
Abstract
Background
Angiogenesis inhibition research is a cutting edge area in angiogenesis-dependent disease therapy, especially in cancer therapy. Recently, studies on anti-angiogenic peptides have provided promising results in the field of cancer treatment.
Methods
A non-redundant dataset of 135 anti-angiogenic peptides (positive instances) and 135 non anti-angiogenic peptides (negative instances) was used in this study. Also, 20% of each class were selected to construct an independent test dataset (see Additional files 1, 2). We proposed an effective machine learning based R package (AntAngioCOOL) to predict anti-angiogenic peptides. We have examined more than 200 different classifiers to build an efficient predictor. Also, more than 17,000 features were extracted to encode the peptides.
Results
Finally, more than 2000 informative features were selected to train the classifiers for detecting anti-angiogenic peptides. AntAngioCOOL includes three different models that can be selected by the user for different purposes; it is the most sensitive, most specific and most accurate. According to the obtained results AntAngioCOOL can effectively suggest anti-angiogenic peptides; this tool achieved sensitivity of 88%, specificity of 77% and accuracy of 75% on the independent test set. AntAngioCOOL can be accessed at https://cran.r-project.org/.
Conclusions
Only 2% of the extracted descriptors were used to build the predictor models. The results revealed that physico-chemical profile is the most important feature type in predicting anti-angiogenic peptides. Also, atomic profile and PseAAC are the other important features.
Similar content being viewed by others


Background
Angiogenesis is the process of formation of new blood vessels from pre-existing vessels to make a supply of nutrients and a waste disposal pathway [1]. Angiogenesis is a normal and fundamental physiological process in growth and development [2,3,4]. However, it is a vital event in cancer progression—transition of tumor from a benign state to a malignant one—and spread of a tumor (metastasis) [5,6,7]. Nowadays, decreasing or inhibiting angiogenesis is a cutting edge research area in cancer therapy which also plays a key role in other angiogenesis-dependent disease therapy [1, 8,9,10,11,12,13,14,15].
Besides other therapeutic peptides, recognition of the anti-angiogenic peptides has stimulated great interest among researchers in the cancer treatment field during recent years [16,17,18,19,20,21,22,23]. However, there are very rare studies in computational detection of ant-angiogenic peptides [8].
In this paper, we have proposed an efficient machine learning based R package to detect anti-angiogenic peptides, namely AntAngioCOOL. Five types of features have been used to encode peptides in order to predict anti-angiogenic ones. According to the obtained results, AntAngioCOOL reached to a satisfactory performance in anti-angiogenic peptide prediction on a benchmark non-redundant independent test dataset.
Methods
Dataset
We have used the gold standard dataset that has been recently published [8]. After removing redundant peptides, this dataset contained 135 anti-angiogenic peptides (positive instances) and 135 non anti-angiogenic peptides (negative instances). Also, a 20% of each class was selected to construct an independent test dataset (see Additional file 1).
Features
The following subsections provide a brief description for each peptide feature. Moreover, Table 1 demonstrates the distribution of the features that have been used to encode each peptide.
Pseudo amino acid composition
We used pseudo amino acid composition (PseAAC) which has been used effectively in predicting cell penetrating peptides [21]. Unlike the simple amino acid composition, PseAAC considers the sequence-order information of the peptide. Interested readers may refer to [24] for further information on PseAAC.
k-mer composition
k-mer composition shows the fraction of all possible subsequences with length k in the given peptide. Also, the reduced amino acid alphabet proposed by Zahiri et al. [25] has been applied to compute another k-mer composition: the 20 alphabet of amino acids have been reduced to a new alphabet with size 8 according to 544 physicochemical and biochemical indices extracted from AAIndex database [26] (C1 = {A, E}, C2 = {I, L, F, M, V}, C3 = {N, D, T, S}, C4 = {G}, C5 = {P}, C6 = {R, K, Q, H}, C7 = {Y, W}, C8 = {C}). We have computed k-mer compositions for k = 2, 3, 4 for each peptide.
Physico-chemical profile
In order to compute this feature type, 544 different physico-chemical indices were extracted from AAIndex [26]. To remove redundancies, a subset of indices with correlation coefficient less than 0.8 and greater than − 0.8 were selected, which resulted in 191 non-redundant physico-chemical indices.
This feature type has been extracted for 5 amino acids of N-termini (5-NT) and C-termini (5-CT). Finally, each peptide has been encoded as a 10 × 191-dimensional feature vector as below:
where \(PC_{j}^{i}\) is the value of the jth physico-chemical index for the ith amino acid of the peptide (for i = \(1, \ldots ,5\) in the 5-CT and i = \(6, \ldots ,10\) in 5-NT)
Atomic profile
A 50-dimensional feature vector has been used to encode each peptide according to its atomic properties as below:
where \(AC_{1}^{i}\) through \(AC_{5}^{i}\) represent the frequency of five types of atoms: C, H, N, O, S in the ith amino acid of the peptides (for i = \(1, \ldots ,5\) in the 5-CT and i = \(6, \ldots ,10\) in 5-NT). For details of atomic composition for each 20 natural amino acid see [17].
Machine learning method
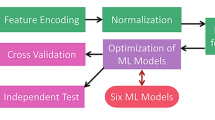
To build a powerful anti-angiogenic peptide predictor, 227 different classifiers (see Additional file 1) in the caret package [27] were examined. Finally, the three best classifiers (those with best sensitivity, specificity and accuracy) were selected to be included in the AntAngioCOOL package. Figure 1 provides a schematic representation of the proposed method.

Schematic representation of the proposed method (AntAngioCOOL) for anti-angiogenic peptide prediction
Evaluation parameters for the prediction performance
The training dataset was used to train the classifier, and then the classifier was evaluated using the test data. The predictions made for the test instances were used to compute the following performance measures:
where, TP and TN are the number of correctly predicted anti-angiogenic peptides and non anti-angiogenic peptides, respectively. Similarly, FP and FN are the number of non anti-angiogenic peptides and anti-angiogenic peptides wrongly predicted as anti-angiogenic peptides and non anti-angiogenic peptides, respectively.
Results
Preprocessing
To remove non-informative features, which can lead to reducing the computational cost without losing the prediction performance, nearZeroVar function from caret package [27] was utilized. This function eliminates those features that have one unique value (i.e. are zero variance features) or features with both of the following characteristics: they have very few unique values relative to the number of samples and the ratio of the frequency of the most common value to the frequency of the second most common value is large. nearZeroVar was applied to the extracted features using its default parameters. Interestingly, less than 2% of the extracted features (2343 out of 175,062) were selected as informative ones to construct the prediction models (see Additional file 1 for more details).
Prediction performance
The performance results of the 227 classifiers with accuracy > 50% in the independent test set have been shown in Additional file 1: Figures S1–S3. We have selected the three best classifiers to be included in the AntAngioCOOL package (Fig. 2): the most sensitive classifier (rpartCost with 88% sensitivity), the most accurate classifier (PART with 75% accuracy) and the classifier with the highest specificity (DeepBoost with 77% specificity). Availability of these three classifiers can help biologists with different questions in mind; e.g. having a list of candidate peptides, what is the narrow list of confident anti-angiogenic peptides or what is the more extended sub-list of candidate anti-angiogenic peptides that contains almost real anti-angiogenic peptides.

Prediction performance of the three selected classifiers among 227 classifiers to be included in AntAngioCOOL package in the test dataset
Discussion
Physico-chemical profile is the most important feature type
Physico-chemical profile is the main feature type in the final selected set of features which is 82% of the final features (Fig. 3a). Interestingly, almost all physico-chemical profile features were selected (1909 out of 1910). Dipeptide and tripeptide compositions are the other important feature types that comprise 9% (200 features) and 4% (101 features) of the final features, respectively. Moreover, Fig. 3b shows the percentage of each feature type that was selected as a subset of the final features.

Feature importance according to their contribution in final selected features (informative features). a Square chart of the distribution of different feature types in the final important features. b Inner layer of this sunburst chart shows the distribution of different feature types in the primary extracted features (in log scale). The outer layer shows the proportion of each feature type selected as informative features
Sequence-order information is useful for anti-angiogenic peptide prediction
As the Fig. 3b shows, in addition to the physico-chemical profile, a considerable percentage of the atomic profile (91.3%) and all the PseAAC features were selected as informative features (Additional file 2 for more details). One of the important common aspects of these three feature types is that they take the sequence-order information of the peptide into account. Therefore, this results stress out that the sequence-order information is an effective factor in anti-angiogenic peptide prediction.
Dipeptide is the most important feature among k-mer composition features
One of the interesting obtained results is that 43.1% of dipeptides were selected as informative features while for tripeptides and quadpeptides there are very small number of informative features for predicting anti-angiogenic peptides: 101 out of 8512 (1.2%) and 32 out of 164,096 (0.02%), respectively. So, dipeptide composition is the most important k-mer composition in anti-angiogenic peptide prediction.
Comparison with the current state-of-the-art methods
The proposed method has been trained and tested with the same data used for AntiAngioPred [8]. Results reveal that AntAngioCOOL has a higher accuracy (77% vs. 75%) and considerable higher sensitivity (88% vs. 65%). Therefore, AntAngioCOOL package can be used more effectively in anti-angiogenic peptide prediction, especially when one is interested in detecting almost anti-angiogenic peptides (in the cost of having some false positives) in a given list of peptides.
Conclusion
In this study an R package (AntAngioCOOL) was proposed to predict anti-angiogenic peptides. AntAngioCOOL exploits five descriptor types for a peptide of interest to perform the prediction including: PseAAC, k-mer composition, k-mer composition (reduced alphabet), physico-chemical profile and atomic profile. After removing the non-informative descriptors, only 2% of the extracted descriptors were used to build the predictor models. AntAngioCOOL includes three different models that can be selected by the user.
The results disclosed that physico-chemical profile is the most important feature type. Also, atomic profile and PseAAC are the other important features. Therefore, it can be concluded that sequence-order information plays a critical role in anti-angiogenic peptide prediction. In addition, according to the results dipeptide has the most contribution in anti-angiogenic peptide prediction among the k-mer composition features.
Abbreviations
- PseAAC:
-
pseudo amino acid composition
References
Mukherjee S, Patra CR. Therapeutic application of anti-angiogenic nanomaterials in cancers. Nanoscale. 2016. https://doi.org/10.1039/c5nr07887c.
Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2010;468:824–8. https://doi.org/10.1038/nature09557.
Simons M, Bonow RO, Chronos NA, Cohen DJ, Giordano FJ, Hammond HK, et al. Clinical trials in coronary angiogenesis: issues, problems, consensus: an expert panel summary. Circulation. 2000;102:E73–86.
Prior BM, Yang HT, Terjung RL. What makes vessels grow with exercise training? J Appl Physiol. 2004;97:1119–28. https://doi.org/10.1152/japplphysiol.00035.2004.
Adair TH, Montani J-P. Overview of angiogenesis. San Rafael: Morgan & Claypool Life Sciences; 2010.
Birbrair A, Zhang T, Wang Z-M, Messi ML, Mintz A, Delbono O. Pericytes at the intersection between tissue regeneration and pathology. Clin Sci. 2015;128:81–93. https://doi.org/10.1042/CS20140278.
Birbrair A, Zhang T, Wang Z-M, Messi ML, Olson JD, Mintz A, et al. Type-2 pericytes participate in normal and tumoral angiogenesis. Am J Physiol Cell Physiol. 2014;307:C25–38. https://doi.org/10.1152/ajpcell.00084.2014.
Ettayapuram Ramaprasad AS, Singh S, Gajendra PSR, Venkatesan S, Brem S, Cotran R, et al. AntiAngioPred: a server for prediction of anti-angiogenic peptides. PLoS ONE. 2015;10:e0136990. https://doi.org/10.1371/journal.pone.0136990.
Stegmann TJ, Hoppert T, Schneider A, Gemeinhardt S, Köcher M, Ibing R, et al. Induction of myocardial neoangiogenesis by human growth factors. A new therapeutic approach in coronary heart disease. Herz. 2000;25:589–99.
Stegmann TJ. FGF-1: a human growth factor in the induction of neoangiogenesis. Expert Opin Investig Drugs. 1998;7:2011–5. https://doi.org/10.1517/13543784.7.12.2011.
Folkman J. Angiogenic therapy of the human heart. Circulation. 1998;97:628–9.
Gonzalez-Perez RR, Rueda BR. Tumor angiogenesis regulators, 1st edn. CRC Press; 2013. https://www.crcpress.com/Tumor-Angiogenesis-Regulators/Gonzalez-Perez-Rueda/p/book/9781466580978.
Folkman J, Klagsbrun M. Angiogenic factors. Science. 1987;235:442–7.
Spill F, Guerrero P, Alarcon T, Maini PK, Byrne HM. Mesoscopic and continuum modelling of angiogenesis. 2014. https://doi.org/10.1007/s00285-014-0771-1.
Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A, et al. Glioblastoma stem-like cells give rise to tumour endothelium. Nature. 2010;468:829–33. https://doi.org/10.1038/nature09624.
Soliman MS, Cano MD, Karagiannis ED, Bakir BH, Popel AS, Gehlbach PL. In vitro evaluation of predicted antiangiogenic peptides in human retinal endothelial cells. Invest Ophthalmol Vis Sci. 2008;49:4594.
Kumar R, Chaudhary K, Singh Chauhan J, Nagpal G, Kumar R, Sharma M, et al. An in silico platform for predicting, screening and designing of antihypertensive peptides. Sci Rep. 2015;5:12512. https://doi.org/10.1038/srep12512.
Gautam A, Chaudhary K, Kumar R, Sharma A, Kapoor P, Tyagi A, et al. In silico approaches for designing highly effective cell penetrating peptides. J Transl Med. 2013;11:74. https://doi.org/10.1186/1479-5876-11-74.
Rajput A, Gupta AK, Kumar M, Miller M, Bassler B, Garsin D, et al. Prediction and analysis of quorum sensing peptides based on sequence features. PLoS ONE. 2015;10:e0120066. https://doi.org/10.1371/journal.pone.0120066.
Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Raghava GPS, et al. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE. 2013;8:e73957. https://doi.org/10.1371/journal.pone.0073957.
Chen L, Chu C, Huang T, Kong X, Cai Y-D. Prediction and analysis of cell-penetrating peptides using pseudo-amino acid composition and random forest models. Amino Acids. 2015;47:1485–93. https://doi.org/10.1007/s00726-015-1974-5.
Sanders WS, Johnston CI, Bridges SM, Burgess SC, Willeford KO. Prediction of cell penetrating peptides by support vector machines. PLoS Comput Biol. 2011;7:e1002101. https://doi.org/10.1371/journal.pcbi.1002101.
Wang X, Wang J, Lin Y, Ding Y, Wang Y, Cheng X, et al. QSAR study on angiotensin-converting enzyme inhibitor oligopeptides based on a novel set of sequence information descriptors. J Mol Model. 2011;17:1599–606. https://doi.org/10.1007/s00894-010-0862-x.
Chou KC. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins. 2001;43:246–55.
Zahiri J, Mohammad-Noori M, Ebrahimpour R, Saadat S, Bozorgmehr JH, Goldberg T, et al. LocFuse: human protein–protein interaction prediction via classifier fusion using protein localization information. Genomics. 2014;104:496–503.
Kawashima S, Pokarowski P, Pokarowska M, Kolinski A, Katayama T, Kanehisa M. AAindex: amino acid index database, progress report 2008. Nucleic Acids Res. 2008;36:D202–5. https://doi.org/10.1093/nar/gkm998.
Kuhn M. Building predictive models in R using the caret package. J Stat Softw. 2008;28(5):1–26. https://doi.org/10.18637/jss.v028.i05.
Authors’ contributions
JZ conceived the idea. ID and PS refined the proposed workflow. BK, JZ, RS, AY and MK performed the data analysis. JZ, GM, AY and BK worked on the interpretation of the results. JZ and GM wrote the first draft of the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The data supporting the conclusions of this article is provided as additional files.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
The authors received no specific funding for this work.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1.
Supplementary Materials and Methods. Train and test datasets; 227 different classifiers.
Additional file 2.
Supplementary Results. Results of feature selection and feature importance analysis.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zahiri, J., Khorsand, B., Yousefi, A.A. et al. AntAngioCOOL: computational detection of anti-angiogenic peptides. J Transl Med 17, 71 (2019). https://doi.org/10.1186/s12967-019-1813-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-019-1813-7