
Abstract
Drug resistance remains a significant challenge in cancer treatment. Recently, the interactions among various cell types within the tumor microenvironment (TME) have deepened our understanding of the mechanisms behind treatment resistance. Therefore, this review aims to synthesize current research focusing on infiltrating cells and drug resistance suggesting that targeting the TME could be a viable strategy to combat this issue. Numerous factors, including inflammation, metabolism, senescence, hypoxia, and angiogenesis, contribute to drug resistance could be a viable strategy to combat this issue. Overexpression of STAT3 is commonly associated with drug-resistant cancer cells or stromal cells. Current research often generalizes the impact of stromal cells on resistance, lacking specificity and statistical robustness. Thus, future research should take notice of this issue and aim to provide high-quality evidence. Despite the existing limitations, targeting the TME to overcome therapy resistance hold promising and valuable potential.
Similar content being viewed by others
Introduction
Drug resistance is a persistent and complex problem that diminishes the effectiveness of treatments and jeopardizes patient outcomes in cancer management [1]. Despite numerous studies dedicated to addressing drug resistance, the outcomes of these efforts have not yet reached a satisfactory level [2, 3]. In recent years, advancements in single-cell analysis, proteomics, genomics, and transcriptomics have facilitated a more detailed exploration of the specific mechanisms of drug resistance for many medicines [4, 5]. Simultaneously, there has been a growing focus on studying the tumor microenvironment (TME), which includes both infiltrating cells and noncellular components [6]. In addition to tumor cells, immune and stromal cells significantly influence the development of tumor [7]. Of these, infiltrated immune cells usually comprise T cells, B cells, tumor-associated macrophages (TAMs), dendritic cells (DCs), and myeloid-derived suppressor cells (MDSCs), among others. Non-immune cells, including tumor-associated fibroblasts (TAFs), mesenchymal stem cells (MSCs), and bone marrow stromal cells (BMSCs), also constitute a significant portion of the stromal cells in the TME [8, 9]. Researchers have gained critical insights by recognizing that tumor development is contingent on the entire TME, not solely on tumor cells [10]. This realization has led to the identification of numerous therapeutic targets and the subsequent development of novel drugs [11]. Moreover, the interactions between different cell types in the TME provide researchers with a more profound insight into the mechanisms of treatment resistance. For instance, TAMs could induce chemoresistance in cancer through regulating glucose metabolism [12]. In prostate cancer, inhibiting PI3K has been shown to enhance the anti-tumor function of CD8 + T cells, thereby transforming “cold” tumors into immunotherapy-responsive cancers [13]. Therefore, we aim to synthesize current research focusing on infiltrating cells and drug resistance. By highlighting both the exciting findings and gaps, we envision to pave the way for overcoming drug resistance through strategies targeting tumor-infiltrating cells in the TME.
Tumor-infiltrating cells regulate immunotherapy resistance
Immune cells in immunotherapy resistance
Immunotherapy has demonstrated a significant capacity to manage cancer and provide survival advantages to patients with carcinoma [11, 14]. However, the development of drug resistance hampers the effectiveness of treatment in clinical settings. Various factors influence the response of immunotherapy, such as inflammation [15], senescence [16], hypoxia [17], and so on in the TME. CD8 + T cells occupy a pivotal role in cancer cell management during immunotherapy, and their activity is subject to regulation by many factors [18]. For instance, AXL inhibited the antitumor activity of cytotoxic CD8 + T cells by regulating CD103 + dendritic cells (DCs) migration, T cell priming, and exhaustion in the TME [19]. In pancreatic ductal adenocarcinoma (PDAC), CCL2 enhanced monocyte infiltration and reduced CD8 + T cell infiltration, whereas inhibiting CCL2 expression and neutralizing monocytes could improve immune checkpoint blockade (ICB) therapy efficacy [20]. PTEN is a well-known tumor suppressor gene and is involved in the regulation of drug response [22]. In PTEN loss prostate carcinoma, the PI3Kα/β/δ inhibitor BAY1082439 can enhance CD8 + T cell-mediated immunity, making ICB more effective by promoting activation of the IFNα/IFNγ pathway, increasing β2-microglobulin expression, and boosting secretion of CXCL10/CCL5 [13]. For non-small cell lung cancer (NSCLC), TME with PTEN loss or low expression also exhibited resistance to anti-PD-L1 treatment. Mechanistically, PTEN loss in the TME promoted the differentiation of CD4 + lymphocytes into Tregs through regulating TGFβ and CXCL10 expression [23]. Tregs can create an immune-suppressed TME, reducing the effectiveness of ICB therapy [24]. In cancer patients with liver metastasis, Tregs play a role in systemic immune suppression, leading to resistance against anti-PD-1 therapy. Strategies aimed at depleting or destabilizing Tregs could potentially overcome this resistance and enhance the efficacy of anti-PD-1 therapy [25]. In a cervical cancer study, the NAT10/ac4C/FOXP1 axis in cancer cells modulated reprogramming glycolytic metabolism (increased glycolysis and lactic acid secretion) in the TME. This altered metabolism led to a higher infiltration of Tregs, causing immune suppression and resistance to ICB therapy [26]. Knockdown NAT10 expression significantly improved the efficiency of anti-PD-L1 therapy. In EGFR mutant NSCLC, upregulation of IL-6 was shown to inhibit the anti-tumor function of T and natural killer (NK) cells, while blocking IL-6 could enhance the efficacy of anti-PD-1 therapy [27]. As a new treatment in clinical practice, CAR T cell therapy has acquired wide attention [28, 29]. A few studies reported the role of immune cells in CAR T cell therapy resistance. Venet et al. [30] identified that inhibiting HLA-DR expression in monocyte would result in anti-CD19 CAR T cells failure. Furthermore, the interaction between bone marrow stromal cells (BMSCs) and CAR T cells could reduce resistance against CAR T cells by suppressing apoptosis [31]. The evidence presented underscores the importance of interactions between CD8 + T cells and other T cell subtypes in influencing the efficacy of ICB therapy, emphasizing the need for further research to classify specific T cell subtypes in studies focused on immunotherapy resistance.
In terms of DCs, Vilgelm et al. [32] identified that loss of DCs was the main reason for the resistance of ICB therapy through suppressing the anti-tumor function of CD8 + T cell and CD4 + T cell response. Similarly, a combination of CD40 agonist and ICB therapy effectively induced complete tumor regressions by modulating T cells dependent on CD40 + DCs [33]. Additionally, tumor-secreted miR424 inhibits DC-mediated T cell activation, leading to ICB resistance, which can be overcome by blocking miR424 [34]. DCs have a primary role in antigen presentation, influencing the adaptive immune response and protecting against immunotherapy resistance. NK cells typically exhibit anti-tumor function, but can be modulated by factors like SNORD46 and IGSF8, affecting their cytotoxicity and interaction with cancer cells. In malignant B cells, NK cell cytotoxicity can be suppressed by disrupted ligand binding [35, 36]. In malignant B cells, NK cell cytotoxicity was significantly suppressed by N-glycan-disrupted ligand binding to NK receptors [37]. Of this process, SPPL3 depressed N-glycosylation, which could be recovered by suppressing B3GNT2 expression. Regulation of SPPL3 or B3GNT2 expression maybe a promising way to improve CAR NK cell therapy. Lymphoid cells generally exhibit anti-tumor functions, with exceptions like Tregs. Among these, CD8 + T cells are key effectors in ICB therapy targeting tumor cells, while NK cells play a similar role in CAR therapy. The emergence of resistance to immunotherapy is often due to impaired quantity and function of these cells. Therefore, therapies directly targeting these cells can significantly improve resistance to immunotherapy.
Various myeloid cells play a significant role in influencing the effectiveness of immunotherapy [38]. Of these, TAMs are the main myeloid cells in TME, which have several subtypes [39]. It is usually deemed as a promoter of immunotherapy resistance [40]. M1-TAMs exhibit proinflammatory and phagocytic properties, contributing to anti-tumor responses [41]. Conversely, M2-TAMs display anti-inflammatory characteristics and are involved in wound healing, tumor development, and immune suppression [42]. Platten et al. [43] found that it was innate rather than adaptive immune factors that could predict the response to immunotherapy. Mechanistically, TAMs induced ICB resistance by modulating various pathways, such as the PD-L1/PD-1/CD80 axis, leading to T cell suppression and activation of Tregs in the TME. TAMs also inhibited the anti-tumor function of CD4 + T cells and pro-inflammatory responses via BMP7/P-SMAD1/MAPK14 axis, causing anti-PD1 therapy resistance [44]. Another TAM study found that TREM2 + TAMs were associated with the exhaustion of CD8 + infiltrating lymphocytes and poor recurrence-free survival, while anti-TREM2 mAb therapy could enhance the activation of T cells and sensitize the ICB therapy [45]. Thus, the authors believed that anti-TREM2 mAb therapy may be an alternative treatment for patients with ICB failure. Similarly, JMJD6 in TAMs suppressed TAMs M2 polarization by STAT3/IL-10 axis, inducing anti-PD-1 therapy resistance. In further results, blocking JMJD6 would enhance the efficacy of anti-PD-1 therapy, which identified the importance that exploring the function of different subtypes of TAMs in ICB resistance [46]. Consisted with the above study, M2 TAMs defined by authors created an immune suppressive TME by regulating SPP1/CD44/PI3K/AKT/HIF-1α/CA9 pathway in glioma, promoting the progression of cancer cell and ICB resistance [40]. These findings warrant further investigation into the distinct roles of various TAMs subtypes. Certain TAM subtypes have been recognized for their protective functions in carcinogenesis43. Furthermore, multiple studies have indicated that TAMs can influence the response to ICB through interactions with T cells [43]. Therefore, future research efforts should prioritize unraveling the regulatory mechanisms governing the interplay between TAMs subtypes and T cells, particularly in the context of immunotherapy resistance.
In a study on melanoma, it was found that STAT3 inhibitors could reduce the infiltration of TAMs and MDSCs, thereby enhancing CD8 + T cells in the TME. Further experiments showed that combining a STAT3 inhibitor with anti-PD-1 therapy could overcome resistance to anti-PD-1 monotherapy [47]. Similarly, MDSCs could be recruited through the IKKβ/ARID1A/NF-κB axis, leading to an immune-suppressed TME in prostate cancer. ICB therapy could be re-sensitized when blocking the axis using anti-NF-κB antibody or targeting CXCR2 [48]. MDSCs enhanced pro-angiogenic activity in the TME by secreting BV8, a protein that supports VEGF-independent tumor angiogenesis [49]. Inhibiting BV8 coul reduce MDSC recruitment to the TME, boost cytotoxic T cell efficacy, and overcome ICB resistance33 [50]. These studies suggest that MDSCs may contribute to the promotion of ICB resistance by modulating T cells. In terms of neutrophils, IL17 expression in another PDAC study promoted tumor-related neutrophil (TAN) infiltration and extracellular traps but inhibited CD8 + T cell infiltration. Inhibiting IL17 or neutrophils significantly boosted ICB therapy effectiveness [21]. Disrupting PADI4-dependent NETosis, which phenocopied IL17 neutralization, also impacted ICB therapy sensitivity. Various studies on TANs have indicated that their presence in the TME can lead to resistance to immunotherapy, but inhibiting TAN infiltration or NET formation can restore immunotherapy efficiency [51,52,53,54]. While most myeloid cells may contribute to immunotherapy resistance, exceptions like M1 TAMs exist. These studies emphasize the importance of exploring the specific functions of each immune cell subtype and identifying reliable predictors to determine the need for additional treatments in immunotherapy. Understanding the interactions between CD8 + T cells and other immune cells is crucial for enhancing the efficacy of immunotherapy.
Non-immune cells in immunotherapy resistance
As a stromal cell, TAFs significantly affect the efficiency of immunotherapy [55, 56]. In a PDAC study, authors found that MEK and STAT3 expressed in TAFs induced an immunosuppressive TME by inhibiting recruitment of activated/memory T cells, which caused immunotherapy resistance [57]. Specifically, MEKi + STAT3i treatment in PKT mice reduced proinflammatory and myofibroblastic TAF phenotypes, while increasing mesenchymal-like TAFs. This shift in TAF plasticity drived M2-to-M1 reprogramming of TAMs and enhanced CD8 + T cell trafficking with specific transcriptional activities. These effects were dependent on TAFs, as TAF-targeted MEK1/STAT3 silencing reduced inflammation and myeloid infiltration in vivo. Then, the authors added MEK and STAT3 inhibitors to immunotherapy (Nivolumab) in vivo, which exhibited significantly better clinical benefit compared with using monotherapy alone. Additionally, MFAP5, which encoded microfibril-associated protein 5, influenced extracellular matrix components and functions by modulating TAFs [58]. Bai et al. [59] reported that MFAP5 knockout in TAFs enhanced cytotoxic T cell infiltration through the RCN2/ERK/STAT1 axis by downregulating HAS2 and CXCL10 expression, and remodeled the matrix, synergistically enhancing immunochemotherapy effects in PDAC. In a separate PDAC investigation, TAFs expressing LRRC15 were exclusively located around the tumor tissue and absent from normal pancreatic tissue. The presence of LRRC15 + TAFs was found to be statistically associated with a poor response to immunotherapy [60]. In lung and colorectal cancers, cancer cells increase PD-L1 expression by activating the WNT/b-catenin pathway in bone marrow-derived TAFs, resulting in resistance to immunotherapy [61]. This resistance can potentially be reduced by using WNT inhibitors. In breast cancer, the TAFS/TAF-S1 cluster showed a positive correlation with Tregs, indicating a poor response to immunotherapy [62]. Adenosine is generated by ATP that undergoes rapid stepwise dephosphorylation by ectonucleotidases and can stimulus A2A receptor and A2B receptor to modulate immune activity [63]. In colorectal cancers, CD73 expression in TAFs was mediated by the A2A and A2B pathways. Targeted inhibition of these two pathways has been demonstrated to attenuate immune suppression and augment antitumor immunity in tumors with a high TAFs densit [64].
The evidence presented highlights the importance of TAFs in immunotherapy. Regulating the infiltration of different TAFs is a significant strategy to improve the effectiveness of immunotherapy [65]. Additionally, IFN-γ activated endothelial cells have been shown to suppress CD8 + T cell-mediated immune responses by increasing the expression of PD-L1 and PD-L2 [66]. Lin et al. [67] reported that the number of PD-L1-positive circulating tumor endothelial cells exhibited an incremental trend with prolonged exposure to anti-PD-1 immunotherapy, correlating with a diminished efficacy in progression-free survival in NSCLC. Furthermore, research has demonstrated that BMSCs can induce senescence in CAR T cells through the modulation of IDO-1 activity, IFNγ, and IL-2 release, ultimately weakening the anti-tumor response [68]. These findings emphasize the involvement of non-immune cells in immunotherapy and suggest the need for further exploration. Understanding how stromal cells influence immune cells is crucial, as they can either transform into pro-tumor subtypes that recruit suppressive factors like regulatory T cells or inhibit the recruitment and functions of cytotoxic T cells by secreting pro-tumor factors. The first involves their transformation into pro-tumor subtypes, which leads to the recruitment of suppressive immune regulatory factors like regulatory T cells [57, 62]. The second approach is inhibiting the recruitment and anti-tumor functions of cytotoxic T cells through the secretion of pro-tumor factors or other influences [68]. Given the essential role of cytotoxic T cells in immunotherapy, it is important to focus on researching stromal cells to directly impact cytotoxic T cell function, rather than using indirect methods. Figure 1 illustrates the results of immunotherapy resistance. Figure 1 shows the results of the immunotherapy resistance. Table 1 provides the key references of this part.

The evolution of infiltration cells contributes to immunotherapy resistance. Exhibiting the interaction between various infiltration cells in tumor microenvironment. Various factors promoted the formation of immunotherapy resistance microenvironment, such as EMT activation, angiogenesis, and metabolism reprogramming. EMT. Tregs: regulatory T cells; MDSC: myeloid-derived suppressor cell; EMT: Epithelial–mesenchymal transition
Tumor-infiltrating cells induce the resistance of targeted therapy
Immune cells in the resistance of targeted therapy
Targeted therapy, long employed in clinical practice, has extended the survival of cancer patients [69, 70]. However, the emergence of drug resistance has significantly undermined patient outcomes [71, 72]. Recently, researchers have focused on the role of infiltration cells in targeted resistance. TAMs have been found to play a key role in influencing the effectiveness of targeted therapy [73]. For example, in breast cancer, TAMs secreting IL-8 can induce resistance to lapatinib, which can be overcome by using IL-8 inhibitors [74]. In myeloma, the interaction between TAMs and cancer cells, facilitated by iron, can lead to TAMs acquiring a pro-tumor phenotype and inducing resistance to bortezomib [75]. The STAT3 pathway was a core pathway to regulate targeted therapy in TAMs [76]. Mechanistically, TAM-derived exosomes in NSCLC attenuated the anti-tumor function of gefitinib by upregulating the AKT, ERK1/2 and STAT3 pathways, decreasing tumor cell apoptosis. In ovarian cancer, upregulation of STAT3 by PARP inhibitors can polarize TAMs, leading to resistance to PARP blockade [77]. Similarly, overexpression of STAT3 can promote adaptive resistance to gefitinib by inducing M2 TAM polarization in lung cancer [78]. In addition to TAMs, other immune cells are correlated with targeted therapy resistance. NSCLC cell generated IL4/34, activating MDSCs to secrete IL10 and ARG1. These secreting factors suppressed CD8 + T cell function and enhanced the pro-tumor function of Treg cells, ultimately leading to osimertinib resistance [79]. Anti-VEGFR2 therapy facilitated N2-like neutrophil polarization, resulting in CD8 + T cell exhaustion. This exhaustion caused the anti-VEGFR2 therapy resistance in breast cancer [80]. Additionally, in certain cases of breast cancer, the interaction between HLA-G and KIR2DL4 can result in resistance to trastuzumab. Inhibiting this signaling pathway can restore NK cell cytotoxicity and enhance sensitivity to trastuzumab [81]. Based on these results, immune cells within TME markedly affect the efficacy of targeted therapies. However, the absence of identified key regulatory cell types for specific targeted treatments impedes the translation of these results into clinical practice.
Non-immune cells in the resistance of targeted therapy
Senescence can lead to targeted therapy resistance [82, 83]. In melanoma, aged dermal fibroblasts released neutral lipids, particularly ceramides, that enhanced lipid uptake in melanoma cells via upregulated FATP2 expression, leading to resistance to BRAF/MEK inhibitors [84]. Inhibiting FATP2 expression restored melanoma cell sensitivity to these inhibitors, indicating that FATP2 may be a promising therapeutic target. Additionally, PARP inhibitors can induce senescent-TAFs that produce a senescence-associated secretory phenotype (SASP) and become resistant to PARP inhibitor therapy in ovarian cancer. Bepotastine, which blocks the SASP, can reverse drug resistance by inhibiting the histamine H1 receptor-induced NF-κB pathway [85]. Furthermore, STAT3 has been implicated in the resistance of non-immune cells to targeted treatments. In neuroendocrine tumors, TAFs can stimulate cancer cells to upregulate STAT3 expression, leading to resistance to everolimus [86].
In NSCLC, TAFs, extracted from epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors-resistance tumor tissues, could produce tryptophan metabolite kynurenine which enhanced the transcription of AHR, inducing the activation of the PI3K/AKT and MEK/ERK pathways. These two pathways created an EGFR tyrosine kinase inhibitors-resistance TME, which could be countered by AHR inhibitor [87]. In gastric cancer, TAFs activate the NF-κB pathway in response to lactic acid secreted by cancer cells. This activation leads to suppression of reactive oxygen species generated by anlotinib in cancer cells through the NF-κB/BDNF/TRKB/KEAP1/NRF2 pathway, promoting anlotinib resistance [88]. In colorectal cancer, TAFs activate the IL6/IL8-JAK2 signaling pathway to promote BRD4 phosphorylation, resulting in resistance to BET inhibitors [89]. TAFs modulated by the TGF-beta pathway also impact the response to cetuximab in head and neck cancer [90]. Additionally, in breast cancer, TAFs contribute to resistance to lapatinib by activating the PI3K/AKT/MTOR and antiapoptotic pathways [91]. Overall, TAFs play a role in inducing resistance to various types of targeted therapies through mechanisms such as senescence and the STAT3 pathway.
In a recent study on lapatinib, researchers found that the interaction between MSCs and TAFs promoted resistance to lapatinib in HER2-positive breast cancer by modulating the PEAK1/INHBA/PI3K/AKT pathway [92]. Similarly, in the bone marrow microenvironment of chronic myeloid leukemia, overexpression of IGFBP-6 or SHH induced an inflammatory microenvironment, facilitating the transition from MSCs to TAFs and conferring resistance to dasatinib [93]. These findings highlight the complex interplay between MSCs and TAFs, suggesting that regulating MSCs to control the resistance-promoting activities of TAFs could be a promising therapeutic strategy. Additionally, in studies on multiple myeloma, MSCs were found to contribute to bortezomib resistance through the CXCL13 pathway [94]. Similarly, in gastrointestinal stromal tumors, MSCs facilitated drug resistance by secreting TGF-β2, which activated the PI3K/AKT pathway and led to poorer patient survival outcomes [95]. Researchers have also discovered that BMSCs play a role in drug resistance. For example, BMSCs induced imatinib resistance in chronic myeloid leukemia by creating a hypoxic tumor microenvironment [96]. Furthermore, BMSCs were found to secrete MMP2, which modified HAPLN1 and activated the NF-κB pathway, resulting in bortezomib resistance in multiple myeloma [97]. In lung adenocarcinoma, BMSCs secreted leptin and IGFBP2 in a hypoxic tumor microenvironment, activating the IGF-1R pathway and leading to erlotinib resistance [98]. BMSCs-induced hypoxia was also observed in the multiple myeloma. Specifically, BMSCs secreted small extracellular vesicles that carried miR-140-5p and miR-28-3p to create a hypoxia TME, resulting in bortezomib resistance [99]. This association between hypoxia, angiogenesis, and the generation of stem cells underscores the multifaceted role of BMSCs in drug resistance mechanisms [100]. Endothelial cells play a central role in angiogenesis and have been shown to impact the effectiveness of targeted therapy [101]. ECs interacted with TAMs to promote angiogenesis in glioblastomas by upregulating the CYP4A/20-HETE/PI3K/AKT pathway [102]. FLA-16, a flavonoid, enhanced vascular normalization by inhibiting CYP4A-mediated VEGF and TGF-β expression through the PI3K/AKT pathway in TAMs and ECs, thus overcoming resistance to anti-VEGF treatment. The mechanism of drug resistance was also reported in another study [103]. Shi et al. [103] demonstrated that tumor cells secreted IGF1, activating IGF1R on ECs and leading to vascular remodeling. This remodeled vasculature supported the proliferation of BRAFV600E kinase inhibitor-resistant tumor cells. Pericytes, in addition to endothelial cells, are also important in angiogenesis, as they secrete TSP-1 and TGFb1 to counteract the effects of BRAFV600E kinase inhibitors [104, 105]. These findings highlight the complex interactions between different stromal cells that warrant further investigation. The infiltration of various cell types can lead to resistance to targeted therapies, emphasizing the importance of identifying specific cellular targets for each therapy to overcome drug resistance. Figure 2 illustrates the specific mechanisms of targeted therapy resistance, while Table 2 provides key references for this section.

The evolution of infiltration cells contributes to resistance against targeted therapy. The picture specifically describes how different cells (including immune and non-immune cells) generate a therapy-resistant microenvironment via CD4 + and CD8 + T cells inhibition, activating and recruiting regulatory T Cells, and angiogenesis upregulation. MDSC: myeloid-derived suppressor cell; Treg: regulatory T cell; ROS: reactive oxygen species; TAM: Tumor-associated macrophage
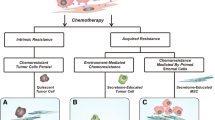
Tumor-infiltrating cells influence chemoresistance
Immune cells in chemoresistance
Chemotherapy, as a primary treatment for many cancers, has greatly improved the outlook for cancer patients. However, the development of chemoresistance hinders the intended survival benefits for patients [106, 107]. In recent years, there have been significant advancements in our understanding of the mechanisms underlying chemoresistance in the TME [108, 109]. Similar to immunotherapy, the activation of STAT3 has been found to influence chemoresistance. For example, STAT3 activation triggered by CCL5 secreted by TAMs can increase the expression of the transcription factor NANOG, leading to chemoresistance [110]. TAM-generated TGF-β1 activated hepatic leukemia factor (HLF) in cancer cells, inducing ferroptosis resistance in triple-negative breast cancer cells by stimulating gamma-glutamyltransferase 1 (GGT1). In response, these cancer cells release IL-6, which promoted further secretion of TGF-β1 by TAMs through the JAK2/STAT3 pathway and finally facilitated cancer progression [111]. Moreover, MDSCs recruited by anaerobic pseudomonas can release IL-23, activating the STAT3-EMT pathway and promoting EMT in tumor cells [112]. In addition to STAT3 regulation, TAMs also influence chemoresistance through various other mechanisms. For instance, oxaliplatin inhibits the differentiation of MDSCs into anti-tumorigenic M1-TAMs, leading to increased migration and invasion of tumor cells [113]. Triptolide inhibits M2-TAM polarization through the PI3K/AKT/NF-kB signaling pathway, thereby suppressing invasion and migration of drug-resistant ovarian cancer cells [114]. The enhanced iron transport in TAMs contributes to tumor progression and chemoresistance [75]. Furthermore, M2-TAMs inhibit TRAF5-mediated necrotic apoptosis in colorectal cancer cells by increasing METTL3-mediated RNA N6-methyladinosine (m6A) modification levels, thereby preventing necrosis [115]. In the context of T cells, colorectal cancer cells were found to secrete CCL20, which subsequently recruited Tregs via the FOXO1/CEBPB/NF-κB signaling pathway, thereby enhancing chemoresistance [116]. As for TANs, IL-1β released by cancer cells treated with chemotherapy was observed to attract TANs and form NETs, ultimately contributing to chemoresistance [117]. Notably, integrin-αvβ1 and metalloproteinase 9, both proteins associated with NETs, were capable of activating TGF-β to induce epithelial-to-mesenchymal transition in breast cancer cells, leading to chemoresistance and metastasis. Furthermore, various studies have shown a positive correlation between TANs or NETs and chemoresistance [54, 118, 119]. NK cells also play a role in chemoresistance, with examples such as IL15-activated NK cells effectively targeting cisplatin-resistant neuroblastoma, thereby overcoming chemoresistance [120]. Moreover, therapy involving helminth-derived Taenia crassiceps combined with 5-fluorouracil was found to enhance NK cell recruitment and cytotoxic activity, ultimately improving chemotherapy efficacy [121]. These findings underscore the significance of NK cells in combating chemoresistance. In contrast to the impact of immunotherapy, T cells do not seem to play a significant role in chemotherapy, as evidenced by studies highlighting distinct treatment mechanisms. When compared to resistance to immunotherapy, TAMs, NK cells, MDSCs, and TANs exhibit similar functions in mediating chemoresistance. Notably, two studies have specifically addressed the role of M2-TAMs in chemoresistance, serving as valuable references for future investigations.
Non-immune stromal cells in chemoresistance
Non-immune stromal cells within the TME play a role in chemoresistance [122]. For example, TAFs secrete the lncRNA CCAL, which interacts with the mRNA-stabilizing protein human antigen R to activate the Wnt/β-catenin pathway in colorectal cancer cells, leading to chemoresistance [123]. Similar to regulating the CXCR4/Wnt/β-catenin pathway, CXCL12, derived from TAFs, could trigger EMT in epithelial ovarian cancer cells to resist cisplatin [124]. Exosomes facilitate communication between different cell types, with TAFs secreting exosomes that contribute to the TME. These exosomes may contain miR-522, which inhibits ferroptosis in cancer cells by targeting ALOX15, thereby promoting chemoresistance in gastric cancer [106]. In prostate cancer, TAFs secrete exosomal miR-423-5p, which promotes chemoresistance by targeting GREM2 through the TGF-β signaling pathway [125]. Moreover, in pancreatic ductal adenocarcinoma (PDAC), the knockout of IL-6 in α-smooth muscle actin (αSMA) + TAFs can enhance the effectiveness of gemcitabine treatment and improve survival rates [126]. Metastatic lymph TAFs have been shown to regulate the p38 and JNK signaling pathways by secreting PI16, ultimately reducing cisplatin-induced apoptosis in esophageal squamous cell cancer cells [127].
Similar to TAFs, MSCs also play a role in modulating cisplatin resistance. When treated with leptin, MSCs can upregulate TGF-β, leading to increased expression of autophagy-related genes such as ATG7, ATG5, and beclin1, ultimately inducing cisplatin resistance in osteosarcoma cells [128]. Additionally, MSCs in the tumor microenvironment of glioma can migrate to glioma cells through guidance from angiogenic cytokines. These migrated MSCs can enhance FOXS1 expression in glioma cells by secreting IL-6, triggering EMT and promoting resistance to temozolomide [129]. Downregulation of METTL3 in MSCs would lead to an increase in AKT protein levels which promoted MSCs adipogenesis, resulting in chemoresistance in acute myeloid leukemia cells [130]. MSCs can also interact with leukemia blasts, activating ABC transporters and facilitating the transition of acute myeloid leukemia cells into a more chemoresistant subset [131]. By secreting CXCL13, MSCs can upregulate the expression of BTK, NF-κB, BCL-2, and MDR-1 mRNA and protein, thereby enhancing the resistance of multiple myeloma cells to bortezomib [94]. The findings suggest that mesenchymal stem cells (MSCs) can play a role in promoting chemoresistance, including in endothelial cells. In glioblastoma, CCBE1, an extracellular matrix protein, stimulated hyper-angiogenesis and partial endothelial-to-mesenchymal transition in human microvascular ECs via the VEGFC/RHO pathway, conferring temozolomide resistance to cancer cells. Mechanistically, SP1 upregulated CCBE1 expression in temozolomide-resistant cells. CCBE1, aided by CAVIN1, was then secreted into the TME, promoting VEGFC maturation through the VEGFR2/VEGFR3/RHO pathway in vascular ECs, thus enhancing abnormal angiogenesis in temozolomide-resistant tumors [132]. The transformation of endothelial cells into mesenchymal stem cell-like cells was also observed in another study on glioblastoma, where the C-MET/WNT/β-catenin/MRP1 pathway was implicated in inducing temozolomide resistance [133]. Two separate studies reported an increase in angiogenesis in chemoresistant tumor tissue. Bone marrow-derived mesenchymal stem cells (BMSCs) were found to induce bortezomib resistance through the generation of HAPLN1 and MMP2, which activated NF-κB signaling [97]. Disrupting gap junctions between acute myeloid leukemia cells and BMSCs using carbenoxolone was shown to impair energy metabolism in tumor cells and reduce chemotherapy resistance [134]. The complex regulatory pathways through which non-immune stromal cells impact chemotherapy resistance present challenges for clinical experimentation. Moreover, the lack of classification of these cell subtypes in the studies mentioned detracts from their overall value. Specific mechanisms are detailed in Fig. 3, while Table 3 provides key references on chemoresistance.

Chemotherapy-induced hypoxia, vascular damage, and chronic inflammation are associated with the development of chemoresistance. Various infiltration cells in the TME play crucial roles in contributing to chemoresistance. Various factors promoted the formation of chemoresistance microenvironment, such as STAT3 pathway activation, EMT activation, and TAFs secreted pro-tumor exosomes. MDSCs: myeloid-derived suppressor cell; Tregs: regulatory T cells; MSCs: mesenchymal stem cells; BMSCs: bone marrow stromal cells; TAFs: Tumor-associated fibroblasts.; TAM: Tumor-associated macrophage
Tumor-infiltrating cells in radiotherapy resistance
Radiation therapy (RT) is an essential treatment for almost all types of cancer [135]. However, the challenge of radio-resistance remains, leading to issues such as treatment failure, tumor spread, cancer recurrence, and ultimately, a poor prognosis [136, 137]. In oral squamous cell carcinoma, cancer cells thriving in an acidic microenvironment acquire characteristics of EMT and develop resistance to radiotherapy [138]. Radiotherapy works by directly damaging and killing tumor cells through DNA damage and releasing neoantigens into the immune system. In colorectal cancer, the combination of PD-L1 + immune cells and budding nucleus β-catenin + tumor cells can create a niche lesion for cancer stem cells, resulting in resistance to neoadjuvant chemoradiotherapy [139]. The inhibition of the DNA damage repair pathway with AZD6738 has been shown to enhance the effectiveness of radioimmunotherapy for hepatocellular carcinoma by recruiting CD8 + T cell infiltration [140]. In liver cancer, inhibiting MELK promotes TAMs M1 polarization and CD8 + T cell infiltration, while blocking TAMs M2 polarization through the secretion of STAT3-derived CCL2 enhances RT efficiency [141]. Similarly, the expression of TMEM160 in colorectal cancer cells can reduce RT sensitivity and CD8 + T cell activity by downregulating PD-L1 degradation, with downregulation of TMEM160 expression leading to the restoration of RT sensitivity [142]. Various studies have also highlighted that the suppression in number and function of CD8 + T cells is a significant factor in RT resistance, with the activation of CD8 + T cells being able to overcome this resistance [143,144,145]. In terms of Tregs, glioblastoma models treated with RT tend to recruit CD103 + Tregs, resulting in RT resistance and a decrease in CD8 + T cell activity [146]. Depletion of CD103 + Tregs has been shown to restore the efficacy of RT. The role of T cells is crucial in immunotherapy, chemoresistance, and RT. In breast cancer, researchers have found that mebendazole can enhance NK cell-mediated cytotoxicity against cancer cells, thereby improving the efficacy of RT [147]. Another study indicated that combination of RT and injection exogenous NK cells could significantly improve RT efficiency in vitro and in vivo [148]. In terms of TAMs, it was observed that miR-143-3p released by esophageal squamous cancer cells promoted TAM M2 polarization, resulting in RT resistance [149]. Furthermore, Ma et al. [150]. reported that engineered M1 macrophage-derived exosomes could enhance cancer cell DNA damage, polarize M2 macrophages into M1 phenotypes, and recover T cell anti-tumor function, consequently improving RT efficiency. Similarly, TANs and NETs would lead to RT resistance [151, 152]. For example, GLUT1 expression in TANs could facilitate glucose uptake, reducing RT efficiency in lung cancer, while inhibiting GLUT1 could restore RT sensitivity by decreasing glucose uptake in TANs [153]. Overall, RT resistance is influenced by various factors, and understanding the molecular mechanisms of radiation tolerance and its interaction with the tumor microenvironment has the potential to enhance the effectiveness of radiation therapy [148]. It is important to recognize that RT resistance is often associated with RT-induced alterations in the immune microenvironment. Anti-tumor lymphoid cells such as T cells and NK cells have the potential to overcome this RT resistance, presenting a promising approach to improving RT efficacy. Therapeutically, modulating the quantity and function of immune cells within the tumor microenvironment through pharmacological interventions or supplementing therapy with exogenous immune cells could enhance RT outcomes.
Furthermore, stromal cells in the TME can also affect RT resistance. TAFs have been identified as key mediators of RT resistance through various mechanisms [154]. For example, TAF-conditioned media has been shown to enhance RT resistance in NSCLC cells by activating the SMAD3/ITGA6/PI3K/AKT pathway [154, 155]. In esophageal squamous-cell carcinoma, TAF-derived collagen type 1 promotes DNA damage repair and induces RT resistance, while cancer cell-secreted CXCL1 further exacerbates this resistance [156]. Interestingly, the reciprocal interaction between cancer cell-secreted CXCL1 and TAFs leads to increased RT resistance through the CXCL1-CXCR2 pathway. In nasopharyngeal carcinoma, TAFs have been found to reduce irradiation-induced DNA damage and promote RT resistance via the IL-8/NF-κB pathway [157]. To solve TAF-induced RT resistance, Jian et al. [158]. created a nanoplatform to clear TAFs and senescent TAFs (which also could induce RT resistance), obviously improving RT resistance. In terms of MDSCs, RT promoted MDSCs infiltration and YTHDF2 expression, resulting to RT resistance [159]. Mechanistically, knockout YTHDF2 in myeloid cell would change MDSC differentiation and attenuate MDSC infiltration and function by activating the NF-κB pathway, recovering RT sensitivity of cancer cells. In pancreatic cancer, tumor cells secrete lactate, promoting the infiltration of an immunosuppressive MDSC phenotype, leading to resistance to RT [160]. Of these, HIF-1α was the essential of lactate-induced resistance by mediating the GPR81/mTOR/HIF-1α/STAT3 pathway, while inhibiting lactate generation or HIF-1α depletion in MDSCs would recover T cell anti-tumor function and thus overcome RT resistance. Similarly, several studies also found that the pro-tumor MDSC subtype facilitated RT resistance by interacting with cancer cells or immune cells [161,162,163]. Regarding ECs, depression of DAB2IP in breast cells would induce RT resistance by promoting angiogenesis [164]. Moreover, reducing STAT3 in breast cancer cells with decreased DAB2IP expression has been found to enhance RT sensitivity by suppressing angiogenesis. This research suggests that stromal cells have the ability to influence cell differentiation and modify the TME by promoting processes like angiogenesis or metabolic changes through interactions with cancer cells, such as repairing RT-induced DNA damage, ultimately leading to RT resistance [159, 160]. While some studies have indicated that certain stromal cells can differentiate into anti-cancer subtypes, there is a lack of research on the specific classification and functions of these subtypes, indicating a need for further investigation.
Antiandrogen therapy resistance is widely observed in clinical practice. Sawyers et al. [165] discovered that NRG1 in TAF supernatant activated HER3 on prostate cancer cells, leading to antiandrogen resistance. Inhibiting the NRG1/HER3 axis could resensitize tumors to androgen deprivation therapy both in vitro and in vivo. Further investigation is needed to fully understand the intricate mechanisms at play. Table 4 in this study serves as a comprehensive reference guide of this section.
Conclusion and perspective
The significant challenge of anti-tumor drug resistance poses a considerable obstacle to effective cancer management and the successful implementation of precision medicine. Therapies targeting the TME may offer a valuable strategy to mitigate drug resistance, with factors such as inflammation, metabolism, senescence, hypoxia, and angiogenesis playing key roles in modulating drug resistance through interactions among various cell types. STAT3 and its pathways are crucial in different therapies and cell types within the TME, with evidence showing that STAT3 enhances ICB resistance and cell polarization, while its inhibition can reverse drug resistance. In chemotherapy, inhibiting STAT3 expression can alleviate chemoresistance, and in targeted therapy, overexpression of STAT3 is linked to drug-resistant cancer or stromal cells. Given the potential of targeting STAT3 to combat cancer therapy resistance, future research should focus on this pathway in the context of TME drug resistance. Clinically, STAT3 expression could be used as an indicator to predict response to adjuvant therapy, and STAT3 inhibitors show promise as effective anti-resistance treatments in cancer management.
Distinct subtypes such as CD8 + T cells, Tregs, and CD4 + T cells are defined within T cells, enabling focused research on their individual roles in therapy resistance. However, clear definitions of subtypes for many immune and non-immune cell types remain uncertain. While some studies have attempted to classify these subtypes, widespread acceptance and application have not been achieved. Furthermore, current research often emphasizes the overall impact of stromal cells on therapy resistance, rather than specific subtypes, leading to reduced statistical robustness and specificity in these studies. Current evidence highlights the anti-tumor functions of specific subsets of stromal cells, such as TAFs and TAMs [166]. Accordingly, future research should address this issue by aiming to provide high-quality evidence. Despite existing limitations, we assert that approaches targeting the TME to overcome therapy resistance hold promising and valuable potential.
Data availability
No datasets were generated or analysed during the current study.
References
Gao YY, Yang WC, Ashby CR Jr., Hao GF. Mapping cryptic binding sites of drug targets to overcome drug resistance. Drug Resist Updates: Reviews Commentaries Antimicrob Anticancer Chemother. 2023;67:100934.
Li D, Yu Q, Wu R, Tuo Z, Zhu W, Wang J et al. Chronobiology of the Tumor Microenvironment: implications for therapeutic strategies and circadian-based interventions. Aging Dis 2024.
Li D, Wang J, Tuo Z, Yoo KH, Yu Q, Miyamoto A et al. Natural products and derivatives in renal, urothelial and testicular cancers: targeting signaling pathways and therapeutic potential. Phytomedicine: international journal of phytotherapy and phytopharmacology 2024.
Cerella C, Dicato M, Diederich M. Enhancing personalized immune checkpoint therapy by immune archetyping and pharmacological targeting. Pharmacol Res. 2023;196:106914.
Gottesman MM, Robey RW, Ambudkar SV. New mechanisms of multidrug resistance: an introduction to the Cancer Drug Resistance special collection. Cancer Drug Resist. 2023;6:590–5.
Karlsson M, Zhang C, Méar L, Zhong W, Digre A, Katona B et al. A single-cell type transcriptomics map of human tissues. Sci Adv 2021;7.
Khalaf K, Hana D, Chou JT-T, Singh C, Mackiewicz A, Kaczmarek M. Aspects of the Tumor Microenvironment involved in Immune Resistance and Drug Resistance. Front Immunol 2021;12.
Paul S, Sinha S, Kundu CN. Targeting cancer stem cells in the tumor microenvironment: an emerging role of PARP inhibitors. Pharmacol Res. 2022;184:106425.
Xin Q, Ma H, Wang H, Zhang XD. Tracking tumor heterogeneity and progression with near-infrared II fluorophores. Explor (Beijing). 2023;3:20220011.
Kim M, Lee NK, Wang CJ, Lim J, Byun MJ, Kim TH, et al. Reprogramming the tumor microenvironment with biotechnology. Biomater Res. 2023;27:5.
Feng D, Li D, Wu R, Han P. Scientific Advancements in Drug Development and Trials for Urothelial Carcinoma: Insights From the 2023 ASCOGU Cancers Symposium. Aging Dis 2023.
Shi Q, Shen Q, Liu Y, Shi Y, Huang W, Wang X et al. Increased glucose metabolism in TAMs fuels O-GlcNAcylation of lysosomal Cathepsin B to promote cancer metastasis and chemoresistance. Cancer cell 2022;40:1207-22.e10.
Qi Z, Xu Z, Zhang L, Zou Y, Li J, Yan W et al. Overcoming resistance to immune checkpoint therapy in PTEN-null prostate cancer by intermittent anti-PI3Kα/β/δ treatment. Nat Commun 2022;13.
Pang K, Shi ZD, Wei LY, Dong Y, Ma YY, et al. Research progress of therapeutic effects and drug resistance of immunotherapy based on PD-1/PD-L1 blockade. Drug Resist Updat. 2023 Jan;66:100907.
Johnson RP, Ratnacaram CK, Kumar L, Jose J. Combinatorial approaches of nanotherapeutics for inflammatory pathway targeted therapy of prostate cancer. Drug Resist Updat. 2022 Sep;64:100865.
Xing S, Hu K, Wang Y. Tumor Immune Microenvironment and Immunotherapy in Non-Small Cell Lung Cancer: Update and New Challenges. Aging Dis 2022;13:1615-32.
Zhang F, Wang X, Hu H, Yang Y, Wang J, Tang Y, et al. A hypoxia related long non-coding RNA signature could accurately predict survival outcomes in patients with bladder cancer. Bioengineered. 2021;12:3802–23.
Yu Q, Zhang F, Feng D, Li D, Xia Y, Gan MF. An inflammation-related signature could predict the prognosis of patients with kidney renal clear cell carcinoma. Front Genet. 2022;13:866696.
Im K, Choi YJ, Kim DH, Kim D-S, Ban K, Ji W, et al. AXL receptor tyrosine kinase inhibition improves the anti-tumor effects of CD8 + T cells by inducing CD103 + dendritic cell-mediated T cell priming. Biochem Biophys Res Commun. 2023;680:7–14.
Li X, He G, Liu J, Yan M, Shen M, Xu L, et al. CCL2-mediated monocytes regulate immune checkpoint blockade resistance in pancreatic cancer. Int Immunopharmacol. 2022;106:108598.
Zhang Y, Chandra V, Riquelme Sanchez E, Dutta P, Quesada PR, Rakoski A et al. Interleukin-17-induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer. J Exp Med 2020;217.
Park M, Bang C, Yun WS, Jeong YM. Low-molecular-weight fucoidan inhibits the proliferation of melanoma via Bcl-2 phosphorylation and PTEN/AKT pathway. Oncol Res. 2023;32:273–82.
Exposito F, Redrado M, Houry M, Hastings K, Molero-Abraham M, Lozano T, et al. PTEN loss confers resistance to Anti-PD-1 therapy in Non-small Cell Lung Cancer by Increasing Tumor Infiltration of Regulatory T Cells. Cancer Res. 2023;83:2513–26.
Zhong J, Guo J, Zhang X, Feng S, Di W, Wang Y, Zhu H. The remodeling roles of lipid metabolism in colorectal cancer cells and immune microenvironment. Oncol Res. 2022;30:231–42.
Lee JC, Mehdizadeh S, Smith J, Young A, Mufazalov IA, Mowery CT et al. Regulatory T cell control of systemic immunity and immunotherapy response in liver metastasis. Sci Immunol 2020;5.
Chen X, Hao Y, Liu Y, Zhong S, You Y, Ao K et al. NAT10/ac4C/FOXP1 promotes malignant progression and facilitates immunosuppression by reprogramming glycolytic metabolism in Cervical Cancer. Adv Sci 2023;10.
Patel SA, Nilsson MB, Yang Y, Le X, Tran HT, Elamin YY, et al. IL6 mediates suppression of T- and NK-cell function in EMT-associated TKI-resistant EGFR-mutant NSCLC. Clin Cancer Res. 2023;29:1292–304.
Qian K, Li G, Zhang S, Fu W, Li T, Zhao J et al. CAR-T‐cell products in solid tumors: Progress, challenges, and strateg ies. Interdisciplinary Med 2024;2.
Wang C, Wang C, Liu H, Zhao S, Qiu J, Li P et al. Immune function assessing of TIM3/CD28-modified CD19 CAR‐T cells and g eneral CD19 CAR‐T cells through a high‐throughput single‐cell microarr ay platform. Interdisciplinary Med 2023;2.
Bourbon E, Sesques P, Gossez M, Tordo J, Ferrant E, Safar V, et al. HLA-DR expression on monocytes and outcome of anti-CD19 CAR T-cell therapy for large B-cell lymphoma. Blood Adv. 2023;7:744–55.
Holthof LC, van der Schans JJ, Katsarou A, Poels R, Gelderloos AT, Drent E, et al. Bone marrow mesenchymal stromal cells can render multiple myeloma cells resistant to Cytotoxic Machinery of CAR T Cells through inhibition of apoptosis. Clin Cancer Res. 2021;27:3793–803.
Kumar A, Ramani V, Bharti V, de Lima Bellan D, Saleh N, Uzhachenko R et al. Dendritic cell therapy augments antitumor immunity triggered by CDK4/6 inhibition and immune checkpoint blockade by unleashing systemic CD4 T-cell responses. J Immunother Cancer 2023;11.
Morrison AH, Diamond MS, Hay CA, Byrne KT, Vonderheide RH. Sufficiency of CD40 activation and immune checkpoint blockade for T cell priming and tumor immunity. Proceedings of the National Academy of Sciences 2020;117:8022-31.
Zhao X, Yuan C, Wangmo D, Subramanian S. Tumor-secreted extracellular vesicles regulate T-Cell Costimulation and can be manipulated to induce tumor-specific T-Cell responses. Gastroenterology 2021;161:560 – 74.e11.
Zhang Y, Zhao Z, Huang LA, Liu Y, Yao J, Sun C et al. Molecular mechanisms of snoRNA-IL-15 crosstalk in adipocyte lipolysis and NK cell rejuvenation. Cell metabolism 2023;35:1457-73.e13.
Li Y, Wu X, Sheng C, Liu H, Liu H, Tang Y et al. IGSF8 is an innate immune checkpoint and cancer immunotherapy target. Cell 2024;187:2703-16.e23.
Zhuang X, Woods J, Ji Y, Scheich S, Mo F, Rajagopalan S, et al. Functional genomics identifies N-acetyllactosamine extension of complex N-glycans as a mechanism to evade lysis by natural killer cells. Cell Rep. 2024;43:114105.
Chu X, Zhang Y, Cheng S. Heterogeneity of tumor-infiltrating myeloid cells in era of single-cell genomics. Chin J Cancer Res. 2022;34:543–53.
Kim J, Kim M, Yong SB, Han H, Kang S, Lahiji SF, et al. Engineering TGF-β inhibitor-encapsulated macrophage-inspired multi-functional nanoparticles for combination cancer immunotherapy. Biomater Res. 2023;27:136.
You G, Zheng Z, Huang Y, Liu G, Luo W, Huang J, et al. scRNA-seq and proteomics reveal the distinction of M2‐like macrophages between primary and recurrent malignant glioma and its critical role in the recurrence. CNS Neurosci Ther. 2023;29:3391–405.
Lolo FN, Rius C, Casas-Tintó S. Elimination of classically-activated macrophages in tumor-conditioned medium by alternatively-activated macrophages. Biology open. 2017;6:1897–903.
Drakes ML, Stiff PJ. Regulation of Ovarian Cancer Prognosis by Immune cells in the Tumor Microenvironment. Cancers (Basel) 2018;10.
Aslan K, Turco V, Blobner J, Sonner JK, Liuzzi AR, Núñez NG et al. Heterogeneity of response to immune checkpoint blockade in hypermutated experimental gliomas. Nat Commun 2020;11.
Cortez MA, Masrorpour F, Ivan C, Zhang J, Younes AI, Lu Y et al. Bone morphogenetic protein 7 promotes resistance to immunotherapy. Nat Commun 2020;11.
Binnewies M, Pollack JL, Rudolph J, Dash S, Abushawish M, Lee T et al. Targeting TREM2 on tumor-associated macrophages enhances immunotherapy. Cell Rep 2021;37.
Chen S, Wang M, Lu T, Liu Y, Hong W, He X, et al. JMJD6 in tumor-associated macrophage regulates macrophage polarization and cancer progression via STAT3/IL-10 axis. Oncogene. 2023;42:2737–50.
Kim TW, Kim Y, Keum H, Jung W, Kang M, Jon S. Combination of a STAT3 inhibitor with anti-PD-1 immunotherapy is an effective treatment regimen for a vemurafenib-resistant melanoma. Mol Ther Oncolytics. 2022;26:1–14.
Li N, Liu Q, Han Y, Pei S, Cheng B, Xu J et al. ARID1A loss induces polymorphonuclear myeloid-derived suppressor cell chemotaxis and promotes prostate cancer progression. Nat Commun 2022;13.
Shojaei F, Wu X, Zhong C, Yu L, Liang XH, Yao J, et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature. 2007;450:825–31.
Benguigui M, Vorontsova A, Timaner M, Levin S, Haj-Shomaly J, Deo A et al. Bv8 blockade sensitizes Anti-PD1 therapy resistant tumors. Front Immunol 2022;13.
Xie P, Yu M, Zhang B, Yu Q, Zhao Y, Wu M, et al. CRKL dictates anti-PD-1 resistance by mediating tumor-associated neutrophil infiltration in hepatocellular carcinoma. J Hepatol. 2024;81:93–107.
Xie Y, Zhou T, Li X, Zhao K, Bai W, Hou X, et al. Targeting ESE3/EHF with Nifurtimox inhibits CXCR2(+) neutrophil infiltration and overcomes pancreatic Cancer Resistance to Chemotherapy and Immunotherapy. Gastroenterology. 2024;167:281–97.
Liu YJ, Li JP, Han M, Li JX, Ye QW, Lin ST, et al. IFIT1 + neutrophil is a causative factor of immunosuppressive features of poorly cohesive carcinoma (PCC). J Transl Med. 2024;22:580.
Mousset A, Bellone L, Gaggioli C, Albrengues J. NETscape or NEThance: tailoring anti-cancer therapy. Trends Cancer 2024.
Van den Bossche V, Zaryouh H, Vara-Messler M, Vignau J, Machiels JP, Wouters A, et al. Microenvironment-driven intratumoral heterogeneity in head and neck cancers: clinical challenges and opportunities for precision medicine. Drug Resist Updat. 2022 Jan;60:100806.
Liu YJ, Zeng SH, Zhang W, Li JP, Yin Y, Zhuang YW, et al. USP51/ZEB1/ACTA2 axis promotes mesenchymal phenotype in gastric cancer and is associated with low cohesion characteristics. Pharmacol Res. 2023;188:106644.
Datta J, Dai X, Bianchi A, De Castro Silva I, Mehra S, Garrido VT, et al. Combined MEK and STAT3 inhibition uncovers stromal plasticity by enriching for Cancer-Associated fibroblasts with mesenchymal stem cell-like features to overcome Immunotherapy Resistance in Pancreatic Cancer. Gastroenterology. 2022;163:1593–612.
Wang Y, Wang R, Li B, Huang Z, Zhao S, Chen S, et al. Cancer-associated fibroblasts in the invasive tumour front promote the metastasis of oral squamous cell carcinoma through MFAP5 upregulation. Gene. 2023;876:147504.
Duan Y, Zhang X, Ying H, Xu J, Yang H, Sun K, et al. Targeting MFAP5 in cancer-associated fibroblasts sensitizes pancreatic cancer to PD-L1-based immunochemotherapy via remodeling the matrix. Oncogene. 2023;42:2061–73.
Dominguez CX, Müller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, et al. Single-cell RNA sequencing reveals stromal evolution into LRRC15 + myofibroblasts as a determinant of patient response to Cancer Immunotherapy. Cancer Discov. 2020;10:232–53.
Huang T, Li F, Cheng X, Wang J, Zhang W, Zhang B et al. Wnt inhibition sensitizes PD-L1 blockade therapy by overcoming bone marrow-derived myofibroblasts-mediated Immune Resistance in Tumors. Front Immunol 2021;12.
Kieffer Y, Hocine HR, Gentric G, Pelon F, Bernard C, Bourachot B, et al. Single-cell analysis reveals fibroblast clusters linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020;10:1330–51.
Tokano M, Matsushita S, Takagi R, Yamamoto T, Kawano M. Extracellular adenosine induces hypersecretion of IL-17A by T-helper 17 cells through the adenosine A2a receptor. Brain Behav Immun Health. 2022;26:100544.
Yu M, Guo G, Huang L, Deng L, Chang C-S, Achyut BR et al. CD73 on cancer-associated fibroblasts enhanced by the A2B-mediated feedforward circuit enforces an immune checkpoint. Nat Commun 2020;11.
Feng DC, Zhu WZ, Wang J, Li DX, Shi X, Xiong Q, et al. The implications of single-cell RNA-seq analysis in prostate cancer: unraveling tumor heterogeneity, therapeutic implications and pathways towards personalized therapy. Mil Med Res. 2024;11:21.
Rodig N, Ryan T, Allen JA, Pang H, Grabie N, Chernova T, et al. Endothelial expression of PD-L1 and PD-L2 down-regulates CD8 + T cell activation and cytolysis. Eur J Immunol. 2003;33:3117–26.
Zhang L, Zhang X, Liu Y, Zhang T, Wang Z, Gu M, et al. PD-L1 + aneuploid circulating tumor endothelial cells (CTECs) exhibit resistance to the checkpoint blockade immunotherapy in advanced NSCLC patients. Cancer Lett. 2020;469:355–66.
Towers R, Trombello L, Fusenig M, Tunger A, Baumann AL, Savoldelli R, et al. Bone marrow-derived mesenchymal stromal cells obstruct AML-targeting CD8(+) clonal effector and CAR T-cell function while promoting a senescence-associated phenotype. Cancer Immunol Immunother. 2024;73:8.
Nardone V, Romeo C, D’Ippolito E, Pastina P, D’Apolito M, Pirtoli L, et al. The role of brain radiotherapy for EGFR- and ALK-positive non-small-cell lung cancer with brain metastases: a review. Radiol Med. 2023;128:316–29.
Xiang H, Xin L, Ye J, Xu L, Zhang H, Zhang S, Liu Y. A multicenter study on efficacy of dual-target neoadjuvant therapy for HER2-positive breast cancer and a consistent analysis of efficacy evaluation of neoadjuvant therapy by Miller-Payne and RCB pathological evaluation systems (CSBrS-026). Chin J Cancer Res. 2023;35:702–12.
Nishal S, Jhawat V, Gupta S, Phaugat P. Utilization of kinase inhibitors as novel therapeutic drug targets: a review. Oncol Res. 2022;30:221–30.
Liu H, Chen X, Jia Y, Chen H, Wang X, Liu G, Luo Y. Facing inevitable PARPis resistance: Mechanisms and therapeutic strate gies for breast cancer treatment. Interdisciplinary Medicine;1.
Kim D, An L, Moon J, Maymi VI, McGurk AI, Rudd BD, et al. Ccr2 + monocyte-derived macrophages Influence trajectories of Acquired Therapy Resistance in Braf-Mutant Melanoma. Cancer Res. 2023;83:2328–44.
Ahmed S, Mohamed HT, El-Husseiny N, El Mahdy MM, Safwat G, Diab AA et al. IL-8 secreted by tumor associated macrophages contribute to lapatinib resistance in HER2-positive locally advanced breast cancer via activation of Src/STAT3/ERK1/2-mediated EGFR signaling. Biochim et Biophys Acta (BBA) - Mol Cell Res 2021;1868.
Camiolo G, Barbato A, Giallongo C, Vicario N, Romano A, Parrinello NL et al. Iron regulates myeloma cell/macrophage interaction and drives resistance to bortezomib. Redox Biol 2020;36.
Yuan S, Chen W, Yang J, Zheng Y, Ye W, Xie H et al. Tumor–associated macrophage–derived exosomes promote EGFR–TKI resistance in non–small cell lung cancer by regulating the AKT, ERK1/2 and STAT3 signaling pathways. Oncol Lett 2022;24.
Ding L, Wang Q, Martincuks A, Kearns MJ, Jiang T, Lin Z et al. STING agonism overcomes STAT3-mediated immunosuppression and adaptive resistance to PARP inhibition in ovarian cancer. J Immunother Cancer 2023;11.
Lu J, Li J, Lin Z, Li H, Lou L, Ding W et al. Reprogramming of TAMs via the STAT3/CD47-SIRPα axis promotes acquired resistance to EGFR-TKIs in lung cancer. Cancer Lett 2023;564.
Wang C, Fei K, Liu L, Duan J, Wang Z, Li S, et al. Abnormal activation of NF-κB and MAPK signaling pathways affect osimertinib resistance and influence the recruitment of myeloid‐derived suppressor cells to shape the immunosuppressive tumor immune microenvironment. Thorac cancer. 2023;14:1843–56.
Zhang Z, Yang C, Li L, Zhu Y, Su K, Zhai L et al. γδT Cell-IL17A-Neutrophil Axis drives immunosuppression and confers breast Cancer Resistance to High-Dose Anti-VEGFR2 therapy. Front Immunol 2021;12.
Zheng G, Guo Z, Li W, Xi W, Zuo B, Zhang R et al. Interaction between HLA-G and NK cell receptor KIR2DL4 orchestrates HER2-positive breast cancer resistance to trastuzumab. Signal Transduct Target Therapy 2021;6.
Li D, Yu Q, Wu R, Tuo Z, Wang J, Ye L, et al. Interactions between oxidative stress and senescence in cancer: mechanisms, therapeutic implications, and future perspectives. Redox Biol. 2024;73:103208.
Wang J, Wei J, Inuzuka H. Aging and cancer hallmarks as therapeutic targets. Acta materia Med 2023;2.
Alicea GM, Rebecca VW, Goldman AR, Fane ME, Douglass SM, Behera R, et al. Changes in aged fibroblast lipid metabolism induce age-dependent Melanoma Cell Resistance to targeted therapy via the fatty acid transporter FATP2. Cancer Discov. 2020;10:1282–95.
Jin P, Li X, Xia Y, Li H, Li X, Yang ZY, et al. Bepotastine sensitizes ovarian Cancer to PARP inhibitors through suppressing NF-κB-Triggered SASP in Cancer-Associated fibroblasts. Mol Cancer Ther. 2023;22:447–58.
Amin T, Viol F, Krause J, Fahl M, Eggers C, Awwad F, et al. Cancer-Associated fibroblasts induce proliferation and therapeutic resistance to Everolimus in neuroendocrine tumors through STAT3 activation. Neuroendocrinology. 2023;113:501–18.
Feng H, Cao B, Peng X, Wei Q. Cancer-associated fibroblasts strengthen cell proliferation and EGFR TKIs resistance through aryl hydrocarbon receptor dependent signals in non-small cell lung cancer. BMC Cancer 2022;22.
Jin Z, Lu Y, Wu X, Pan T, Yu Z, Hou J, et al. The cross-talk between tumor cells and activated fibroblasts mediated by lactate/BDNF/TrkB signaling promotes acquired resistance to anlotinib in human gastric cancer. Redox Biol. 2021;46:102076.
Wang W, Tang Y-A, Xiao Q, Lee WC, Cheng B, Niu Z et al. Stromal induction of BRD4 phosphorylation results in chromatin remodeling and BET inhibitor resistance in Colorectal Cancer. Nat Commun 2021;12.
Yegodayev KM, Novoplansky O, Golden A, Prasad M, Levin L, Jagadeeshan S et al. TGF-Beta-activated Cancer-Associated fibroblasts limit Cetuximab Efficacy in Preclinical models of Head and Neck Cancer. Cancers 2020;12.
Zervantonakis IK, Poskus MD, Scott AL, Selfors LM, Lin J-R, Dillon DA et al. Fibroblast–tumor cell signaling limits HER2 kinase therapy response via activation of MTOR and antiapoptotic pathways. Proceedings of the National Academy of Sciences 2020;117:16500-8.
Hamalian S, Güth R, Runa F, Sanchez F, Vickers E, Agajanian M, et al. A SNAI2-PEAK1-INHBA stromal axis drives progression and lapatinib resistance in HER2-positive breast cancer by supporting subpopulations of tumor cells positive for antiapoptotic and stress signaling markers. Oncogene. 2021;40:5224–35.
Cambria D, Longhitano L, La Spina E, Giallongo S, Orlando L, Giuffrida R et al. IGFBP-6 alters mesenchymal stromal cell phenotype driving Dasatinib Resistance in Chronic myeloid leukemia. Life 2023;13.
Zhang G, Miao F, Xu J, Wang R. Mesenchymal stem cells from bone marrow regulate invasion and drug resistance of multiple myeloma cells by secreting chemokine CXCL13. Bosnian J Basic Med Sci 2019.
Zhao Y, Weng Z, Zhou X, Xu Z, Cao B, Wang B, Li J. Mesenchymal stromal cells promote the drug resistance of gastrointestinal stromal tumors by activating the PI3K-AKT pathway via TGF-β2. J Transl Med 2023;21.
Li X, Xiao Y, Wang X, Huang R, Wang R, Deng Y, et al. Connexin 43-modified bone marrow stromal cells reverse the imatinib resistance of K562 cells via Ca2+-dependent gap junction intercellular communication. Chin Med J. 2023;136:194–206.
Mark C, Warrick J, Callander NS, Hematti P, Miyamoto S. A Hyaluronan and Proteoglycan Link protein 1 Matrikine: role of Matrix Metalloproteinase 2 in multiple myeloma NF-κB activation and drug resistance. Mol cancer Research: MCR. 2022;20:1456–66.
Wang F, Zhang L, Sai B, Wang L, Zhang X, Zheng L, et al. BMSC-derived leptin and IGFBP2 promote erlotinib resistance in lung adenocarcinoma cells through IGF-1R activation in hypoxic environment. Cancer Biol Ther. 2019;21:61–71.
Zhang H, Du Z, Tu C, Zhou X, Menu E, Wang J. Hypoxic bone marrow stromal cells secrete mir-140-5p and mir-28-3p target SPRED1 to confer drug resistance in multiple myeloma. Cancer Res 2023.
Li DX, Feng DC, Shi X, Wu RC, Chen K, Han P. Identification of endothelial-related molecular subtypes for bladder cancer patients. Front Oncol. 2023;13:1101055.
Chen X, Yang G, Guo X, Zhang J, Sun W, Liu D, et al. DJ-1/FGFR-1 signaling pathway contributes to Sorafenib Resistance in Hepatocellular Carcinoma. Oxidative Med Cell Longev. 2022;2022:1–20.
Wang C, Li Y, Chen H, Zhang J, Zhang J, Qin T, et al. Inhibition of CYP4A by a novel flavonoid FLA-16 prolongs survival and normalizes tumor vasculature in glioma. Cancer Lett. 2017;402:131–41.
Xu G, Luo Y, Wu W, Liu X, Yu X, Bao Y, et al. The evolution of Acquired Resistance to BRAF(V600E) kinase inhibitor is sustained by IGF1-Driven tumor vascular remodeling. J Invest Dermatol. 2022;142:445–58.
Li DX, Yu QX, Zeng CX, Ye LX, Guo YQ, Liu JF, et al. A novel endothelial-related prognostic index by integrating single-cell and bulk RNA sequencing data for patients with kidney renal clear cell carcinoma. Front Genet. 2023;14:1096491.
Ngo MT, Sarkaria JN, Harley BAC. Perivascular stromal cells instruct Glioblastoma Invasion, Proliferation, and therapeutic response within an Engineered Brain Perivascular Niche Model. Adv Sci 2022;9.
Zhang H, Deng T, Liu R, Ning T, Yang H, Liu D et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol Cancer 2020;19.
Lv L, Shi Y, Deng Z, Xu J, Ye Z, He J, et al. A polymeric nanocarrier that eradicates breast cancer stem cells and delivers chemotherapeutic drugs. Biomater Res. 2023;27:133.
To KKW, Cho WCS. Exosome secretion from hypoxic cancer cells reshapes the tumor microenvironment and mediates drug resistance. Cancer Drug Resist. 2022;5:577–94.
Zhang S, Li N, Wang F, Liu H, Zhang Y, Xiao J, et al. Characterization of the tumor microenvironment and identification of spatially predictive biomarkers associated with beneficial neoadjuvant chemoradiotherapy in locally advanced rectal cancer. Pharmacol Res. 2023;197:106974.
Ma J, Shayiti F, Ma J, Wei M, Hua T, Zhang R, et al. Tumor-associated macrophage‐derived CCL5 promotes chemotherapy resistance and metastasis in prostatic cancer. Cell Biol Int. 2021;45:2054–62.
Li H, Yang P, Wang J, Zhang J, Ma Q, Jiang Y et al. HLF regulates ferroptosis, development and chemoresistance of triple-negative breast cancer by activating tumor cell-macrophage crosstalk. J Hematol Oncol 2022;15.
Gu J, Lv X, Li W, Li G, He X, Zhang Y et al. Deciphering the mechanism of Peptostreptococcus anaerobius-induced chemoresistance in colorectal cancer: the important roles of MDSC recruitment and EMT activation. Front Immunol 2023;14.
Liu Z, Xie Y, Xiong Y, Liu S, Qiu C, Zhu Z, et al. TLR 7/8 agonist reverses oxaliplatin resistance in colorectal cancer via directing the myeloid-derived suppressor cells to tumoricidal M1-macrophages. Cancer Lett. 2020;469:173–85.
Le F, Yang L, Han Y, Zhong Y, Zhan F, Feng Y et al. TPL inhibits the Invasion and Migration of Drug-resistant ovarian Cancer by targeting the PI3K/AKT/NF-κB-Signaling pathway to inhibit the polarization of M2 TAMs. Front Oncol 2021;11.
Lan H, Liu Y, Liu J, Wang X, Guan Z, Du J, Jin K. Tumor-Associated macrophages promote oxaliplatin resistance via METTL3-Mediated m6A of TRAF5 and necroptosis in Colorectal Cancer. Mol Pharm. 2021;18:1026–37.
Wang D, Yang L, Yu W, Wu Q, Lian J, Li F et al. Colorectal cancer cell-derived CCL20 recruits regulatory T cells to promote chemoresistance via FOXO1/CEBPB/NF-κB signaling. J Immunother Cancer 2019;7.
Mousset A, Lecorgne E, Bourget I, Lopez P, Jenovai K, Cherfils-Vicini J et al. Neutrophil extracellular traps formed during chemotherapy confer treatment resistance via TGF-β activation. Cancer Cell 2023;41:757 – 75.e10.
Shukrun R, Baron S, Fidel V, Shusterman A, Sher O, Kollender N, et al. Suggested role for neutrophil extracellular trap formation in ewing sarcoma immune microenvironment. Cancer Sci. 2024;115:36–47.
Dickey EM, Bianchi A, Amirian H, Hosein PJ, Faustman D, Brambilla R, Datta J. Transmembrane TNF-TNFR2 signaling as a critical immunoregulatory node in pancreatic cancer. Oncoimmunology. 2024;13:2326694.
Jacob M, Wiedemann S, Brücher D, Pieper NM, Birkhold M, Särchen V, et al. Increased MCL1 dependency leads to new applications of BH3-mimetics in drug-resistant neuroblastoma. Br J Cancer. 2023;129:1667–78.
Mendoza-Rodríguez MG, Medina-Reyes D, Sánchez-Barrera CA, Fernández-Muñoz KV, García-Castillo V, Ledesma-Torres JL, et al. Helminth-derived molecules improve 5-fluorouracil treatment on experimental colon tumorigenesis. Biomed Pharmacother. 2024;175:116628.
Frerichs LM, Frerichs B, Petzsch P, Köhrer K, Windolf J, Bittersohl B et al. Tumorigenic effects of human mesenchymal stromal cells and fibroblasts on bladder cancer cells. Front Oncol 2023;13.
Deng X, Ruan H, Zhang X, Xu X, Zhu Y, Peng H, et al. Long noncoding RNA CCAL transferred from fibroblasts by exosomes promotes chemoresistance of colorectal cancer cells. Int J Cancer. 2019;146:1700–16.
Zhang F, Cui JY, Gao HF, Yu H, Gao FF, Chen JL, Chen L. Cancer-associated fibroblasts induce epithelial-mesenchymal transition and cisplatin resistance in ovarian cancer via CXCL12/CXCR4 axis. Future Oncol (London England). 2020;16:2619–33.
Shan G, Gu J, Zhou D, Li L, Cheng W, Wang Y, et al. Cancer-associated fibroblast-secreted exosomal mir-423-5p promotes chemotherapy resistance in prostate cancer by targeting GREM2 through the TGF-β signaling pathway. Exp Mol Med. 2020;52:1809–22.
McAndrews KM, Chen Y, Darpolor JK, Zheng X, Yang S, Carstens JL, et al. Identification of functional heterogeneity of Carcinoma-Associated fibroblasts with distinct IL6-Mediated Therapy Resistance in Pancreatic Cancer. Cancer Discov. 2022;12:1580–97.
Liang L, Zhang X, Su X, Zeng T, Suo D, Yun J et al. Fibroblasts in metastatic lymph nodes confer cisplatin resistance to ESCC tumor cells via PI16. Oncogenesis 2023;12.
Feng H, Zhang Q, Zhao Y, Zhao L, Shan B. Leptin acts on mesenchymal stem cells to promote chemoresistance in osteosarcoma cells. Aging. 2020;12:6340–51.
Xue B-z, Xiang W, Zhang Q, Wang H-f, Zhou Y-j, Tian H et al. CD90low glioma-associated mesenchymal stromal/stem cells promote temozolomide resistance by activating FOXS1-mediated epithelial-mesenchymal transition in glioma cells. Stem Cell Res Ther 2021;12.
Zp P, Wang B, Hou Dy Y, Rl W, Xt X, Wh. Huang Hf. METTL3 mediates bone marrow mesenchymal stem cell adipogenesis to promote chemoresistance in acute myeloid leukaemia. FEBS Open Bio. 2021;11:1659–72.
Boutin L, Arnautou P, Trignol A, Ségot A, Farge T, Desterke C, et al. Mesenchymal stromal cells confer chemoresistance to myeloid leukemia blasts through Side Population functionality and ABC transporter activation. Haematologica. 2020;105:987–9998.
Wang M, Xia D, Xu D, Yin Y, Xu F, Zhang B, et al. Neovascularization directed by CAVIN1/CCBE1/VEGFC confers TMZ-resistance in glioblastoma. Cancer Lett. 2024;582:216593.
Huang M, Zhang D, Wu JY, Xing K, Yeo E, Li C et al. Wnt-mediated endothelial transformation into mesenchymal stem cell-like cells induces chemoresistance in glioblastoma. Sci Transl Med 2020;12.
Kouzi F, Zibara K, Bourgeais J, Picou F, Gallay N, Brossaud J, et al. Disruption of gap junctions attenuates acute myeloid leukemia chemoresistance induced by bone marrow mesenchymal stromal cells. Oncogene. 2019;39:1198–212.
Zerella MA, Zaffaroni M, Ronci G, Dicuonzo S, Rojas DP, Morra A, et al. A narrative review for radiation oncologists to implement preoperative partial breast irradiation. Radiol Med. 2023;128:1553–70.
Shaikh MH, Dawson A, Prokopec SD, Barrett JW, Khan PYFZ. Loss of LRP1B expression drives acquired chemo and radio-resistance in HPV-positive head and neck cancer. Oral Oncol. 2023;146:106580.
Zhang R, Chen S, Yang Z, Zhang N, Guo K, Lv K, et al. Actin polymerization inhibition by targeting ARPC2 affects intestinal stem cell homeostasis. Burns Trauma. 2023;11:tkad038.
de Bem, Prunes B, Nunes JS, da Silva VP, Laureano NK, Gonçalves DR, Machado IS, et al. The role of tumor acidification in aggressiveness, cell dissemination and treatment resistance of oral squamous cell carcinoma. Life Sci. 2022;288:120163.
Takahashi H, Watanabe H, Hashimura M, Matsumoto T, Yokoi A, Nakagawa M, et al. A combination of stromal PD-L1 and tumoral nuclear β-catenin expression as an indicator of colorectal carcinoma progression and resistance to chemoradiotherapy in locally advanced rectal carcinoma. J Pathol Clin Res. 2022;8:458–69.
Sheng H, Huang Y, Xiao Y, Zhu Z, Shen M, Zhou P et al. ATR inhibitor AZD6738 enhances the antitumor activity of radiotherapy and immune checkpoint inhibitors by potentiating the tumor immune microenvironment in hepatocellular carcinoma. J Immunother Cancer 2020;8.
Tang B, Zhu J, Shi Y, Wang Y, Zhang X, Chen B et al. Tumor cell-intrinsic MELK enhanced CCL2-dependent immunosuppression to exacerbate hepatocarcinogenesis and confer resistance of HCC to radiotherapy. Mol Cancer 2024;23.
Dai X, Wu Z, Ruan R, Chen J, Huang C, Lei W et al. TMEM160 promotes tumor immune evasion and radiotherapy resistance via PD-L1 binding in colorectal cancer. Cell Communication Signal 2024;22.
Osipov A, Blair AB, Liberto J, Wang J, Li K, Herbst B, et al. Inhibition of focal adhesion kinase enhances antitumor response of radiation therapy in pancreatic cancer through CD8 + T cells. Cancer Biology Med. 2021;18:206–14.
Beck JD, Diken M, Suchan M, Streuber M, Diken E, Kolb L et al. Long-lasting mRNA-encoded interleukin-2 restores CD8(+) T cell neoantigen immunity in MHC class I-deficient cancers. Cancer Cell 2024;42:568 – 82.e11.
Li M, Xiao J, Song S, Han F, Liu H, Lin Y, et al. PREX2 contributes to radiation resistance by inhibiting radiotherapy-induced tumor immunogenicity via cGAS/STING/IFNs pathway in colorectal cancer. BMC Med. 2024;22:154.
van Hooren L, Handgraaf SM, Kloosterman DJ, Karimi E, van Mil LWHG, Gassama AA, et al. CD103 + regulatory T cells underlie resistance to radio-immunotherapy and impair CD8 + T cell activation in glioblastoma. Nat cancer. 2023;4:665–81.
Choi HS, Ko YS, Jin H, Kang KM, Ha IB, Jeong H et al. Mebendazole increases anticancer activity of Radiotherapy in Radiotherapy-Resistant Triple-negative breast Cancer cells by enhancing natural killer cell-mediated cytotoxicity. Int J Mol Sci 2022;23.
Ames E, Canter RJ, Grossenbacher SK, Mac S, Smith RC, Monjazeb AM, et al. Enhanced targeting of stem-like solid tumor cells with radiation and natural killer cells. Oncoimmunology. 2015;4:e1036212.
Gao LR, Zhang J, Huang N, Deng W, Ni W, Xiao Z, Liu M. Tumor-derived exosomal mir-143-3p induces macrophage M2 polarization to cause Radiation Resistance in locally advanced esophageal squamous cell carcinoma. Int J Mol Sci 2024;25.
Ma X, Yao M, Gao Y, Yue Y, Li Y, Zhang T, et al. Functional Immune Cell-Derived exosomes Engineered for the trilogy of Radiotherapy Sensitization. Advanced science (Weinheim, Baden-Wurttemberg. Germany). 2022;9:e2106031.
Wang B, Su X, Zhang B, Pan S. GSK484, an inhibitor of peptidyl arginine deiminase 4, increases the radiosensitivity of colorectal cancer and inhibits neutrophil extracellular traps. J Gene Med. 2023;25:e3530.
Guo J, Guo J, Cheng B, Gong M, Sun X, Zhang H, Ma J. Ozone enhances the efficacy of radiation therapy in esophageal cancer. J Radiat Res 2024.
Ancey PB, Contat C, Boivin G, Sabatino S, Pascual J, Zangger N, et al. GLUT1 expression in Tumor-Associated neutrophils promotes Lung Cancer Growth and Resistance to Radiotherapy. Cancer Res. 2021;81:2345–57.
Domogauer JD, de Toledo SM, Howell RW, Azzam EI. Acquired radioresistance in cancer associated fibroblasts is concomitant with enhanced antioxidant potential and DNA repair capacity. Cell Commun Signal. 2021;19:30.
Han F, Chen S, Zhang K, Zhang K, Wang M, Wang P. Single-cell transcriptomic sequencing data reveal aberrant DNA methylation in SMAD3 promoter region in tumor-associated fibroblasts affecting molecular mechanism of radiosensitivity in non-small cell lung cancer. J Transl Med. 2024;22:288.
Yang X, Chen X, Zhang S, Fan W, Zhong C, Liu T, et al. Collagen 1-mediated CXCL1 secretion in tumor cells activates fibroblasts to promote radioresistance of esophageal cancer. Cell Rep. 2023;42:113270.
Huang W, Zhang L, Yang M, Wu X, Wang X, Huang W, et al. Cancer-associated fibroblasts promote the survival of irradiated nasopharyngeal carcinoma cells via the NF-κB pathway. J Experimental Clin cancer Research: CR. 2021;40:87.
Jian C, Wu T, Wang L, Gao C, Fu Z, Zhang Q, Shi C. Biomimetic Nanoplatform for Dual-targeted clearance of activated and senescent Cancer-Associated fibroblasts to improve Radiation resistance in breast Cancer. Small. 2024;20:e2309279.
Wang L, Dou X, Chen S, Yu X, Huang X, Zhang L et al. YTHDF2 inhibition potentiates radiotherapy antitumor efficacy. Cancer Cell 2023;41:1294 – 308.e8.
Yang X, Lu Y, Hang J, Zhang J, Zhang T, Huo Y, et al. Lactate-modulated immunosuppression of myeloid-derived suppressor cells contributes to the Radioresistance of Pancreatic Cancer. Cancer Immunol Res. 2020;8:1440–51.
Guan L, Nambiar DK, Cao H, Viswanathan V, Kwok S, Hui AB, et al. NFE2L2 mutations enhance Radioresistance in Head and Neck Cancer by modulating Intratumoral myeloid cells. Cancer Res. 2023;83:861–74.
Yamamoto S, Kato M, Takeyama Y, Azuma Y, Yukimatsu N, Hirayama Y, et al. Irradiation plus myeloid-derived suppressor cell-targeted therapy for overcoming treatment resistance in immunologically cold urothelial carcinoma. Br J Cancer. 2023;128:2197–205.
Liang H, Deng L, Hou Y, Meng X, Huang X, Rao E, et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat Commun. 2017;8:1736.
Zhang X, Wang Q, Zhang R, Kong Z. DAB2IP-knocking down resulted in radio-resistance of breast cancer cells is associated with increased hypoxia and vasculogenic mimicry formation. Int J Radiat Biol. 2023;99:1595–606.
Zhang Z, Karthaus WR, Lee YS, Gao VR, Wu C, Russo JW et al. Tumor Microenvironment-Derived NRG1 promotes Antiandrogen Resistance in prostate Cancer. Cancer Cell 2020;38:279 – 96.e9.
Caramelo B, Zagorac S, Corral S, Marqués M, Real FX. Cancer-associated fibroblasts in bladder Cancer: Origin, Biology, and Therapeutic opportunities. Eur Urol Oncol. 2023;6:366–75.
Acknowledgements
We appreciate Figdraw (https://www.figdraw.com) and Chengdu Basebiotech Co.,Ltd for their assistance in drawing and data process.
Funding
Dechao Feng: This research was funded by Chinese Scholarship Council (grant no. 202206240086); Yubo Yang: Program for Talents of Chongqing University Three Gorges Hospital (Grant No. ZX-50010101-2022-347); Wuran Wei: a regional innovation cooperation project of Sichuan Province (Grant No. 23QYCX0136); Mang Ke: Zhejiang Province Public Welfare Technology Application Research Project in China (TGY23H160090, LGF21H160029); Taizhou Science and Technology Project, Zhejiang Province (20ywb12); Traditional Chinese medicine of Zhejiang province science and technology plan project (2020ZB293).The funders had no role in the study design, data collection or analysis, preparation of the manuscript, or the decision to publish.
Author information
Authors and Affiliations
Contributions
DXL, FLS, RCW, and QXY proposed the project, conducted the literature search, interpreted the data, and wrote the manuscript; WRW, WCC,SH and DCF supervised the project and interpreted the data. All authors reviewed and edited the manuscript.
Corresponding authors
Ethics declarations
Ethical approval and consent to participate
Not applicable.
Consent for publication
All authors concur with publishing the study at the present version. The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, D., Shao, F., Yu, Q. et al. The complex interplay of tumor-infiltrating cells in driving therapeutic resistance pathways. Cell Commun Signal 22, 405 (2024). https://doi.org/10.1186/s12964-024-01776-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-024-01776-7