
Abstract
Background
Tissue-resident memory T (TRM) cells can reside in the tumor microenvironment and are considered the primary response cells to immunotherapy. Heterogeneity in functional status and spatial distribution may contribute to the controversial role of TRM cells but we know little about it.
Methods
Through multiplex immunofluorescence (mIF) (CD8, CD103, PD-1, Tim-3, GZMB, CK), the quantity and spatial location of TRM cell subsets were recognized in the tissue from 274 patients with NSCLC after radical surgery. By integrating multiple machine learning methods, we constructed a TRM-based spatial immune signature (TRM-SIS) to predict the prognosis. Furthermore, we conducted a CD103-related gene set enrichment analysis (GSEA) and verified its finding by another mIF panel (CD8, CD103, CK, CD31, Hif-1α).
Results
The density of TRM cells was significantly correlated with the expression of PD-1, Tim-3 and GZMB. Four types of TRM cell subsets was defined, including TRM1 (PD-1−Tim-3−TRM), TRM2 (PD-1+Tim-3−TRM), TRM3 (PD-1−Tim-3+TRM) and TRM4 (PD-1+Tim-3+TRM). The cytotoxicity of TRM2 was the strongest while that of TRM4 was the weakest. Compare with TRM1 and TRM2, TRM3 and TRM4 had better infiltration and stronger interaction with cancer cells. The TRM-SIS was an independent prognostic factor for disease-free survival [HR = 2.43, 95%CI (1.63–3.60), P < 0.001] and showed a better performance than the TNM staging system for recurrence prediction. Furthermore, by CD103-related GSEA and mIF validation, we found a negative association between tumor angiogenesis and infiltration of TRM cells.
Conclusions
These findings reveal a significant heterogeneity in the functional status and spatial distribution of TRM cells, and support it as a biomarker for the prognosis of NSCLC patients. Regulating TRM cells by targeting tumor angiogenesis may be a potential strategy to improve current immunotherapy.
Similar content being viewed by others

Introduction
Despite advances in improvements in diagnostics and treatment, lung cancer remains the leading cause of cancer-related mortality worldwide, with approximately 61% (early-stage) ~ 34% (local-advanced) of 5-year survival rate, and non-small cell lung cancer (NSCLC) counts for over 80% of all lung cancer cases [1]. About 22% (stage I) ~ 68% (stage III) patients with operable NSCLC suffer from recurrence within 5 years, mainly in the first 2 years, indicating treatment failure in a considerable number of cases [2, 3]. The tumor microenvironment (TME), containing cancer cells, multifarious immune cells, vascular, mesenchymal cells and non-cell components, contributes greatly to the development of NSCLC. Consequently, there is an urgent need to improve the understanding of the TME of NSCLC for better patient stratification and recognizing novel targets to improve therapies.
Tissue-resident memory T cells (TRM) are a special population of immune cells that persistently reside in barrier tissues (skin, intestine and lung), lymphoid organs as well as solid tumors, patrol their microenvironment, and rapidly respond to alarm signals [4, 5]. TRM cells are characterized by their expression of CD103 integrin, mediating the adhesion of intraepithelial T-cells to epithelial cells [6]. Emerging evidence has linked the quantity of TRM cells, mostly CD103+CD8+TRM (referred to by “TRM” in this study unless otherwise noted), to better response to immunotherapy and better prognosis of patients with lung cancer [7,8,9]. However, there are also different opinions that TRM present a immunoregulatory characterization and exert a positive impact on tumor progression [10,11,12]. Of note, deep sequencing of TRM in human lung cancer has revealed that TRM appears to be functionally exhausted to varying degrees, expressing multiple immune checkpoints, especially PD-1 and Tim-3, which suggests that TRM cells are highly heterogeneous and may be a potential target of immune checkpoint blockade [8, 13, 14]. Nonetheless, heterogeneity of TRM functional subsets remains unknown.
Spatial architecture of the TME is increasingly considered extremely important to orchestrate tumor immunity and therapeutic response [15]. It has been recognized that CD8+ T cells in the invasive margin (IM) were more abundant but more dysfunctional than those in the tumor center (TC) [16, 17]. Likewise, a higher density of T cells and their different prognostic effects were observed in the stroma compartment compared to the epithelium compartment in NSCLC [18,19,20]. Additionally, intercellular interactions, as indicated by the spatial distance between cells, have received increasing attention. Some parameters, such as the nearest neighboring distance [21, 22] and proximity score (also known as an effective score or mixing score) [17, 23, 24], have been proposed to quantify this spatial interaction. Previous studies, including ours, have found that the poor spatial interaction between intratumoral CD8+T cells and cancer cells was correlated with lymph node metastases, recurrence and worse survival in patients with NSCLC [17, 25, 26]. Recently, approaches to comprehending the spatial architecture of the TME are transitioning from traditional single-color immunohistochemistry (IHC) to multiplex IHC/multiplex immunofluorescence (mIF), enabling the quantification and localization of multiple cell phenotypes simultaneously at single-cell resolution [27]. In this context, exploring the spatial architecture of TRM functional subsets should not be absent.
Herein, we determined the density and spatial localization of CD103+CD8+ TRM subsets with different immune checkpoint coexpression patterns through mIF in a cohort of patients with operable NSCLC, and revealed their significant heterogeneity.
Materials and methods
Patient cohort and sample collection
Formalin-fixed, paraffin-embedded (FFPE) tissue specimens and clinical information were retrospectively collected from NSCLC patients who underwent radical surgery at Shandong Cancer Hospital & Institute (SCHI, Shandong, China) between 2014 and 2018. Patients treated with preoperative antitumor therapy were excluded from this cohort. The final analysis included only lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) because of the small number of patients with other histological types (n = 5). This study had the approval of the Institutional Ethics Committee (SDTHEC2022007013).
Tissue microarray construction and mIF staining
Tissue microarrays (TMA) were constructed from FFPE tissues as described in our previous studies [17, 26]. mIF staining was conducted with Akoya-Opal-Kit (Akoya Biosciences, U.S.A.; cat. NEL871001KT) following a previously described protocol [17, 26]. Briefly, 3 μm slides obtained from the TMA blocks were subjected to a deparaffinization using xylene and a rehydration through a graded ethanol series. Then, an antigen retrieval was performed using microwave incubation with citric acid buffer or EDTA buffer. Each slide underwent multiple rounds of staining, which involved antigen blocking with 1% BSA, reaction with specific primary antibodies and corresponding secondary antibodies, and covalent binding of tyramide signal amplification (TSA) fluorophores. Then antibody complexes were removed by the subsequent antigen retrieval before a new staining round. After all rounds of staining, the slides were counterstained with DAPI and scanned using the Vectra Polaris Scanner System (PerkinElmer, U.S.A.). Detailed information on the antibodies and reagents used can be found in Additional file 1: Table S1.
Automatic image processing and immune-related feature definition
Multispectral images were processed in the Inform software 2.4.8 (PerkinElmer, U.S.A.) following a previously described procedure [17, 26]. In brief, after multispectral images unmix into single-spectral images, tissue segmentation into the epithelium compartment and stroma compartment, cell segmentation and phenotype identification, single-marker phenotypes and single-cell two-dimensional coordinates can be generated. Both the trained tissue segmentation classifier and the cell phenotype classifier had an accuracy of more than 98%. At least two investigators, including an experienced pathologist, independently reviewed all images.
To reveal the characteristics of TRM cells, we calculated the number of all types of cell phenotypes of interest (Additional file 1: Table S2) and intercellular Euclidean distances. The cell density was determined by normalizing the corresponding cell count to the total cell counts (cell / 1000 cells) using the following formula:
where A represents one cell phenotype, and n corresponds to the number of cells. To assess the spatial characteristics of TRM, we established three distinct spatial features. The infiltration score (InS) was defined as the ratio of cell count between the epithelial and stromal compartment, reflecting the level of immune cell infiltration, whose formula was:
The mean nearest neighboring distance (mNND) from A cells to cancer cells was defined as the mean distance between each A cell and its nearest neighboring cancer cell, which was negatively related to the interplay intensity between them. The formula of mNND was:
where C cell represents the cancer cell, and dmin (Ai ~ C) represents the minimum distance from the i-th A cell to its nearest neighboring cancer cell. The cancer-cell proximity score (CCPS) of A cells was defined as the average number of A cells within a predefined radius of all cancer cells. The formula of mNND was:
in which r represents a predefined radius, and \(n\left({C}_{i}\stackrel{r}{\to }A\right)\) represents the number of A cells within r-μm radius of the i-th cancer cell. A higher CCPS of A cells indicated more effective A cells interplayed with cancer cells.
Prognostic signature construction
The entire cohort was randomly divided into a training set and a testing set at a ratio of 7: 3 according to the 10 events per variable (10 EPV) principle. Firstly, in the training set, we performed an importance evaluation of candidate immune-related features through multiple machine learning algorithms, including Least Absolute Shrinkage and Selection Operator (LASSO) regression algorithm (tenfold cross-validations), eXtreme Gradient Boosting (XGBoost) algorithm (1000-time repetitions) and Random Forest (RF) algorithm. The intersection of their respective top 10 important features was selected to construct a TRM-based spatial immune signature (TRM-SIS) in the Cox regression model:
where in \({HR}_{{X}_{i}}\) is the HR for prognostic feature Xi selected by the above procedure.
Verification of partial results in a public cohort
To verify our results on CD103 coexpression with checkpoints and partially validate our prediction model, we constructed an external cohort by collecting samples from The Cancer Genome Atlas (TCGA) database based on the same inclusion criteria as the SCHI-NSCLC cohort. We followed the principles of the TCGA Research Network to process the data and retained only those cases with complete mRNA expression information and clinical prognostic information [28].
Gene Set Enrichment Analysis (GSEA)
To determine the CD103-related biological pathways, we collected a suitable Gene Expression Omnibus (GEO) dataset (GSE10245) and conducted GSEA in official GSEA software (V.4.3.2) with default parameters. Samples of the GEO dataset were divided into two groups evenly based on the median standardized counts of CD103 mRNA expression. Gene sets in “C5: GOBP” collection from the Human Molecular Signatures Database (MSigDB) were applied. A gene set would be considered statistically significant if the absolute value of its normalized enrichment score (NES) was over 1.00 and its false discovery rate (FDR) q-value was below 0.05.
Statistical Analyses
The Mann–Whitney U tests for two independent samples and Kruskal–Wallis tests for multiple samples were used to analyze the differences in immune-related features and clinicopathological characteristics. Spearman rank correlation coefficients were used to evaluate the correlation among these variables. Disease-free survival (DFS) was defined as the time from the first day of radical surgery to a progression event or last follow-up. To evaluate the predictive performance of the TRM-SIS, the areas under the curve (AUCs) of time-dependent receiver operating characteristic (ROC) curves were applied. The survival analyses were conducted by Kaplan–Meier plot and log-rank test between patients with high TRM-SIS and low TRM-SIS. We used the univariate and multivariate Cox proportional hazard regression models (backward elimination) to estimate the independent prognostic value of the TRM-SIS. A two-sided P value less than 0.05 was considered statistically significant. All statistical results were reviewed by at least two investigators independently. All statistical analyses were carried out in R 4.2.3 and SPSS 26.0 (IBM, U.S.A.).
Results
Demographic characteristics of the NSCLC cohort
A total of 274 patients with stage IA-IIIB NSCLC meeting the inclusion criteria were eventually enrolled, among which 108 cases (39.4%) relapsed during the follow-up. The median follow-up time was 65.9 months. The majority of the cases were male (65.0%), with stage I disease (55.3%) and LUAD (65.3%) (Additional file 1: Table S3).
Quality control evaluation of mIF
To ensure that the mIF project we developed was effective and stable, we conducted a quality control (QC) evaluation through a QC tool from the manufacturer (Akoya Biosciences, U.S.A.). It demonstrated an acceptable heterogeneity of signal intensity across multispectral images, low-level crosstalk among spectral channels and accurate unmixing from noise, indicating a good performance of the mIF project (Additional file 1: Figure S1, S2, Additional file 1: Table S4).
TRM cells possess better infiltration and stronger interaction with cancer cells than Tnon-RM cells
Based on previous results from single-cell sequencing [8, 13, 14], we employed mIF to identify cytotoxic T cells (CD8+ cells), TRM cells (CD103+CD8+ cells), Tnon-RM cells (CD8+CD103− cells), immune checkpoints (PD-1, Tim-3), cytotoxic indicator (GZMB) and cancer cells (CK+ cells) (Fig. 1A). As mentioned earlier, three spatial parameters were defined to describe the spatial distribution of cells (Fig. 1B, C). TRM accounted for more than half of CD103+ cells both in TC and in IM (58.87% and 53.21%, respectively), indicating that cytotoxic T cells were the primary source of CD103 expression (Fig. 1C). TRM cells predominated cytotoxic T cells in TC (60.50%) but vice versa in IM (36.55%) (Fig. 1D). Compared with Tnon-RM, TRM cells possessed higher InS in TC (P < 0.001), shorter mNND to cancer cells in both TC (P < 0.001) and IM (P < 0.001), and higher CCPS in IM (P < 0.001), reflecting better infiltration and stronger interaction with cancer cells of TRM (Fig. 1E–G).

Multiple immunofluorescence (mIF) identifies TRM and their spatial infiltration features in patients with NSCLC. A The merged and single-stained images for the panel of mIF. B Diagram illustrating the calculation of cell density (Dens), infiltration score (InS), mean nearest neighboring distance (mNND) and cancer-cell proximity score (CCPS). Line a ~ s represent the nearest distance from each immune cell to its neighboring cancer cell when calculating mNND. C Proportion of CD8+CD103+ cells (TRM cells) in total CD103+ cells. D Proportion of CD8+CD103+ cells (TRM cells) TRM in total CD8+ cells. E–G Differences of spatial infiltration features between TRM and Tnon-RM. Statistical significance was calculated using the Mann–Whitney U test
TRM subsets defined by immune checkpoint coexpression patterns presented discrepant functional states and spatial architecture
Close interaction between T cells and cancer cells can induce the exhaustion of T cells [29]. The expression of immune checkpoints could reflect the functional state of T cells [14]. We found that CD103+CD8+ cells showed significant positive correlations with PD-1+CD8+ cells, Tim-3+CD8+ cells and GZMB+CD8+ cells (Fig. 2A–F). Considering the specific phenotype of TRM in GZMB production, we compared the cytotoxic capacity among different CD8+T cell subsets by the proportion of GZMB+ cells [5, 30]. The proportion of GZMB+ cells reached its highest in PD-1+Tim-3−TRM and lowest in PD-1+Tim-3+TRM, where it was still much higher than that in Tnon-RM subsets, indicating a considerable heterogeneity of cytotoxicity among CD8+ T-cell subsets with different immune checkpoint expression patterns (Fig. 2G, H). Consequently, based on the different immune checkpoint expression patterns, we divide TRM into four subgroups: PD-1−Tim-3−TRM (TRM1), PD-1+Tim-3−TRM (TRM2), PD-1−Tim-3+TRM (TRM3), and PD-1+Tim-3+TRM (TRM4). In TC, four subsets accounted for 40.21%, 9.52%, 25.04% and 25.23% of TRM, respectively. In IM, there was more TRM1 (51.23%) and less TRM4 (15.45%) than TC (Fig. 2I). Afterward, we compared the spatial features in pairs among four TRM subsets. The InS of TRM1 and TRM2 were significantly lower than those of TRM3 and TRM4. Notably, the mNND to cancer cells progressively decreased from TRM1 to TRM4 whereas the CCPS progressively increased from TRM1 to TRM4 (Fig. 2J–L). These findings evidenced spatial and functional heterogeneity in TRM cell subsets.

Definition of different TRM cell subsets in patients with NSCLC. A–F Close positive correlation of CD103 and immune-related molecules (PD-1, Tim-3 and GZMB) on CD8+T cells. The correlation was calculated using the Spearman rank correlation coefficient. G–H Discrepant cytotoxic abilities among different TRM cell subsets defined by different immune checkpoint coexpression patterns. I Composition of four types of TRM subsets. J–L Differences of spatial features among TRM subsets. Statistical significance was calculated using the Kruskal–Wallis test. *P < 0.05; **P < 0.01; ***P < 0.001
Construction of TRM-based spatial immune signature by integration of multiple machine learning methods
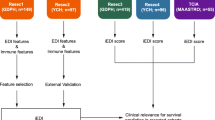
The entire cohort (n = 274) was randomly divided into a training set (n = 192) and testing set (n = 82), and no clinicopathological factors were significantly different between them (Additional file 1: Table S5; Additional file 2: Suppl_data1). For a comprehensive assessment of TRM, we constructed the TRM-SIS via integrating three machine learning algorithms, including LASSO, XGBoost and Random Forest. We firstly assessed and ranked the importance of all candidate TRM-related features by the three algorithms, respectively (Fig. 3A–C). In order to select the robust features, we took the intersection features of the three sets of top 10 important features as the variables for constructing the TRM-SIS and finally screened out six important features: density of CD103+ cells in TC, density of TRM3 in IM, InS of Tnon-RM in TC, InS of TRM1 in IM, InS of TRM4 in IM and mNND from TRM to cancer cells in TC (Fig. 3D, E). Their cut-off values were determined using X-tile software (Yale University, U.S.A.) [31]. The TRM-SIS was calculated for each patient according to the linear combination of these features weighted by their coefficients of the univariate Cox analyses: TRM-SIS = [ln0.41 × Dens(CD103) in TC] + [ln0.51 × Dens(TRM3) in IM] + [ln0.41 × InS(Tnon-RM) in TC] + [ln0.36 × InS(TRM1) in IM] + [ln2.04 × InS(TRM4) in IM] + [ln0.49 × mNND(TRM ~ CK) in TC].

Construction of immune scoring system by integrating multiple machine learning methods. A–C The importance feature maps show the top 10 important features in LASSO model, XGBoost model and Random Forest model. D Venn diagram demonstrates the six immune features shared by the three models: density of CD103+ cells in TC, density of TRM3 in IM, infiltration score of Tnon-RM in TC, infiltration score of TRM1 in IM, infiltration score of TRM4 in IM and mean nearest neighboring distance from TRM to cancer cells in TC. E Univariate Cox analyses show the prognostic relevance of the six selected features. F–H Time-dependent ROC curves and AUC values of TRM-SIS system and TNM staging system for prediction of recurrence risk at 1, 3 and 5 years in the training set, testing set and entire cohort. The TRM-SIS system presents better performance than the TNM staging system for recurrence prediction. I–K Survival analyses based on the TRM-SIS in the training set, testing set and entire cohort. Statistical significance was calculated using the log-rank test. AUC, area under the curve; DFS, disease-free survival; ROC, receiver operating characteristic
The effectiveness of the TRM-SIS was evaluated and validated through time-dependent ROC analyses. The AUC values for 1-, 3-, and 5-year DFS were as follows: in the training set, 0.823, 0.794, and 0.742, respectively; in the testing set, 0.687, 0.691, and 0.690, respectively; and in the entire cohort, 0.776, 0.730, and 0.693, respectively. Notably, the TRM-SIS system manifested better predictive ability than the TNM staging system (Fig. 3F–H). In survival analyses, NSCLC patients with high TRM-SIS presented worse prognosis compared with those with low TRM-SIS (mDFS: 33.07 months vs. not reached, P < 0.001 in the training set; mDFS: 67.97 months vs. not reached, P = 0.048 in the testing set; mDFS: 58.93 months vs. not reached, P < 0.001 in the entire cohort, respectively) (Fig. 3I–K; Additional file 1: Table S6). As shown in univariate and multivariate Cox models, TRM-SIS [HR = 2.43, 95%CI (1.63–3.60), P < 0.001] and tumor stage [stage II vs. stage I: HR = 1.85, 95%CI (1.15–2.98), P = 0.011; stage III vs. stage I: HR = 3.10, 95%CI (1.99–4.83), P < 0.001] were independent prognostic factors for DFS (Table 1).
To improve our ability to stratify patients, we integrated TRM-SIS and tumor stage to predict prognosis of patients. The AUC values for 1-, 3-, and 5-year DFS were as follows: in the training set, 0.820, 0.810, and 0.780, respectively; in the testing set, 0.757, 0.703, and 0.711, respectively; and in the entire cohort, 0.770, 0.739, and 0.730, respectively, which indicated better performance (Additional file 1: Figure S3). Collectively, our findings show that spatial features of specific TRM cell subsets may act as a biomarker for the prognosis of patients with NSCLC.
Validation of the clinical value of CD103 in NSCLC
To verify our results to some extent, we collected 244 eligible cases with stage I-III NSCLC from TCGA database (Additional file 1 Table S7; Additional file 2: Suppl_data2). Similarly, the expression of CD103 was significantly correlated with that of PD-1, Tim-3 and GZMB (Additional file 1: Figure S4A-C). Further, the survival analyses showed that patients in the high CD103 expression group had longer DFS despite no statistical significance (mDFS: not reached vs. not reached, P = 0.170) and significantly longer OS (mOS: 65.1 months vs. 31.7 months, P = 0.002) compared with those in the low CD103 expression group (Additional file 1: Figure S4D, E).
Tumor angiogenesis was unfavorable to TRM cells
Lung TRM cells have been found to exhibit plasticity in transportation and retention in virus-infected mouse models[32, 33]. For a better understanding of potential regulatory mechanisms and the relevant biological pathways of TRM in the tumor microenvironment, we performed a GSEA using 20 suitable samples with operable NSCLC from the GSE10245 dataset (Additional file 2: Suppl_data3). The 20 tumor samples were divided into high and low CD103 expression groups according to the median normalized count of ITGAE (coding CD103 protein). Among a total of 463 gene sets which were significantly enriched between two groups, 371 upregulated gene sets in the low CD103 expression group accounted for the largest proportion (371/463, 80.13%). As shown in the top 20 pathways related to the low expression of CD103, most of them involved the extracellular stromal microenvironment, especially angiogenesis (Fig. 4A). Figure 4B demonstrated the three main angiogenesis-related pathways enriched in the low CD103 expression group: “GOBP_POSITIVE_REGULATION_OF_ENDOTHELIAL_CELL_PRO-LIFERATION” (|NES|= 2.14, FDR q-value = 0.001), “GOBP_VASCULAR_ENDOTHELIAL-_GROWTH_FACTOR_ RECEPTOR_SIGNALING_PATHWAY” (|NES|= 2.11, FDR q-value = 0.001) and “GOBP_ENDO-THELIAL_CELL_PROLIFERATION” (|NES|= 2.05, FDR q-value = 0.002). A close relationship and high overlapping rate between the above three angiogenesis-related pathways enriched in the low CD103 expression group were found (Fig. 4C). Specifically, through leading-edge analysis, a total of 23 genes were found to be significant in at least two pathways (23/40, 57.50%), and VEGFC, NRP1 as well as ITGB3 overlapped in all three pathways. Furthermore, we conducted a correlation analysis and found that ITGAE was negatively correlated with VEGFC (r = − 0.47, P = 0.036), NRP1 (r = − 0.37, P = 0.107) and ITGB3 (r = − 0.34, P = 0.140) (Fig. 4D). For gene sets positively correlated with CD103, detailed enrichment profiles and overlapped genes among the top three pathways were also depicted (Additional file 1: Figure S5).

Tumor angiogenesis may impede the infiltration of TRM cells. A Gene set map showing the top 20 biological pathways enriched in the low ITGAE (coding CD103) expression group in gene set enrichment analysis (GSEA), mostly related to angiogenesis or immunity. B Detailed enrichment profiles for the three main angiogenesis-correlated pathways in GSEA. C Gene overlapping rate among the main three angiogenesis-correlated pathways in GSEA. D Correlation among the expression of CD103, overlapped genes (NRP1, VEGFC, ITGB3), and hypoxia indicator (HIF1A). E The merged and single-stained images for the panel of multiplex immunofluorescence. F Correlation among TRM cells, cancer microvessels (CMVs) (indicated by CD31), and hypoxia (indicated by Hif-1α). The correlation was calculated using the Spearman rank correlation coefficient. *P < 0.05; **P < 0.01; ***P < 0.001
Given the results of GSEA, it is rational to assume that tumor angiogenesis impeded TRM. Based on this assumption, we performed another panel of mIF staining in the SCHI-NSCLC cohort to investigate the effect of cancer microvessels (CMVs) on the density and spatial architecture of TRM cells (Fig. 4E). CD31 was used as an indicator of CMVs’ endothelium, while Hif-1α as an indicator of cellular hypoxia. In the correlation analyses, we found that the CMVs were negatively correlated with the density of TRM cells (r = − 0.32, P < 0.001 in TC; r = − 0.44, P < 0.001 in IM), the InS of TRM cells (r = − 0.28, P < 0.001 in TC; r = − 0.38, P < 0.001 in IM) and the CCPS of TRM cells (r = − 0.13, P = 0.039 in TC; r = − 0.37, P < 0.001 in IM) while positively with the mNND from TRM cells to cancer cells (r = 0.25, P < 0.001 in TC) (Fig. 4F). In contrast, no significant correlation was observed between CMVs and Tnon-RM cells (Additional file 1: Figure S6). Interestingly, the density and spatial architecture of TRM were not associated with Hif-1α (Fig. 4F). Moreover, compared with the low TRM-SIS group, the high TRM-SIS group had a significantly higher density of CMVs in IM (P = 0.006) (Additional file 1: Figure S7A, B). Survival analysis also showed that high density of CMVs (P = 0.054 in TC; P = 0.007 in IM) and high level of hypoxia (P = 0.018 in TC; P = 0.006 in IM) were associated with poorer prognosis in NSCLC patients (Additional file 1: Figure S7C–F).
Discussion
In this study, using mIF, we deciphered the spatial architecture of CD103+CD8+ TRM functional subsets in the TME of 274 patients with NSCLC. Our results demonstrated that the TRM was a population of cells with significant heterogeneity in their functional state and spatial distribution.
To the best of our knowledge, this is the first study to reveal the spatial distribution of TRM subsets. Utilizing mIF to detect multiple protein markers simultaneously, we can characterize TRM subsets more precisely than the traditional immunohistochemistry method. More importantly, it can provide unreported spatial information that cannot be revealed through dissociative techniques such as single-cell RNA sequencing or flow cytometry. Our results confirmed the previous findings histologically that CD103 expression was linked to the magnitude of CD8+T cell infiltration and cytotoxic T cell responses in human lung cancer [7, 8]. A few studies have demonstrated that CD103+TRM cells are more abundant in the epithelial compartment of prostate cancer and CD39+TRM cells are closer to cancer cells in melanoma, which provides partial support for our results [34, 35].
As previously mentioned, the role of TRM in tumorigenesis and prognosis remains controversial. However, few studies have focused on the internal heterogeneity of TRM cells. We divided TRM cells into four subsets based on different expression patterns of immune checkpoints (PD-1 and Tim-3) and found a significant heterogeneity among them. Specifically, TRM2 had the strongest cytotoxicity but the lowest cell density, while TRM4 had the weakest cytotoxicity. TRM3 and TRM4 had a higher InS, shorter mNND to cancer cells and higher CCPS than TRM1 and TRM2. More importantly, TRM4 was associated with an unfavorable prognosis of NSCLC patients. These results suggested that TRM4 may be in a terminally dysfunctional state, TRM2 in a pre-dysfunctional state, and TRM3 in an intermediate state. The great heterogeneity among TRM functional subsets is one of the reasons for the inconsistent prognostic relevance of TRM cells and the tumor progression after TRM adoptive transfer [7,8,9,10,11,12]. Based on current understanding of the PD-(L)1 axis as an immune brake, the superior functionality of TRM2 was unexpected [36]. Likewise, the transcriptome profiling of human lung cancer also showed that PD-1+TRM possessed superior antitumor activity, suggesting potentially unknown mechanisms contributing to activation of T cells [13]. We have to underline that although our results presented a gradient from TRM1 to TRM4, it does not mean that the four TRM subsets are in a progressive evolutionary relationship. Through temporal single-cell tracing, Liu et al. thought that pre-dysfunctional T cells were unlikely to be derived from terminally dysfunctional cells [37]. In addition, Tim3+PD1+CD8+T cells did not respond well to anti-PD-(L)1 immunotherapy [38, 39]. Unfortunately, as a monotherapy, the Tim-3 blockade did not show substantial clinical benefit. However, in mouse models of melanoma, colorectal cancer and acute myeloid leukemia, combined blocking of Tim-3 and PD-(L)1 pathways restored T-cell response better than blocking PD-(L)1 pathway alone [40]. Several phase I clinical trials combining Tim-3 and PD-(L)1 monoclonal antibody have shown modest antitumor activity in lung cancer (NCT03099109; NCT02608268) [41, 42], and a bispecific antibody targeting both Tim-3 and PD-1 (R07121661) has also entered phase I clinical trials (NCT03708328). Therefore, improving the postoperative outcomes of NSCLC patients with high infiltration of TRM4 needs further studies.
In the setting of the most stringent selection criteria to control the overfitting and the overcomplexity of our predictive model, which was to take the intersection of LASSO, XGBoost and RF results, the TRM-related features we selected could be more representative [43]. Six features were screened out in all three methods and used to construct a TRM-SIS (Fig. 5). Importantly, TRM-SIS and tumor stage were independent prognostic factors for DFS. Thus, the model combining TRM-SIS and tumor stage showed a better performance in patient stratification in our cohort. A clinical implication behind our model is that perioperative treatment or increased monitoring may be worth considering in patients with high TRM-SIS due to their high risk of recurrence. Although patients receiving perioperative treatment were not included in our model, the inclusion of these patients in predictive models could be of importance in further studies.

Proposed model of TRM-based spatial immune signature predicting DFS of patients with non-small cell lung cancer. Higher density of CD103+ cells in TC (A), higher density of TRM3 cells in IM (B), higher infiltration score of Tnon-RM cells in TC (C), higher infiltration score of TRM1 cells in IM (D), lower infiltration score of TRM4 cells in IM (E) and higher mean nearest neighboring distance from TRM cells to cancer cells in TC (F) are associated with longer disease-free survival (DFS)
Considering that the intratumoral TRM cells are removed with surgery, the reason why TRM-SIS can affect the long-term prognosis of postoperative patients remains unclear. The potential explanation may be that it acts as a biomarker for the tumor with a “hot” immune environment or identifying patient with a long-term systemic antitumor immunity. Once a tumor is removed after radical surgery, immune surveillance may rebuild and take advantage. Intriguingly, previous studies have found that lung TRM could migrate to the draining lymph nodes and enhance immune responses of the nodes in virus-infected mouse models, suggesting a potential TRM storage pool in draining lymph nodes and a plasticity for TRM cells retention [32, 33]. More efforts are needed to investigate the retention and transportation of intratumoral TRM, which will help in the development of new antitumor targets.
Cancer vasculature is a vital pipeline to transport immune cells into malignant tissue [44]. Through GSEA, we found tumor angiogenesis may play a pivotal role in the regulation of TRM. Furthermore, utilizing mIF to quantify CMVs and TRM, we verified the results of GSEA at the level of cellular pathology, which strengthened the evidence that CMVs may act as an immune barrier [45]. Notably, we found that CMVs were significantly negatively correlated with the infiltration of TRM cells but not with that of Tnon-RM cells, suggesting that the inhibition of CD8+T by CMVs may be selective. Moreover, although our previous study demonstrated a significant positive correlation between Hif-1α and CMVs [17], no significant correlation between Hif-1α and reduced infiltration of TRM cells was observed in this study, suggesting that there might be an angiogenesis-independent pathway for tumor hypoxia on TRM cells. Vignali et al. found that tumor hypoxia drove CD39-dependent T-cell dysfunction to limit antitumor immunity [46]. Given the coexpression of CD39 and CD103 in some TRM [47], it is reasonable to speculate that Hif-1α can promote a part of CD103+TRM via an angiogenesis-independent pathway. Of note, the negative association between CMVs and TRM appears to be statistically more significant in IM than in TC, suggesting angiogenesis could be one of the mechanisms for local progression in NSCLC.
CMVs are generally thought abnormal. CMVs have been found to hinder CD8+T cell trafficking through the upregulation of apoptotic ligands, like Fas-L, and the downregulation of adhesion molecules, such as E-selectin and VCAM-1 [44, 48]. Importantly, antiangiogenic agents could induce the normalization of CMVs and therefore were considered one of the wonderful partners for immunotherapy. For advanced NSCLC, the ABCP regimen (atezolizumab-bevacizumab-carboplatin-paclitaxel) and its analogous regimen (immunotherapy-antiangiogenic agents-chemotherapy) have conferred benefits in overall survival [49]. Two recent phase 2 clinical trials reported promising benefits and acceptable toxicity of neoadjuvant anti-PD-1 antibody combined with antiangiogenic agent in patients with resectable NSCLC [50, 51]. Considering the high level of CMVs in the high TRM-SIS group, antiangiogenic therapy in TRM-restricted patients may be a promising option to improve outcomes. Thus, it is crucial to clarify the impact of tumor vascular normalization strategies on TRM for promoting immunotherapy.
Some limitations of this study have to be acknowledged. Firstly, this is a single-center, retrospective study, although we validated some of our results in a public cohort. Secondly, although the expression of CD103 has been observed in the majority of TRM cells, CD103−TRM also exist [52]. Hence, identification through our phenotypic markers might underestimate the size of the TRM population. Additionally, the lack of tumor adjacent lung tissue or lung tissue from non-cancer patients as control makes it hard to determine whether the features of TRM cells in this study are tumor-specific [53, 54]. Lastly, after clarifying the significance of the spatial architecture of TRM for the prognosis of NSCLC, its relevance for therapeutic efficacy was not elaborated due to the lack of a suitable cohort, which is exactly the orientation requiring more effort.
In summary, this study highlights the significant heterogeneity of TRM cells in their functional state and spatial distribution, which could be a prognostic biomarker for patients with NSCLC. Tumor angiogenesis may inhibit the infiltration of TRM cells, suggesting that targeting tumor angiogenesis to promote TRM cells may be a potential strategy to improve current immunotherapy.
Availability of data and materials
All data and materials are published in the manuscript, supplementary materials on the journal website or available from corresponding author upon reasonable request.
References
Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73(1):17–48.
Lou F, Sima CS, Rusch VW, Jones DR, Huang J. Differences in patterns of recurrence in early-stage versus locally advanced non-small cell lung cancer. Ann Thorac Surg. 2014;98(5):1755–60.
Shin DW, Cho JH, Yoo JE, Cho J, Yoon DW, Lee G, et al. Conditional survival of surgically treated patients with lung cancer: a comprehensive analyses of overall, recurrence-free, and relative survival. Cancer Res Treat. 2021;53(4):1057–71.
Mami-Chouaib F, Blanc C, Corgnac S, Hans S, Malenica I, Granier C, et al. Resident memory T cells, critical components in tumor immunology. J Immunother Cancer. 2018;6(1):87.
Okła K, Farber DL, Zou W. Tissue-resident memory T cells in tumor immunity and immunotherapy. J Exp Med. 2021;218(4): e20201605.
Corgnac S, Boutet M, Kfoury M, Naltet C, Mami-Chouaib F. The emerging role of CD8(+) tissue resident memory T (T-RM) cells in antitumor immunity: a unique functional contribution of the CD103 integrin. Front Immunol. 2018;9:1904.
Corgnac S, Malenica I, Mezquita L, Auclin E, Voilin E, Kacher J, et al. CD103(+)CD8(+) T(RM) cells accumulate in tumors of Anti-PD-1-responder lung cancer patients and are tumor-reactive lymphocytes enriched with Tc17. Cell Rep Med. 2020;1(7): 100127.
Ganesan AP, Clarke J, Wood O, Garrido-Martin EM, Chee SJ, Mellows T, et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat Immunol. 2017;18(8):940–50.
Djenidi F, Adam J, Goubar A, Durgeau A, Meurice G, de Montpréville V, et al. CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J Immunol. 2015;194(7):3475–86.
Anz D, Mueller W, Golic M, Kunz WG, Rapp M, Koelzer VH, et al. CD103 is a hallmark of tumor-infiltrating regulatory T cells. Int J Cancer. 2011;129(10):2417–26.
Reading JL, Gálvez-Cancino F, Swanton C, Lladser A, Peggs KS, Quezada SA. The function and dysfunction of memory CD8(+) T cells in tumor immunity. Immunol Rev. 2018;283(1):194–212.
Gabriely G, da Cunha AP, Rezende RM, Kenyon B, Madi A, Vandeventer T, et al. Targeting latency-associated peptide promotes antitumor immunity. Sci Immunol. 2017;2(11):eaaj1738.
Clarke J, Panwar B, Madrigal A, Singh D, Gujar R, Wood O, et al. Single-cell transcriptomic analysis of tissue-resident memory T cells in human lung cancer. J Exp Med. 2019;216(9):2128–49.
van der Leun AM, Thommen DS, Schumacher TN. CD8(+) T cell states in human cancer: insights from single-cell analysis. Nat Rev Cancer. 2020;20(4):218–32.
Fu T, Dai LJ, Wu SY, Xiao Y, Ma D, Jiang YZ, et al. Spatial architecture of the immune microenvironment orchestrates tumor immunity and therapeutic response. J Hematol Oncol. 2021;14(1):98.
Steele KE, Tan TH, Korn R, Dacosta K, Brown C, Kuziora M, et al. Measuring multiple parameters of CD8+ tumor-infiltrating lymphocytes in human cancers by image analysis. J Immunother Cancer. 2018;6(1):20.
Yang G, Cai S, Hu M, Li C, Yang L, Zhang W, et al. Functional status and spatial architecture of tumor-infiltrating CD8+ T cells are associated with lymph node metastases in non-small cell lung cancer. J Transl Med. 2023;21(1):320.
Al-Shibli KI, Donnem T, Al-Saad S, Persson M, Bremnes RM, Busund LT. Prognostic effect of epithelial and stromal lymphocyte infiltration in non-small cell lung cancer. Clin Cancer Res. 2008;14(16):5220–7.
Donnem T, Hald SM, Paulsen EE, Richardsen E, Al-Saad S, Kilvaer TK, et al. Stromal CD8+ T-cell density—a promising supplement to tnm staging in non-small cell lung cancer. Clin Cancer Res. 2015;21(11):2635–43.
Peng H, Wu X, Zhong R, Yu T, Cai X, Liu J, et al. Profiling tumor immune microenvironment of non-small cell lung cancer using multiplex immunofluorescence. Front Immunol. 2021;12: 750046.
Väyrynen JP, Haruki K, Väyrynen SA, Lau MC, Dias Costa A, Borowsky J, et al. Prognostic significance of myeloid immune cells and their spatial distribution in the colorectal cancer microenvironment. J Immunother Cancer. 2021;9(4): e002297.
Gartrell RD, Enzler T, Kim PS, Fullerton BT, Fazlollahi L, Chen AX, et al. Neoadjuvant chemoradiation alters the immune microenvironment in pancreatic ductal adenocarcinoma. Oncoimmunology. 2022;11(1):2066767.
Keren L, Bosse M, Marquez D, Angoshtari R, Jain S, Varma S, et al. A structured tumor-immune microenvironment in triple negative breast cancer revealed by multiplexed ion beam imaging. Cell. 2018;174(6):1373–87.
Jia K, Chen Y, Sun Y, Hu Y, Jiao L, Ma J, et al. Multiplex immunohistochemistry defines the tumor immune microenvironment and immunotherapeutic outcome in CLDN18.2-positive gastric cancer. BMC Med. 2022;20(1):223.
Peng H, Wu X, Liu S, He M, Xie C, Zhong R, et al. Multiplex immunofluorescence and single-cell transcriptomic profiling reveal the spatial cell interaction networks in the non-small cell lung cancer microenvironment. Clin Transl Med. 2023;13(1): e1155.
Yang L, Zhang W, Sun J, Yang G, Cai S, Sun F, et al. The role of spatial interplay patterns between PD-L1-positive tumor cell and T cell in recurrence of locally advanced non-small cell lung cancer. Cancer Iimmunol Immun. 2023;72(7):2015–27.
Tan WCC, Nerurkar SN, Cai HY, Ng HHM, Wu D, Wee YTF, et al. Overview of multiplex immunohistochemistry/immunofluorescence techniques in the era of cancer immunotherapy. Cancer Commun. 2020;40(4):135–53.
Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018;173(2):400-16.e11.
Lopez de Rodas M, Schalper KA. Tumour antigen-induced T cell exhaustion - the archenemy of immune-hot malignancies. Nat Rev Clin Oncol. 2021;18(12):749–50.
Hombrink P, Helbig C, Backer RA, Piet B, Oja AE, Stark R, et al. Programs for the persistence, vigilance and control of human CD8(+) lung-resident memory T cells. Nat Immunol. 2016;17(12):1467–78.
Camp RL, Dolled-Filhart M, Rimm DL. X-tile: a new bio-informatics tool for biomarker assessment and outcome-based cut-point optimization. Clin Cancer Res. 2004;10(21):7252–9.
Beura LK, Wijeyesinghe S, Thompson EA, Macchietto MG, Rosato PC, Pierson MJ, et al. T cells in nonlymphoid tissues give rise to lymph-node-resident memory T cells. Immunity. 2018;48(2):327–38.
Stolley JM, Johnston TS, Soerens AG, Beura LK, Rosato PC, Joag V, et al. Retrograde migration supplies resident memory T cells to lung-draining LN after influenza infection. J Exp Med. 2020;217(8): e20192197.
Zhou Q, Ou Y, Dai X, Chen X, Wu S, Chen W, et al. Prevalence of tumour-infiltrating CD103(+) cells identifies therapeutic-sensitive prostate cancer with poor clinical outcome. Brit J Cancer. 2023;128(8):1466–77.
Attrill GH, Owen CN, Ahmed T, Vergara IA, Colebatch AJ, Conway JW, et al. Higher proportions of CD39+ tumor-resident cytotoxic T cells predict recurrence-free survival in patients with stage III melanoma treated with adjuvant immunotherapy. J Immunother Cancer. 2022;10(6): e004771.
Chamoto K, Yaguchi T, Tajima M, Honjo T. Insights from a 30-year journey: function, regulation and therapeutic modulation of PD1. Nat Rev Immunol. 2023;23(10):682–95. https://doi.org/10.1038/s41577-023-00867-9.
Liu B, Hu X, Feng K, Gao R, Xue Z, Zhang S, et al. Temporal single-cell tracing reveals clonal revival and expansion of precursor exhausted T cells during anti-PD-1 therapy in lung cancer. Nat Cancer. 2022;3(1):108–21.
Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501.
Klapholz M, Drage MG, Srivastava A, Anderson AC. Presence of Tim3(+) and PD-1(+) CD8(+) T cells identifies microsatellite stable colorectal carcinomas with immune exhaustion and distinct clinicopathological features. J Pathol. 2022;257(2):186–97.
Wolf Y, Anderson AC, Kuchroo VK. TIM3 comes of age as an inhibitory receptor. Nat Rev Immunol. 2020;20(3):173–85.
Harding JJ, Moreno V, Bang YJ, Hong MH, Patnaik A, Trigo J, et al. Blocking TIM-3 in treatment-refractory advanced solid tumors: a phase Ia/b Study of LY3321367 with or without an Anti-PD-L1 antibody. Clin Cancer Res. 2021;27(8):2168–78.
Curigliano G, Gelderblom H, Mach N, Doi T, Tai D, Forde PM, et al. Phase I/Ib clinical trial of sabatolimab, an Anti-TIM-3 antibody, alone and in combination with spartalizumab, an Anti-PD-1 antibody, in advanced solid tumors. Clin Cancer Res. 2021;27(13):3620–9.
Cuocolo R, Caruso M, Perillo T, Ugga L, Petretta M. Machine learning in oncology: a clinical appraisal. Cancer Lett. 2020;481:55–62.
Mazzone M, Bergers G. Regulation of blood and lymphatic vessels by immune cells in tumors and metastasis. Annu Rev Physiol. 2019;81:535–60.
Zhang T, Jia Y, Yu Y, Zhang B, Xu F, Guo H. Targeting the tumor biophysical microenvironment to reduce resistance to immunotherapy. Adv Drug Deliver Rev. 2022;186: 114319.
Vignali PDA, DePeaux K, Watson MJ, Ye C, Ford BR, Lontos K, et al. Hypoxia drives CD39-dependent suppressor function in exhausted T cells to limit antitumor immunity. Nat Immunol. 2023;24(2):267–79.
Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med. 2019;25(8):1251–9.
van der Woude LL, Gorris MAJ, Halilovic A, Figdor CG, de Vries IJM. Migrating into the tumor: a roadmap for T cells. Trends Cancer. 2017;3(11):797–808.
Socinski MA, Nishio M, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, et al. IMpower150 final overall survival analyses for atezolizumab plus bevacizumab and chemotherapy in first-line metastatic nonsquamous NSCLC. J Thorac Oncol. 2021;16(11):1909–24.
Zhao J, Zhao L, Guo W, Wang S, Tao X, Li L, et al. Efficacy, safety, and biomarker analysis of neoadjuvant camrelizumab and apatinib in patients with resectable NSCLC: a phase 2 clinical trial. J Thorac Oncol. 2023;18(6):780–91.
Okage K, Shimada Y, Yoh K, Wakabayashi M, Fukutani M, Furuya H, et al. Pembrolizumab and ramucirumab neoadjuvant therapy for PD-L1-positive stage IB-IIIA lung cancer (EAST ENERGY). J Clin Oncol. 2023;41(16_suppl):8509.
Sun H, Sun C, Xiao W, Sun R. Tissue-resident lymphocytes: from adaptive to innate immunity. Cell Mol Immunol. 2019;16(3):205–15.
Gao P, Wang H, Li H, Shu L, Han Z, Li S, et al. miR-21-5p inhibits the proliferation, migration, and invasion of glioma by targeting S100A10. J Cancer. 2023;14(10):1781–93.
Dai X, Ye L, Li H, Dong X, Tian H, Gao P, et al. Crosstalk between microglia and neural stem cells influences the relapse of glioblastoma in GBM immunological microenvironment. Clin Immunol. 2023;251: 109333.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the National Natural Science Foundation of China [82373424; 82172866], the Natural Science Foundation of Shandong Province [ZR2021LZL005; ZR2019LZL019], the Department of Science & Technology of Shandong Province [2021CXGC011102], and the Start-up fund of Shandong Cancer Hospital [2020PYA04].
Author information
Authors and Affiliations
Contributions
GY: conceptualization, methodology, data curation, software, formal analysis, visualization, writing- original draft preparation. SC: methodology, data curation, validation. MH: investigation. CL: investigation. ly: methodology, investigation. WZ: methodology, validation. JS: resources, methodology, supervision. FS: Investigation. LX: resources, funding acquisition, writing- reviewing and editing. XS: Project administration, supervision, funding acquisition, writing- reviewing and editing.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Review Committee of Shandong Cancer Hospital (SDTHEC2022007013) and complied with the provisions of the Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
All authors declare that they have no known competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1
. Information of regents used in multiplex immunofluorescence test. Table S2. Scheme of cell phenotypes in multiplex immunofluorescence detection. Table S3. Clinicopathological characteristics of 274 NSCLC patients enrolled in this study. Table S4. The ratio of the 90% signal level to the 10% signal level. Table S5. Clinicopathological characteristics of 274 NSCLC patients in the training set and testing set. Table S6. Discrepancy of the clinicopathological characteristics between patients in high-risk group and low-risk group. Table S7. Clinicopathological characteristics of 244 NSCLC patients in the TCGA validation cohort. Figure S1. Evaluating heterogeneity of signal intensity of CD8 (A), CD103 (B), PD-1 (C), Tim-3 (D), GZMB (E) and CK (F), DAPI (G) and autofluorescence (H) across multiplex immunofluorescence (mIF) images. Figure S2. Evaluating crosstalk between spectral channels in eight randomly selected mIF images. Figure S3. Predicted probability of the recurrence in resectable NSCLC by integrating TRM-SIS and tumor stage. Figure S4. Validation of the clinical value of CD103 in the TCGA cohort of NSCLC. A–C Close correlation of ITGAE (coding CD103) and PDCD1 (coding PD-1), HAVCR2 (coding Tim-3) and GZMB (coding GZMB). Figure S5. Gene set enrichment analysis (GSEA) positively correlated with CD103. Figure S6. Correlation among Tnon-RM cells, cancer microvessels (CMVs) (indicated by CD31), and hypoxia (indicated by Hif-1α). Figure S7. Clinical relevance of cancer microvessels and hypoxia.
Additional file 2:
Source data for the spatial immune signature construction. Suppl_data1. Source data for the spatial immune signature construction. Suppl_data2. Source data for the TCGA validation cohort. Suppl_data3. GEO accession codes of samples in the GSEA.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, G., Cai, S., Hu, M. et al. Spatial features of specific CD103+CD8+ tissue-resident memory T cell subsets define the prognosis in patients with non-small cell lung cancer. J Transl Med 22, 27 (2024). https://doi.org/10.1186/s12967-023-04839-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-023-04839-4