzed to assess their biological relevance
We defined CAD and non-CAD phenotypes using ICD-9, ICD-10, and OPCS-4 codes. Refer to Additional file 1: Table S1 for a comprehensive list of diagnoses and health statuses considered associated with the CAD phenotype. Moreover, at this stage, kinship estimates are computed to identify related individuals, who are then removed before performing association tests, calculating PRS scores, and applying ML-based feature selection methods across the CV iterations. Imputation and quality control were performed to generate the initial set of single nucleotide polymorphisms (SNPs), which are genetic variations in a population at specific positions in the DNA sequence (Fig. 1B–C). Imputation refers to the process of filling in missing data in a dataset, while quality check refers to removing markers and individuals with low quality or that are unlikely to contribute to association analysis, thus increasing the power and accuracy of downstream analyses. Quality control on the genetic markers included checking for markers that were in linkage disequilibrium (LD), which can lead to redundant information, and markers that had high levels of missing data or low minor allele frequency (MAF). Markers that failed these quality control criteria were removed from the analysis. Association tests between CAD and known risk factors were performed and principal component analysis (PCA) was applied to genotype data to uncover population structure (Fig. 1D–E). The steps illustrated in Fig. 1F–K shows the machine learning framework which aims to compare PRS with standard ML and feature selection algorithms. In more details, three feature selection strategies, encompassing filter-based and embedded methods, were used in combination with three classification algorithms (Fig. 1G). These methods were then compared against three PRS strategies (Fig. 1H–I). Machine learning helps identify new risk loci for complex diseases, selecting genetic variants that, when combined, yield higher prediction accuracy compared to PRS-based approaches. To guarantee a fair comparison, PRS was defined within the training set and evaluated on an independent test set, preventing bias. The computational framework offers a more accurate assessment, enabling informed comparisons between ML and PRSs. 10-folds cross validation was used to more robustly estimates prediction performance of all tested methods (Fig. 1F), while the Area Under the ROC Curve (AUC) statistics was used to as main accuracy metric (Fig. 1K). Subsets of SNPs that are most informative for the identification of the CAD phenotype, which were selected through ML-driven feature selection algorithms, were further analyzed to assess their biological relevance, by using the tool Functional Mapping and Annotation (Fig. 1L). For further details on the methodology used in this study, please refer to the “Materials and Methods” section.
Population characteristics and association between CAD and known risk factors
We examined the UKB population characteristics and associations between CAD patients and known risk factors across the entire UK Biobank population. Baseline characteristics of CAD are summarized in Table 1 by sex, age and commonly used risk factors. CAD participants’ average age is 60.33 years, and non-CAD controls’ average is 56.24 years. Triglycerides are slightly higher in CAD cases, as expected [22].
LDL and total cholesterol values are higher in non-CAD cases, likely due to statin treatment in CAD patients, which reduces both LDL and total cholesterol levels. Associations between known risk factors and CAD cases were then estimated through logistic regression analysis. The logistic regression model also included basic covariates (e.g., sex and age), ethnic background, genotype batch, assessment center, and PCs adjusting for population structure. Figure 2 shows odds ratios and 95% confidence intervals from multivariable ordinal regression, indicating the association between cardiovascular risk factors and CAD cases. HDL is a protective factor, while LDL and cholesterol show no increased risk, likely due to statin treatment in CAD-diagnosed individuals [23, 24]. Principal component analysis (PCA) was applied to genotype data to uncover population structure and use the PCs as covariates (Additional file 1: Fig. S2) for subsequent analyses.

Odds ratios of traditional cardiovascular risk factors. A larger odds ratio indicates a stronger association between the risk factor and CAD. The red color is used to indicate significant associations (p < 0.05). The vertical line at x = 1 indicates an odds ratio of 1, in which case there is "no effect". For the calculation of odds ratios pertaining to traditional cardiovascular risk factors, we considered the complete set of UK Biobank individuals or non-CAD, which consisted of 467,215 and 35,290 individuals for the respective categories
Additional file 1: Fig. S3 includes the SHAP summary plot, which shows the contribution or the importance of each feature, including risk factors, batches, and PCs, on the CAD risk and their effect on the single predictions. Shapley values help detect risk factors, batches, or PCs affecting predictions. Features are ranked by their ability to improve predictions, with age and sex having mild contributions. Genotype batch and assessment center show no contributions to CAD status, while vascular heart problem diagnosis has a high positive contribution. However, it should be acknowledged that the lack of diagnosis is not necessarily associated with non-CAD cases. Additional file 1: Fig. S4 shows correlation and VIF analysis outcomes, assessing multicollinearity between predictor variables in a CAD status regression model. High VIF score variables were removed, specifically total cholesterol and ethnic background, represented by LDL/HDL levels and the first 20 PCs. The final set of covariates was used for genotype-CAD association analysis, generating summary statistics for CAD-relevant single-nucleotide variants. It should be noted that summary statistics are systematically computed within each generated training dataset in a cross-fold validation framework, as they are used in one of the feature selection strategies to select risk loci evaluated in this study and to build PRSs. This aligns with the machine learning framework, where feature selection and model training must be implemented within a training set and evaluated within an unseen test set to avoid data leakage [25].
Using PRS and ML methods for CAD prediction based on panels of genetic variants
Predicting phenotypes with panels of genetic variants captures cumulative effects, reducing noise and false positives for accurate predictions. To this end, we first applied various ML strategies, foregoing the use of feature selection, to establish baseline models for CAD prediction utilizing panels of genetic variants. We then compared the predictive performance of these machine learning models to those achieved using PRS-based models. ML-based models were trained on genomic variants as well as on known risk factors to determine the extent to which genomic variants can enhance the predictive power of risk factor-based models. It should also be noted that PRS-based predictions start from the analysis of a pre-selected set of genetic variants. However, in contrast to the ML approach, variants are selected based on association tests, which evaluates each feature individually and selects the ones with the highest statistical significance. In each employed method, the feature selection process commences with a refined set of SNPs, achieved by applying a Minor Allele Frequency (MAF) filter of 0.01 and an R2 threshold value of 0.1 for linkage disequilibrium (LD) to the entire pool of imputed variants (7.87 million). Then, ten fold cross-validation was used to assess model stability and mitigate overfitting by generating 10 training and test data sets. These sets were used to build and evaluate predictive models of CAD based on PRS (PLINK-PRS, LDpred2 and lassosum), and standard ML algorithms coupled with FS approaches. Figure 3 shows the area under the receiver operating characteristic (ROC) curve (AUC) computed on 10 test sets by using CAD-based classification models covering PRS-based strategies (Fig. 3A) and standard ML algorithms working with genotype data (Fig. 3B) or known risk factors for CAD (Fig. 3C). Notably, when excluding risk factors and other relevant covariates, the best methods among PRSs is lassosum, which achieves an AUC of about 0.55. Lassosum uses penalized regression (LASSO) in its approach to PRS calculation. This implies that the best performances for the PRS calculation are achieved by reducing or even eliminating some of the genetic variants selected with association (or univariate) tests. It is also possible to observe that standard ML methods are not able to improve the performance of PRSs. Indeed, the best performing method is LASSO, which also achieves an AUC of about 0.55. However, PRS-based methods exhibit a higher variance on the test sets. This may result from PRS-based predictions being more prone to overfitting compared to the ML approach. Figure 3C shows ML models trained with risk factors achieve high accuracy (AUC > 75%), with LASSO models achieving the best accuracy. PRS and standard ML algorithms do not improve accuracy. SVM with a Gaussian kernel had the lowest performance, as it struggles to model high-dimensional genetic data effectively. In the case of high-dimensional genetic data, the number of dimensions is too high, and the manifold becomes too complex for the kernel to model effectively, resulting in poor prediction performance.

Classification performance obtained by using PRS, standard classification algorithms and known CAD risk factors. Bar-plots showing the AUC values computed with tenfold cross-validation. Error bars are used to assess model stability, while the different subplots aim to highlight the performance of PRS methods, and ML approaches using genotype data or known risk factors as predictors. A The AUC values obtained by combining PRS scores with logistic regression. B The AUC values obtained by using standard machine learning algorithms. C The AUC values obtained by using known risk factors as input to standard machine learning algorithms
Predicting CAD susceptibility via ML-driven feature selection
Adding genomic variants to traditional risk factors can improve the accuracy of predicting complex diseases such CAD. To this end, different ML-driven feature selection strategies were implemented to assess the effectiveness of various ML-based feature selection techniques in enhancing the accuracy and optimizing the panel size of genomic variants for CAD susceptibility prediction based on genotype SNPs data. Feature selection is important in selecting panels of genetic variants because it helps in reducing the dimensionality of data by selecting a subset of relevant and informative variants. This not only improves the interpretability and understanding of the data, but also enhances the accuracy and performance of predictive models, as it reduces the potential for overfitting and biases. Additionally, feature selection helps in identifying the most important variants that contribute to the phenotype, providing insights into the underlying biological mechanisms. Three different ML algorithms were implemented. The first is based on Random Forest-based feature selection. In this approach, a Random Forest classifier is trained on the data, and the importance of each feature is calculated based on its contribution to the classifier's performance. By considering then top k features based on their importance, it is possible to select a subset of the most informative features for further analysis or to train a predictive model with improved performance. We then implemented a second feature selection strategy using the mRMR algorithm, which can identify a subset of genotype data that is both highly relevant to a phenotype of interest and distinct from other features. It measures both the relevance of each feature with the phenotype and their mutual dependencies and selects a subset of features that maximizes the relevance while minimizing redundancy. Finally, a feature selection strategy based on the result of a standard case/control association analysis using Fisher's exact test implemented in PLINK association analysis was used. The methods generate a score for each variant's contribution to CAD prediction, and different cut-offs were applied to select the top features. These cut-offs were explored to determine the influence of the number of selected features on prediction performance. The feature selection strategy was implemented for each training set of 10-CV and used to train three classification models based on RFs, SVM, and LASSO. Figure 4 shows the performance of ML-driven feature selection algorithms based on AUC values computed with classification models trained with subsets of selected genomic variants (top k) and known risk factors. It is possible to observe that RF-based models systematically outperform LASSO and SVM-based models. Notably, feature selection based on RF classifiers lead to CAD prediction models with an AUC value close to 0.8 (Fig. 4C). This result is achieved by using the top 50 features. Moreover, SVM and LASSO do not perform better than a classification model trained on known risk factors (red dash-dot line) and that SVM-based classifiers achieve high AUC values only when considering large set of features. Finally, we observed a plateau in the classification performance when using the top 50 features selected by the employed feature selection algorithms, possibly indicating that a relatively small set of genetic variants is sufficient for improving risk factor-based models. Figure 5 compares different classification models for CAD prediction, including those based solely on risk factors, all genetic variants (without feature selection), the best performing PRS, which is based on the lassosum, and genomic variants selected through feature selection. Notably, feature selection effectively selects genomic variants enhancing risk factor-based models, with the top 50 variants performing comparably across methods. PRS integration does not increase accuracy, and the best PRS combined with risk factors yields slightly lower performance than top 50 RF-selected features with risk factors.

Evaluating the accuracy of models that utilize both genotype SNPs and risk factors in predicting CAD susceptibility through various machine learning techniques. Bar-plots showing the AUC values computed with tenfold cross-validation. Error bars are used to assess model stability, while the different subplots aim to highlight the performance of three feature selection strategies. Selected features were systematically evaluated with three different classification algorithms: RF, SVM and LASSO. Each classifier was trained with selected genotypes, known risk factors and PCs. A The AUC values obtained by using GWAS-driven feature selection. B The AUC values obtained by using mRMR-based feature selection. C The AUC values obtained by RF-based feature selection and by selecting the top 50 features. The red dash-dot line represents the classification accuracy achieved by using known CAD risk factors

Comparing the accuracy of different classification models, feature selection techniques and predictors. Bar-plots showing the AUC values computed with tenfold cross-validation by using two main classification algorithms (RF and LASSO) and different sets of features (or predictors). Feature sets include known risk factors, all SNPs, PRS, and the top 50 genomic variants selected by RF, mRMR and GWAS results. Moreover, classification models were trained with both genotype data and a combination of genotype data and risk factor. Classifiers annotated with top PRS are trained with the best performing PRS method, which is lassosum. Error bars are used to assess model stability
Evaluating the stability of feature selection algorithms
ML-driven feature selection strategies were evaluated within a 10-CV framework. We therefore sought to evaluate how frequently the same features or genomic variants are selected across different training sets. Figure 6A displays the consistency of feature selection by depicting the percentage of overlap in the top K features selected across multiple runs of cross-validation. GWAS-based feature selection, which expected to be more robust than RF-based feature selection, since it uses a filtering method for feature selection, it provides the most stable results only when considering the top 10 features. However, when reaching the plateau in the classification performance, which corresponds to the selection of the top 50 features, all feature selection methods exhibit a similar level of model stability.

Feature selection stability and selection of the most stable genomic variants. A Bar-plots showing the reliability of the feature selection process by visualizing the extent to which the same features were chosen repeatedly across multiple iterations of cross-validation, shown as a percentage of overlap in the top K selected features. B Features that are selected more than 5 times across 10-CV and total number of selected features across the same runs. C Genomic variants that are consistently recognized as significant by different feature selection techniques and are deemed to have a significant impact on the results by all methods used are selected in order to define a small set of novel SNPs for CAD risk prediction
Figure 6B includes detailed information on the number of features that are always selected across the multiple runs of cross-validation. Our subsequent goal was to identify genomic variants that are consistently selected as important by each feature selection strategy and are deemed relevant across all methods. To this end, we identified genomics variants that appear in the top 50 genomic features at least five times, and that are selected across all methods. Figure 6C reports that 6 genomics variants are consistently selected as important by the feature selection strategies.
Assessing the biological relevance of ML-driven variants for predicting CAD
A literature review was conducted in early 2023 to underscore the significance of the selected genes, and the association between genomic variants and genes was determined using FUMA software version 1.5.2. We identified the top 6 genetic variants that were selected by all feature selection methods and used FUMA [21] to map these genetic variants to their corresponding genes (see Table 2). Selected loci have been described in recent CAD-related GWASs, and the mechanism of action for several of them has been linked either with lipid levels (LDLR, SORT1, and LPA) or with molecular changes within the vascular wall implicated in atherosclerosis (CDKN2A/B, FES/FURIN, PHACTR1) (Additional file 1: Tables S2 and S3). LDLR and SORT1 loci have been associated with changes in low-density lipoprotein (LDL) levels [26, 27], whereas LPA is also associated with lipoprotein (a) levels [28]. On the other hand, the 9p21 locus, harboring ANRIL and CDKN2A/B genes, is one of the strongest genetic associations for CAD identified through GWAS. ANRIL is a long non-coding RNA that regulates gene expression, cell proliferation, senescence, apoptosis, extracellular matrix remodeling, and inflammation [29]. ANRIL exerts its effects through endothelial cell function, macrophage polarization, and VSMC phenotypic transition. ANRIL also regulates plaque stability and is involved in thrombogenesis, vascular remodeling or repair, and plaque stability through its regulation of the tumor suppressor genes, CDKN2A/B. In contrast, the 15q26.1 locus harbors two genes, FES and FURIN, which have been shown to regulate the migration of monocytes and vascular smooth muscle cells and monocyte‐endothelial adhesion, respectively [30, 31]. Finally, the genetic risk locus on chromosome 6p24, which contains PHACTR1 and EDN1 genes, is associated with multiple vascular diseases, including CAD, migraine headache, coronary calcification, hypertension, fibromuscular dysplasia, microvascular angina, and arterial dissection [32, 33]. The expected mechanism of action involves the regulation of vascular smooth muscle cell proliferation and vasoconstriction, as well as the promotion of natriuresis and lower systemic blood pressure through the opposing effects of the ET-A and ET-B receptors. Altogether, this evidence supports that that the ML-selected variants capture widely the mechanistic aspects of the disease etiology, including the risk that arises from lipid levels, inflammation, and vascular biology.
Conclusion
Our study highlights the significant advantages of using machine learning (ML) and feature selection in GWAS for identifying key genomic variants related to CAD. Unlike traditional association analysis, which tests one SNP at a time, and polygenic risk scores (PRS) that may rely on a broad set of SNPs, our approach enhances the development of risk prediction models that rely on a compact panel of genomic variants. Specifically, we demonstrated how feature selection can play a crucial role in identifying a compact set of highly predictive SNPs. This focused approach could significantly simplify the clinical validation and application of genetic variants for disease risk prediction. Furthermore, our research tackles critical methodological challenges encountered when utilizing machine learning (ML)-based approaches for the discovery of disease-associated genomic variants, including feature selection, data leakage, and cross-validation. Cross-validation was also employed in evaluating methods for building PRSs since it contributes to assessing their robustness. Our study demonstrates that their performance remains comparable to those achieved by ML-based approaches, even though PRS utilizes a larger set of SNPs. Our findings are supported by earlier studies aimed at employing advanced data mining algorithms to select more effective genomic variants [34]. Notably, employing feature selection with RF-based classifiers resulted in CAD prediction models achieving an AUC value near 0.8, comparable to that of PRS models. However, unlike PRS, this approach utilizes a smaller panel of genomic variants. Furthermore, six genomic variants were consistently identified as significant by all feature selection methods, exhibiting crucial associations with lipid levels, inflammation, and vascular biology, which are key aspects of CAD etiology. However, our study also found that incorporating PRS did not result in an increase in accuracy, and the best-performing PRS achieved similar performance to ML-driven models. Overall, this study emphasizes the significance of employing machine learning and feature selection to identify panels of genetic variants, rather than single variants, for predicting complex diseases like CAD. It also proposes a computational protocol that should be utilized when comparing ML-based approaches with PRS-based methods for disease risk prediction. In future research, the proposed protocol can be expanded to include initial steps focusing specifically on rare genetic variants. Such an approach could leverage recent statistical methods designed to analyze associations between rare functional variants and common diseases [35,36,37]. Ultimately, this would contribute to improving the accuracy of CAD risk prediction [38, 39].
Materials and methods
Data collection, phenotype extraction and quality control (QC) at the individual and phenotype levels
We collected more than 300000 individuals with CAD and non CAD phenotypes from the intial set of 500,000 individuals in the UK Biobank (ID Application: 58990). The R package ukbpheno v1.0 [40] was used for phenotype extraction, while QCs were done using PLINK software v1.9 and v2.0 [41, 42]. CAD patients were then selected based on a combination of self-reported UKB statuses, standard medical codes (ICD9, ICD10, OPCS4), diagnosed conditions, and various combinations of these criteria. A complete list of ICD9, ICD10 and OPCS4 codes, along with their description is provided in Additional file 1 Table S1. Then, only white participants from the UK Biobank were selected to create a more homogenous population sample. Exclusions were made for those individuals identified with sex chromosome aneuploidy, and participants who had withdrawn their consent. Importantly, we calculated kinship coefficients to filter out related individuals, aiming to maintain a dataset of unrelated participants only, which resulted in a final cohort of 337,110 individuals—23,559 with CAD and 313,551 classified as non-CAD. This selected group of individuals was utilized for the machine learning analysis, including conducting association tests and calculating polygenic risk scores. We also selected information on CAD-associated risk factors (diabetes, blood pressure, vascular/heart problems diagnosed by doctor, smoking, BMI, CRP, Triglycerides, HDL, LDL and the total cholesterol), basic covariates (e.g., sex and age), ethnic background, genotype batch and assessment center. This information all together can help address better genome-wide association analysis for the detection of CAD risk loci.
Genotype-level QC using imputed genotype data
Genetic analyses were done using UKBB imputed genotypes from ~ 96 M SNPs [43], see Additional file 1: Fig. S1. Autosomal markers were extracted solely from SNPs with an imputation information metric (INFO) greater than 0.3 to ensure high-quality imputed genotypes [43]. Proportions [44, 45]. Next, we filtered the genotype data based on marker minor allele frequency and missingness per marker. The MAF (--maf) filter was set to 0.01 (MAF < 1%), in order to remove rare variants, while the option --geno 0.2 removed markers with high rates of missing data. The value 0.2 indicates that a marker will be removed if more than 20% of the genotypes are missing. SNPs with minor allele count less than 100 were also removed. Similarly, we also removed individuals with > 20% missing genotypes (--mind 0.2). Next, we filtered our data based on Hardy–Weinberg equilibrium (HWE) (--hwe 1E-25). It removed markers that deviate significantly from the expected HWE The value 1E-25 indicates the p-value threshold for HWE deviation. The pre-processing of genotype was used to conduct exploratory analysis by using PCA and correlation analysis between risk factors, covariates, PCA and the CAD status. We finally generated a total number of 7,874,484 SNPs with genotyping rate 0.99.
Principal component analysis, multi-collinearity and SHAP values
We assessed population stratification through Principal Component Analysis (PCA) to identify and control the effects of hidden population structure on genotype–phenotype association tests [46]. Moreover, since principal components (PCs) will be considered in the association tests to account for hidden population structures by associating SNPs with the CAD phenotype, we aimed to determine if they also correlate with known risk factors and basic covariates (e.g., age, sex, ethnic background). This helps assess multicollinearity and identify a set of variables with lower correlation. Our PCA analysis initially included the entire participant set to verify the effective encapsulation of population structure. Subsequently, PCA was recalculated for each iteration of cross-fold validation on the selected set of unrelated, white individuals for the machine learning analysis. However, we first applied PCA to the whole set of genotypes including 487,408 participants and used the first 20 PCs along with known risk factors and basic covariates (e.g., age, sex, ethnic background) to assess multicollinearity and identify a set of variables with lower correlation. If high correlation is found, those variables will be removed. The preference is to retain the PCs as they encapsulate the population structure effectively. We evaluated multicollinearity using correlation analysis and variance inflation factor analysis (VIF). The corr() method in Pandas was used to compute a correlation matrix displaying the pair-wise relationships between variables, while VIF [47] was used to measure the degree of multicollinearity between predictor variables in a regression model predicting the CAD status with basic covariates (e.g., sex and age), CAD-risk factors, ethnic background, genotype batch, assessment center, and PCs adjusting for population structure. The VIF was performed in R by using the car package and it generates a VIF value for each predictor. Differently from the correlation analysis, VIF quantifies the extent of correlation between one predictor and the other predictors (PCs and phenotypic information) in a model predicting the CAD status. It is calculated as the ratio of the variance of an estimated regression coefficient to the variance that would be expected if the predictor (or clinical) variable were uncorrelated with all other predictor (or clinical) variables in the model. Clinical variables with a high VIF value were removed. The resulting set of risk factors, clinical variables and the first 20 PCs was used to compute association statistics and model machine learning-based classifiers using polygenic risk scores or genomics variants selected by the implemented ML-driven feature selection strategies. Since principal components are also used as covariates in the machine learning framework and to adjust for population structure in GWAS which are then used to compute PRs, PCA was also systematically applied to each training set generated by the tenfold cross-validation. Moreover, we also performed SHAP (SHapley Additive exPlanations) analysis [48], which is a method used in machine learning for explaining the output of a model by attributing the contribution of each feature to the prediction. In the context of this research on CAD prediction, SHAP values were used to better understand the contribution of each risk factor, genetic variant and covariate to the prediction of the CAD status.
Machine learning and feature selection algorithms
Different feature selection strategies were compared to assess ML effectiveness against traditional association tests. The study also aimed to evaluate if ML-based models using PRS from new training datasets outperformed those developed through feature selection, by using cross validation. It should be noted that computing polygenic risk score (PRS) within a training and validation framework (e.g., cross validation) is important when comparing PRS performance with ML performance, since it allows for an assessment of the stability and generalizability of the PRS model. In cross-validation, the original dataset is divided into multiple folds, and the PRS model is estimated and evaluated on each fold. This helps to ensure that the PRS model is not overfitting to the training data and can provide an estimate of the expected performance of the model when applied to independent datasets. By evaluating the PRS model within a cross-validation framework helps ensure that PRS-based predictions are properly compared with those obtained by using models generated through ML and feature selection algorithms. To compute PRS, association testing was first performed to generate summary statistics and estimate SNP effects by using the PLINK software. This step is re-computed with each training set to guarantee a fair comparison with the ML approach. Since the phenotype in the present data sets is dichotomous (CAD/non-CAD cases), a logistic regression was performed by using sex, age, the genotyping batch, the assessment center, the first 20 PCs and CAD risk factors as additional covariates [49]. It should be noted that the PCA was repeated for each training set generated by 10CV. The estimated effects were then used to compute PRS-based scores or rank SNPs for a filter-based feature selection strategy. Three different genome-wide PRS strategies, PLINK-PRS, LDpred2 [50], and lassosum [51], were computed and used as input for logistic regression-based classifiers to distinguish CAD from non-CAD cases. In this research, we methodically examine three techniques for feature selection in ML models: (1) applying univariate test statistics with PLINK; (2) implementing the Maximum Relevance Minimum Redundancy (mRMR) algorithm [16], aimed at discovering predictive and uncorrelated features; (3) using Random Forest (RF) feature importance scores to capture complex interactions. The objective of these methods is to pinpoint a concise and efficient set of single-nucleotide polymorphisms (SNPs) for incorporation into machine learning models that classify individuals with CAD. Each adopted feature selection method delivers a final ranking score for the selected variants. The top k variants (with k + 5, 10, 15, 20, 50, 100, 200, 300, and 500) were then used as input for several ML-based classifiers, including Logistic Regression (LR), Lasso (LA), Support Vector Machine (SVM) and Random Forest-based (RF). The entire machine learning-based pipeline was constructed using Python and Scikit-learn. We used the following configurations for our chosen machine learning methods:
LR with a maximum iteration parameter (max_iter) set to 1000, and ‘liblinear’ as the solver. RF classifiers with 500 trees, setting the maximum number of features considered for making the best split at 35. SVM for classification purposes, utilizing the radial basis function (rbf) as the kernel method to allow for non-linear classification. Each of these configurations was designed to cater to different requirements: while the Random Forest provided a quadratic classifier and the LR and Lasso as linear one, the Support Vector Machine offered a non-linear classification model.
Preparation of the training set for CAD prediction and the implemented evaluation strategy
The study aims to improve CAD prediction models using genomic variants and machine learning-based feature selection. By employing a supervised learning approach, we randomly selected 35,000 non-CAD individuals from UKB to guarantee a balanced training dataset. Repeated sampling verified a consistent distribution of CAD-associated risk factors, confirming random sampling as an effective method to avoid unbalanced classification problems. After randomly selecting a set of individual labels as non-CAD, we applied a MAF cut-off value and LD pruning to reduce the number of SNPs for subsequent ML- and PRS-based analyses. We selected a very low minor allele frequency (MAF < 0.01) and used the linkage disequilibrium, which assesses the non-random association of alleles at different loci in each population, to remove SNPs so that no pair within 500 kbs had squared allele count correlation (r2) greater than 0.1. In more detail, a windows size of 500 kb, a step size of 25 and an r2 greater than 0.1 were used to drastically reduce the number of SNPs to 177,017 variants. At this stage the training and validation dataset retrieved from the UKB includes 63,216 (34,056 cases, 27,295 controls) individuals. To evaluate the performance of machine learning (ML) models applied to genotypic data and or clinical variables, we divided the computed dataset into training and test sets using a tenfold cross-validation approach. The samples were then stratified based on phenotype (CAD) and sex, and the proportion of CAD/non-CAD cases for males and females was preserved in each fold. The performance of each algorithm was evaluated using AUC-ROC, which assesses the model's ability to distinguish between positive and negative samples. High AUC-ROC values indicate accurate identification with minimal false positives. To assess stability, feature selection frequency across datasets and methods was recorded. Features consistently selected were deemed stable, biologically relevant, and contributed to a reliable and generalizable predictive model.
References
Mavaddat N, Michailidou K, Dennis J, et al. Polygenic risk scores for prediction of breast cancer and breast cancer subtypes. Am J Hum Genet. 2019;104:21–34.
Guseynova NT. Application of DNA-based molecular genetic markers for plant identification. Adv Biol Earth Sci. 2023;8:2.
Deloukas P, Kanoni S, Willenborg C, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45:25–33.
Tcheandjieu C, Zhu X, Hilliard AT, et al. Large-scale genome-wide association study of coronary artery disease in genetically diverse populations. Nat Med. 2022;28:1679–92.
Wald NJ, Old R. The illusion of polygenic disease risk prediction. Genet Med. 2019;21:1705–7.
De La Vega FM, Bustamante CD. Polygenic risk scores: a biased prediction? Genome Med. 2018;10:100.
Ayoub A, McHugh J, Hayward J, et al. Polygenic risk scores: improving the prediction of future disease or added complexity? Br J Gen Pract. 2022;72:396–8.
Bermingham ML, Pong-Wong R, Spiliopoulou A, et al. Application of high-dimensional feature selection: evaluation for genomic prediction in man. Sci Rep. 2015;5:10312.
Romagnoni A, Jégou S, Van Steen K, et al. Comparative performances of machine learning methods for classifying crohn disease patients using genome-wide genotyping data. Sci Rep. 2019;9:10351.
Nicholls HL, John CR, Watson DS, et al. Reaching the end-game for GWAS: machine learning approaches for the prioritization of complex disease loci. Front Genet. 2020;11:350.
Fortino V, Wisgrill L, Werner P, et al. Machine-learning-driven biomarker discovery for the discrimination between allergic and irritant contact dermatitis. Proc Natl Acad Sci USA. 2020;117:33474–85.
Fortino V, Kinaret PAS, Fratello M, et al. Biomarkers of nanomaterials hazard from multi-layer data. Nat Commun. 2022;13:3798.
Fortino V, Scala G, Greco D. Feature set optimization in biomarker discovery from genome-scale data. Bioinformatics. 2020;36:3393–400.
Fortino V, Kinaret P, Fyhrquist N, et al. A robust and accurate method for feature selection and prioritization from multi-class OMICs data. PLoS ONE. 2014;9: e107801.
Tadist K, Najah S, Nikolov NS, et al. Feature selection methods and genomic big data: a systematic review. J Big Data. 2019;6:79.
Peng H, Long F, Ding C. Feature selection based on mutual information: criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans Pattern Anal Mach Intell. 2005;27:1226–38.
Elliott J, Bodinier B, Bond TA, et al. Predictive accuracy of a polygenic risk score-enhanced prediction model vs a clinical risk score for coronary artery disease. JAMA. 2020;323:636–45.
Baker E, Escott-Price V. Polygenic risk scores in alzheimer’s disease: current applications and future directions. Front Digit Health. 2020;2:14.
Ho DSW, Schierding W, Wake M, et al. Machine learning SNP based prediction for precision medicine. Front Genet. 2019;10:267.
Nogueira S, Sechidis K, Brown G. On the stability of feature selection algorithms. J Mach Learn Res. 2017;18:6345–98.
Watanabe K, Taskesen E, van Bochoven A, et al. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8:1826.
O’Sullivan JW, Raghavan S, Marquez-Luna C, et al. Polygenic risk scores for cardiovascular disease: a scientific statement from the American heart association. Circulation. 2022;146:e93–118.
Natarajan P, Young R, Stitziel NO, et al. Polygenic risk score identifies subgroup with higher burden of atherosclerosis and greater relative benefit from statin therapy in the primary prevention setting. Circulation. 2017;135:2091–101.
Howe LJ, Dudbridge F, Schmidt AF, et al. Polygenic risk scores for coronary artery disease and subsequent event risk amongst established cases. Hum Mol Genet. 2020;29:1388–95.
Varoquaux G, Cheplygina V. Machine learning for medical imaging: methodological failures and recommendations for the future. npj Digital Med. 2022;5:48.
Kjolby M, Nielsen MS, Petersen CM. Sortilin, encoded by the cardiovascular risk gene SORT1, and its suggested functions in cardiovascular disease. Curr Atheroscler Rep. 2015;17:496.
Ference BA, Ginsberg HN, Graham I, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. evidence from genetic, epidemiologic, and clinical studies. a consensus statement from the european atherosclerosis society consensus panel. Eur Heart J. 2017;38:2459–72.
Kronenberg F, Mora S, Stroes ESG, et al. Lipoprotein (a) in atherosclerotic cardiovascular disease and aortic stenosis: a european atherosclerosis society consensus statement. Eur Heart J. 2022;43:3925–46.
Holdt LM, Teupser D. Long noncoding RNA ANRIL: lnc-ing genetic variation at the chromosome 9p21 locus to molecular mechanisms of atherosclerosis. Front Cardiovasc Med. 2018;5:145.
Karamanavi E, McVey DG, van der Laan SW, et al. The FES gene at the 15q26 coronary-artery-disease locus inhibits atherosclerosis. Circ Res. 2022;131:1004–17.
Yang X, Yang W, McVey DG, et al. FURIN expression in vascular endothelial cells is modulated by a coronary artery disease-associated genetic variant and influences monocyte transendothelial migration. J Am Heart Assoc. 2020;9: e014333.
Gupta RM. Causal gene confusion: the complicated EDN1/PHACTR1 locus for coronary artery disease. Arterioscler Thromb Vasc Biol. 2022;42:610–2.
Gupta RM, Hadaya J, Trehan A, et al. A genetic variant associated with five vascular diseases is a distal regulator of endothelin-1 gene expression. Cell. 2017;170:522-533.e15.
Selvaraj MS, Li X, Li Z, et al. Whole genome sequence analysis of blood lipid levels in > 66,000 individuals. Nat Commun. 2022;13:5995.
Li Z, Li X, Liu Y, et al. Dynamic scan procedure for detecting rare-variant association regions in whole-genome sequencing studies. Am J Hum Genet. 2019;104:802–14.
Li X, Li Z, Zhou H, et al. Dynamic incorporation of multiple in silico functional annotations empowers rare variant association analysis of large whole-genome sequencing studies at scale. Nat Genet. 2020;52:969–83.
Li X, Quick C, Zhou H, et al. Powerful, scalable and resource-efficient meta-analysis of rare variant associations in large whole genome sequencing studies. Nat Genet. 2023;55:154–64.
Hasbani NR, Westerman KE, Kwak SH, et al. Type 2 diabetes modifies the association of CAD genomic risk variants with subclinical atherosclerosis. Circ Genom Precis Med. 2023;16: e004176.
Fiziev PP, McRae J, Ulirsch JC, et al. Rare penetrant mutations confer severe risk of common diseases. Science. 2023;380:1131.
Yeung MW, van der Harst P, Verweij N. ukbpheno v1.0: an R package for phenotyping health-related outcomes in the UK biobank. STAR Protoc. 2022. https://doi.org/10.1016/j.xpro.2022.101471.
Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Chang CC, Chow CC, Tellier LC, et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7.
Bycroft C, Freeman C, Petkova D, et al. The UK biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–9.
Wigginton JE, Cutler DJ, Abecasis GR. A note on exact tests of Hardy-Weinberg equilibrium. Am J Hum Genet. 2005;76:887–93.
Graffelman J, Moreno V. The mid p value in exact tests for Hardy-Weinberg equilibrium. Stat Appl Genet Mol Biol. 2013;12:433–48.
Price AL, Patterson NJ, Plenge RM, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9.
Akinwande MO, Dikko HG, Samson A. Variance inflation factor: as a condition for the inclusion of suppressor variable(s) in regression analysis. Open J Stat. 2015;05:754–67.
Lundberg S, Lee S-I. A unified approach to interpreting model predictions. arXiv 2017.
Hill A, Loh P-R, Bharadwaj RB, et al. Stepwise distributed open innovation contests for software development: acceleration of genome-wide association analysis. Gigascience. 2017;6:1–10.
Vilhjálmsson BJ, Yang J, Finucane HK, et al. Modeling linkage disequilibrium increases accuracy of polygenic risk scores. Am J Hum Genet. 2015;97:576–92.
Mak TSH, Porsch RM, Choi SW, et al. Polygenic scores via penalized regression on summary statistics. Genet Epidemiol. 2017;41:469–80.
Acknowledgements
This research has been conducted using the UK Biobank Resource under the application number [58990]. This work uses data provided by patients and collected by the NHS as part of their care and support. Part of the computational analyzes were performed on servers provided by UEF Bioinformatics Center, University of Eastern Finland, Finland.
Funding
This work was supported by the Academy of Finland [grant agreements 336275 and 332510] (V.F.), the Jane and Aatos Erkko Foundation (V.F.), Sydantutkimussaatio (V.F.), the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (Grant No. 802825 to M.U.K), Finnish Foundation for Cardiovascular Research (M.U.K) and the Sigrid Juselius Foundation (M.U.K).
Author information
Authors and Affiliations
Contributions
Conceptualization: VF; Methodology: Z.A. and V.F.; Data preparation and data curation: Z.A., M.M.; Software implementation: Z.A. ; Investigation: M.M., M.U.K., V.F.; Writing—Original Draft: V.F.; Writing-review and editing: Z.A., M.M., M.U.K., V.F.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The research involving genotype data from the UK Biobank was conducted under the ethical approval granted to the UK Biobank by the North West Multi-centre Research Ethics Committee (MREC), operating under the Research Tissue Bank (RTB) approval. Consequently, no separate ethical clearance was necessary for this study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Dr. Alireza Zourmand is now affiliated with Häme University of Applied Sciences Hämeenlinna, Finland.
Supplementary Information
Additional file 1: Table S1.
List of CAD-related phenotypes that were used to select CAD cases within the UK Biobank cohort over a 12 year follow-up period. Table S2. Association statistics between the identified loci and CAD in various cohort-based studies. Table S3. List of other major CVD GWAS associations by using the selected ML-driven loci. Figure S1. Summary of GWAS QC and initial pre-processing. UKBB raw imputation genotypes consisted of 97 million markers, that were subjected to QCs steps shown here. Figure S2. Principal component analysis applied to genotype data to uncover population structure and use the PCs as covariates. A Variance explained by the first 20 PCs. B Scatter plots that display the variance explained by the first 6 PCs. Different colors are associated to different ethnic groups in order to visually show the population structure among the individual of the UK Biobank cohort. Figure S3. SHAP summary plot. This plot shows the SHAP values for each feature and observation (dots). Each dot has three characteristics: vertical location shows what feature it is depicting; color shows whether that feature was high or low for that row of the dataset; horizontal location shows whether the effect of that value caused a higher or lower prediction. Figure S4. Correlation-based analysis comparing risk factors, principal components and ethnicity. (A) Standard correlation analysis showing associations between risk factors and PCs and ethnic group and PCs. (B-C) Variance inflation factor (VIF) to measure of the amount of multicollinearity in a set of multiple regression variables. (B) VIF results before (B) and after removing (C) cholesterol and ethnicity.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Alireza, Z., Maleeha, M., Kaikkonen, M. et al. Enhancing prediction accuracy of coronary artery disease through machine learning-driven genomic variant selection. J Transl Med 22, 356 (2024). https://doi.org/10.1186/s12967-024-05090-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-024-05090-1