
Abstract
Background
Patients with heart failure with reduced ejection fraction (HFrEF) and central sleep apnea (CSA) are at a very high risk of fatal outcomes.
Objective
To test whether the circulating miRNome provides additional information for risk stratification on top of clinical predictors in patients with HFrEF and CSA.
Methods
The study included patients with HFrEF and CSA from the SERVE-HF trial. A three-step protocol was applied: microRNA (miRNA) screening (n = 20), technical validation (n = 60), and biological validation (n = 587). The primary outcome was either death from any cause, lifesaving cardiovascular intervention, or unplanned hospitalization for worsening of heart failure, whatever occurred first. MiRNA quantification was performed in plasma samples using miRNA sequencing and RT-qPCR.
Results
Circulating miR-133a-3p levels were inversely associated with the primary study outcome. Nonetheless, miR-133a-3p did not improve a previously established clinical prognostic model in terms of discrimination or reclassification. A customized regression tree model constructed using the Classification and Regression Tree (CART) algorithm identified eight patient subphenotypes with specific risk patterns based on clinical and molecular characteristics. MiR-133a-3p entered the regression tree defining the group at the lowest risk; patients with log(NT-proBNP) ≤ 6 pg/mL (miR-133a-3p levels above 1.5 arbitrary units). The overall predictive capacity of suffering the event was highly stable over the follow-up (from 0.735 to 0.767).
Conclusions
The combination of clinical information, circulating miRNAs, and decision tree learning allows the identification of specific risk subphenotypes in patients with HFrEF and CSA.
Similar content being viewed by others
Introduction
Patients with symptomatic chronic heart failure (CHF) show a high prevalence of sleep-disordered breathing (SDB) [1]. In particular, central sleep apnea (CSA) can be found in up to 40% of patients with HF with reduced ejection fraction (HFrEF) [2]. Patients with HFrEF and CSA represent a population at very high risk of adverse outcomes [3, 4]. Additional efforts focused on risk stratification are imperative in order to facilitate care and optimal treatment allocation. In this context, the retrospective analysis of biosamples from the SERVE-HF (Treatment of Sleep-Disordered Breathing with Predominant Central Sleep Apnea by Adaptive Servo Ventilation in Patients with Heart Failure) trial employing state-of-the-art molecular and computational tools constitute an outstanding platform to develop novel biological markers [5,6,7].
In the last decade, microRNAs (miRNAs), single-stranded small RNA molecules of 19–25 nucleotides in length that regulate gene expression at a posttranscriptional level, have gained great attention as biomarkers with potential clinical application [8]. The noninvasive quantification of miRNA profiles in different bodily fluids has been reported to be highly sensitive, robust and cost-effective for the clinical management of different pathological conditions, including HF [9]. In this context, the combination of miRNA profiling and clinical data using machine learning (ML) algorithms has recently become an innovative approach to define patient subphenotypes [10]; and consequently, a novel strategy towards precision medicine [11].
The number of studies in the field of miRNAs and SDB is limited [12]. Furthermore, no previous studies have attempted to use miRNAs for risk stratification in the field of CSA. Here, we hypothesized that the circulating miRNome may aid in risk stratification of the patients with HFrEF and CSA. Therefore, the aim of the current study is to investigate the potential of plasma miRNAs as biomarkers of adverse outcomes in this patient population, specifically, whether these transcripts can improve the accuracy of a clinical prognostic model previously described in the same study population [13].
Methods
Study design
The SERVE-HF trial investigated the effects of adding adaptive servo ventilation (ASV) (AutoSet CS, ResMed) vs. guideline-based medical treatment alone on survival and cardiovascular outcomes in patients with HFrEF and predominantly CSA. The SERVE-HF trial was an international, multicenter, randomized, parallel-group and event-driven study (clinical trial identifier: NCT00733343). Information about the trial design, procedures, outcomes, and results have been previously reported [5, 6]. The trial was conducted in accordance with the Good Clinical Practice guidelines and principles of the 2002 Declaration of Helsinki. Institutional Review Board approval was obtained, and all patients signed an informed consent form to participate in the study.
Study patients
Enrolled patients had HF with a left ventricular ejection fraction (LVEF) ≤ 45%, New York Heart Association (NYHA) class ≥ II, and predominant CSA (apnea–hypopnea index [AHI] ≥ 15 events per hour, with > 50% central events and a central AHI of ≥ 10 events per hour). Patients were advised to use the ASV device for at least 5 h per night, 7 days per week.
This sub-study includes 587 patients available for miRNA analysis (289 in the control group and 298 in the ASV group). Plasma samples were collected at the time of randomization and stored at − 80 C for later analysis.
Outcome
The primary outcome in the time-to-event analysis was the first event of the composite of death from any cause, a life-saving cardiovascular intervention, or an unplanned hospitalization for worsening chronic heart failure, with the latter two end-point events being assessed by the end-point review committee. Life-saving cardiovascular interventions included heart transplantation, implanting a long-term ventricular assist device, resuscitation after sudden cardiac arrest, or appropriate shock for ventricular arrhythmia in patients with an implantable cardioverter-defibrillator. Previous results from the SERVE-HF trial showed that the incidence of the primary outcome did not differ significantly between the control and ASV groups [6].
microRNA quantification
MiRNA quantification was performed blinded to clinical variables. The results were merged with the baseline clinical data. In the screening phase, miRNA expression profiles were assessed using the HTG EdgeSeq miRNA Whole Transcriptome Assay (miRNA WTA) (HTG Molecular, Tucson, AZ, USA). This technique is a multiplexed, digital expression assay that combines quantitative nuclease protection (qNPA) with next-generation sequencing (NGS) to quantify miRNA expression without RNA extraction. The HTG assay includes probes for 2083 distinct human miRNAs from miRBase v20 [14] as well as probes for 13 housekeeping mRNAs, 5 negative process controls, and one positive process control. Sequencing was performed on the Illumina MiSeq platform.
For validation, total RNA was isolated from 150 µL of plasma using the miRNeasy Serum/Plasma Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Synthetic Caenorhabditis elegans cel-miR-39-3p (1.6 × 108 copies/µL) (Qiagen) was added to each sample as a quality control for the RNA isolation procedure. RNA was stored at – 80 ºC. Reverse transcription was performed using the Reverse Transcription TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems®, Darmstadt, Germany). The reverse transcription reaction was then diluted with water (1:3 ratio). The expression of miRNAs which met the selection criteria were analyzed by quantitative RT-PCR (RT‐qPCR) with specific TaqMan miRNA assays (Applied Biosystems®). Amplification was performed using the ViiA 7 Real-Time PCR System (Applied Biosystems®). Relative miRNA expression was calculated by first exporting the raw amplification data to the LinRegPCR (11.0) software which calculates the initial concentration (N0) of a given miRNA per sample [15, 16], expressed in arbitrary fluorescence units. Relative miRNA levels (RQ) were then calculated as: RQ = N0[miRNA]/N0[miR-486-5p], using miR-486-5p as an internal standard [7]. Relative expression levels were log-transformed for statistical analysis.
Statistical analysis
All analyses were performed with R software version 4.0.3 (the R foundation for Statistical Computing). The two-tailed significance level was set at p < 0.05. Continuous variables were expressed as mean ± standard deviation (SD) and median (interquartile range), and categorical variables were expressed as frequencies (percentages). Comparisons of baseline characteristics and miRNA levels between study groups were carried out using non-parametric Wilcoxon test for continuous variables and Fisher’s exact test for categorical variables. Correlations between continuous variables (baseline characteristics and miRNA variables) were assessed with Spearman rank correlation coefficients.
Unadjusted and adjusted associations of miRNA levels with primary outcome were assessed using Cox regression models. Associated crude and adjusted hazard ratios (HR) with their 95% confidence intervals were presented as HR (95% CI). Three models were constructed using the clinical risk factors identified by Ferreira et al. [13]. Model 1 included ASV, age, and sex. Model 2 included variables from model 1 plus systolic blood pressure (SBP) < 120 mmHg, diabetes, diuretics, cardiac device, and 6-min walking distance, in addition to atrial fibrillation. Model 3 included variables from model 2 and log(NT-proBNP). The goodness of fit of the models was assessed by calculating Harrell’s c-index. The prognostic capacity of each miRNA to predict the event on top of clinical model during follow-up was assessed by calculating the increase in c-index, the continuous Net Reclassification Improvement (cNRI) and the Integrated Discrimination Improvement (IDI). cNRI and IDI were calculated using the survIDINRI package of the R software [17].
The customized regression tree model was constructed using the Classification and Regression Trees (CART) algorithm [18] and implementing HR as splitting criteria (the flowchart for the customized-CART algorithm is shown in Additional file 1: Figure S1). A bagging procedure (based on 1000 iterations) was used for variable selection and error measurement [19]. The goal of this model was to split the population into risk groups aiming to predict the average risk of having the event, rather predicting whether or not the participants will suffer the studied event over a period of time. In this context, standard confusion matrix and similar error measures do not apply. We use incidence rates (IR) of event per 100 patients/year to summarize the absolute risk. HR were used for representing relative risks within each final node. Kaplan–Meier curves were used to illustrate differences between groups in the observed time-to-event. As measures of classification accuracy, we considered: (1) the incremental area under the cumulative/dynamic receiver-operating characteristic curve (iAUC) between 3 months and 5 years of follow-up [20] of the ordinal risk represented by a hierarchization of end nodes; and (2) the incidence rate variation index (IRV) defined by \({IRV=(1/N){\sum }_{i=1}^{f}n}_{i}|{IR}_{i}-IR|\), where IR is the incidence rate of the population, and IRi and ni, the incidence rate and the sample size on the i-th end node, respectively (\(1\le i\le f)\), where \(f\) stands for the number of terminal nodes. The obtained results were employed in an informed stepwise Cox regression model. R statistical environment (www.r-project.org) was also used for these statistical analyses including the survival [21], rpart [22], and nsROC [23] packages.
Results
Patient characteristics
Patient characteristics are shown in Table 1. The mean age was 69.5 ± 9.8 years and 89.8% were male. The mean ± SD follow-up time was of 3.1 ± 1.8 years, similar to the SERVE-HF biomarker sub-study (Additional file 1: Table S1). Patients with the primary outcome during the follow-up were older, predominately male, and showed a poor clinical and biochemical profile. ASV and control groups were well-balanced (Additional file 1: Table S2), except for central apnea index/total AHI [45% (19–67) vs. 53% (24–76), p-value = 0.018] and for the use of antiarrhythmic drugs, which was higher in the ASV group (21.5% vs. 12.8%, p-value = 0.006). The incidence of the primary outcome did not significantly differ between the ASV group and the control group, with event rates of 56.7% and 49.1%, respectively.
MicroRNA sequencing and technical validation
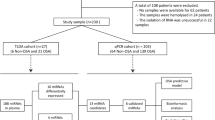
MiRNA sequencing was performed in 10 cases and 10 matched controls (Fig. 1A). Patient characteristics are depicted in Additional file 1: Table S3. Four samples did not pass the quality control check and were discarded from subsequent analysis. MiRNA candidates were selected according to the following criteria: ≥ 1.25-fold differential expression, test significance cutoff of p-value less than 0.1, and previously described stable expression in plasma samples [24] in order to ensure their detectability in further analyses. Five miRNAs met these criteria: miR-106b-3p, miR-133a-3p, miR-335-5p, miR-501-3p and miR-532-5p (Fig. 1B). Levels of all miRNAs were downregulated in cases compared to controls.

microRNA screening and selection of candidates. A Screening phase: Heatmap showing the unsupervised hierarchical clustering in 10 cases and 10 matched controls. The heat map illustrates the levels of plasma microRNAs. Each column represents a patient. Each row represents a microRNA. Red spectra represent increasing expression, while blue spectra represent decreasing expression (see color scale on the right side of the map); B Screening phase: Volcano plot for each microRNA after comparison of cases and controls. The log10 (Fold Change) indicates the mean expression level for each microRNA. Each dot represents one microRNA. In green, microRNA candidates that fulfil the selection criteria. In this phase, microRNA expression profiles were assessed using the HTG EdgeSeq miRNA Whole Transcriptome Assay (miRNA WTA) (HTG Molecular, Tucson, AZ, USA); C Technical validation phase: Dot plot of microRNA expression validated in 30 cases and 30 matched controls. Comparisons of microRNA levels were performed using non-parametric Wilcoxon test. In this phase, microRNA expression profiles were assessed using RT-qPCR. Relative quantification was performed using miR-486-5p for normalization. Relative expression levels were log-transformed for statistical analysis
To corroborate the sequencing results, we evaluated the expression levels of the five miRNAs by RT-qPCR in a sample set of 30 cases and 30 matched controls, including the samples used for miRNA sequencing (Additional file 1: Table S4). In this technical validation, four samples did not pass the quality control check and were discarded for further analysis. We retained miRNAs detected in at least 80% of samples, with a test significance cutoff of p-value less than 0.1, showing the same relationship between cases and controls as in the screening phase. All miRNAs were expressed in 100% of the samples. Three out of five miRNAs (miR-106b-3p, miR-335-5p and miR-532-5p) did not show statistical differences between the study groups (Fig. 1C) and were therefore discarded for further biological validation. In concordance with the sequencing findings, the RT-qPCR analysis showed that plasma levels of miR-133a-3p and miR-501-3p were downregulated in the event group (Fig. 1C).
Biomarker performance
We then quantified miR-133a-3p and miR-501-3p in the whole population using RT-qPCR. No differences in the levels of both miRNAs were observed between treatment groups (control vs. ASV) (Additional file 1: Table S2). During follow-up, a higher proportion of patients with a reported event below the median was observed for miR-133a-3p (59.2% vs. 46.9%, p-value = 0.004) (Table 2). No differences were observed for miR-501-3p (Additional file 1: Table S5). In the multivariate analyses, miR-133a-3p was inversely associated with the risk of experiencing the primary outcome in model 1 and model 2 (Table 3). However, this association was attenuated after adjusting for NT-proBNP levels [HR (95% CI 0.96 (0.92–1.00), p = 0.061)] (model 3). Similar results were observed when both treatment groups were analyzed separately (Table 3). No association was observed between miR-501-3p and the outcome (Table 3). Furthermore, no meaningful correlations were observed between miRNA levels and clinical variables (rho < 0.3) (Additional file 1: Figure S2).
The c-index for both miRNAs was low (0.561 for miR-133a-3p and 0.515 for miR-501-3p) (Additional file 1: Table S6). To further explore the potential role of plasma miRNAs as biomarkers, we evaluated the effect of adding the miRNAs to a clinical prognostic model previously described by our group [13], including the prevalence of atrial fibrillation based on the association with CSA [25]. The addition of miR-133a-3p to models 1 and 2 allowed a significant reclassification of the patients (Table 4). However, the addition of miR-133a-3p did not allow the reclassification of the patients when NT-proBNP was part of the clinical model (model 3) (Table 4).
Based on previous findings of our group [7], we next evaluated whether the plasma levels of miRNAs could define specific groups of patients at specific risk of adverse outcomes. To this end, we constructed customized regression tree models based on the CART algorithm using HRs derived from Cox regression models as splitting criteria. The variables that composed the model previously constructed in the SERVE-HF cohort [13], age, treatment group allocation (ASV or control), male sex, SBP < 120 mmHg, diabetes, diuretic, cardiac device and 6 min walk distance and NT‐proBNP, in addition to atrial fibrillation, were considered in the stratification process (Fig. 2A). The circles represent the entire follow-up, and the different colors represent the probability of the patients in the node to be free-of-events. NT-proBNP was the most relevant predictor. Other clinical variables that entered at the second level were diabetes, 6 min walk distance, and SBP. MiR-133a-3p also entered at the second level of the regression tree, redefining a very low-risk group: those patients with log (NT-proBNP) ≤ 6 pg/mL (cutoff for miR-133a-3p = 1.5 arbitrary units).

Decision tree machine learning approach. A Decision trees calculated using the Classification and Regression Trees (CART) algorithm in the whole study sample. Predictors considered in the analysis were age, treatment group allocation (ASV or control), male sex, SBP < 120 mmHg, diabetes, diuretics, cardiac device and 6 min walk distance, NT‐proBNP, atrial fibrillation, in addition to the microRNA candidate: miR-133a-3p. The results are presented in a binary decision tree that was constructed by splitting a node into two child nodes repeatedly. Generation of novel nodes was based on the selected predictors and cutoffs. Incidence rates (IR) of events per 100 patients/year, number of patients per node and hazard ratios (HR) for the eight final nodes defined by the regression tree model including microRNAs are included. The length of each color in the bands is proportional to the percentage of the total time that patients are submitted to the risk range; B Kaplan–Meier curves illustrated differences among nodes in the observed time-to-event outcome. Patients at risk for each subgroup of patients identified are displayed; C Incremental area under the cumulative/dynamic ROC curve (iAUC) of the ordinal risk of the final nodes. MicroRNA expression profiles were assessed using RT-qPCR. Relative quantification was performed using miR-486-5p for normalization. Relative expression levels were log-transformed for statistical analyses
Survival Kaplan–Meier curves for the final nodes defined by the regression tree model are shown in Fig. 2B. Four subgroups of patients were identified: low risk (node 6 which included miR-133a-3p; reference), intermediate risk (nodes 7, 8, 9 and 10; HR from 2.039 to 4.002), high risk (nodes 11 and 12; HR from 6.055 to 7.079) and very high risk (node 13; HR = 11.957). The iAUC between 3 months and 5 years was 0.758 (0.724 to 0.788) (Fig. 2C). The overall predictive capacity was highly stable over the entire follow-up period (from 0.735 to 0.767). The number of participants with and without an event and the performance metrics at different time points are displayed in Additional file 1: Tables S7, S8.
Discussion
CSA has been described in up to 40% of CHF patients receiving optimal medication [26]. CSA is associated with impaired cardiac function, poor prognosis, and high risk of death in patients with HF [1]; and has therefore been proposed as a marker of HF severity. Novel prognostic approaches are imperative to improve the clinical management of patients with HFrEF and CSA.
Here, we investigated the circulating miRNome in HFrEF and CSA in order to identify novel tools to improve decision-making. Our first analysis suggests that individual miRNAs are poor biomarkers for risk stratification in a heterogeneous cohort of patients with HFrEF and CSA. Patients with low plasma levels of miR-133a-3p were at a higher risk of adverse outcomes. However, the multivariable analysis showed that the association of this miRNA with the outcome was attenuated after adjusting for NT-proBNP levels. In line with this, miR-133a-3p did not improve the prognostic model based on clinical variables when NT-proBNP was included. Due to the weak correlation between miR-133a-3p and NT-proBNP (rho = − 0.167), it seems that miR-133a-3p does not recapitulate the information provided by NT-proBNP, but that NT-proBNP is simply a better biomarker. These results support previous investigations that suggest at best, only modest improvement in prognostication of novel biomarkers in addition to clinical variables and/or natriuretic peptides [27].
We next hypothesized that miRNAs may be useful to define specific subphenotypes of patients with HFrEF and CSA. To explore this hypothesis, we used decision tree learning based on the CART algorithm, a technique that takes into account high-level interactions of predictors and outcomes and defines subgroups of patients (Additional file 1: Figure S1). These results suggest that miR-133a-3p may serve as a complement to the clinical attributes NT-proBNP, diabetes, 6 min walk distance, and SBP. In particular, miR-133a-3p defines a subphenotype with a very low risk for the clinical outcome that would not have been identified without the inclusion of this circulating miRNA. Importantly, the regression tree did not select relevant clinical variables such as age, sex, or atrial fibrillation, which highlights the prognostic value of miR-133a-3p on top of the information already collected in the electronic health record. Plasma levels of miR-133a-3p appear to be informative when medical history alone does not explain the full complexity of the prognosis.
The final nodes of the decision tree model defined eight subgroups of patients with characteristic clinical and molecular patterns and variable levels of risk. Therefore, the tree may provide a framework to efficiently make clinical decisions, such as adjusting therapeutic decisions and follow-up strategies. For instance, we defined a subgroup of patients with a low-risk status (logNT-proBNP levels ≤ than 6 pg/mL and miR-133a-3p levels ≥ than 1.5 arbitrary units, IR100 = 5.3). Conversely, we identified a subphenotype of very high-risk patients with HFrEF and CSA who may benefit most from intensive monitoring and care (logNT-proBNP levels higher > 8 pg/mL and SBP < 120 mmHg, IR100 = 72.9). In summary, our study suggests that risk assessment in patients with HFrEF and CSA could be performed in a more personalized manner. Further investigations on the benefit of individualizing decision-making according to the patient subphenotypes are essential. These findings highlight the potential of advanced statistical approaches for prognostic enrichment.
ML tailored for biomarker discovery has recently emerged as an alternative to traditional statistical approaches, which are often limited to between-group comparisons and/or linear relationships [28]. Despite their proven value as mechanism-based clinical stratification biomarkers, the exploration of miRNAs in the context of ML is still in its infancy. Nevertheless, we have previously described several combinations of clinical variables and plasma miRNAs that allow the identification of specific clinical subphenotypes in heterogeneous diseases such as coronary artery disease [10], cardiometabolic disease [29], and end-stage renal disease [7], demonstrating the validity of decision tree learning. Interestingly, other subclasses of non-coding RNAs, such as circular RNAs (circRNAs), are not useful in the same context [30], which may indicate the higher value of miRNAs to define specific subgroups of patients over other non-coding transcripts when using decision tree models. Our results are also in line with studies showing that the integration of the cell-free miRNA signature with electronic health data, using ML approaches, constitute an innovative approach to define specific clinical patterns and further develop clinical tools [31].
Although the results are promising, some limitations must be acknowledged. First, this is an exploratory study based on a post hoc analysis in a subpopulation of the SERVE-HF trial. Future verification of the findings should be performed in a prospective multicenter study. The specificity of the SERVE-HF cohort limits this step. Nevertheless, the similarities between our study sample set and the entire SERVE-HF population suggest generalizability of the findings to patients with the characteristics of the clinical trial. Second, owing to the epidemiological factors associated with CSA and with HFrEF, relatively few women were recruited into the study. Third, to date, no standardized method has been established for the detection and quantification of circulating miRNAs in plasma/serum samples, making it difficult to compare expression profiles generated by different quantification strategies. Indeed, the reproducibility of the results obtained by different platforms, e.g., microarrays, RNA sequencing, digital droplet PCR or RT-qPCR, is still a challenge, as demonstrated in the technical validation phase of the current investigation in which we validated two out of five candidates. Fourth, the causal association between plasma miRNAs and the outcome is unclear. Mechanistically, miR-133a-3p is a muscle-specific miRNA that has been implicated in cardiac development, cardiac protection, and regeneration [32], which is supported by our observation that patients with higher cardiovascular risk have lower levels of miR-133a-3p. How higher levels of miR-133a-3p mediate protective responses in patients with low NT-proBNP levels warrants further in vivo and in vitro studies.
Conclusions
In conclusion, we constructed a simple decision tree model to analyze the risk of adverse outcomes in patients with HFrEF and CSA. This approach emerges as a powerful strategy to improve the clinical assessment in specific subgroups of patients by integrating molecular information, i.e., circulating miRNAs and clinical predictors.
Data availability
The data that supports the findings of this study are available upon reasonable request from the corresponding author.
Abbreviations
- HFrEF:
-
Heart failure with reduced ejection fraction
- CSA:
-
Central sleep apnea
- miRNA:
-
MicroRNA
- SERVE-HF:
-
Treatment of sleep-disordered breathing with predominant central sleep apnea by adaptive servo ventilation in patients with heart failure
- CART:
-
Classification and regression trees
- CHF:
-
Chronic heart failure
- SDB:
-
Sleep-disordered breathing
- ASV:
-
Adaptive servo ventilation
- LVEF:
-
Left ventricular ejection fraction
- ML:
-
Machine learning
- NYHA:
-
New York Heart Association
- AHI:
-
Apnea–hypopnea index
- qNPA:
-
Quantitative nuclease protection
- SD:
-
Standard deviation
- cNRI:
-
Continuous net reclassification improvement
- SBP:
-
Systolic blood pressure
- IDI:
-
Integrated discrimination improvement
- iAUC:
-
Incremental area under the cumulative/dynamic receiver-operating characteristic curve
- IRV:
-
Incidence rate variation index
References
Cowie MR, Linz D, Redline S, Somers VK, Simonds AK. Sleep disordered breathing and cardiovascular disease: JACC state-of-the-art review. J Am Coll Cardiol. 2021;78:608–24. https://doi.org/10.1016/j.jacc.2021.05.048.
Grimm W, Sosnovskaya A, Timmesfeld N, Hildebrandt O, Koehler U. Prognostic impact of central sleep apnea in patients with heart failure. J Card Fail. 2015;21:126–33. https://doi.org/10.1016/J.CARDFAIL.2014.10.017.
Javaheri S, Shukla R, Zeigler H, Wexler L. Central sleep apnea, right ventricular dysfunction, and low diastolic blood pressure are predictors of mortality in systolic heart failure. J Am Coll Cardiol. 2007;49:2028–34. https://doi.org/10.1016/J.JACC.2007.01.084.
Bitter T, Westerheide N, Prinz C, Hossain MS, Vogt J, Langer C, et al. Cheyne-Stokes respiration and obstructive sleep apnoea are independent risk factors for malignant ventricular arrhythmias requiring appropriate cardioverter-defibrillator therapies in patients with congestive heart failure. Eur Heart J. 2011;32:61–74. https://doi.org/10.1093/EURHEARTJ/EHQ327.
Cowie MR, Woehrle H, Wegscheider K, Angermann C, D’Ortho MP, Erdmann E, et al. Rationale and design of the SERVE-HF study: treatment of sleep-disordered breathing with predominant central sleep apnoea with adaptive servo-ventilation in patients with chronic heart failure. Eur J Heart Fail. 2013;15:937–43. https://doi.org/10.1093/EURJHF/HFT051.
Cowie MR, Woehrle H, Wegscheider K, Angermann C, d’Ortho M-P, Erdmann E, et al. adaptive servo-ventilation for central sleep apnea in systolic heart failure. N Engl J Med. 2015;373:1095–105. https://doi.org/10.1056/NEJMOA1506459.
de Gonzalo-Calvo D, Martínez-Camblor P, Bär C, Duarte K, Girerd N, Fellström B, et al. Improved cardiovascular risk prediction in patients with end-stage renal disease on hemodialysis using machine learning modeling and circulating microribonucleic acids. Theranostics. 2020;10:8665–76. https://doi.org/10.7150/THNO.46123.
Blanco-Domínguez R, Sánchez-Díaz R, de la Fuente H, Jiménez-Borreguero LJ, Matesanz-Marín A, Relaño M, et al. A novel circulating MicroRNA for the detection of acute myocarditis. N Engl J Med. 2021;384:2014–27. https://doi.org/10.1056/NEJMOA2003608.
Wong LL, Zou R, Zhou L, Lim JY, Phua DCY, Liu C, et al. Combining circulating MicroRNA and NT-proBNP to detect and categorize heart failure subtypes. J Am Coll Cardiol. 2019;73:1300–13. https://doi.org/10.1016/J.JACC.2018.11.060.
de Gonzalo-Calvo D, Vilades D, Martínez-Camblor P, Vea À, Nasarre L, Sanchez Vega J, et al. Circulating microRNAs in suspected stable coronary artery disease: a coronary computed tomography angiography study. J Intern Med. 2019. https://doi.org/10.1111/joim.12921.
de Gonzalo-Calvo D, Vea A, Bär C, Fiedler J, Couch LS, Brotons C, et al. Circulating non-coding RNAs in biomarker-guided cardiovascular therapy: a novel tool for personalized medicine? Eur Heart J. 2019;40:1643–50. https://doi.org/10.1093/eurheartj/ehy234.
Pinilla L, Barbé F, de Gonzalo-Calvo D. MicroRNAs to guide medical decision-making in obstructive sleep apnea: a review. Sleep Med Rev. 2021. https://doi.org/10.1016/J.SMRV.2021.101458.
Ferreira JP, Duarte K, Woehrle H, Cowie MR, Wegscheider K, Angermann C, et al. Biomarkers in patients with heart failure and central sleep apnoea: findings from the SERVE-HF trial. ESC Heart Fail. 2020;7:503–11. https://doi.org/10.1002/EHF2.12521.
Kozomara A, Birgaoanu M, Griffiths-Jones S. MiRBase: From microRNA sequences to function. Nucleic Acids Res. 2019;47:D155–62. https://doi.org/10.1093/nar/gky1141.
De Ronde MWJ, Ruijter JM, Lanfear D, Bayes-Genis A, Kok MGM, Creemers EE, et al. Practical data handling pipeline improves performance of qPCR-based circulating miRNA measurements. RNA. 2017;23:811–21. https://doi.org/10.1261/RNA.059063.116.
Ruijter JM, Ramakers C, Hoogaars WMH, Karlen Y, Bakker O, van den Hoff MJB, et al. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009;37: e45. https://doi.org/10.1093/nar/gkp045.
Uno H, Tian L, Cai T, Kohane IS, Wei LJ. A unified inference procedure for a class of measures to assess improvement in risk prediction systems with survival data. Stat Med. 2013;32:2430–42. https://doi.org/10.1002/SIM.5647.
Breiman L, Friedman J, Olshen R et al. Classification and regression trees. The Wadsworth Statistics/Probability Series. 1983.
Breiman L. Bagging predictors. Mach Learn. 1996;24:123–40.
Martínez-Camblor P, Bayón GF, Pérez-Fernández S. Cumulative/dynamic ROC curve estimation. J Stat Comput Simul. 2016;86:3582–94. https://doi.org/10.1080/00949655.2016.1175442.
Therneau T, Grambsch P. Modeling survival data: extending the cox model. New York: Springer; 2000.
Therneau T, Atkinson B, Ripley B. Recursive partitioning and regression trees. R Package Version 41–15 2019. https://www.scirp.org/reference/referencespapers.aspx?referenceid=3251272 (Accessed 21 Nov 2022).
Pérez-Fernández S, Martínez-Camblor P, Filzmoser P. Nsroc: an r package for non-standard roc curve analysis. The R Journal. 2018;10:55–77.
Belmonte T, Mangas A, Calderon-Dominguez M, Quezada-Feijoo M, Ramos M, Campuzano O, et al. Peripheral microRNA panels to guide the diagnosis of familial cardiomyopathy. Transl Res. 2020;218:1–15. https://doi.org/10.1016/j.trsl.2020.01.003.
Tavares L, Lador A, Valderrábano M. Sleep apnea and atrial fibrillation: role of the cardiac autonomic nervous system. Method Debakey Cardiovasc J. 2021;17:49–52. https://doi.org/10.14797/ZYUT2951.
Oldenburg O, Lamp B, Faber L, Teschler H, Horstkotte D, Töpfer V. Sleep-disordered breathing in patients with symptomatic heart failure: a contemporary study of prevalence in and characteristics of 700 patients. Eur J Heart Fail. 2007;9:251–7. https://doi.org/10.1016/J.EJHEART.2006.08.003.
Demissei BG, Cotter G, Prescott MF, Felker GM, Filippatos G, Greenberg BH, et al. A multimarker multi-time point-based risk stratification strategy in acute heart failure: results from the RELAX-AHF trial. Eur J Heart Fail. 2017;19:1001–10. https://doi.org/10.1002/EJHF.749.
Badimon L, Robinson EL, Jusic A, Carpusca I, deWindt LJ, Emanueli C, et al. Cardiovascular RNA markers and artificial intelligence may improve COVID-19 outcome: a position paper from the EU-CardioRNA cost action CA17129. Cardiovasc Res. 2021;117:1823–40. https://doi.org/10.1093/cvr/cvab094.
de Gonzalo-Calvo D, Vilades D, Martínez-Camblor P, Vea À, Ferrero-Gregori A, Nasarre L, et al. Plasma microRNA profiling reveals novel biomarkers of epicardial adipose tissue: a multidetector computed tomography study. J Clin Med. 2019;8:780. https://doi.org/10.3390/jcm8060780.
Vilades D, Martínez-Camblor P, Ferrero-Gregori A, Bär C, Lu D, Xiao K, et al. Plasma circular RNA hsa_circ_0001445 and coronary artery disease: Performance as a biomarker. FASEB J. 2020;34:4403–14. https://doi.org/10.1096/FJ.201902507R.
Hu X, Liao S, Bai H, Wu L, Wang M, Wu Q, et al. Integrating exosomal microRNAs and electronic health data improved tuberculosis diagnosis. EBioMedicine. 2019;40:564–73. https://doi.org/10.1016/J.EBIOM.2019.01.023.
Gangwar RS, Rajagopalan S, Natarajan R, Deiuliis JA. Noncoding RNAs in cardiovascular disease: pathological relevance and emerging role as biomarkers and therapeutics. Am J Hypertens. 2018;31:150–65. https://doi.org/10.1093/ajh/hpx197.
Acknowledgements
The original SERVE-HF trial (ClinicalTrials.gov number, NCT00733343) was sponsored by ResMed. We thank Sarah Cushman for English language editing.
Funding
Open Access funding enabled and organized by Projekt DEAL. TT and CB acknowledge funding by the German Research Foundation (DFG, TRR267 Project-ID 403584255-TRR 267). DdGC has received financial support from Instituto de Salud Carlos III (Miguel Servet 2020: CP20/00041) co-funded by the European Union. This work is supported by Instituto de Salud Carlos III (PI20/00577), co-funded by the European Union, IRBLleida—Fundació Dr. Pifarré, CERCA Programme/Generalitat de Catalunya. CIBERES (CB07/06/2008) is an initiative of the Instituto de Salud Carlos III.
Author information
Authors and Affiliations
Contributions
Conception and design: DdGC, FZ, WK, TT, CB. Data acquisition: All authors. Statistical analysis: KD, PMC. Data analysis and interpretation: DdGC, CB. Manuscript drafting: DdGC, TB, CB. Review & editing: All authors. Final approval of the submitted version: All authors. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
TT is a founder and shareholder of Cardior Pharmaceuticals GmbH (outside the topic of this paper). CB and TT have filed patents on the use of circulating RNAs for diagnosis and prognosis of cardiovascular disease. WK reports advisory board fees from AstraZeneca, Novartis, Amgen, Pfizer, The Medicines Company, DalCor, Kowa, Corvidia, OMEICOS, Daiichi-Sankyo, Novo Nordisk, New Amsterdam Pharma, TenSixteen Bio, Esperion, Genentech; lecture fees from Bristol-Myers Squibb, Novartis, Amgen, Berlin-Chemie, Sanofi and AstraZeneca; grants and non-financial support from Abbott, Roche Diagnostics, Beckmann, and Singulex, all outside the submitted work. FZ reports personal fees from Applied Therapeutics, Bayer, Boehringer, BMS, CVRx, Cardior, Cereno pharmaceutical, Cellprothera, CEVA, Merck, Novartis, NovoNordisk, Owkin, Pfizer, Servier, having stock options at G3Pharmaceutical and equities at Cereno pharmaceutical, Cardiorenal, Eshmoun Clinical research and being the founder of Cardiovascular Clinical Trialists. CEA reports membership in the ServeHF Steering committee, and received consulting fees and honoraria from ResMed, in context with the current work. She further reports grant support, personal fees and/or non-financial support from Abbott, Astra-Zeneca, Boehringer Ingelheim & Eli Lilly Company, Biotronik, Novo Nordisk, Novartis, Thermo Fisher and Vifor, all outside of the submitted work. Members of the ServeHF Steering committee, received consulting fees and honoraria from ResMed, in context with current work
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Baseline characteristics in patients included and not included in the current investigation. Table S2. Baseline characteristics and miRNA levels according to study groups. Table S3. Baseline characteristics according to the primary outcome (Screening). Table S4. Baseline characteristics according to the primary outcome (technical validation). Table S5. Comparison of baseline characteristics according to median value of miR-501-3p. Table S6. C-index for miRNAs and NT-proBNP. Table S7. Number of participants with event (Positive) within time points (1, 3 and 5 years), and those without event. Table S8. Classification metrics at different time points. Figure S1. Flow-diagram for the customized-CART algorithm. Figure S2. Correlation coefficient maps between microRNA expression levels and baseline clinical characteristics. Correlations between continuous variables were assessed with Spearman rank correlation coefficients, represented as rho values.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
de Gonzalo-Calvo, D., Martinez-Camblor, P., Belmonte, T. et al. Circulating miR-133a-3p defines a low-risk subphenotype in patients with heart failure and central sleep apnea: a decision tree machine learning approach. J Transl Med 21, 742 (2023). https://doi.org/10.1186/s12967-023-04558-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-023-04558-w