
Abstract
Podocytes are differentiated postmitotic cells which cannot be replaced after podocyte injury. The mechanism of podocyte repopulation after injury has aroused wide concern. Parietal epithelial cells (PECs) are heterogeneous and only a specific subpopulation of PECs has the capacity to replace podocytes. Major progress has been achieved in recent years regarding the role and function of a subset of PECs which could transdifferentiate toward podocytes. Additionally, several factors, such as Notch, Wnt/ß-catenin, Wilms’ tumor-1, miR-193a and growth arrest-specific protein 1, have been shown to be involved in these processes. Finally, PECs serve as a potential therapeutic target in the conditions of podocyte loss. In this review, we discuss the latest observations and concepts about the recruitment of podocytes from PECs in glomerular diseases as well as newly identified mechanisms and the most recent treatments for this process.
Similar content being viewed by others

Podocytes are terminally differentiated cells and generally do not replicate, presenting a major obstacle to their restoration [1,2,3,4]. Replacing lost podocytes is a therapeutic opportunity to limit and reverse glomerular scarring and proteinuria. Previous studies have demonstrated that some glomerular parietal epithelial cells (PECs) act as progenitors of podocytes in healthy glomeruli and follow a decreased number of podocytes under healthy and disease conditions [5]. Romagnani’s group was the first to characterize progenitor cells of the Bowman’s capsule, providing novel insight into glomerular physiology [6,7,8]. In Bowman’s capsule, they isolated and characterized CD24+CD133+ PECs as multipotent progenitor cells. These cells could be triggered to generate mature and functional tubular cells in human. Several signaling pathways regulate PEC proliferation and differentiation toward podocytes [9]. In this review, we summarize the current progress about the roles and functions, involved mechanisms and potential therapeutic targets for podocyte recruitment from PECs.
Introduction of podocyte injury
Podocytes maintain the glomerular filtration barrier. Podocyte injury causes proteinuria and terminally glomerulosclerosis. The critical factor preventing podocyte injury may be the lack of regenerative capacity in podocytes.
Some podocyte progenitors were activated in the human Bowman capsule after podocyte injury (Fig. 1). PECs are the well known source for podocyte progenitors. Besides, the cell of renin lineage (CoRL) is a new-found candidate for the podocyte progenitor.

Schema representing the fate of PECs and cells of renin lineage as podocyte progenitors. Both of them could acquire podocyte-like qualities. PECs: parietal epithelial cells
A current study [10] suggests the CoRL transdifferentiate to podocytes in acute FSGS, 5/6 nephrectomy-induced chronic kidney disease, and uninephrectomy models. However, only podocyte loss, rather than a decrease in nephron mass, triggers CoRL of juxtaglomerular origin to move to the injury site.
Interestingly, the podocyte depletion paradigm have raised concerns these years. Puelles et al. [11] collected kidneys at autopsy from Caucasian American adults, and they suggested that older age and hypertension are associated with podocyte depletion, which is more obviously when both older age and hypertension are present.
Above all, podocyte injury was a complex outcome for defending cell death under different pathological conditions. These postmitotic cells has limited capacity for regeneration. PECs and CoRL were candidates for the podocyte progenitor after podocyte injury.
Introduction of PECs
PECs in glomerular development
PECs emerge late during nephrogenesis. Presumptive PECs and podocytes share a common phenotype until the S-shaped body stage after the mesenchymal to epithelial differentiation. Between the late S-shaped body and capillary loop stage, some PECs constitute Bowman’s capsule. The others upregulate podocyte-specific genes, gradually differentiating into podocytes in mouse [5]. Once PECs differentiate into podocytes, they lose their ability to proliferate and amplify [5].
The function of PECs
PECs serve important roles in the process of replacing injured podocytes. PECs and podocytes derived from the common mesenchymal progenitors and ultimately develop different phenotypes during glomerulogenesis [12]. Studies have identified CD133+CD24+ cells located in Bowman’s capsule that were capable of podocyte and tubular differentiation in vitro [6, 8]. Some PECs coexpress proteins unique to both podocytes and PECs [13,14,15,16]. Lasagni et al. demonstrated that PEC differentiation into podocytes can be enhanced by the glycogen synthase kinases 3-α and 3-β (GSK3) inhibitor BIO (6-bromo-indirubin-3’-oxime) both in vivo and in vitro. Furthermore, BIO could increase retinoic acid (RA) binding to its specific RA response elements (RARE) and strengthen PEC sensitivity to the differentiation effect of endogenously produced RA.
PECs show hierarchical differentiation based on their location. At urinary pole, PECs express CD133 and CD24 without podocyte markers (nestin, complement receptor-1, and podocalyxin), which were defined as CD133+CD24+PDX− PECs; In the same way, they were defined as CD133−CD24−PDX+ PECs in the vascular pole and CD133+CD24+PDX+ PECs in the rest of regions [8]; In the case of progressive podocyte depletion, PECs begin to express podocyte proteins, including those related to focal segmental glomerular sclerosis (FSGS), aging nephropathy, and membranous nephropathy [13, 14]. PECs expressing CD133 and CD24 can alleviate renal damage by promoting tubular regeneration and podocyte substitution. They can also induce glomerular injury, such as crescent formation and glomerulosclerosis [17, 18]. Several studies have shown that the invasion of activated PECs contribute to FSGS [19]. Adhesion of the glomerular tuft to Bowman’s capsule occurs in the early stage of FSGS development, serving as a bridge for PEC migration [20]. Hence, detecting activated PECs could be an auxiliary diagnostic method for early FSGS.
Studies have shown that PECs play an important role during normal glomerular development and normal function under healthy and disease conditions [9, 21]. First, PECs are progenitors of podocytes. Second, the intracellular tight junctions of PECs can restrict glomerular filtrate to the urinary space. Third, PECs can possibly synthesize and repair the basement membrane of Bowman’s capsule [21].
Although PECs are important for health and disease, in some disease states, the reaction of PECs directly contributes to the deterioration of glomerular function. On the one hand, the abnormal proliferation of PECs could lead to crescentic glomerulonephritis and collapsing glomerulopathy. Proliferation of PECs leads to a substantial increase in cell number within crescents in murine nephrotoxic serum nephritis and collapsing glomerulopathy [17]. On the other hand, PECs are involved in glomerular scarring [22]. It is currently believed that extracellular matrix accumulation likely arises from activated PECs, and podocyte injury is the mechanism of segmental scarring in FSGS [23, 24]. The functions of PECs are summarized in Table 1.
PECs can transdifferentiate toward podocytes
Under physiological conditions
In juveniles, PECs can migrate to the vascular pole and differentiate into mature podocytes. Appel et al. [25] generated triple-transgenic juvenile mice that allow irreversible and specific labeling of PECs through administration of doxycycline for 14 days. They observed that all genetically labeled cells coexpressed podocyte marker proteins. Wanner et al. [26] genetically labeled PECs with membrane-tagged enhanced green fluorescent protein (mG) in inducible hPODXL1.rtTA;tetO-Cre;mT/mG mice exposed to doxycycline from embryonic day (E) 8.5 to postnatal day (P) 28. Interestingly, mG-labeled podocytes were observed in mouse kidney sections after postnatal kidney development. Additionally, the PEC-derived podocytes were measurable in P1 kidneys.
Interestingly, the number of PEC-derived podocytes gradually increased with age in animals [27]. In the mature kidney, PECs can serve as precursor cells to differentiate into mature podocytes and supplements to podocyte deletion. PECs can be divided into three subtypes according to their location as mentioned above. Injection of CD133+CD24+PDX− cells, but not CD133−CD24−PDX+ or CD133+CD24+PDX+ cells, into mice with Adriamycin-induced nephropathy reduced proteinuria and improved chronic glomerular damage [8]. Then, Kietzmann et al. established an immortalized polyclonal human PEC line [28]. These researchers observed that human PECs highly expressed PEC-specific markers but did not express or weakly expressed podocyte-specific markers. Using a preclinical model of FSGS, Schneider et al. showed that the podocyte density was lower in aged mice than in young mice. However, the percentage of activated PECs was higher in aged mice [27]. Additionally, the percentage of phosphorylated ERK (pERK)-stained PECs was highest in aged FSGS mice, suggesting that phosphorylated pERK might be a potential mechanism needing further exploration.
In summary, PECs can migrate to the vascular pole and differentiate into mature podocytes in juvenile kidneys. Additionally, PECs can serve as precursor cells to differentiate into podocytes and supplements to podocyte deletion in mature kidneys. However, is the replacement of podocytes by PECs sufficient in aged kidneys? Podocyte numbers were lower in aged mice than young mice. The capacity for PEC-to-podocyte transition was reduced with aging. This is a multifactorial process, and the cell fate of PECs is multivariable and in need of thorough study.
Under pathophysiological conditions
Podocyte injury and deletion, which lead to PEC activation and matrix secretion, are key factors in glomerular sclerosis [29,30,31]. Many glomerular diseases, such as FSGS and diabetic nephropathy (DN), are associated with podocyte injury.
FSGS is characterized by initial injury to podocytes, with secondary activation in the neighboring glomerular PECs. Smeets et al. demonstrated the function of PEC-producing matrix proteins by utilizing PEC-reporter mice, which contribute to the segmental scarring process [19]. These researchers demonstrated that PECs were activated following primary podocyte injury in mice. The extracellular matrix proteins produced by activated PECs result in thickening of the Bowman’s basement membrane. Similar observations in human biopsies have also been reported [32]. Thus, the mechanisms of segmental scarring in FSGS may be activated PECs followed by podocyte injury.
Glomerular PECs can serve as the progenitor niche of podocytes, which involved in the regression of DN [9]. Activated PECs are increased in patients with DN, especially in advanced stages. Podocyte populations can regenerate, which is linked to the regression of DN; concurrent reversal of DN is an attainable goal. PECs could be a progenitor cell population for the restoration of podocytes in DN [33, 34]. In the late phase of diabetes, the endothelium is terrible injured, which might lead to leakage of plasma and subsequently induce PECs activation [9].
Interestingly, the remission of podocyte loss is closely related to the differentiation of PECs into podocytes [33, 35, 36]. When many podocytes are lost, PECs can migrate to the capillary loops, and phenotypic changes occur. PECs are activated in the early stage of disease, which promotes glomerular sclerosis. During the recovery period, they differentiate into podocytes, replenish injured podocytes and promote disease repair.
After podocyte injury, the number of podocyte can be partially or completely recovered without podocyte proliferation. There are two major candidate progenitor sources for podocyte: PECs and CoRL. Circulating and bone marrow derived progenitors seemed to be out of this role. Some studies [8, 25] strongly supported that some PECs served as podocyte progenitors, while some studies [37, 38] suggested a different paradigm. They showed that the cells co-expressing PECs and podocytes specific markers at the Bowman’s capsule is that they originate from migrating cells of podocyte, not PECs. The expression of podocyte-specific proteins in PECs could be associated with protein degradation [39]. We propose a model that PECs progenitors mainly located in the tubular pole, and CoRL primarily situated in the vascular pole. However, their functions are not limited by their anatomic situations.
Finally, one might be puzzled that whether the replacement of PECs-derived podocytes is sufficient after podocyte loss? Although a subtype of PECs might differentiate into podocytes, the magnitude of regeneration may not fully replace with the depletion of podocyte [40]. The fact that the decreased number of podocyte in disease suggest that the ability of the potential podocyte progenitors to restore podocyte loss is quite restricted. We assumed that mechanism of transition are multi-factorial. When the podocyte injury is persistent and severe, the regeneration is not observed, and the matrix production and hyperplastic cellular lesion do occur, which finally lead to glomerulosclerosis and FSGS.
Mechanism involved in the transition of PECs to podocytes
The mechanisms involved in the process of PEC-to-podocyte transition are partially understood, and observations are continually emerging [28, 41]. A better understanding of these factors could provide novel horizons for clinical therapeutics. We sum up the major signals as follows.
Notch signaling
The Notch receptor family is evolutionarily conserved [42, 43]. Upon ligand binding, the Notch receptor initiates a series of proteolytic cleavage events, ultimately inducing the synthesis of Notch target genes.
Notch signaling regulates cell differentiation in kidney development [44]. Blockade of Notch signaling significantly alleviated PEC hyperplasia in a mouse model of FSGS [45]. Additionally, inhibition of Notch signaling suppressed the migration and mesenchymal phenotypic transition of cultured PECs, indicating that Notch-regulated mesenchymal phenotypic transformation and cell migration could replenish podocyte deficiency [46].
The podocyte loss-induced Notch activation in PECs requires further research, and TGF-β may be a candidate [45]. Properly regulating Notch expression might provide a novel strategy for the treatment of renal disease.
Wnt/β-catenin signaling
Wnt/β-catenin signaling is a widely recognized pathway that mediates cell proliferation and differentiation, inflammation, angiogenesis, tumorigenesis and fibrosis. The activity of Wnt/β-catenin is necessary for lineage specification during the late stages of nephrogenesis [47, 48], and the Wnt pathway is activated in injured glomeruli. Activated Wnt/β-catenin signaling leads to decreased podocyte differentiation markers and increased PEC-specific markers, whereas β-catenin deletion promotes podocyte marker expression [49, 50].
Thus, the Wnt/β-catenin signal is likely involved in the transition from PECs to podocytes. The specific mechanisms require further investigation.
Wilms’ tumor 1 (WT1)
Emerging studies have demonstrated that WT1 mutations are associated with complex developmental syndromes involving the kidney [51,52,53]. As a transcription factor, WT1 is required for normal renal development [52]. In adults, WT1 expression is extremely high in renal podocytes and lower in PECs [54]. WT1 null mice are unable to form kidneys [55], and WT1 mutations lead to a number of human renal diseases [56, 57].
Upregulated WT1 serves important roles in the differentiation of PECs toward podocytes [58, 59]. In addition, WT1 inhibits Wnt/β-catenin signaling [60], which is considered a prerequisite for the differentiation of PECs to podocytes.
MiR-193a
MicroRNAs are noncoding RNAs that are approximately 21 nucleotides long and play an important role in RNA silencing by regulating mRNA degradation and protein translation. MicroRNAs are widely involved in glomerular disease.
Gebeshuber et al. [61] suggested that upregulated miR-193a in mice decreased WT1 and markers of podocytes, leading to FSGS. Leonie et al.[28] generated a human immortalized PEC line that highly expressed PEC-specific markers. Additionally, down-regulating the expression of miR-193a in human PECs resulted in transdifferentiation toward podocyte, accompanied by increased levels of podocyte markers and decreased levels of PEC markers. Interestingly, miR-193a is widely expressed in human and mouse crescents. Inhibiting the expression of miR-193a in a rodent model of nephrotoxic nephritis could decreased proteinuria and crescent formation. In addition, a luciferase assay suggested a putative interaction between miR-193a and apolipoprotein L1 (APOL1) [62]. APOL1 expression and downregulation of miR-193a coincided with the expression of podocyte markers during the transition [62], and APOL1 and miR-193a share a reciprocal feedback relationship.
Thus, miR-193a represents a master switch that modifies the expression of PEC and podocyte markers in vitro and might also be relevant in vivo.
Growth arrest-specific protein 1 (Gas1)
Gas1 is a pleiotropic protein with multi-functions, including antiproliferative and proapoptotic activities. In kidney, Gas is expressed during nephrogenesis, and its expression is regulated by WT1 [63, 64]. Diabetes favors a decrease in Gas1 expression and increased progenitor cell markers as well as WT1 in Bowman’s capsule cells [65]. In addition, Gas1 deficiency in renal injury in the early stages of diabetes promotes the activation and proliferation of PECs, and they differentiate into podocytes [65]. Hence, Gas1 might be a novel regulator of renal regeneration in diabetes.
Above all, the Notch signaling, Wnt/β-catenin signaling, WT1, miR-193 and Gas1 were involved in the transition of PECs to podocytes. The relationship among these regulators are often neglected, which should be pay great attention: (1) β-catenin and WT1. β-catenin and WT1 are main master regulators which exhibits opposite functions in podocyte injury. Kim and colleague identified a gene, CXXC5, as a novel WT1 transcriptional target [66]. WT1 negatively controls Wnt/β-catenin pathway via CXXC5 in nephrogenesis. As study went deeper, Zhou et al. showed that β-catenin could target WT1 for ubiquitin-regulated degradation of protein, and the decreased of WT1 further in turn activated β-catenin [67]. The balance between β-catenin and WT1 could determine the state of podocyte. (2) Notch and WT1. Asfahani et al. suggested that loss of WT1 in mature podocytes regulated podocyte Notch activation, which control early events in WT1-associated glomerulosclerosis [68]. (3) Gas1 and WT1. Gas 1 is expressed in nephrogenesis and could be regulated by the transcription factor WT1.
The regeneration of terminally differentiated podocytes is a novel frontier. Accumulating evidence suggested that mechanisms involved in the process of PECs to podocyte transition are complex. The interplay among these stimulating factors for the recruitment of podocytes from PECs still need further research. An improved understanding of the mechanism is beneficial for the development of targeted drug in clinic.
PECs as a potential therapeutic target
Numerous studies have shown that targeting PECs might have some therapeutic value [7, 69, 70]. Administering retinoids to animal models of membranous nephropathy or FSGS increased the amount of PECs, which express podocyte proteins [16]. Thus, retinoids can enhance the progenitor capacity of PECs under certain conditions. Vitamin D, which is expressed in PECs, participating in the differentiation of PECs to podocytes in vitro [18, 71]. Additionally, Notch inhibitors can modulate glomerular regeneration in animal models of FSGS, which influences glomerulosclerosis and proteinuria [72].
Interestingly, the Angiotensin-converting enzyme inhibitors (ACEi) also had potential therapeutic effects. ACEi, a well-known drug for hypertension and proteinuria, could reduce the proliferation of PECs in animal models of human immune deficiency-associated nephropathy (Tg26 mice). Additionally, ACEi enhanced the ability of PECs to turn into progenitor cells in Munich Wistar Frömter rats (which develop progressive glomerular injury) [37, 73].
Prednisone limits podocyte loss by increasing regeneration by augmenting the number of podocyte progenitors in experimental FSGS mouse model [15]. And the effects were accompanied by increasing p-ERK expression.
Other potential therapeutic goals cannot be ignored, which include CXCL12 [74], epidermal growth factor (EGF) and its receptor (EGFR) [75, 76], and amino acid transporter 2 (LAT2) [77].
CXCL12 [74] is highly expressed in normal glomerular podocytes. Suppression of CXCL12 could activate PECs that integrate into glomeruli, express podocyte specific markers, and interdigitate with existing cells.
EGFR is ubiquitously expressed in PECs and podocytes. In addition, EGFR was found to be specifically expressed in human glomerulonephritis, with proliferation and dedifferentiation of these cells. In a mouse model of RPGN, EGFR deficiency in podocytes significantly alleviated RPGN and prevented renal failure and death [75].
LAT2 is upregulated in PECs and podocytes in advance of the crescent formation as well as in the crescent lesion. Seven days after LAT inhibitor administration, the crescent formation of CGN was remarkably alleviated.
Conclusion
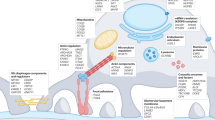
Increasing attention has been given to the biological role of PECs in health and disease as a result of PEC lineage tracing technology in animals, the exploration of specific cell markers, the isolation and identification of cultured PECs. Based on these advances, a better understanding of the potential role of activated PECs is urgent (Fig. 2).

Diagram outlining the fate of podocyte under physiological and pathophysiological conditions. Meanwhile, PECs and cells of renin lineage could differentiation into new podocytes. In addition, PECs could serve as a potential therapeutic goal in podocyte injury. PECs: parietal epithelial cells
First, replacing lost podocytes is a treating target to curb and reverse proteinuria and glomerular scarring. A subpopulation of PECs could compensate podocyte loss. However, how to identify and apply these specific PECs has been a challenge.
Second, PEC activation is manifested in both normal and disease conditions. Understanding what triggers and regulates PEC-to-podocyte transition is important and might promote repair and reduce the progression of glomerular disease. More studies are needed to improve the theoretical basis. Besides, the number of activated PECs were inadequate to fully replace the podocyte loss. The actually cues and mechanisms were worthy to be studied further.
Third, the PEC-to-podocyte transition lays the foundation for pharmacologic strategies, which aimed at accelerating podocyte, and thereby, glomerular regeneration. Novel therapeutics aiming to reduce podocyte loss or enhance PEC-to-podocyte transition might prevent glomerular disease. Further studies were needed to find new therapeutic targets for these glomerular diseases.
Fourth, PECs and CoRL are two candidates for podocyte progenitor after podocyte loss. PECs progenitors located mainly at the tubular pole of the glomerulus, while the CoRL mainly at the vascular pole. We should explore the differences between them upon podocyte injury.
Finally, there were compelling opinions that support the role of PECs as adult podocyte progenitors. We need to be cautious that there might be two-way processes that are occurred, where PECs differentiate into podocytes, and vice versa.
Availability of data and materials
Not applicable.
References
Reiser J, Altintas MM. Podocytes. F1000Res. 2016. https://doi.org/10.12688/f1000research.7255.1.
Endlich N, Siegerist F, Endlich K. Are podocytes motile? Pflugers Arch. 2017;469(7–8):951–7. https://doi.org/10.1007/s00424-017-2016-9.
Endlich K, Kliewe F, Endlich N. Stressed podocytes-mechanical forces, sensors, signaling and response. Pflugers Arch. 2017;469(7–8):937–49. https://doi.org/10.1007/s00424-017-2025-8.
Campbell KN, Tumlin JA. Protecting podocytes: a key target for therapy of focal segmental glomerulosclerosis. Am J Nephrol. 2018;47(1):14–29. https://doi.org/10.1159/000481634.
Smeets B, Moeller MJ. Parietal epithelial cells and podocytes in glomerular diseases. Semin Nephrol. 2012;32(4):357–67. https://doi.org/10.1016/j.semnephrol.2012.06.007.
Sagrinati C, Netti GS, Mazzinghi B, et al. Isolation and characterization of multipotent progenitor cells from the Bowman’s capsule of adult human kidneys. J Am Soc Nephrol. 2006;17(9):2443–56. https://doi.org/10.1681/asn.2006010089.
Romagnani P. Parietal epithelial cells: their role in health and disease. Contrib Nephrol. 2011;169:23–36. https://doi.org/10.1159/000313943.
Ronconi E, Sagrinati C, Angelotti ML, et al. Regeneration of glomerular podocytes by human renal progenitors. J Am Soc Nephrol. 2009;20(2):322–32. https://doi.org/10.1681/asn.2008070709.
Su H, Chen S, He FF, Wang YM, Bondzie P, Zhang C. New insights into glomerular parietal epithelial cell activation and its signaling pathways in glomerular diseases. Biomed Res Int. 2015;2015: 318935. https://doi.org/10.1155/2015/318935.
Pippin JW, Kaverina NV, Eng DG, et al. Cells of renin lineage are adult pluripotent progenitors in experimental glomerular disease. Am J Physiol Renal Physiol. 2015;309(4):F341-358. https://doi.org/10.1152/ajprenal.00438.2014.
Puelles VG, Cullen-McEwen LA, Taylor GE, et al. Human podocyte depletion in association with older age and hypertension. Am J Physiol Renal Physiol. 2016;310(7):F656-f668. https://doi.org/10.1152/ajprenal.00497.2015.
Gupta S, Verfaillie C, Chmielewski D, et al. Isolation and characterization of kidney-derived stem cells. J Am Soc Nephrol. 2006;17(11):3028–40. https://doi.org/10.1681/asn.2006030275.
Ohse T, Vaughan MR, Kopp JB, et al. De novo expression of podocyte proteins in parietal epithelial cells during experimental glomerular disease. Am J Physiol Renal Physiol. 2010;298(3):F702-711. https://doi.org/10.1152/ajprenal.00428.2009.
Zhang J, Hansen KM, Pippin JW, et al. De novo expression of podocyte proteins in parietal epithelial cells in experimental aging nephropathy. Am J Physiol Renal Physiol. 2012;302(5):F571-580. https://doi.org/10.1152/ajprenal.00516.2011.
Zhang J, Pippin JW, Krofft RD, Naito S, Liu ZH, Shankland SJ. Podocyte repopulation by renal progenitor cells following glucocorticoids treatment in experimental FSGS. Am J Physiol Renal Physiol. 2013;304(11):F1375-1389. https://doi.org/10.1152/ajprenal.00020.2013.
Zhang J, Pippin JW, Vaughan MR, et al. Retinoids augment the expression of podocyte proteins by glomerular parietal epithelial cells in experimental glomerular disease. Nephron Exp Nephrol. 2012;121(1–2):e23-37. https://doi.org/10.1159/000342808.
Smeets B, Uhlig S, Fuss A, et al. Tracing the origin of glomerular extracapillary lesions from parietal epithelial cells. J Am Soc Nephrol. 2009;20(12):2604–15. https://doi.org/10.1681/asn.2009010122.
Smeets B, Angelotti ML, Rizzo P, et al. Renal progenitor cells contribute to hyperplastic lesions of podocytopathies and crescentic glomerulonephritis. J Am Soc Nephrol. 2009;20(12):2593–603. https://doi.org/10.1681/asn.2009020132.
Smeets B, Kuppe C, Sicking EM, et al. Parietal epithelial cells participate in the formation of sclerotic lesions in focal segmental glomerulosclerosis. J Am Soc Nephrol. 2011;22(7):1262–74. https://doi.org/10.1681/asn.2010090970.
Smeets B, Stucker F, Wetzels J, et al. Detection of activated parietal epithelial cells on the glomerular tuft distinguishes early focal segmental glomerulosclerosis from minimal change disease. Am J Pathol. 2014;184(12):3239–48. https://doi.org/10.1016/j.ajpath.2014.08.007.
Shankland SJ, Anders HJ, Romagnani P. Glomerular parietal epithelial cells in kidney physiology, pathology, and repair. Curr Opin Nephrol Hypertens. 2013;22(3):302–9. https://doi.org/10.1097/MNH.0b013e32835fefd4.
Eng DG, Sunseri MW, Kaverina NV, Roeder SS, Pippin JW, Shankland SJ. Glomerular parietal epithelial cells contribute to adult podocyte regeneration in experimental focal segmental glomerulosclerosis. Kidney Int. 2015;88(5):999–1012. https://doi.org/10.1038/ki.2015.152.
Lim BJ, Yang JW, Do WS, Fogo AB. Pathogenesis of focal segmental glomerulosclerosis. J Pathol Transl Med. 2016;50(6):405–10. https://doi.org/10.4132/jptm.2016.09.21.
Chan GC, Eng DG, Miner JH, et al. Differential expression of parietal epithelial cell and podocyte extracellular matrix proteins in focal segmental glomerulosclerosis and diabetic nephropathy. Am J Physiol Renal Physiol. 2019;317(6):F1680-f1694. https://doi.org/10.1152/ajprenal.00266.2019.
Appel D, Kershaw DB, Smeets B, et al. Recruitment of podocytes from glomerular parietal epithelial cells. J Am Soc Nephrol. 2009;20(2):333–43. https://doi.org/10.1681/asn.2008070795.
Wanner N, Hartleben B, Herbach N, et al. Unraveling the role of podocyte turnover in glomerular aging and injury. J Am Soc Nephrol. 2014;25(4):707–16. https://doi.org/10.1681/asn.2013050452.
Schneider RR, Eng DG, Kutz JN, Sweetwyne MT, Pippin JW, Shankland SJ. Compound effects of aging and experimental FSGS on glomerular epithelial cells. Aging. 2017;9(2):524–46. https://doi.org/10.18632/aging.101176.
Kietzmann L, Guhr SS, Meyer TN, et al. MicroRNA-193a regulates the transdifferentiation of human parietal epithelial cells toward a podocyte phenotype. J Am Soc Nephrol. 2015;26(6):1389–401. https://doi.org/10.1681/asn.2014020190.
Merchant ML, Barati MT, Caster DJ. Proteomic analysis identifies distinct glomerular extracellular matrix in collapsing focal segmental glomerulosclerosis. J Am Soc Nephrol. 2020;31(8):1883–904. https://doi.org/10.1681/asn.2019070696.
McNicholas BA, Eng DG, Lichtnekert J, Rabinowitz PS, Pippin JW, Shankland SJ. Reducing mTOR augments parietal epithelial cell density in a model of acute podocyte depletion and in aged kidneys. Am J Physiol Renal Physiol. 2016;311(3):F626-639. https://doi.org/10.1152/ajprenal.00196.2016.
Kim S, Kim YH, Choi KH, Jeong HJ. Glomerular epithelial CD44 expression and segmental sclerosis in IgA nephropathy. Clin Exp Nephrol. 2016;20(6):871–7. https://doi.org/10.1007/s10157-015-1222-z.
Fatima H, Moeller MJ, Smeets B, et al. Parietal epithelial cell activation marker in early recurrence of FSGS in the transplant. Clin J Am Soc Nephrol. 2012;7(11):1852–8. https://doi.org/10.2215/cjn.10571011.
Pichaiwong W, Hudkins KL, Wietecha T, et al. Reversibility of structural and functional damage in a model of advanced diabetic nephropathy. J Am Soc Nephrol. 2013;24(7):1088–102. https://doi.org/10.1681/asn.2012050445.
Andeen NK, Nguyen TQ, Steegh F, Hudkins KL, Najafian B, Alpers CE. The phenotypes of podocytes and parietal epithelial cells may overlap in diabetic nephropathy. Kidney Int. 2015;88(5):1099–107. https://doi.org/10.1038/ki.2015.273.
Xu Z, Fan J. Islet transplantation promotes podocyte regeneration in a model of diabetic nephropathy. Turkish J Med Sci. 2017;47(6):1925–30. https://doi.org/10.3906/sag-1704-102.
Migliorini A, Angelotti ML, Mulay SR, et al. The antiviral cytokines IFN-α and IFN-β modulate parietal epithelial cells and promote podocyte loss: implications for IFN toxicity, viral glomerulonephritis, and glomerular regeneration. Am J Pathol. 2013;183(2):431–40. https://doi.org/10.1016/j.ajpath.2013.04.017.
Benigni A, Morigi M, Rizzo P, et al. Inhibiting angiotensin-converting enzyme promotes renal repair by limiting progenitor cell proliferation and restoring the glomerular architecture. Am J Pathol. 2011;179(2):628–38. https://doi.org/10.1016/j.ajpath.2011.04.003.
Darisipudi MN, Kulkarni OP, Sayyed SG, et al. Dual blockade of the homeostatic chemokine CXCL12 and the proinflammatory chemokine CCL2 has additive protective effects on diabetic kidney disease. Am J Pathol. 2011;179(1):116–24. https://doi.org/10.1016/j.ajpath.2011.03.004.
Guhr SS, Sachs M, Wegner A, et al. The expression of podocyte-specific proteins in parietal epithelial cells is regulated by protein degradation. Kidney Int. 2013;84(3):532–44. https://doi.org/10.1038/ki.2013.115.
Kaverina NV, Eng DG, Miner JH, Pippin JW, Shankland SJ. Parietal epithelial cell differentiation to a podocyte fate in the aged mouse kidney. Aging. 2020;12(17):17601–24. https://doi.org/10.18632/aging.103788.
Roeder SS, Stefanska A, Eng DG, et al. Changes in glomerular parietal epithelial cells in mouse kidneys with advanced age. Am J Physiol Renal Physiol. 2015;309(2):F164-178. https://doi.org/10.1152/ajprenal.00144.2015.
Siebel C, Lendahl U. Notch signaling in development, tissue homeostasis, and disease. Physiol Rev. 2017;97(4):1235–94. https://doi.org/10.1152/physrev.00005.2017.
Hori K, Sen A, Artavanis-Tsakonas S. Notch signaling at a glance. J Cell Sci. 2013;126(Pt 10):2135–40. https://doi.org/10.1242/jcs.127308.
Mukherjee M, Fogarty E, Janga M, Surendran K. Notch signaling in kidney development, maintenance, and disease. Biomolecules. 2019. https://doi.org/10.3390/biom9110692.
Ueno T, Kobayashi N, Nakayama M, et al. Aberrant Notch1-dependent effects on glomerular parietal epithelial cells promotes collapsing focal segmental glomerulosclerosis with progressive podocyte loss. Kidney Int. 2013;83(6):1065–75. https://doi.org/10.1038/ki.2013.48.
Sweetwyne MT, Susztak K. For better or worse: a niche for Notch in parietal epithelial cell activation. Kidney Int. 2013;83(6):988–90. https://doi.org/10.1038/ki.2013.66.
Merkel CE, Karner CM, Carroll TJ. Molecular regulation of kidney development: is the answer blowing in the Wnt? Pediatr Nephrol. 2007;22(11):1825–38. https://doi.org/10.1007/s00467-007-0504-4.
Lin Y, Liu A, Zhang S, et al. Induction of ureter branching as a response to Wnt-2b signaling during early kidney organogenesis. Dev Dyn. 2001;222(1):26–39. https://doi.org/10.1002/dvdy.1164.
Kato H, Susztak K. Repair problems in podocytes: Wnt, Notch, and glomerulosclerosis. Semin Nephrol. 2012;32(4):350–6. https://doi.org/10.1016/j.semnephrol.2012.06.006.
Kato H, Gruenwald A, Suh JH, et al. Wnt/β-catenin pathway in podocytes integrates cell adhesion, differentiation, and survival. J Biol Chem. 2011;286(29):26003–15. https://doi.org/10.1074/jbc.M111.223164.
Zhou H, Kajiyama H, Tsuji T, et al. Urinary exosomal Wilms’ tumor-1 as a potential biomarker for podocyte injury. Am J Physiol Renal Physiol. 2013;305(4):F553-559. https://doi.org/10.1152/ajprenal.00056.2013.
Yeger H, Baumal R, Harason P, Phillips MJ. Lectin histochemistry of Wilms’ tumor. Comparison with normal adult and fetal kidney. Am J Clin Pathol. 1987;88(3):278–85. https://doi.org/10.1093/ajcp/88.3.278.
Aldrink JH, Heaton TE, Dasgupta R, et al. Update on Wilms tumor. J Pediatr Surg. 2019;54(3):390–7. https://doi.org/10.1016/j.jpedsurg.2018.09.005.
Miller-Hodges E. Clinical aspects of WT1 and the kidney. Methods Mol Biol. 2016;1467:15–21. https://doi.org/10.1007/978-1-4939-4023-3_2.
Kreidberg JA, Sariola H, Loring JM, et al. WT-1 is required for early kidney development. Cell. 1993;74(4):679–91. https://doi.org/10.1016/0092-8674(93)90515-r.
Sun LZ, Wang HY, Li M, et al. Clinical and pathological features and mutational types of WT1 mutation-associated nephropathy. Zhonghua Er Ke Za Zhi. 2018;56(10):769–74. https://doi.org/10.3760/cma.j.issn.0578-1310.2018.10.010.
Hastie ND. Wilms’ tumour 1 (WT1) in development, homeostasis and disease. Development. 2017;144(16):2862–72. https://doi.org/10.1242/dev.153163.
Shankland SJ, Pippin JW, Duffield JS. Progenitor cells and podocyte regeneration. Semin Nephrol. 2014;34(4):418–28. https://doi.org/10.1016/j.semnephrol.2014.06.008.
Kabgani N, Grigoleit T, Schulte K, et al. Primary cultures of glomerular parietal epithelial cells or podocytes with proven origin. PLoS ONE. 2012;7(4): e34907. https://doi.org/10.1371/journal.pone.0034907.
Kim MK, McGarry TJ, Broin PO, Flatow JM, Golden AA, Licht JD. An integrated genome screen identifies the Wnt signaling pathway as a major target of WT1. Proc Natl Acad Sci USA. 2009;106(27):11154–9. https://doi.org/10.1073/pnas.0901591106.
Gebeshuber CA, Kornauth C, Dong L, et al. Focal segmental glomerulosclerosis is induced by microRNA-193a and its downregulation of WT1. Nat Med. 2013;19(4):481–7. https://doi.org/10.1038/nm.3142.
Kumar V, Vashistha H, Lan X, et al. Role of apolipoprotein L1 in human parietal epithelial cell transition. Am J Pathol. 2018;188(11):2508–28. https://doi.org/10.1016/j.ajpath.2018.07.025.
Spagnuolo R, Corada M, Orsenigo F, et al. Gas1 is induced by VE-cadherin and vascular endothelial growth factor and inhibits endothelial cell apoptosis. Blood. 2004;103(8):3005–12. https://doi.org/10.1182/blood-2003-07-2459.
Kann M, Bae E, Lenz MO, et al. WT1 targets Gas1 to maintain nephron progenitor cells by modulating FGF signals. Development. 2015;142(7):1254–66. https://doi.org/10.1242/dev.119735.
Luna-Antonio BI, Rodriguez-Muñoz R, Namorado-Tonix C, Vergara P, Segovia J, Reyes JL. Gas1 expression in parietal cells of Bowman’s capsule in experimental diabetic nephropathy. Histochem Cell Biol. 2017;148(1):33–47. https://doi.org/10.1007/s00418-017-1550-z.
Kim MS, Yoon SK, Bollig F, et al. A novel Wilms tumor 1 (WT1) target gene negatively regulates the WNT signaling pathway. J Biol Chem. 2010;285(19):14585–93. https://doi.org/10.1074/jbc.M109.094334.
Zhou L, Li Y, He W, et al. Mutual antagonism of Wilms’ tumor 1 and β-catenin dictates podocyte health and disease. J Am Soc Nephrol. 2015;26(3):677–91. https://doi.org/10.1681/asn.2013101067.
Asfahani RI, Tahoun MM, Miller-Hodges EV, et al. Activation of podocyte notch mediates early Wt1 glomerulopathy. Kidney Int. 2018;93(4):903–20. https://doi.org/10.1016/j.kint.2017.11.014.
Romoli S, Angelotti ML, Antonelli G, et al. CXCL12 blockade preferentially regenerates lost podocytes in cortical nephrons by targeting an intrinsic podocyte-progenitor feedback mechanism. Kidney Int. 2018;94(6):1111–26. https://doi.org/10.1016/j.kint.2018.08.013.
Rizzo P, Novelli R, Rota C, et al. The role of angiotensin II in parietal epithelial cell proliferation and crescent formation in glomerular diseases. Am J Pathol. 2017;187(11):2441–50. https://doi.org/10.1016/j.ajpath.2017.07.004.
Wang Y, Borchert ML, DeLuca HF. Identification of the vitamin D receptor in various cells of the mouse kidney. Kidney Int. 2012;81(10):993–1001. https://doi.org/10.1038/ki.2011.463.
Lasagni L, Ballerini L, Angelotti ML, et al. Notch activation differentially regulates renal progenitors proliferation and differentiation toward the podocyte lineage in glomerular disorders. Stem Cells. 2010;28(9):1674–85. https://doi.org/10.1002/stem.492.
Yadav A, Vallabu S, Kumar D, et al. HIVAN phenotype: consequence of epithelial mesenchymal transdifferentiation. Am J Physiol Renal Physiol. 2010;298(3):F734-744. https://doi.org/10.1152/ajprenal.00415.2009.
Massa F, Schedl A. Awakening the Bowman: inhibition of CXCL12 signaling activates parietal epithelial cells. Kidney Int. 2018;94(6):1042–4. https://doi.org/10.1016/j.kint.2018.09.011.
Flamant M, Bollée G, Hénique C, Tharaux PL. Epidermal growth factor: a new therapeutic target in glomerular disease. Nephrol Dial Transpl. 2012;27(4):1297–304. https://doi.org/10.1093/ndt/gfs030.
Bollée G, Flamant M, Schordan S, et al. Epidermal growth factor receptor promotes glomerular injury and renal failure in rapidly progressive crescentic glomerulonephritis. Nat Med. 2011;17(10):1242–50. https://doi.org/10.1038/nm.2491.
Kurayama R, Ito N, Nishibori Y, et al. Role of amino acid transporter LAT2 in the activation of mTORC1 pathway and the pathogenesis of crescentic glomerulonephritis. Lab Invest. 2011;91(7):992–1006. https://doi.org/10.1038/labinvest.2011.43.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the National Natural Science Foundation of China (82100763), Fundamental Research Funds for the Central Universities (2042021kf0150 and 2042020kf0137), the Zhongnan Hospital of Wuhan University Science, Technology and Innovation Seed Fund Project (cxpy2020027 and znpy2019036), the Excellent Doctor (Post), Zhongnan Hospital of Wuhan University (ZNYB2020009), the Hubei Province Health and Family Planning Scientific Research Project (WJ2019MB103), the Zhongnan Hospital of Wuhan University Science, Technology and Innovation Seed Fund (znpy2017044), the Clinical Research Project for Wu Jieping Medical Foundation (320.6750.19089-58), and the Research Fund from Medical Sci-Tech Innovation Platform of Zhongnan Hospital, Wuhan University (PTXM2020028).
Author information
Authors and Affiliations
Contributions
CY conceived and designed the study. CY and LN wrote the review article. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
We confirm that the manuscript has been read and approved by all named authors and that there are no other persons who satisfied the criteria for authorship but are not listed. We further confirm that the order of authors listed in the manuscript has been approved by all of us.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ni, L., Yuan, C. & Wu, X. The recruitment mechanisms and potential therapeutic targets of podocytes from parietal epithelial cells. J Transl Med 19, 441 (2021). https://doi.org/10.1186/s12967-021-03101-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-021-03101-z