
Abstract
Background
The human gut microbiota (GM) is involved in the pathogenesis of hypertension (HTN), and could be affected by various factors, including sex and geography. However, available data directly linking GM to HTN based on sex differences are limited.
Methods
This study investigated the GM characteristics in HTN subjects in Northwestern China, and evaluate the associations of GM with blood pressure levels based on sex differences. A total of 87 HTN subjects and 45 controls were recruited with demographic and clinical characteristics documented. Fecal samples were collected for 16S rRNA gene sequencing and metagenomic sequencing.
Results
GM diversity was observed higher in females compared to males, and principal coordinate analysis showed an obvious segregation of females and males. Four predominant phyla of fecal GM included Firmicutes, Bacteroidetes, Actinobacteria and Proteobacteria. LEfSe analysis indicated that phylum unidentified_Bacteria was enriched in HTN females, while Leuconostocaceae, Weissella and Weissella_cibaria were enriched in control females (P < 0.05). Functionally, ROC analysis revealed that Cellular Processes (0.796, 95% CI 0.620 ~ 0.916), Human Diseases (0.773, 95% CI 0.595 ~ 0.900), Signal transduction (0.806, 95% CI 0.631 ~ 0.922) and Two-component system (0.806, 95% CI 0.631 ~ 0.922) could differentiate HTN females as effective functional classifiers, which were also positively correlated with systolic blood pressure levels.
Conclusions
This work provides evidence of fecal GM characteristics in HTN females and males in a northwestern Chinese population, further supporting the notion that GM dysbiosis may participate in the pathogenesis of HTN, and the role of sex differences should be considered.
Trial registration Chinese Clinical Trial Registry, ChiCTR1800019191. Registered 30 October 2018 – Retrospectively registered, http://www.chictr.org.cn/.
Similar content being viewed by others

Background
As a common and chronic medical condition, hypertension (HTN) has become a global health issue, accounting for approximately 10.8 million deaths worldwide [1]. HTN is a complex and modifiable risk factor for cardiovascular diseases (CVDs) and stroke, while a diverse range of endogenous and environmental factors contributes to both HTN onset and progression [2,3,4]. Although an increased risk of HTN has been demonstrated to be associated with approximately 900 genetic loci, only < 6% of the variance in systolic blood pressure (BP) could be explained by common genetic risk variants [5]. Given its complexity and heterogeneity, the elucidation of HTN pathogenesis remains challenging.
Growing evidence has revealed the potential role of the gut microbiota (GM) in host homeostasis and multiple physiological processes [6], and suggests associations between GM and various diseases including atherosclerotic CVDs [7]. As the longest organ, the gastrointestinal tract is mainly involved in the absorption of nutrients and ions which greatly impacts BP [8], and a substantial amount of work has supported the role of GM as a potential factor in BP regulation or even causal determinants of HTN pathogenesis [2, 9,10,11]. There is evidence of gastrointestinal pathophysiology in animal HTN models, in which fecal transplantation from HTN subjects increases BP in germ-free mice [12]. In addition, GM dysregulation is also associated with various metabolic diseases and HTN-related risk factors, such as obesity, hyperlipidemia and diabetes mellitus [13]. Overall, current data strongly indicate that GM may play an important role in HTN pathogenesis [12, 14,15,16].
Due to the novel concept of GM-influenced HTN pathogenesis [2], it is critical to investigate GM alterations in HTN subjects in different regions with different genetic background and dietary habits [2, 16,17,18,19]. Moreover, obesity, hyperlipidemia and diabetes could result in GM alterations [20], and these variations should also be taken into consideration. Of note, few studies have investigated the associations between GM and HTN subjects based on sex differences. Given that sex is important in BP regulation [2, 21, 22], we investigated the GM characteristics of female and male HTN subjects, respectively, and the associations between GM characteristics and BP levels were discussed.
Methods
Study participants and study protocol
From July 2018 to June 2020, we recruited 205 subjects from our outpatient clinics at Honghui Hospital, Xi’an Jiaotong University, China. All selected subjects were aged between 18 and 80 years old, and were able to provide written informed consent. Subjects with one of the following conditions were excluded: ① pregnant or lactating, or with chronic diseases including cancers, inflammation or surgical history in the alimentary tract, or serious systematic dysfunctions; ② taking any medications that may disrupt their original GM, such as fiber supplements, probiotics or prebiotics within 6 weeks, or antimicrobial drugs within 6 months before GM sampling [2, 23]; ③ taking any anti-inflammatory agents, acid-suppressing agents, immunosuppressants, or anti-HTN medication, which seem to modulate GM [2, 24, 25].
This study was approved by the Ethics Committee of Honghui Hospital, Xi’an Jiaotong University (Protocol Number: 201801022, approved January 8th, 2018), and all subjects provided written informed consent before enrollment. HTN was diagnosed if systolic BP (SBP) ≥ 130 mmHg and/or diastolic BP (DBP) ≥ 80 mmHg; subjects with normal BP (SBP < 120 mmHg and DBP < 80 mmHg) served as controls [26, 27]. The study flow is shown in Fig. 1.

Study flow diagram. BMI body mass index; WC waist circumference; BP blood pressure; FBG fasting blood glucose; TG triglyceride; TC total cholesterol; LDL-C low-density lipoprotein cholesterol; HDL-C high-density lipoprotein cholesterol; non-HDL-C non-high-density lipoprotein cholesterol; GM gut microbiota
Measurements of demographic and clinical parameters
On their first visit, all medical information was recorded for the recruited subjects. Body weight and height were measured without shoes and with light clothing to the nearest 0.1 cm or to the nearest 0.1 kg, respectively. Body mass index (BMI) was calculated as weight in kilograms divided by the square of height in meters. Waist circumference (WC) was measured midway between the lower rib margin and the iliac crest in the standing position with a non-expandable tape to the nearest 0.1 cm. BP was assessed using a medical electronic sphygmomanometer (OMRON HEM-7130 professional portable blood pressure monitor, OMRON, Dalian, China) on the left arm positioned at heart level in a seated position with palm face up. The subjects were required to rest for at least 3 ~ 5 min before BP measurement, and BP was measured in duplicate at a 1 ~ 2-min interval. The average of the 2 readings was calculated. Mean arterial pressure (MAP) was calculated using the formula [(2 × diastolic BP) + systolic BP]/3 [16].
Peripheral venous blood samples were obtained in the morning after an overnight (at least 8 h) fast. Levels of fasting plasma glucose (FPG), serum triglyceride (TG), total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C) and non-high-density lipoprotein cholesterol (non-HDL-C) were measured using an automatic biochemical analyzer (Cobas c701, Roche, Mannheim, Germany). All instruments were calibrated regularly.
Fecal sample collection and DNA extraction
We followed the standard protocols of fecal sample collection and processing [28]. Briefly, fresh fecal samples were collected from each participant at home, and temporarily stored in foam boxes with frozen cold packs. After immediate transportation to the Clinical Laboratory at Honghui Hospital, Xi’an Jiaotong University within 6 h, fecal samples were stored at − 80 °C until further processing.
Samples were sent in cold-chain with dry ice to Novogene Co., Led. (Beijing, China) for subsequent procedures. Genomic DNA was extracted from feces using the QIAamp Fast DNA Stool Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. After the evaluation of genomic DNA concentration and quality, DNA samples, greater than 1 µg and with an OD value between 1.8 ~ 2.0, were qualified for further analysis.
16S rRNA gene amplification, sequencing and analysis
The V3 ~ V4 hypervariable regions of the 16S rRNA gene were amplified using specific primers (338F: 5ʹ-ACTCCTACGGGAGGCAGCAG-3ʹ; 806R: 5ʹ-GGACTACHVGGGTWTCTAAT-3ʹ) with barcodes, and all PCRs were performed with Phusion® High Fidelity PCR Master Mix (New England Biolabs, Ipswich, MA, USA). After mixing the PCR products, the mixture was purified using a GeneJET™ Gel Extraction Kit (Thermo Scientific, Waltham, MA, USA). Sequencing libraries were generated using the NEB Next® Ultra™ DNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, MA, USA) following the manufacturer’s recommendations, and library quality was assessed on the Qubit® 2.0 Fluorometer (Thermo Scientific, Waltham, MA, USA). Finally, the prepared libraries were sequenced on an Illumina HiSeq platform (Illumina NovaSeq 6000, PE150, Illumina, San Diego, CA, USA).
UPARSE software (Uparse v7.0.1001, http://drive5.com/uparse/) [29] and Quantitative Insights Into Microbial Ecology (QIIME) software (v1.7.0) [30] were introduced for sequencing analysis. Acquired high-quality clean reads with ≥ 97% similarity were de novo clustered into the same operational taxonomic units (OTUs), and the representative sequence for each OTU was screened and used to annotate taxonomic information based on the RDP classifier [31]. After OTUs with annotation were produced, microbial diversity was assessed using QIIME software (v1.7.0) [30]. Alpha-diversity analysis was performed based on 4 indices, including Shannon, Chao1, Simpson and abundance coverage-based estimator (ACE). Beta-diversity of GM composition was estimated using the unweighted UniFrac method to calculate the distances between samples, and then visualized by principal coordinates analysis (PCoA). Linear discriminant analysis (LDA) effect size (LEfSe) algorithm with an LDA score threshold of 2 (on a log10 scale) was applied to identify the enriched and significant bacteria in each group, with a P value < 0.05.
Metagenomic sequencing and analysis
After DNA extraction and quality control, a total of 1 µg DNA per sample was used for library preparation. Sequencing libraries were generated using the NEBNext® Ultra™ DNA Library Prep Kit (New England Biolabs, Ipswich, MA, USA) following the manufacturer’s recommendations. Briefly, the qualified DNA was fragmented to a size of 350 bp by sonication, and the acquired DNA fragments were end-polished, A-tailed, and ligated with the full-length adaptor for Illumina sequencing. Then PCR amplification was performed, and PCR products were purified using the AMPure XP system. Then, libraries were initially quantified using a Qubit® 2.0 Fluorometer (Thermo Scientific, Waltham, MA, USA) and diluted to 2 ng/µL. Finally, libraries were analyzed using an Agilent2100 Bioanalyzer for size distribution, and then quantified using real-time PCR to ensure the quality (effective concentration > 3 nM). After clustering, the prepared libraries were sequenced on an Illumina HiSeq platform (Illumina NovaSeq 6000, PE250, Illumina, San Diego, CA, USA).
Short Oligonucleotide Analysis Package 2 (SOAP2) software (v2.04, http://soap.genomics.org.cn/soapdenovo.html) and Bowtie2.2.4 software were used for raw data processing, and the remaining Scaftigs were used for subsequent analysis. Genes were predicted on Scaftigs (≥ 500 bp) using MetaGeneMark (prokaryotic GeneMark.hmm v2.10), and a non-redundant gene catalog was constructed with CD-HIT (v4.5.8) software. For information on the abundance of genes, clean reads were realigned to the gene catalog (Unigenes) using Bowtie2.2.4 software. Genes with more than 2 mapped reads were deemed to be present in a sample [32]. The abundance of genes was calculated by counting the number of reads and normalizing based on gene length. Unigenes were aligned to the KEGG database (Release 73.1, Version: 2018.01, with animal and plant genes removed) for gene functional annotation using DIAMOND software (version 0.9.9), and GM functions were evaluated and compared across groups in the present study.
Statistical analysis
Statistical analyses and figure constructions were conducted using the SPSS PASW v23 (IBM SPSS Inc., Chicago, IL), R platform v4.0.2 (R Foundation, Vienna, Austria), GraphPad Prism v5.01 (GraphPad Software Inc., San Diego, CA, USA) and MedCalc v19.0.4 (MedCalc Software Bvba, Ostend, Belgium) software. Quantitative variables are presented as mean ± standard deviation (SD), and the normal distribution of quantitative variables was assessed by the Shapiro-Wilk test. The t-test was used for comparisons of the demographic and clinical parameters between groups, depending on the homogeneity of variance. The comparisons of GM diversities between groups were assessed using the Wilcoxon rank-sum test. The top 10 GM taxa in each level (phylum, class, order, family, genus and species) sorted by higher relative abundances were identified, and the significant differences between groups were assessed using the Wilcoxon rank-sum test. The “PerformanceAnalytics” package in R was utilized for Spearman’s correlation analysis between any two demographic or clinical parameters of the subjects. In addition, Spearman’s correlation test was used to analyze the correlations between GM composition/KEGG functions and BP levels. A P value < 0.05 was considered statistically significant, whereas a P value between 0.05 and 0.1 was considered a tendency. Receiver operator characteristic (ROC) curve analysis was performed, and GM functions with an area under the curve (AUC) ≥ 0.700 were selected for presentation.
Results
Demographic and clinical characteristics of the study subjects
As described above, 87 HTN subjects and 45 controls were finally recruited in this study. Table 1 shows the demographic and clinical characteristics of the study subjects grouped by sex and BP levels. With the exception of SBP, DBP and MAP (P < 0.05), the other parameters showed no significances between HTN and control females, as well as HTN and control males. Figure 2 shows the correlations between any two characteristics of the enrolled females and males.

Correlation analysis between demographic and clinical characteristics of the study subjects. Correlation analysis between demographic and clinical characteristics of the enrolled females (A) and males (B), respectively. Bar plots present the distribution of each parameter, and the scatter plots show the distribution for each two parameters. The numbers in this figure indicate the correlation coefficients of each two parameters by Spearman method in R, and the red dots and asterisks indicate the degrees of statistical significances, •P < 0.1, *P < 0.05, **P < 0.01, ***P < 0.001
16S rRNA gene sequencing analysis of gut microbiota in the study subjects
After 16S rRNA gene sequencing, the species accumulation curve of GM in Fig. 3A and the rarefaction curves in Fig. 3B showed a plateau of species richness, indicating that the sample numbers and sequencing depth herein covered enough information for the following analysis. Finally, we acquired a total of 1653 unique OTUs (ranging from 208 to 416 per sample), and please refer to Fig. 3C ~ 3E for details.

The basic information of the 16S rRNA gene sequencing. The species accumulation curve of gut microbiota detected in HTN and control subjects in this study (A). The line indicates the averaged accumulated increase of detected OTUs vs. number of samples. The box-plots show the 25th, 50th and 75th percentile at each sample size. The rarefaction curve of the number of sequence reads and their corresponding number of OTUs in females and males in this study (B). Venn diagrams of observed OTU numbers in different comparison groups: females vs. males (C), HTN group vs. control group in females (D) and males (E), respectively. Every circle depicts the number of unique OTUs observed in one group. Overlapping OTUs shared by two groups are represented in the areas of intersection among corresponding circles
Initially, we evaluated the α-diversities of GM in females and males, which were significantly lower in males compared with females (Fig. 4A). Moreover, the PCoA model for β-diversity analysis revealed a segregation between females and males (Fig. 4B). Therefore, the study subjects should be divided into female and male groups in the following analysis. Then, we conducted the α- and β-diversity analyses between the HTN and control subjects in females and males, respectively. However, the differences of GM diversities were not significant between either HTN vs. control females (Fig. 4C, D), or HTN vs. control males (Fig. 4E, F).

The diversity analysis of gut microbiota in the study subjects. Violin plots of α-diversity analysis in different comparison groups: females vs. males (A), HTN group vs. control group in females (C) and males (E), respectively. Each plot represents one index of the α-diversity distribution, including Shannon, Chao1, Simpson and ACE indices, for each comparison groups. Wilcoxon rank-sum test was used for the analysis of significant differences between different comparison groups. #P < 0.1, *P < 0.05, **P < 0.01. Plots of principal coordinate analysis (PCoA) based on the OTU level in different comparison groups: females vs. males (B), HTN group vs. control group in females (D) and males (F), respectively. Each square/circle indicates one sample. The distance between samples represents the GM similarity or differences in the samples, and the PCoA analysis was conducted with unweighted UniFrac method
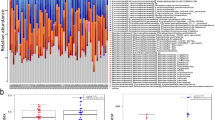
The differences of GM composition between HTN and control subjects were taxonomically evaluated at six different levels, including phylum, class, order, family, genus and species, and the top 10 GM taxa with higher relative abundances were summarized in each level (Fig. 5). Consistent with previous results, the GM taxa were mostly included in four predominant phyla, which were Firmicutes, Bacteroidetes, Actinobacteria and Proteobacteria. The relative abundances of Firmicutes, Bacteroidetes, Clostridia, Bacteroidia, Clostridiales, Bacteroidales, Ruminococcaceae, Lachnospiraceae and Faecalibacterium were greater than 0.100 at respective taxonomic levels in HTN and control females. Of note, the unidentified_Bacteria abundance was greater in HTN females, whereas the relative abundances of Bacteroidia, Bacteroidales, Leuconostocaceae, Weissella and Weissella_cibaria were lower in HTN females compared with control females (Fig. 5A). LEfSe analysis indicated that unidentified_Bacteria was enriched in HTN females; while, Leuconostocaceae, Weissella and Weissella_cibaria were enriched in control females (Fig. 6A). In contrast, no significant differences of GM composition were noted between HTN and control males except for Erysipelotrichia, which was enriched in HTN males (Figs. 5B and 6B). Furthermore, the correlations between GM taxa and SBP/DBP/MAP levels were investigated in females and males, respectively, and data were showed in Additional file 1: Tables S1 and S2 in the Additional files.

Relative abundances and comparative analysis of the taxonomic composition of gut microbiota in the study subjects. Relative abundances and comparative analysis of the taxonomic composition of gut microbiota in the enrolled females (A) and males (B), respectively. Bar plots show the relative abundances of the top 10 taxa at respective levels, including phyla, class, orders, family, genera and species, in HTN and control females and males. Each component of the cumulative bar chart indicates a phylum, a class, an order, a family, a genus or a species, respectively. The taxa with significant difference between groups are presented using Wilcoxon rank-sum test. #P < 0.1, *P < 0.05, **P < 0.01

Linear discriminant analysis (LDA) effect size (LEfSe) analysis of gut microbiota taxa in the study subjects. LDA scores indicate differentially represented taxa in HTN and control groups in females (A) and males (B), respectively. The logarithmic threshold for discriminative features was set to 2.0
Metagenomic sequencing analysis of gut microbiota
After the preliminary analysis based on the 16S rRNA gene sequencing, we conducted further analysis of GM functions by performing matagenomic sequencing. A total of 36 HTN subjects (24 females and 12 males) and 18 controls (9 females and 9 males) were randomly selected for metagenomic analysis. Their demographic and clinical characteristics were demonstrated in Table 2 grouped by sex and BP levels.
After GM gene and functional annotation aligned to the KEGG database, the functional changes in the microbial community were evaluated. GM functions showed different patterns between HTN and control subjects in females and males, as presented in Fig. 7. Our results revealed that Cellular Processes and Human Diseases represented the enrichment in Level 1 KEGG functions in HTN females (P < 0.05), as well as Metabolism, Environmental Information Processing and Organismal System (P < 0.1, Fig. 7A). In addition, the relative abundance of Signal transduction in Level 2 and Two-component system in Level 3 were increased in HTN females compared with controls (P < 0.05, Fig. 7B, C). Please refer to Fig. 7D ~ 7 F for detailed information about male subjects. Notably, the relative abundances of Metabolism, Environmental Information Processing, Cellular Processes, Human Diseases, Carbohydrate metabolism, Amino acid metabolism, Membrane transport, Cellular community – prokaryotes, ABC transporters and Quorum Sensing were enriched in females compared to males (P < 0.05); please refer to Additional file 1: Figure S1 in the additional files for details. These data further supported our previous strategy of separation of female and male subjects in GM analysis.

Relative abundances and comparative analysis of the annotated KEGG functions of gut microbiota in the study subjects. Relative abundances and comparative analysis of the annotated KEGG functions of GM at three levels in HTN and control groups in females (A–C) and males (D–F), respectively. The KEGG functions with significant differences between groups are presented with Wilcoxon rank-sum test. #P < 0.1, *P < 0.05, **P < 0.01
To explore the implications of altered GM functions in HTN pathogenesis, Spearman’s correlation analysis was introduced to evaluate the correlations between GM functions and BP levels. Significantly positive correlations of Cellular Processes, Human Diseases, Signal transduction and Two-component system with SBP were found in females (P < 0.05, Fig. 8A). Besides, the correlation of Human Diseases with MAP was also noted (P < 0.05, Fig. 8B). To further explore the altered GM functions that could facilitate the identification of HTN subjects, ROC curve analysis was introduced herein (Fig. 8C, D). The AUCs of GM functions in Level 1 that could differentiate HTN females from controls included Cellular Processes (0.796, 95% CI 0.620 ~ 0.916), Human Diseases (0.773, 95% CI 0.595 ~ 0.900), Environmental Information Processing (0.718, 95% CI 0.534 ~ 0.860), Organismal Systems (0.718, 95% CI 0.534 ~ 0.860) and Metabolism (0.704, 95% CI 0.520 ~ 0.849). Besides, Signal transduction (0.806, 95% CI 0.631 ~ 0.922), Two-component system (0.806, 95% CI 0.631 ~ 0.922), Carbohydrate metabolism (0.722, 95% CI 0.539 ~ 0.863), Metabolism of cofactors and vitamins (0.718, 95% CI 0.534 ~ 0.860), and Amino acid metabolism (0.708, 95% CI 0.525 ~ 0.853) in Level 2 and 3 could effectively distinguish HTN females from controls.

Exploration of the altered KEGG functions of gut microbiota in the enrolled HTN females. Correlations of altered KEGG functions of gut microbiota with SBP (A) and MBP (B) levels in the enrolled females. The horizontal axis represents the relative abundances of KEGG functions, and the vertical axis represents different BP levels. Data was analyzed and plotted with Spearman method in R. Receiver operator characteristic (ROC) curve analysis of identification of HTN females from controls based on KEGG functions of gut microbiota (C, D)
Discussion
Hypertension (HTN) is a multifactorial and complicated condition [3, 4], and growing evidence suggests a novel role of GM in HTN onset and progression [16, 33, 34]. The adult GM consists trillions of microorganisms dominated by phyla Firmicutes, Bacteroidetes, Actinobacteria and Proteobacteria, and maintains the gut immunity and whole-body homeostasis [35]; dysbiosis or imbalance in the GM community may have detrimental effects on health [36]. Several demographic factors, such as age, BMI, sex, ethnicity and culture, geographic location and socioeconomic environment, could influence both GM and BP levels [2, 15, 21, 24, 37,38,39], and it is important to consider these confounding factors in GM-related studies on HTN. Although biological sex could shape the host GM [40,41,42], available data directly linking GM to HTN based on sex differences are limited [21]. Therefore, residents with similar dietary habits living in Shaanxi Province, China, were recruited to minimize the regional differences of GM composition in this study; and the differences of GM characteristics between female and male subjects were analyzed. Of note, an obvious segregation of GM diversity between females and males was found, supporting a respective analysis for female and male subjects in the subsequent GM analysis.
GM has been implicated in the pathogenesis of HTN by means of influencing sodium intake, production of certain metabolites, low-grade inflammation, etc. [37, 43]. Epidemiological data have linked salt and fiber intake with changes in BP levels, suggesting a connection between gut and HTN [3, 4, 44,45,46,47]. High sodium intake could reduce the relative abundances of certain beneficial taxa such as Lactobacillus spp, and GM-produced metabolites may also influence sodium absorption [48]. Besides, an increased consumption of dietary fiber, which could modulate GM as well as be fermented by GM, has been indicated to decrease BP levels [45, 46, 48, 49]. The potent mechanism may involve the production of short-chain fatty acids (SCFAs), such as acetate, butyrate and propionate, which are mainly derived from the fermentation of indigestible carbohydrates and consumption of protein or peptide [15, 49,50,51,52]. GM-produced SCFAs are rapidly absorbed in the colon and taken up by the liver or enter the circulation, and serve as precursors or substrates in various physiological processes [53,54,55]. In rodent models, SCFAs were reported to bind to G-protein-coupled receptors involved in the regulation of vasoreactivity and BP levels [56,57,58]. The negative correlations between the abundances of butyrate-producing bacteria and BP levels have been observed in obese pregnant women [59], and fiber and acetate supplementation could improve GM dysbiosis and increase the relative abundances of certain bacteria that may play a protective role in HTN [49]. In summary, SCFAs and a range of SCFA-producing taxa could play potent roles in maintaining GM homeostasis and BP levels [33, 60,61,62]. It is known that Bacteroidetes phylum members could produce high levels of acetate and propionate, whereas certain species in Firmicutes may produce high amounts of butyrate [63,64,65]. Consistent with these data, our results showed that the relative abundances of Bacteroidia and Bacteroidales were greater than 0.10 at respective taxonomic levels, and were lower in HTN females compared with controls. Besides, Leuconostocaceae, Weissella and Weissella_cibaria were also enriched in control females. As Firmicutes phylum members, Leuconostocaceae, Weissella and Weissella_cibaria are producers of SCFAs, and Weissella cibaria may have antihypertensive and antioxidant effects in spontaneously hypertensive rats models [66, 67], indicating their potential for HTN prevention and further supporting our results herein.
Ruminococcaceae is involved in intestinal epithelium maintenance as it is inversely correlated with intestinal permeability [68,69,70], and its abundance was found diminished in elderly HTN patients [71]. Lower abundance of Ruminococcus may associate with HTN [72,73,74], and a particular reduction in Ruminococcus was also found in SD rats with minocycline-induced programmed HTN [75]. However, Kim SR et al. reported a higher abundance of Ruminococcus torques in HTN, which was significantly associated with SBP [15]; while, Dan et al. showed increased Ruminococcaceae and decreased Ruminococcus in HTN subjects [76]. In this study, Ruminococcus_bromii, as a member of Firmicutes and Ruminococcus, was positively correlate with DBP. In other words, certain inconsistent results exist, which may be due to variations in genetics, sex, diet and lifestyle, geographical differences or other unknown factors [37]. Moreover, these data implicated a more important role of individual GM taxon than phyla in BP regulation [21].
GM-related studies indicate that GM could exert potential influences on various diseases [35, 77,78,79,80], and certain GM characteristics could be utilized as non-invasive biomarkers for early diagnosis in clinical practice [81, 82]. As one of the most prevalent CVDs and a leading risk factor of other CVDs, HTN is certainly accompanied by GM alterations [83,84,85]. Exploration of the altered KEGG functions of GM in HTN may help illustrate its functional roles in HTN pathogenesis, and might provide a new perspective on the interpretation of HTN and additional auxiliary diagnosis in the future [86]. In this study, we randomly selected HTN and control subjects from the recruited females and males, who also lacked significant differences in the majority of these demographic and clinical characteristics, for metagenomic sequencing analysis. The relative abundances of Human Diseases, Signal transduction and Two-component system were increased in HTN females compared with controls, positively correlated with increased SBP and MAP levels, and contributed to the effective identification of HTN females from controls. Besides, our data indicated that the altered GM functions differed between HTN females and males compared to respective controls, further supporting the notion that females and males should be separately analyzed in GM-related analysis. In terms of sex differences in this study, we found higher α-diversities of GM in females, as well as an obvious segregation in β-diversity between females and males, and the GM composition and functions were also different between female and male subjects, which could be contributed by genetic and epigenetic factors, sex steroid milieu, gonadal status, dietary and lifestyle-related factors, etc. [40, 87,88,89]. Specifically, previous data indicated that the primary sex steroid hormones, such as estrogen, progesterone and testosterone, may participate in the regulation of GM diversity, composition and function [38, 40, 89]. For example, it was reported that estrogen could promote the growth of certain beneficial bacteria such as Lactobacillus and Bifidobacterium, which were also considered to improve the cardiovascular health [18, 89,90,91]. Moreover, sex differences also exist in lifestyle-related factors, such as dietary preferences, sex*diet interactions and physical activity levels, which may influence the GM characteristics and HTN onset and progression differently in females and males [18, 19, 42]. The present study was conducted in northwestern China, and regional variations in diet, lifestyle and environmental factors may potentially contribute to the observed differences in GM characteristics between females and males. It is crucial to note that the specific mechanisms underlying the observed differences herein are multifactorial and complicated, and may not be fully elucidated. Further researches are needed to better reveal the potential causes for the observed sex-based differences in GM, to elucidate the underlying mechanisms, and to facilitate understanding of the implications of GM for health and diseases, including HTN.
Although our data revealed certain GM changes in HTN subjects, it has several limitations. First, this investigation was conducted with a limited sample size due to the practical limitations of data availability. Second, the subjects were recruited in a single hospital and were grouped based on BP levels alone. Third, BP measurement in clinic is used for HTN diagnosis [92] rather than office BP monitoring [9]. Nevertheless, certain confounding factors were taken into consideration herein, such as genetics, geography, sex and treatment-naive recruitment, thus our data were reliable despite some other or unknown factors. Furthermore, evidence directly linking GM and sex differences in BP regulation is rather limited, and the role of GM in sex-dependent HTN is only hypothesized [2, 21, 22, 42]. Our data could provide evidence of fecal GM characteristics in HTN females and males, respectively, which might fill in the gap to a certain degree. However, this is only a cross-sectional clinical study in China, and it is infeasible to draw any definite conclusions about the causal relationships between GM and HTN based on sex differences. Issues regarding the differences in GM profiles of HTN females and males remain to be addressed in future studies [72], and detailed evaluations are needed in larger numbers of treatment-naive HTN patients.
Conclusions
In conclusion, the pathogenesis of HTN is multifactorial and complicated, and evidence suggests that GM may play a novel role in HTN onset and progression. This study provide the first evidence of GM characteristics and alterations in HTN females and males, respectively, in northwestern China, further supporting the theory that GM dysbiosis underlies HTN pathogenesis. Future studies are needed to elucidate the underlying mechanisms and potential therapeutic interventions targeting GM for HTN prevention and management [14].
Availability of data and materials
The data generated and analyzed during the current study are available from the corresponding authors on reasonable request.
Abbreviations
- CVDs:
-
Cardiovascular diseases
- HTN:
-
Hypertension
- BP:
-
Blood pressure
- GM:
-
Gut microbiota
- SBP:
-
Systolic blood pressure
- DBP:
-
Diastolic blood pressure
- BMI:
-
Body mass index
- WC:
-
Waist circumference
- MAP:
-
Mean arterial pressure
- FPG:
-
Fasting plasma glucose
- TG:
-
Triglyceride
- TC:
-
Total cholesterol
- LDL-C:
-
Low-density lipoprotein cholesterol
- HDL-C:
-
High-density lipoprotein cholesterol
- non-HDL-C:
-
Non-high-density lipoprotein cholesterol
- OTUs:
-
Operational taxonomic units
- ACE:
-
Abundance coverage-based estimator
- PCoA:
-
Principal coordinates analysis
- LDA:
-
Linear discriminant analysis
- LEfSe:
-
Linear discriminant analysis effect size
- SD:
-
Standard deviation
- ROC:
-
Receiver operator characteristic
- AUC:
-
Area under the curve
References
GBD 2019 Risk Factors Collaborators. Global burden of 87 risk factors in 204 countries and territories, 1990–2019: a systematic analysis for the global burden of disease study 2019. Lancet. 2020;396:1223–49.
Marques FZ, Jama HA, Tsyganov K, Gill PA, Rhys-Jones D, Muralitharan RR, Muir J, Holmes A, Mackay CR. Guidelines for transparency on gut microbiome studies in essential and experimental hypertension. Hypertension. 2019;74:1279–93.
Mills KT, Bundy JD, Kelly TN, Reed JE, Kearney PM, Reynolds K, Chen J, He J. Global disparities of hypertension prevalence and control: a systematic analysis of population-based studies from 90 countries. Circulation. 2016;134:441–50.
Mills KT, Stefanescu A, He J. The global epidemiology of hypertension. Nat Rev Nephrol. 2020;16:223–37.
Evangelou E, Warren HR, Mosen-Ansorena D, Mifsu B, Pazoki R, Gao H, Ntritsos G, Dimou N, Cabrer CP, Karaman I, et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet. 2018;50:1412–25.
Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–20.
Jie ZY, Xia HH, Zhong SL, Feng Q, Li SH, Liang SS, Zhong HZ, Liu ZP, Gao Y, Zhao H, et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat Commun. 2017;8:12.
Linz D, Wirth K, Linz W, Heuer HOO, Frick W, Hofmeister A, Heinelt U, Arndt P, Schwahn U, Boehm M, Ruetten H. Antihypertensive and laxative effects by pharmacological inhibition of sodium-proton-exchanger subtype 3-mediated sodium absorption in the gut. Hypertension. 2012;60:1560.
Jama HA, Beale A, Shihata WA, Marques FZ. The effect of diet on hypertensive pathology: is there a link via gut microbiota-driven immunometabolism? Cardiovascular Res. 2019;115:1435–47.
Marques FZ, Mackay CR, Kaye DM. Beyond gut feelings: how the gut microbiota regulates blood pressure. Nat Reviews Cardiol. 2018;15:20–32.
Jama HA, Kaye DM, Marques FZ. The gut microbiota and blood pressure in experimental models. Curr Opin Nephrol Hypertens. 2019;28:97–104.
Li J, Zhao FQ, Wang YD, Chen JR, Tao JE, Tian G, Wu SL, Liu WB, Cui QH, Geng B, et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome. 2017;5:19.
Boulange CL, Neves AL, Chilloux J, Nicholson JK, Dumas M-E. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med. 2016;8:42.
Yan QL, Gu YF, Li XC, Yang W, Jia LQ, Chen CM, Han XY, Huang YK, Zhao LZ, Li P, et al. Alterations of the gut microbiome in hypertension. Front Cell Infect Microbiol. 2017;7:9.
Kim S, Goel R, Kumar A, Qi YF, Lobaton G, Hosaka K, Mohammed M, Handberg EM, Richards EM, Pepine CJ, Raizada MK. Imbalance of gut microbiome and intestinal epithelial barrier dysfunction in patients with high blood pressure. Clin Sci. 2018;132:701–18.
Palmu J, Salosensaari A, Havulinna AS, Cheng S, Inouye M, Jain M, Salido RA, Sanders K, Brennan C, Humphrey GC, et al. Association between the gut microbiota and blood pressure in a Population Cohort of 6953 individuals. J Am Heart Association. 2020;9:e016641.
He Y, Wu W, Zheng H-M, Li P, McDonald D, Sheng H-F, Chen M-X, Chen Z-H, Ji G-Y, Zheng Z-D-X, et al. Regional variation limits applications of healthy gut microbiome reference ranges and disease models. Nat Med. 2018;24:1532–5.
Santos-Marcos JA, Haro C, Vega-Rojas A, Alcala-Diaz JF, Molina-Abril H, Leon-Acuña A, Lopez-Moreno J, Landa BB, Tena-Sempere M, Perez-Martinez P, et al. Sex differences in the gut microbiota as potential determinants of gender predisposition to disease. Mol Nutr Food Res. 2019;63:e1800870.
Campaniello D, Corbo MR, Sinigaglia M, Speranza B, Racioppo A, Altieri C, Bevilacqua A. How diet and physical activity modulate gut microbiota: evidence, and perspectives. Nutrients. 2022;14:2456.
Liu X, Li Y, Guo Y, Li L, Yang K, Liu R, Mao Z, Bie R, Wang C. The burden, management rates and influencing factors of high blood pressure in a chinese rural population: the rural diabetes, obesity and lifestyle (RuralDiab) study. J Hum Hypertens. 2018;32:236–46.
Beale AL, Kaye DM, Marques FZ. The role of the gut microbiome in sex differences in arterial pressure. Biol Sex Diff. 2019;10:22.
Colafella KMM, Denton KM. Sex-specific differences in hypertension and associated cardiovascular disease. Nat Rev Nephrol. 2018;14:185–201.
Lau E, Neves JS, Ferreira-Magalhaes M, Carvalho D, Freitas P. Probiotic ingestion, obesity, and metabolic-related disorders: results from NHANES, 1999–2014. Nutrients. 2019;11:1482.
Zhernakova A, Kurilshikov A, Bonder MJ, Tigchelaar EF, Schirmer M, Vatanen T, Mujagic Z, Vila AV, Falony G, Vieira-Silva S, et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science. 2016;352:565–9.
Choi MS, Yu JS, Yoo HH, Kim D-H. The role of gut microbiota in the pharmacokinetics of antihypertensive drugs. Pharmacol Res. 2018;130:164–71.
National Center for Cardiovascular Diseases, Chinese Medical Doctor Association, Hypertension Committee of the Chinese Medical Doctor Association. Chinese society of cardiology, chinese medical association, hypertension committee of cross-straits medicine exchange association. clinical practice guidelines for the management of hypertension in China. Chin J Cardiol. 2022;50:1050–95.
Whelton PK, Carey RM, Aronow WS, Casey DE Jr, Collins KJ, Himmelfarb CD, DePalma SM, Gidding S, Jamerson KA, Jones DW, et al. ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NM CNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults a report of the American college of cardiology/american heart association task force on clinical practice guidelines. Hypertension. 2018;71:1269–324.
Franzosa EA, Morgan XC, Segata N, Waldron L, Reyes J, Earl AM, Giannoukos G, Boylan MR, Ciulla D, Gevers D, et al. Relating the metatranscriptome and metagenome of the human gut. Proc Natl Acad Sci USA. 2014;111:E2329–38.
Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10:996–8.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6.
Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–7.
Qin JJ, Li RQ, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65.
Ma J, Li H. The role of Gut microbiota in atherosclerosis and hypertension. Front Pharmacol. 2018;9:1082.
Qi Y, Kim S, Richards EM, Raizada MK, Pepine CJ. Gut microbiota potential for a unifying hypothesis for prevention and treatment of hypertension. Circul Res. 2017;120:1724–6.
Howitt MR, Garrett WS. A complex microworld in the gut gut microbiota and cardiovascular disease connectivity. Nat Med. 2012;18:1188–9.
Sekirov I, Russell SL, Antunes LC, Finlay BB. Gut microbiota in health and disease. Physiol Rev. 2010;90:859–904.
Yang T, Santisteban MM, Rodriguez V, Li E, Ahmari N, Carvajal JM, Zadeh M, Gong MH, Qi YF, Zubcevic J, et al. Gut dysbiosis is linked to hypertension. Hypertension. 2015;65:1331–40.
de La Cuesta-Zuluag J, Kelley ST, Chen Y, Escobar JS, Mueller NT, Ley RE, McDonald D, Huang S, Swafford AD, Knight R, Thackray VG. Age- and sex-dependent patterns of gut microbial diversity in human adults. Msystems. 2019;4:e00261–00219.
Bowyer RCE, Jackson MA, Le Roy CI, Lochlainn MN, Spector TD, Dowd JB, Steves CJ. Socioeconomic status and the gut microbiome: a TwinsUK cohort study. Microorganisms. 2019;7:17.
Valeri F, Endres K. How biological sex of the host shapes its gut microbiota. Front Neuroendocr. 2021;61:15.
Takagi T, Naito Y, Inoue R, Kashiwagi S, Uchiyama K, Mizushima K, Tsuchiya S, Dohi O, Yoshida N, Kamada K, et al. Differences in gut microbiota associated with age, sex, and stool consistency in healthy japanese subjects. J Gastroenterol. 2019;54:53–63.
Bolnick DI, Snowberg LK, Hirsch PE, Lauber CL, Org E, Parks B, Lusis AJ, Knight R, Caporaso JG, Svanback R. Individual diet has sex-dependent effects on vertebrate gut microbiota. Nat Commun. 2014;5:13.
Richards EM, Pepine CJ, Raizada MK, Kim S. The Gut, its microbiome, and hypertension. Curr Hypertens Rep. 2017;19:36.
Takase H, Sugiura T, Kimura G, Ohte N, Dohi Y. Dietary sodium consumption predicts future blood pressure and incident hypertension in the Japanese normotensive general population. J Am Heart Assoc. 2015;4:e001959.
Streppel MT, Arends LR, van’t Veer P, Grobbee DE, Geleijnse JM. Dietary fiber and blood pressure—a meta-analysis of randomized placebo-controlled trials. Arch Intern Med. 2005;165:150–6.
Whelton SP, Hyre AD, Pedersen B, Yi Y, Whelton PK, He J. Effect of dietary fiber intake on blood pressure: a meta-analysis of randomized, controlled clinical trials. J Hypertens. 2005;23:475–81.
Chen L, He F, Dong YB, Huang Y, Wang CQ, Harshfield GA, Zhu HD. Modest Sodium reduction increases circulating short-chain fatty acids in untreated hypertensives a Randomized, Double-Blind, placebo-controlled trial. Hypertension. 2020;76:73–9.
Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H, Haase S, Mahler A, Balogh A, Marko L, et al. Salt-responsive gut commensal modulates T(H)17 axis and disease. Nature. 2017;551:585–9.
Marques FZ, Nelson E, Chu PY, Horlock D, Fiedler A, Ziemann M, Tan JK, Kuruppu S, Rajapakse NW, El-Osta A, et al. High-Fiber Diet and acetate supplementation change the gut microbiota and prevent the development of hypertension and heart failure in hypertensive mice. Circulation. 2017;135:964–77.
Bartolomaeus H, Balogh A, Yakoub M, Homann S, Marko L, Hoeges S, Tsvetkov D, Krannich A, Wundersitz S, Avery EG, et al. Short-chain fatty acid propionate protects from hypertensive cardiovascular damage. Circulation. 2019;139:1407–21.
Maier TV, Lucio M, Lee LH, VerBerkmoes NC, Brislawn CJ, Bernhardt J, Lamendella R, McDermott JE, Bergeron N, Heinzmann SS, et al. Impact of Dietary Resistant Starch on the human gut Microbiome, Metaproteome, and Metabolome. mBio. 2017;8:e01343–01317.
Yang F, Chen H, Gao Y, An N, Li X, Pan X, Yang X, Tian L, Sun J, Xiong X, Xing Y. Gut microbiota-derived short-chain fatty acids and hypertension: mechanism and treatment. Biomed Pharmacother. 2020;130:110503.
Moore WEC, Cato EP, Holdeman LV. Some current concepts in intestinal bacteriology. Am J Clin Nutr. 1978;31:33-S42.
Wolever TMS, Spadafora P, Eshuis H. Interaction between Colonic acetate and propionate in humans. Am J Clin Nutr. 1991;53:681–7.
Cummings JH, Pomare EW, Branch WJ, Naylor CPE, Macfarlane GT. Short chain fatty acids in human large intestine, portal, hepatic and venouse blood. Gut. 1987;28:1221–7.
Poll BG, Cheema MU, Pluznick JL. Gut microbial metabolites and blood pressure regulation: focus on SCFAsand TMAO. Physiology. 2020;35:275–84.
Muralitharan RR, Jama HA, Xie L, Peh A, Snelson M, Marques FZ. Microbial peer pressure the role of the gut microbiota in hypertension and its complications. Hypertension. 2020;76:1674–87.
Avery EG, Bartolomaeus H, Maifeld A, Marko L, Wiig H, Wilck N, Rosshart SP, Forslund SK, Muller DN. The gut microbiome in hypertension recent advances and future perspectives. Circul Res. 2021;128:934–50.
Gomez-Arango LF, Barrett HL, McIntyre HD, Callaway LK, Morrison M, Nitert MD, Grp ST. Increased systolic and diastolic blood pressure is Associated with altered gut microbiota composition and butyrate production in early pregnancy. Hypertension. 2016;68:974–81.
Koh A, De Vadder F, Kovatcheva-Datchary P, Bäckhed F. From Dietary Fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell. 2016;165:1332–45.
Miyamoto J, Kasubuchi M, Nakajima A, Irie J, Itoh H, Kimura I. The role of short-chain fatty acid on blood pressure regulation. Curr Opin Nephrol Hypertens. 2016;25:379–83.
Canale MP, Noce A, Di Lauro M, Marrone G, Cantelmo M, Cardillo C, Federici M, Di Daniele N, Tesauro M. Gut dysbiosis and western diet in the pathogenesis of essential arterial hypertension: a narrative review. Nutrients. 2021;13:1162.
Gill PA, van Zelm MC, Muir JG, Gibson PR. Short chain fatty acids as potential therapeutic agents in human gastrointestinal and inflammatory disorders. Aliment Pharmacol Ther. 2018;48:15–34.
Macfarlane S, Macfarlane GT. Regulation of short-chain fatty acid production. Proc Nutr Soc. 2003;62:67–72.
Li A, Wang N, Li N, Li B, Yan F, Song Y, Hou J, Huo G. Modulation effect of chenpi extract on gut microbiota in high-fat diet-induced obese C57BL/6 mice. J Food Biochem. 2021;45:e13541.
Piñero F, Vazquez M, Baré P, Rohr C, Mendizabal M, Sciara M, Alonso C, Fay F, Silva M. A different gut microbiome linked to inflammation found in cirrhotic patients with and without hepatocellular carcinoma. Ann Hepatol. 2019;18:480–7.
Tong T, Wang YN, Zhang CM, Kang SG. In vitro and in vivo antihypertensive and antioxidant activities of fermented roots of Allium hookeri. Chin Herb Med. 2021;13:541–8.
Mörkl S, Lackner S, Meinitzer A, Mangge H, Lehofer M, Halwachs B, Gorkiewicz G, Kashofer K, Painold A, Holl AK, et al. Gut microbiota, dietary intakes and intestinal permeability reflected by serum zonulin in women. Eur J Nutr. 2018;57:2985–97.
Kang C, Wang B, Kaliannan K, Wang X, Lang H, Hui S, Huang L, Zhang Y, Zhou M, Chen M, Mi M. Gut microbiota mediates the protective effects of dietary capsaicin against chronic low-grade inflammation and associated obesity induced by high-fat diet. mBio. 2017;8:e00470-17.
Leclercq S, Matamoros S, Cani PD, Neyrinck AM, Jamar F, Stärkel P, Windey K, Tremaroli V, Bäckhed F, Verbeke K, et al. Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc Natl Acad Sci U S A. 2014;111:E4485–93.
Yu Y, Mao G, Wang J, Zhu L, Lv X, Tong Q, Fang Y, Lv Y, Wang G. Gut dysbiosis is associated with the reduced exercise capacity of elderly patients with hypertension. Hypertens Res. 2018;41:1036–44.
Li J, Yang X, Zhou X, Cai J. The role and mechanism of intestinal flora in blood pressure regulation and hypertension development. Antioxid Redox Signal. 2021;34:811–30.
Qin Y, Zhao J, Wang Y, Bai M, Sun S. Specific alterations of gut microbiota in chinese patients with hypertension: a systematic review and Meta-analysis. Kidney Blood Press Res. 2022;47:433–47.
Santisteban MM, Kim S, Pepine CJ, Raizada MK. Brain-gut-bone Marrow Axis: implications for hypertension and related therapeutics. Circ Res. 2016;118:1327–36.
Hsu CN, Chan JYH, Wu KLH, Yu HR, Lee WC, Hou CY, Tain YL. Altered gut microbiota and its metabolites in hypertension of developmental origins: exploring differences between fructose and antibiotics exposure. Int J Mol Sci. 2021;22:2674.
Xie D, Zhang MS, Wang BL, Lin H, Wu EQ, Zhao HH, Li SC. Differential analysis of hypertension-associated intestinal microbiota. Int J Med Sci. 2019;16:872–81.
Fu JY, Bonder MJ, Cenit MC, Tigchelaar EF, Maatman A, Dekens JAM, Brandsma E, Marczynska J, Imhann F, Weersma RK, et al. The gut Microbiome contributes to a substantial proportion of the variation in blood lipids. Circul Res. 2015;117:817–24.
Qin JJ, Li YR, Cai ZM, Li SH, Zhu JF, Zhang F, Liang SS, Zhang WW, Guan YL, Shen DQ, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60.
Lambeth SM, Carson T, Lowe J, Ramaraj T, Leff JW, Luo L, Bell CJ, Shah VO. Composition, diversity and abundance of gut microbiome in prediabetes and type 2 diabetes. J diabetes Obes. 2015;2:1–7.
Org E, Blum Y, Kasela S, Mehrabian M, Kuusisto J, Kangas AJ, Soininen P, Wang Z, Ala-Korpela M, Hazen SL, et al. Relationships between gut microbiota, plasma metabolites, and metabolic syndrome traits in the METSIM cohort. Genome Biol. 2017;18:70.
Ren Z, Li A, Jiang J, Zhou L, Yu Z, Lu H, Xie H, Chen X, Shao L, Zhang R, et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut. 2019;68:1014–23.
Liu B, Lin W, Chen S, Xiang T, Yang Y, Yin Y, Xu G, Liu Z, Liu L, Pan J, Xie L. Gut microbiota as a subjective measurement for auxiliary diagnosis of insomnia disorder. Front Microbiol. 2019;10:1770.
Liu H, Chen X, Hu X, Niu H, Tian R, Wang H, Pang H, Jiang L, Qiu B, Chen X, et al. Alterations in the gut microbiome and metabolism with coronary artery disease severity. Microbiome. 2019;7:68.
Zuo K, Li J, Li K, Hu C, Gao Y, Chen M, Hu R, Liu Y, Chi H, Wang H, et al. Disordered gut microbiota and alterations in metabolic patterns are associated with atrial fibrillation. Gigascience. 2019;8:giz058.
Fernandes R, Viana SD, Nunes S, Reis F. Diabetic gut microbiota dysbiosis as an inflammaging and immunosenescence condition that fosters progression of retinopathy and nephropathy. Biochim Et Biophys Acta-Molecular Basis Disease. 2019;1865:1876–97.
Cho I, Blaser MJ. The human microbiome: at the interface of health and disease. Nat Rev Genet. 2012;13:260–70.
Aagaard K, Petrosino J, Keitel W, Watson M, Katancik J, Garcia N, Patel S, Cutting M, Madden T, Hamilton H, et al. The human Microbiome Project strategy for comprehensive sampling of the human microbiome and why it matters. FASEB J. 2013;27:1012–22.
Haro C, Rangel-Zuniga OA, Alcala-Diaz JF, Gomez-Delgado F, Perez-Martinez P, Delgado-Lista J, Quintana-Navarro GM, Landa BB, Navas-Cortes JA, Tena-Sempere M, et al. Intestinal microbiota is influenced by gender and body Mass Index. PLoS ONE. 2016;11:16.
Yoon K, Kim N. Roles of sex hormones and gender in the gut microbiota. J Neurogastroenterol Motil. 2021;27:314–25.
Siddiqui R, Makhlouf Z, Alharbi AM, Alfahemi H, Khan NA. The gut microbiome and female health. Biology. 2022;11:1683.
Chen L, Liu Y, Tang Z, Song Z, Cao F, Shi X, Xie P, Wei P, Li M. Radix Angelica dahuricae extract ameliorates oestrogen deficiency-induced dyslipidaemia in ovariectomized (OVX) rats by modulating the gut microbiota and bile acid signalling. Phytomedicine. 2022;107:154440.
Yano Y, Niiranen TJ. Gut microbiome over a lifetime and the association with hypertension. Curr Hypertens Rep. 2021;23:15.
Acknowledgements
Not applicable.
Funding
This work was supported by the Youth Program of Xi’an Municipal Health Commission of China, No. 2022qn07; the General Program of Xi’an Municipal Health Commission of China, No. 2023ms11 and No. 2020ms14; the National Natural Science Foundation of China, No. 81702067.
Author information
Authors and Affiliations
Contributions
L-J and G-L searched and reviewed published articles, interpreted data, constructed tables, drafted the article, and made substantial contributions to the conception and design of this study. W-JH, Z-HP, Y-Y, Z-MY, Y-WJ, L-JY and Z-Y conducted the experiments, performed sequencing analysis and constructed the figures and tables. W-JH and W-GD performed statistical analysis. Y-YJ contributed to the conception of the study. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of Honghui Hospital, Xi’an Jiaotong University (Protocol number 201801022) and was registered on the Chinese Clinical Trial Registry (http://www.chictr.org.cn/; registration number ChiCTR1800019191). Informed consent was provided by all recruited subjects prior to the study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
12967_2023_4176_MOESM1_ESM.docx
Additional file 1: Table S1 Correlations between gut microbiota taxa and blood pressure levels in female subjects. Table S2 Correlations between gut microbiota taxa and blood pressure levels in male subjects. Fig. S1 Relative abundances and comparative analysis of the annotated KEGG functions of gut microbiota in female and male subjects. The KEGG functions with significant differences between groups are presented by Wilcoxon rank-sum test. #P < 0.1, *P < 0.05, **P < 0.01.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lv, J., Wang, J., Yu, Y. et al. Alterations of gut microbiota are associated with blood pressure: a cross-sectional clinical trial in Northwestern China. J Transl Med 21, 429 (2023). https://doi.org/10.1186/s12967-023-04176-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-023-04176-6