
Abstract
Mammalian cells responding to specific perturbations of homeostasis can undergo a regulated variant of cell death that elicits adaptive immune responses. As immunogenic cell death (ICD) can only occur in a precise cellular and organismal context, it should be conceptually differentiated from instances of immunostimulation or inflammatory responses that do not mechanistically depend on cellular demise. Here, we critically discuss key conceptual and mechanistic aspects of ICD and its implications for cancer (immuno)therapy.
Similar content being viewed by others


Introduction
All mammalian cells (including normal and neoplastic cells) respond to relatively mild perturbations of homeostasis by activating signal transduction cascades aimed at repairing macromolecular and/or organellar damage and restoring normal cellular functions [1,2,3,4]. When successful, such stress responses fully re-establish cellular homeostasis, hence preserving organismal fitness [5, 6]. Conversely, failed adaptation to stress generally elicits regulated cell death (RCD) as a means to preserve organismal homeostasis in the context of cellular loss [7,8,9].
Importantly, most (if not all) cellular responses to stress are hard-wired to immune signaling [10]. Thus, even when normal cellular functions are ultimately restored, stressed cells pre-alert the immune system of a potential danger by: (1) altering their surface properties, and (2) releasing cytokines, chemokines and so-called damage-associated molecular patterns (DAMPs) [11,12,13]. Generally, these signals support the establishment of an inflammatory response that recruits innate immune effector cells to sites of cellular stress, but per se fail to elicit antigen-specific adaptive immunity [10]. Such an immune engagement, however, serves as a platform for the potential initiation of adaptive immune responses if stressed cells fail to recover homeostasis and ultimately undergo RCD [2, 14]. Whether RCD ultimately promotes or inhibits antigen-specific immune responses depends on several critical determinants [15, 16].
Here, we discuss key determinants of immunogenic cell death (ICD) and provide a brief overview of accumulating data on the prominent implications of ICD for cancer (immuno)therapy.
Core ICD determinants
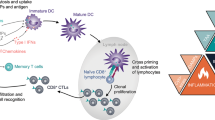
Five core features are required for RCD to elicit antigen-specific immune responses (over mere innate immune signaling coupled to inflammation) of relevance for cancer (immuno)therapy (Fig. 1). As discussed here below, the absence of any of these determinants converts ICD into immunologically silent or even tolerogenic variants of RCD.

Core requirements for the initiation of adaptive immune responses by dying cells. For cell death to drive bona fide adaptive immune responses: (1) cell death must occur in the context of adaptive stress responses; (2) cell death must ultimately occur, as opposed to successful adaptation to stress; (3) dying cells must present antigens that are not covered by thymic tolerance; (4) regulated cell death (RCD) must be accompanied by the emission of endogenous molecules that operate as immunological adjuvants; and (5) microenvironmental conditions must be permissive for antigen-presenting cell (APC) recruitment, maturation and migration to lymph nodes (or other sites of antigen presentation), as well as for cytotoxic T lymphocyte (CTL) infiltration and activation. Depending on which of these conditions is lacking, cell death can drive innate immune signaling coupled with local inflammation, actively promote immunological tolerance and/or result in antigen-specific CTL priming and expansion but no effector immune response. ACD, accidental cell death; DAMP, damage-associated molecular pattern; ICD, immunogenic cell death
Stress
Cell death is not immunogenic when it occurs as an accidental, unregulated process that does not involve adaptation to stress, as in the presence of very harsh physicochemical or mechanic conditions (which can be modeled in experimental settings, but are quite rare in human pathophysiology) [17]. In line with this notion, cancer cells succumbing to a variety of therapeutic agents including selected chemotherapies [18], targeted anticancer agents [19] and radiation therapy (RT) [20] can be successfully used to elicit prophylactic anticancer immunity upon inoculation in immunocompetent, syngeneic hosts. However, the same does not hold true when the same cells are killed instantaneously by freeze-thawing cycles [21, 22]. Interestingly, although accidental cell death (ACD) occurring in the absence of stress responses results in a necrotic morphology that has been consistently associated with inflammation in patients affected by a variety of conditions, rapid ACD may turn out to be considerably less inflammatory than stress-driven regulated instances of necrosis such as necroptosis or pyroptosis [23]. Indeed, many of the immunostimulatory signals underlying inflammatory responses to necrotic cells are actively synthesized during stress responses (e.g., cytokines, chemokines) or released along with (failing) adaptation to stress (e.g., DAMPs) [13].
Death
As mentioned above, cell death must occur for perturbations of cellular homeostasis to ultimately results in adaptive immune responses [15]. Thus, while successful adaptation to stress may still ignite local inflammatory responses, cancer cells must die for their corpses to be efficiently taken up by antigen-presenting cells (APCs), especially dendritic cells (DCs), and processed for antigen presentation [24, 25]. At least in part, this reflects the notion that immature DCs are highly proficient at (macro)pinocytosis, which involves material of subcellular size, but much less so at engulfing entire cells [26]. While macrophages are surely more efficient than DCs at the latter, (1) living cancer cells tend to express high levels of anti-phagocytic molecules such as CD47 [27, 28], but much less so pro-phagocytic molecules such as calreticulin (CALR) [29], on their surface; (2) macrophages generally take up cells and their corpses in an immunologically silent manner [30, 31]; (3) macrophages have limited migratory capacity and hence do not reach lymph nodes and generally are excluded by intratumoral tertiary lymphoid structures (another site of efficient antigen presentation to T cells) [32].
Antigenicity
Cancer (and normal) cells undergoing stress-driven RCD must be sufficiently antigenic to elicit adaptive immune responses [33]. This means that dying cells must express antigens whose cognate T-cell receptor (TCR) has not be purged by the circulating T-cell repertoire during thymic selection [34]. The source of such antigenicity can vary quite considerably as it encompasses (1) pathogen-encoded antigens [15, 35], (2) mutational neoantigens [36], and (3) a large and hitherto poorly recognized panel of non-mutational neoantigens as generated, for instance, by epigenetic alterations resulting in transcriptional shifts [37], alternative splicing events [38], enzymatic and non-enzymatic protein modifications [39], and/or translation of cryptic sequences [40]. Thus, irrespective of antigen source, RCD can be immunogenic in one host but not necessarily in another, simply reflecting interindividual differences in the circulating T-cell repertoire [41]. In the absence of antigenicity, stress-driven RCD causes robust inflammatory reactions that are relevant for a variety of non-malignant disorders [42], but it fails to engage adaptive immune modules.
Adjuvanticity
Similar to prophylactic vaccines against pathogens, ICD requires robust adjuvants to initiate adaptive immune responses [43, 44]. Such adjuvants, which are commonly referred to as DAMPs, are fully endogenous to dying cells and are generally released or exposed on the plasma membrane as a consequence of pre-mortem cellular stress [45]. DAMPs can be broadly classified into three main families: pro-phagocytic signals, immunostimulatory molecules and cytokines/chemokines [46]. The prototypic ICD-associated “eat-me” signal is CALR, an endoplasmic reticulum (ER) chaperone that is exposed on the outer leaflet of the plasma membrane downstream of the integrated stress response (ISR) and consequent phosphorylation of eukaryotic translation initiation factor 2 subunit alpha (EIF2S1, best known as eIF2α) [47, 48]. Common immunostimulatory DAMPs mechanistically linked to ICD encompass ATP, which is actively secreted by an autophagy-dependent mechanism [49], as well as high mobility group box 1 (HMGB1) and annexin A1, both of which appear to be passively released upon nuclear and plasma membrane permeabilization [50,51,52]. Finally, type I interferon (IFN) and C-X-C motif chemokine ligand 10 (CXCL10) have been involved in multiple instances of ICD [53, 54]. Of note, multiple DAMPs operate by binding to pattern recognition receptors (PRRs) expressed on immune cells that originally evolved as part of the host defense from pathogens, such as Toll-like receptor 4 (TLR4), which binds HMGB1 [46], and formyl peptide receptor 1 (FPR1), which binds ANXA1 [52]. Thus, not only defects in DAMP emission, but also lack or dysfunction of cognate PRRs can abolish the immunogenicity of RCD. Importantly, in the absence of adjuvanticity, the stress-driven demise of cells with sufficient antigenicity actively drives DC-dependent immune tolerance [10].
Microenvironment
There is an important microenvironmental component in the elicitation of adaptive immunity by cancer cells undergoing RCD [55]. On the one hand, the microenvironment of dying cells must be permissive for infiltration by APC precursors, their maturation/activation and either their egress to draining lymph nodes or their incorporation into tertiary lymphoid structures for local antigen presentation to T cells [56]. Thus, while in prophylactic experimental settings (that involve the subcutaneous administration of cancer cells exposed to ICD-inducing agents in vitro) the dermis offers a privileged, fully immunocompetent microenvironment for the elicitation of adaptive immunity (provided that all other core ICD determinants are present) [43, 57], the same may not always hold true when RCD occurs within the TME, which is generally dominated by immunosuppressive mechanisms that may interfere with APC functions [58, 59]. On the other hand, antigen specific T cells as efficiently primed by ICD-elicited APCs must have access to their targets and encounter favorable conditions for mediating effector functions [58, 60]. This implies that even in the context of robust T cell priming and clonal expansion, malignant lesions may be protected from immunological eradication as a consequence of stromal exclusion and/or local immunosuppression, for instance upon direct T cell inhibition via CD274 (best known as PD-L1).
Taken together, these observations delineate the key molecular and cellular components of adaptive immune responses elicited by ICD, as opposed to innate immune signaling and inflammation as driven by non-immunogenic RCD variants. Supporting the central relevance of each of these mechanisms, both pathogens and malignant cells have evolved a variety of strategies to either subvert immunogenic stress signaling, RCD, antigenicity and/or adjuvanticity, or condition the microenvironment to suppress ICD initiation or execution [15, 61]. Discussing these strategies in detail, however, goes largely beyond the scope of the present Commentary.
ICD and cancer (immuno)therapy
Accumulating preclinical and clinical data suggest that the induction of ICD is particularly relevant for the efficacy of cancer (immuno)therapy [62].
Preclinical evidence
In a variety of rodent tumor models, ICD signaling has been mechanistically linked to superior responses to clinically relevant therapies, including (but not limited to) chemotherapy based on anthracycline and (some) platinum derivatives [63, 64], targeted anticancer agents specific for epidermal growth factor receptor (EGFR) [19], multitarget tyrosine kinase inhibitors [65], radiation therapy [66, 67], and photodynamic therapy [68]. Specifically, in numerous prophylactic or therapeutic experimental settings involving the aforementioned clinically relevant agents, pharmacological or genetic strategies interrupting stress signaling in cancer cells, DAMP emission therefrom, or DAMP detection by immune cells compromised the emergence of protective anticancer immunity or disease control, respectively [45]. Similar defects in prophylactic or therapeutic disease control have been documented upon the depletion or inhibition of numerous immune effector cells involved in the elicitation of anticancer immunity downstream of ICD, such as DCs [47], interleukin 17 A (IL17A)-producing γδ T cells [69], as well as CD4+ and CD8+ T cells [70]. Importantly though, documenting a drop in treatment efficacy in tumor-bearing mice subjected to pharmacological or genetic strategies that block specific immune functions as compared to their fully immunocompetent counterparts does not necessarily identify bona fide ICD induction [15]. Along similar lines, while a wide panel of bona fide ICD inducers have been shown to synergize (or at least positively interact) with immune checkpoint inhibitors (ICIs) in otherwise ICI-resistant mouse tumor models [71], the formal implication of ICD in these findings remains to be formally elucidated. Indeed, multiple anticancer agents exert therapeutically relevant immunostimulatory effects that are RCD-independent and rather reflect the direct interactions between such agents and vascular, stromal, immunological or microbial components of the local or systemic TME [72]. This latter consideration largely justifies prophylactic vaccination assays as a simple, widely applicable experimental approach to discriminate between bona fide ICD and the RCD-independent derepression of pre-existing (ICI-actionable) adaptive immune responses [15].
Clinical evidence
At least three lines of correlative clinical evidence are available in support of the key relevance of ICD for cancer (immuno)therapy. First, in numerous cohorts of patients with cancer, defects in immunogenic stress signaling, RCD, DAMP emission or DAMP sensing have been shown to have a detrimental impact not only on prognosis in largely unselected patient populations [52], but also on response to ICD-inducing therapeutic agents [51]. Such defects encompass molecular or transcriptional signatures of suboptimal cellular responses to stress (e.g., poor eIF2α phosphorylation) [73], reduced expression levels of specific DAMPs or receptors thereof (e.g., low CALR expression) [74], as well as single-nucleotide polymorphisms associated with limited PRR signaling (in TLR4 or FPR1, for instance) [51, 52].
Second, a considerable fraction of the therapeutic armamentarium currently available for clinical cancer management has been shown to elicit ICD (or other forms of immunostimulation) [75]. Importantly, these approaches have often been developed into clinically efficient therapies in an empirical and immune agnostic manner (i.e., harnessing human cancer xenografts in immunodeficient mice at preclinical stages and developing therapeutic schedules in patients via the maximum tolerated dose paradigm) [76]. Thus, if (ICD-driven) anticancer immunity had relevance for therapeutic outcome, one would expect immunostimulatory agents (including ICD inducers) to be enriched as compared to immunosuppressive (or immunologically neutral) therapies, which currently is the case [10]. Moreover, drug discovery programs have been designed to actively search for ICD inducer and two of such drugs, i.e., lurbinectedin and belantamab mafodotin, have received regulatory approval for use in cancer patients [77, 78].
Third, in line with preclinical findings, a growing number of ICD inducers positively interact with ICIs or other immunotherapeutic approaches in patients with cancer [79, 80]. Notable examples of such successful combinations include (1) nab-paclitaxel plus atezolizumab (an ICI specific for PD-L1), which is currently employed in the management of triple negative breast cancer (TNBC) [81], carboplatin/etoposide plus atezolizumab, which is approved for patients with extensive-stage small cell lung cancer (SCLC) [82], as well nab-paclitaxel/carboplatin plus the programmed cell death 1 (PDCD1, best known as PD-1) blocker pembrolizumab [83].
Altogether, these preclinical and clinical findings suggest that ICD induction plays a major role in the successful control of multiple neoplasms by (immuno)therapy.
Conclusions and future perspectives
In summary, ICD-driven adaptive immunity is mechanistically and conceptually different from both inflammatory reactions driven by non-immunogenic variants of RCD and adaptive immune responses that do not rely on cell stress and death. Importantly, several RCD routines have been characterized in molecular terms and classified based on the mechanistic involvement of specific signal transduction cascades (Table 1) [9]. For instance, apoptosis is currently defined as an RCD variant that is precipitated by the activation of cysteine proteases of the caspase family, while necroptosis involves the activating phosphorylation of receptor interacting serine/threonine kinase 3 (RIPK3) and consequent phosphorylation-dependent oligomerization of the pore-forming protein mixed lineage kinase domain like pseudokinase (MLKL) [84]. That said, once adaptation to stress fails, cells appear to die irrespective of active signaling, largely because of bioenergetic failure and/or irreparable damage to macromolecular structures that underlie cellular homeostasis itself, including (but not limited to) organelles and membranes [9]. The signal transduction cascades elicited during cell death rather seem to determine the kinetic and immunological manifestations of the process, rather than its occurrence sensu stictu [85]. In line with this notion, both pharmacological and genetic interventions targeting so-called “executioners” of cell death invariably delay the cellular demise, but do not prevent it, at least in mammalian systems [9].
Most importantly, the biochemical cascades underlying RCD in its multiple variants are not necessarily linked to its immunogenicity [85]. As a standalone example, apoptotic cell death as precipitated by caspases is normally an immunologically silent event, largely reflecting the ability of caspase 3 (CASP3) to initiate signaling pathways that promote macrophage-mediated efferocytosis in the absence of active immunostimulatory signaling and the overall implication of apoptosis in development and adult tissue homeostasis [86]. However, multiple caspase-dependent instances of RCD that classify as ICD by all definitions have been reported [22, 87]. Thus, the immunogenicity of a specific RCD instance cannot be determined with certainty based on the molecular pathways that precipitate RCD only, as abundantly discussed herein. Indeed, RCD-independent, host-related factors including antigenicity and microenvironmental parameters stand out as critical determinants of RCD immunogenicity [10].
Despite this and other conceptual (and experimental) caveats, ICD stands out as a major, therapeutically actionable process for cancer immuno(therapy). Future efforts will have to focus on identifying novel, clinically useful ICD inducers (irrespective of the RCD mode they impinge on) as well as biomarkers predicting the likelihood of specific neoplastic lesions to elicit adaptive immune responses downstream of ICD in response to treatment. Alongside, it will be important to devise clinically viable strategies to increase the immunogenicity of otherwise immunologically silent RCD variants, and to investigate novel combinatorial regimens combining ICD inducers and immunotherapy in the clinic, with the ultimate goal to facilitate efficient anticancer immunosurveillance. We surmise that ICD induction will occupy an ever more central stage in modern cancer management.
Availability of data and materials
Not applicable.
References
Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. 2020;21:421–38.
Galluzzi L, Yamazaki T, Kroemer G. Linking cellular stress responses to systemic homeostasis. Nat Rev Mol Cell Biol. 2018;19:731–45.
He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93.
Groenendyk J, Agellon LB, Michalak M. Calcium signaling and endoplasmic reticulum stress. Int Rev Cell Mol Biol. 2021;363:1–20.
Hofer SJ, Kroemer G, Kepp O. Autophagy-inducing nutritional interventions in experimental and clinical oncology. Int Rev Cell Mol Biol. 2022;373:125–58.
Miller DR, Cramer SD, Thorburn A. The interplay of autophagy and non-apoptotic cell death pathways. Int Rev Cell Mol Biol. 2020;352:159–87.
Fairlie WD, Tran S, Lee EF. Crosstalk between apoptosis and autophagy signaling pathways. Int Rev Cell Mol Biol. 2020;352:115–58.
Chong SJF, Marchi S, Petroni G, Kroemer G, Galluzzi L, Pervaiz S. Noncanonical cell fate regulation by Bcl-2 proteins. Trends Cell Biol. 2020;30:537–55.
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, et al. Molecular mechanisms of cell death: recommendations of the nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541.
Kroemer G, Galassi C, Zitvogel L, Galluzzi L. Immunogenic cell stress and death. Nat Immunol. 2022;23:487–500.
Wu Chuang A, Kepp O, Kroemer G, Bezu L. Endoplasmic reticulum stress in the cellular release of damage-associated molecular patterns. Int Rev Cell Mol Biol. 2020;350:1–28.
Fucikova J, Moserova I, Urbanova L, Bezu L, Kepp O, Cremer I, Salek C, Strnad P, Kroemer G, Galluzzi L, Spisek R. Prognostic and predictive value of DAMPs and DAMP-Associated processes in Cancer. Front Immunol. 2015;6:402.
Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12:860–75.
Green DR. The coming decade of cell death research: five riddles. Cell. 2019;177:1094–107.
Galluzzi L, Vitale I, Warren S, Adjemian S, Agostinis P, Martinez AB, Chan TA, Coukos G, Demaria S, Deutsch E et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J Immunother Cancer2020,8.
Melero I, Alvarez M, Minute L, Berraondo P. Premortem Tumor stress in Radioimmunotherapy. Trends Cancer. 2020;6:173–4.
Fucikova J, Kepp O, Kasikova L, Petroni G, Yamazaki T, Liu P, Zhao L, Spisek R, Kroemer G, Galluzzi L. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020;11:1013.
Galluzzi L, Humeau J, Buqué A, Zitvogel L, Kroemer G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat Rev Clin Oncol. 2020;17:725–41.
Petroni G, Buqué A, Zitvogel L, Kroemer G, Galluzzi L. Immunomodulation by targeted anticancer agents. Cancer Cell. 2021;39:310–45.
Rodriguez-Ruiz ME, Vitale I, Harrington KJ, Melero I, Galluzzi L. Immunological impact of cell death signaling driven by radiation on the tumor microenvironment. Nat Immunol. 2020;21:120–34.
Minute L, Teijeira A, Sanchez-Paulete AR, Ochoa MC, Alvarez M, Otano I, Etxeberrria I, Bolaños E, Azpilikueta A, Garasa S et al. Cellular cytotoxicity is a form of immunogenic cell death. J Immunother Cancer2020,8.
Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas-Stubbs S, Obeid M, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005;202:1691–701.
Wang Q, Wang Y, Ding J, Wang C, Zhou X, Gao W, Huang H, Shao F, Liu Z. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature. 2020;579:421–6.
Kotsias F, Cebrian I, Alloatti A. Antigen processing and presentation. Int Rev Cell Mol Biol. 2019;348:69–121.
Martinek J, Wu TC, Cadena D, Banchereau J, Palucka K. Interplay between dendritic cells and cancer cells. Int Rev Cell Mol Biol. 2019;348:179–215.
Wang Y, Xiang Y, Xin VW, Wang XW, Peng XC, Liu XQ, Wang D, Li N, Cheng JT, Lyv YN, et al. Dendritic cell biology and its role in tumor immunotherapy. J Hematol Oncol. 2020;13:107.
Logtenberg MEW, Scheeren FA, Schumacher TN. The CD47-SIRPα immune checkpoint. Immunity. 2020;52:742–52.
Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science. 2000;288:2051–4.
Fucikova J, Spisek R, Kroemer G, Galluzzi L. Calreticulin and cancer. Cell Res. 2021;31:5–16.
Martins I, Kepp O, Galluzzi L, Senovilla L, Schlemmer F, Adjemian S, Menger L, Michaud M, Zitvogel L, Kroemer G. Surface-exposed calreticulin in the interaction between dying cells and phagocytes. Ann N Y Acad Sci. 2010;1209:77–82.
Gordon S, Plüddemann A. Macrophage clearance of apoptotic cells: a critical assessment. Front Immunol. 2018;9:127.
Fridman WH, Meylan M, Petitprez F, Sun CM, Italiano A, Sautès-Fridman C. B cells and tertiary lymphoid structures as determinants of tumour immune contexture and clinical outcome. Nat Rev Clin Oncol. 2022;19:441–57.
Vitale I, Sistigu A, Manic G, Rudqvist NP, Trajanoski Z, Galluzzi L. Mutational and antigenic landscape in tumor progression and cancer immunotherapy. Trends Cell Biol. 2019;29:396–416.
Rudqvist NP, Galluzzi L. T cells: friends and foes. Int Rev Cell Mol Biol. 2019;342:xi–xiv.
Zindel J, Kubes P. DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu Rev Pathol. 2020;15:493–518.
Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–8.
Zitvogel L, Perreault C, Finn OJ, Kroemer G. Beneficial autoimmunity improves cancer prognosis. Nat Rev Clin Oncol. 2021;18:591–602.
Haen SP, Löffler MW, Rammensee HG, Brossart P. Towards new horizons: characterization, classification and implications of the tumour antigenic repertoire. Nat Rev Clin Oncol. 2020;17:595–610.
Clement CC, Osan J, Buque A, Nanaware PP, Chang YC, Perino G, Shetty M, Yamazaki T, Tsai WL, Urbanska AM, et al. PDIA3 epitope-driven immune autoreactivity contributes to hepatic damage in type 2 diabetes. Sci Immunol. 2022;7:eabl3795.
Guilloux Y, Lucas S, Brichard VG, Van Pel A, Viret C, De Plaen E, Brasseur F, Lethe B, Jotereau F, Boon T. A peptide recognized by human cytolytic T lymphocytes on HLA-A2 melanomas is encoded by an intron sequence of the N-acetylglucosaminyltransferase V gene. J Exp Med. 1996;183:1173–83.
Caron E, Vincent K, Fortier MH, Laverdure JP, Bramoullé A, Hardy MP, Voisin G, Roux PP, Lemieux S, Thibault P, Perreault C. The MHC I immunopeptidome conveys to the cell surface an integrative view of cellular regulation. Mol Syst Biol. 2011;7:533.
Fuchs Y, Steller H. Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat Rev Mol Cell Biol. 2015;16:329–44.
Chen J, Jin Z, Zhang S, Zhang X, Li P, Yang H, Ma Y. Arsenic trioxide elicits prophylactic and therapeutic immune responses against solid tumors by inducing necroptosis and ferroptosis. Cell Mol Immunol. 2023;20:51–64.
Le Naour J, Sztupinszki Z, Carbonnier V, Casiraghi O, Marty V, Galluzzi L, Szallasi Z, Kroemer G, Vacchelli E. A loss-of-function polymorphism in ATG16L1 compromises therapeutic outcome in head and neck carcinoma patients. Oncoimmunology. 2022;11:2059878.
Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97–111.
Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. 2020;20:95–112.
Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61.
Bezu L, Sauvat A, Humeau J, Leduc M, Kepp O, Kroemer G. eIF2α phosphorylation: a hallmark of immunogenic cell death. Oncoimmunology. 2018;7:e1431089.
Kepp O, Bezu L, Yamazaki T, Di Virgilio F, Smyth MJ, Kroemer G, Galluzzi L. ATP and cancer immunosurveillance. Embo j. 2021;40:e108130.
Kofla G, Radecke C, Frentsch M, Walther W, Stintzing S, Riess H, Bullinger L, Na IK. Conventional amphotericin B elicits markers of immunogenic cell death on leukemic blasts, mediates immunostimulatory effects on phagocytic cells, and synergizes with PD-L1 blockade. Oncoimmunology. 2022;11:2068109.
Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–9.
Vacchelli E, Ma Y, Baracco EE, Sistigu A, Enot DP, Pietrocola F, Yang H, Adjemian S, Chaba K, Semeraro M, et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science. 2015;350:972–8.
Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remédios C, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. 2014;20:1301–9.
Sprooten J, Agostinis P, Garg AD. Type I interferons and dendritic cells in cancer immunotherapy. Int Rev Cell Mol Biol. 2019;348:217–62.
Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell. 2017;168:707–23.
Sautès-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer. 2019;19:307–25.
Liu P, Zhao L, Kepp O, Kroemer G. Crizotinib - a tyrosine kinase inhibitor that stimulates immunogenic cell death. Oncoimmunology. 2019;8:1596652.
Fridman WH, Zitvogel L, Sautès-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. 2017;14:717–34.
Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74.
Galluzzi L, Chan TA, Kroemer G, Wolchok JD, López-Soto A. The hallmarks of successful anticancer immunotherapy. Sci Transl Med. 2018;10:eaat7807.
Rodriguez-Ruiz ME, Buqué A, Hensler M, Chen J, Bloy N, Petroni G, Sato A, Yamazaki T, Fucikova J, Galluzzi L. Apoptotic caspases inhibit abscopal responses to radiation and identify a new prognostic biomarker for breast cancer patients. Oncoimmunology. 2019;8:e1655964.
Decraene B, Yang Y, De Smet F, Garg AD, Agostinis P, De Vleeschouwer S. Immunogenic cell death and its therapeutic or prognostic potential in high-grade glioma. Genes Immun. 2022;23:1–11.
Yamazaki T, Buqué A, Ames TD, Galluzzi L. PT-112 induces immunogenic cell death and synergizes with immune checkpoint blockers in mouse tumor models. Oncoimmunology. 2020;9:1721810.
Liu P, Chen J, Zhao L, Hollebecque A, Kepp O, Zitvogel L, Kroemer G. PD-1 blockade synergizes with oxaliplatin-based, but not cisplatin-based, chemotherapy of gastric cancer. Oncoimmunology. 2022;11:2093518.
Petrazzuolo A, Perez-Lanzon M, Liu P, Maiuri MC, Kroemer G. Crizotinib and ceritinib trigger immunogenic cell death via on-target effects. Oncoimmunology. 2021;10:1973197.
Yamazaki T, Kirchmair A, Sato A, Buqué A, Rybstein M, Petroni G, Bloy N, Finotello F, Stafford L, Navarro Manzano E, et al. Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat Immunol. 2020;21:1160–71.
Zhou H, Tu C, Yang P, Li J, Kepp O, Li H, Zhang L, Zhang L, Zhao Y, Zhang T, et al. Carbon ion radiotherapy triggers immunogenic cell death and sensitizes melanoma to anti-PD-1 therapy in mice. Oncoimmunology. 2022;11:2057892.
Gomes-da-Silva LC, Zhao L, Bezu L, Zhou H, Sauvat A, Liu P, Durand S, Leduc M, Souquere S, Loos F, et al. Photodynamic therapy with redaporfin targets the endoplasmic reticulum and Golgi apparatus. Embo J. 2018;37:e98354.
Ma Y, Aymeric L, Locher C, Mattarollo SR, Delahaye NF, Pereira P, Boucontet L, Apetoh L, Ghiringhelli F, Casares N, et al. Contribution of IL-17-producing gamma delta T cells to the efficacy of anticancer chemotherapy. J Exp Med. 2011;208:491–503.
Bloom AC, Bender LH, Tiwary S, Pasquet L, Clark K, Jiang T, Xia Z, Morales-Kastresana A, Jones JC, Walters I, et al. Intratumorally delivered formulation, INT230-6, containing potent anticancer agents induces protective T cell immunity and memory. Oncoimmunology. 2019;8:e1625687.
Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity. 2016;44:343–54.
Zitvogel L, Galluzzi L, Viaud S, Vétizou M, Daillère R, Merad M, Kroemer G. Cancer and the gut microbiota: an unexpected link. Sci Transl Med. 2015;7:271ps271.
Darini C, Ghaddar N, Chabot C, Assaker G, Sabri S, Wang S, Krishnamoorthy J, Buchanan M, Aguilar-Mahecha A, Abdulkarim B, et al. An integrated stress response via PKR suppresses HER2 + cancers and improves trastuzumab therapy. Nat Commun. 2019;10:2139.
Fucikova J, Becht E, Iribarren K, Goc J, Remark R, Damotte D, Alifano M, Devi P, Biton J, Germain C, et al. Calreticulin expression in human non-small cell lung cancers correlates with increased Accumulation of Antitumor Immune cells and favorable prognosis. Cancer Res. 2016;76:1746–56.
Garg AD, More S, Rufo N, Mece O, Sassano ML, Agostinis P, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: immunogenic cell death induction by anticancer chemotherapeutics. Oncoimmunology. 2017;6:e1386829.
Matias M, Pinho JO, Penetra MJ, Campos G, Reis CP, Gaspar MM. The challenging melanoma landscape: from early drug discovery to clinical approval. Cells. 2021;10:3088.
Montes de Oca R, Alavi AS, Vitali N, Bhattacharya S, Blackwell C, Patel K, Seestaller-Wehr L, Kaczynski H, Shi H, Dobrzynski E, et al. Belantamab Mafodotin (GSK2857916) drives immunogenic cell death and Immune-mediated antitumor responses in vivo. Mol Cancer Ther. 2021;20:1941–55.
Xie W, Forveille S, Iribarren K, Sauvat A, Senovilla L, Wang Y, Humeau J, Perez-Lanzon M, Zhou H, Martinez-Leal JF, et al. Lurbinectedin synergizes with immune checkpoint blockade to generate anticancer immunity. Oncoimmunology. 2019;8:e1656502.
D’Amico L, Menzel U, Prummer M, Müller P, Buchi M, Kashyap A, Haessler U, Yermanos A, Gébleux R, Briendl M, et al. A novel anti-HER2 anthracycline-based antibody-drug conjugate induces adaptive anti-tumor immunity and potentiates PD-1 blockade in breast cancer. J Immunother Cancer. 2019;7:16.
Kepp O, Zitvogel L, Kroemer G. Clinical evidence that immunogenic cell death sensitizes to PD-1/PD-L1 blockade. Oncoimmunology. 2019;8:e1637188.
Sharmni Vishnu K, Win TT, Aye SN, Basavaraj AK. Combined atezolizumab and nab-paclitaxel in the treatment of triple negative breast cancer: a meta-analysis on their efficacy and safety. BMC Cancer. 2022;22:1139.
Mansfield AS, Każarnowicz A, Karaseva N, Sánchez A, De Boer R, Andric Z, Reck M, Atagi S, Lee JS, Garassino M, et al. Safety and patient-reported outcomes of atezolizumab, carboplatin, and etoposide in extensive-stage small-cell lung cancer (IMpower133): a randomized phase I/III trial. Ann Oncol. 2020;31:310–7.
Saito S, Toyokawa G, Momosaki S, Kozuma Y, Shoji F, Yamazaki K, Takeo S. Dramatic response to pembrolizumab with chemotherapy followed by salvage surgery in a lung cancer patient. Thorac Cancer. 2021;12:2217–20.
Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. 2017;18:127–36.
Green DR. Caspase activation and inhibition. Cold Spring Harb Perspect Biol. 2022;14:a041020.
Boada-Romero E, Martinez J, Heckmann BL, Green DR. The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol. 2020;21:398–414.
Zhang R, Neighbors JD, Schell TD, Hohl RJ. Schweinfurthin induces ICD without ER stress and caspase activation. Oncoimmunology. 2022;11:2104551.
Acknowledgements
Not applicable.
Funding
LG is/has been supported (as a PI unless otherwise indicated) by two Breakthrough Level 2 grants from the US DoD BCRP (#BC180476P1; #BC210945), by a Transformative Breast Cancer Consortium Grant from the US DoD BCRP (#W81XWH2120034, PI: Formenti), by a U54 grant from NIH/NCI (#CA274291, PI: Deasy, Formenti, Weichselbaum), by the 2019 Laura Ziskin Prize in Translational Research (#ZP-6177, PI: Formenti) from the Stand Up to Cancer (SU2C), by a Mantle Cell Lymphoma Research Initiative (MCL-RI, PI: Chen-Kiang) grant from the Leukemia and Lymphoma Society (LLS), by a Rapid Response Grant from the Functional Genomics Initiative (New York, US), by startup funds from the Dept. of Radiation Oncology at Weill Cornell Medicine (New York, US), by industrial collaborations with Lytix Biopharma (Oslo, Norway), Promontory (New York, US) and Onxeo (Paris, France), as well as by donations from Promontory (New York, US), the Luke Heller TECPR2 Foundation (Boston, US), Sotio a.s. (Prague, Czech Republic), Lytix Biopharma (Oslo, Norway), Onxeo (Paris, France), Ricerchiamo (Brescia, Italy), and Noxopharm (Chatswood, Australia).OK is supported by the Institut National du Cancer (INCa) and the DIM elicit. GK is supported by the Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR) – Projets blancs; AMMICa US23/CNRS UMS3655; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Fondation pour la Recherche Médicale (FRM); a donation by Elior; Equipex Onc-oPheno-Screen; European Joint Programme on Rare Diseases (EJPRD); Gustave Roussy Odyssea, the European Union Horizon 2020 Projects Oncobiome and Crimson; Fondation Carrefour; Institut National du Cancer (INCa); Institut Universitaire de France; LabEx Immuno-Oncology (ANR-18-IDEX-0001); a Cancer Research ASPIRE Award from the Mark Foundation; the RHU Immunolife; Seerave Foundation; SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); and SIRIC Cancer Research and Personalized Medicine (CARPEM). This study contributes to the IdEx Université de Paris ANR-18-IDEX-0001.
Author information
Authors and Affiliations
Contributions
LG and FM conceived the article. LG wrote the first version of the article with critical input from all authors. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors provided their consent to publication.
Competing interests
LG is/has been holding research contracts with Lytix Biopharma, Promontory and Onxeo, has received consulting/advisory honoraria from Boehringer Ingelheim, AstraZeneca, OmniSEQ, Onxeo, The Longevity Labs, Inzen, Imvax, Sotio, Promontory, Noxopharm, EduCom, and the Luke Heller TECPR2 Foundation, and holds Promontory stock options. OK is a scientific co-founder of Samsara Therapeutics. GK has been holding research contracts with Daiichi Sankyo, Eleor, Kaleido, Lytix Pharma, PharmaMar, Osasuna Therapeutics, Samsara Therapeutics, Sanofi, Sotio, Tollys, Vascage and Vasculox/Tioma. GK has been consulting for Reithera. GK is on the Board of Directors of the Bristol Myers Squibb Foundation France. GK is a scientific co-founder of everImmune, Osasuna Therapeutics, Samsara Therapeutics and Therafast Bio. GK is the inventor of patents covering therapeutic targeting of aging, cancer, cystic fibrosis and metabolic disorders. Among these, patents were licensed to Bayer (WO2014020041-A1, WO2014020043-A1), Bristoll-Myers Squibb (WO2008057863-A1), Osasuna Therapeutics (WO2019057742A1), PharmaMar (WO2022049270A1 and WO2022048775-A1), Raptor Pharmaceuticals (EP2664326-A1), Samsara Therapeutics (GB202017553D0), and Therafast Bio (EP3684471A1). EH is an employee of Sonata Therapeutics. FMM is an employee of Kite Pharma, Inc. All other authors have no conflicts to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Galluzzi, L., Kepp, O., Hett, E. et al. Immunogenic cell death in cancer: concept and therapeutic implications. J Transl Med 21, 162 (2023). https://doi.org/10.1186/s12967-023-04017-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-023-04017-6