
Abstract
Conventional therapies for metastatic cancers have limited efficacy. Recently, cancer therapies targeting noncancerous cells in tumor microenvironments have shown improved clinical outcomes in patients. However, further advances in our understanding of the metastatic tumor microenvironment are required to improve treatment outcomes. Adipocytes are distributed throughout the body, and as a part of the metastatic tumor microenvironment, they interact with cancer cells in almost all organs. Adipocytes secrete various factors that are reported to exert clinical effects on cancer progression, including engraftment, survival, and expansion at the metastatic sites. However, only a few studies have comprehensively examined their impact on cancer cells. In this review, we examined the impact of adipocytes on cancer by describing the adipocyte-secreted factors that are involved in controlling metastatic cancer, focusing on adipokines, such as adiponectin, leptin, visfatin, chemerin, resistin, apelin, and omentin. Adipocyte-secreted factors promote cancer metastasis and contribute to various biological functions of cancer cells, including migration, invasion, proliferation, immune evasion, and drug resistance at the metastatic sites. We propose the establishment and expansion of “adipo-oncology” as a research field to enhance the comprehensive understanding of the role of adipocytes in metastatic cancers and the development of more robust metastatic cancer treatments.
Similar content being viewed by others
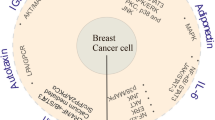

Introduction
With the aging of society, the incidence of cancer is increasing worldwide, and cancer has become the leading cause of death, particularly in developed countries [1, 2]. To date, cancer therapies, including surgery, anticancer drugs, and radiation, have targeted cancer cells and have been effective to a certain extent. However, the effectiveness of conventional therapies is limited, particularly in cases of metastatic cancers.
In recent years, cancer therapies that target noncancerous cells within tumor microenvironments have emerged. Of these, the best-known are therapies targeting immune cells, such as immune checkpoint inhibitor-based therapies, which have markedly improved the outcomes of certain aggressive cancers, including melanoma [3,4,5,6]. Therapies targeting blood vessels are also becoming more common, and vascular-targeted therapies have been incorporated into the standard treatment regimens for some cancers [7,8,9]. Recently, cancer-associated fibroblasts (CAFs), another major host cell type, have been studied for targeted therapies [10]. However, even with the application of these non-neoplastic cell-targeting therapies, a complete cure for all cancers is yet to be achieved. This is partly because our understanding of the microenvironment surrounding cancer cells is incomplete. Despite the wide variety of cell types that comprise the metastatic tumor microenvironment, the function of only few of the resident cells have been examined in terms of cancer progression. Therefore, a better understanding of the metastatic tumor microenvironment is required.
Adipocytes are a significant component of the metastatic tumor microenvironment. They are distributed throughout the body and become part of the metastatic tumor microenvironment by interacting with cancer cells in almost all organs, except the brain. Adipocytes secrete various factors, including cytokines (adipokines), proteases, chemokines, lipokines, vasoactive factors, coagulation regulators, free fatty acids, amino acids, steroids, nucleotides, and extracellular vesicles [11,12,13,14]. Many of these secretory factors exert clinical effects on cancer progression according to previous studies [15,16,17,18]. Although adipocytes are a thus critical factors for understanding the metastatic tumor microenvironment, few studies have comprehensively examined their impact on cancer cells.
We have recently uncovered various roles of adipocytes in cancer progression [18], based on analyses of histopathology, CAF induction, immune evasion, proliferation, and dormancy. In addition, we are actively conducting in vitro, in vivo, and translational studies.
In this review, we systematically examined the impact of adipocytes on metastatic cancer by describing the effect of the factors they secrete on cancer, with a particular focus on adipokines, which are mainly produced by adipocytes (Fig. 1, Table 1).

Adipocyte-derived factors govern engraftment, survival, and progression of metastatic cancers
Adiponectin
Adiponectin is a common adipokine. It was discovered in 1996 as the gene with the most abundant expression in adipose tissue [96]. Adiponectin is now known to be secreted by the muscles, brain, and other tissues, in addition to adipose tissue [96, 97]. The protein is a polypeptide composed of 244 amino acids, and it plays a vital role in glucose and fatty acid metabolism [98, 99]. High adiponectin levels decrease the risk of diabetes [100, 101]. Adiponectin secretion is stimulated by calorie restriction and exercise [102, 103].
Notably, white adipocytes secrete more adiponectins compared to beige or brown adipocytes [104, 105]. However, when white adipose tissues undergo browning due to drug administration, the amount of secreted adiponectin increases [106]. In contrast, knockout (KO) of Zfp423, which negatively regulates Prdm16 expression, tends to decrease adiponectin secretion in browned white adipocytes [107].
The adiponectin receptors ADIPOR1 and ADIPOR2 were first reported in 2003 [108], followed by the discovery of T-cadherin as another adiponectin receptor in 2004 [109]. These receptors are mainly distributed in the muscle tissue and vascular endothelium [110]. ADIPOR1 and ADIPOR2 have been reported to activate AMPK signaling and suppress MAPK, PI3K/AKT, and WNT signaling [108, 110, 111]. In contrast, T-cadherin, considered a non-signaling protein, has been reported to bind adiponectin and induce AMPK phosphorylation in the myocardium [112].
Adiponectin is mainly involved in the inhibition of tumor growth in nasopharyngeal [19], ovarian [20], hepatocellular [21], pancreatic [22], breast [23], colon [24], and prostate cancers [25] as well as in malignant mesotheliomas [26] and glioblastomas [27]. Tumor growth suppression mechanisms include activation of ADIPOR1 signaling, which induces AMPK phosphorylation, attenuation of the β-catenin signaling pathway [22], and activation of MAPK [23], ERK1/2, and AKT signaling [27]. In an in vivo study, adiponectin treatment suppressed transplanted colon tumor growth, and regulated metabolic, inflammatory and cell cycle signaling in colon cancer [28].
To date, adiponectin has not been directly administered to patients with cancer in any trials. Most cancer-related trials involving adiponectin use it as a marker of feasible output from weight loss or a healthy diet [29,30,31,32]. No direct correlation between adiponectin levels and clinical outcomes, such as cancer recurrence or metastasis, has been reported in randomized controlled trials to date. However, partial effects of serum adipocyte level on the density of tumor-infiltrating lymphocytes have been reported in patients with Stage III colon cancer [33]. Notably, abnormal levels of serum adiponectin are associated with shorter progression-free survival in metastatic colorectal cancer [34]. Furthermore, in a randomized clinical trial, a dose–response effect of exercise was observed to increase adiponectin levels and potentially reduce the risk of breast cancer [30].
Leptin
Leptin, a member of the adipokine family of proteins, is secreted by adipocytes. Leptin maintains calorie consumption and is mainly involved in energy storage by storing of triglycerides in the adipose tissues [113, 114]. Leptin overexpression reduces triglyceride concentrations [115, 116]. The leptin receptor was first identified as OB-R in 1995 [117]. Leptin receptors have various isoforms [118], which differ in the length of their intracellular domains. OB-Rfl, which has the longest intracellular domain, can activate the JAK-STAT pathway [118, 119]. In contrast, short isoforms are reported to activate MAPK signaling pathways [120]. Soluble leptin receptors are also present, and their levels correlate with the number of membrane leptin receptors and have been reported to be increased by obesity [121, 122]. Leptin primarily regulates brain function and the levels of brain-derived hormones [123, 124]. Leptin receptors are typically distributed in the central nervous system [125, 126]. However, it is now known that leptin receptors are also distributed in liver cells (hepatocytes), adipocytes, fibroblasts, and endothelial cells [50, 127, 128]. Leptin has been reported to serve as a proliferative factor in tumors derived from multiple organs, such as the lungs [51, 52], liver [53], breast [54, 55], prostate [56, 57], pancreas [58], ovaries [59, 60], brain [61], and colorectum [62], exerting its effects via the JAK/STAT, MEK/ERK1/2, NOTCH, JNK, and/or PI3K/AKT signaling pathways. In an in vivo study, a leptin receptor antagonist prolonged the average survival time of a mouse xenograft model of triple-negative breast cancer cell lines [63]. In addition, overexpression of the leptin receptor has been observed in cancer tissues compared to normal tissues, particularly in cancers with an aggressive phenotype or drug resistance [64,65,66]. Moreover, leptin stimulates cancer cell migration, invasion, CAF induction, and CAF-mediated tumor progression [67], and changes in the polarity of tumor cells [68]. The presence of leptin also stimulates leptin receptor expression in cancer cells [69].
Leptin secretion is higher in beige than in white adipocytes [105]. In white adipocytes, the mRNA production of the leptin gene is approximately twofold higher in subcutaneous adipocytes than in the visceral adipocytes of the major omentum [70].
A Phase 3 clinical trial has reported a correlation between the efficacy of VEGFR inhibitors and blood leptin levels in patients with colorectal cancer [71]. In a preclinical study, PDX prostate cancer growth inhibition has been reported in response to leptin receptor antagonist administration [129]. Leptin may also play a role in hormone therapy resistance, as leptin levels in the blood increase due to hormone therapy for breast cancer [130]. Moreover, a randomized controlled trial reported that aerobic exercise reduced leptin levels and the risk of breast cancer in a dose-dependent manner [30].
Resistin
Resistin is a member of the adipokine family, is an adipocyte-secreted factor whose levels increase with obesity [131]. Resistin induces insulin resistance by inhibiting AMPK phosphorylation [132]. In humans, resistin is primarily secreted by peripheral blood mononuclear cells and other organs, such as pancreatic islet cells [132, 133], whereas in rodents, its main source is adipocytes and other tissues [134]. The structure and function of human resistin also differ from those of murine resistin [132]. A strong correlation between serum resistin levels and insulin resistance has been observed in rodent studies; however, the correlation between serum resistin levels and insulin resistance in human studies is controversial [132, 135, 136]. Human resistin levels correlate strongly with visceral obesity [137]. Single nucleotide polymorphisms in human RETN are associated with altered plasma resistin levels, dyslipidemia, and insulin resistance, particularly in East Asian populations [138]. Animal studies using transgenic mice have reported that obesity strongly suppresses resistin secretion [139]. However, resistin secretion can be controlled by some antidiabetic drugs [139]. Resistin is also reported to be secreted from brown fat, and some antidiabetic drugs increase its secretion [78]. Animal studies in rats have reported that resistin is secreted at higher levels by females than by males and that it is secreted in the stomach, intestinal tract, skeletal muscle, and adipose tissue [79].
CAP1, decorin, ROR1, and TLR4 have been identified as resistin receptors that activate different signaling cascades [80,81,82,83]. Resistin promotes cancer proliferation via AKT and STAT signaling [84, 85], angiogenesis via VEGFR, SAPK/JNK, and NFKB signaling [86, 87], epithelial-mesenchymal transition via the WNT/β-catenin pathway [88], and invasion and metastasis via the WNT/β-catenin, TGFβR, MAPK, pathways [140]. In an animal study, the administration of resistin promoted ovarian tumor growth by regulating micro RNA (miRNA) expression [88].
In a clinical study on postoperative weight loss in patients with breast cancer, weight loss significantly decreased blood resistin levels, but did not significantly affect blood inflammatory cytokines or lipid composition [141]. In in vitro experiments, resistin has been implicated in cancer resistance and an increase in cancer stem cells [85], and has been suggested to be a potential important target for cancer therapy.
Visfatin
Visfatin/nicotinamide phosphoribosyltransferase (NAMPT) is another member of the adipokine family of proteins [142]. Visfatin is a pre-B-cell colony-enhancing factor that promotes the maturation of early B-lineage precursor cells [143]. It is secreted by the visceral and subcutaneous adipocytes [144, 145]. A study using animal tissues reported that visfatin is more abundant in brown adipose tissues than in other types of adipose tissue, whereas in humans, brown and white adipose tissues showed no significant differences in visfatin expression [146]. Visfatin is also secreted by the liver, skeletal muscles, neutrophils, and fetal membranes [147]. Serum visfatin levels are associated with obesity, inflammation, cardiovascular diseases, and endothelial cell dysfunction [89, 90].
Hypersecretion of visfatin is correlated with worse prognosis in breast, endometrial, and renal cell cancers [91,92,93]. Visfatin promotes cell proliferation in endometrial cancer [94], invasiveness of liver cancer [95], and inhibition of apoptosis in breast cancer by activating the PI3K/AKT, MAPK, and ERK1/2 signaling pathways [94]. In an in vivo study in mice, visfatin promoted endometrial tumor growth by stimulating PI3K/AKT and other signaling pathways [94].
Clinical studies have reported significantly lower blood visfatin levels in pediatric patients with leukemia in complete remission [148]. In in vitro experiments, non-small cell lung cancer cell lines that became resistant to cisplatin treatment were reported to have elevated visfatin levels, whereas visfatin KO restored sensitivity to cisplatin [149]. Thus, visfatin inhibitors may contribute to increased drug resistance in patients with lung cancer.
Chemerin
Chemerin is an adipokine that was identified in 1997 [150]. Chemerin is found in the serum, plasma, adipocytes, and the liver [151]. Chemerin, produced mainly by adipocytes and the liver, is a ligand for chemokine-like receptor 1 (CMKLR1), G-protein-coupled receptor 1, and C–C motif chemokine receptor-like 2 [152]. Chemerin has been reported to be expressed during differentiation into brown adipocytes [153] and is abundant in mouse white adipose tissue [151]. Moreover, it promotes the differentiation of bone marrow adipocytes [41]. Inhibition of chemerin secretion by antidiabetic drugs has been reported [42].
In a cohort study of over 7,000 people, chemerin concentration was significantly associated with cancer mortality [43]. In patients with breast cancer, serum chemerin levels were significantly associated with histological grade and Ki67 expression [44]. However, the role of chemerin in tumor growth remains controversial. In an in vitro study, chemerin suppressed the proliferation of ovarian cancer cell lines and could potentially regulate INFα secretion by cancer cells [45]. In addition, chemerin suppressed the viability and invasion of breast cancer cell lines [46]. In an in vivo study, the chemerin analog CG34 significantly stimulated the growth and bioluminescence signals of colorectal cancer xenografts [47]. Monoclonal antibodies targeting chemerin led to reduced lipid storage and diminished renal cancer growth by alleviating the suppression of fatty acid oxidation and ferroptosis induced by chemerin [48]. Furthermore, chemerin overexpression in breast cancer reduced tumor growth by recruiting natural killer cells and T cells in vivo [49]. In addition, chemerin overexpression suppressed hepatocellular carcinoma cell proliferation and tumor metastasis by reducing AKT phosphorylation [154]. In an in vivo study in mice, intraperitoneal chemerin administration suppressed breast tumor growth [46]. In contrast, chemerin has been reported to promote chemotaxis and migration of cutaneous squamous cell carcinoma [155], and its attenuation inhibited renal tumor growth in vivo [48]. These results suggested that chemerin may regulate different functions in cancers derived from different organs.
Currently, no randomized clinical trials of chemerin or its inhibitors in cancer have been reported in PubMed. However, an in vivo study has reported that chemerin inhibitors promoted cancer cell senescence and enhanced the therapeutic effect of cisplatin [156]. Small molecules that selectively inhibit the chemerin receptor CMKLR1 have also been reported to inhibit endometriosis growth [157], and chemerin signaling inhibitors are expected to serve as orthologous cancer therapeutics.
Apelin
In 1998, apelin was identified as a ligand of the human APJ receptor [158]. In addition to adipocytes, apelin is broadly expressed in many organs and tissues, including the brain, kidneys, and heart [159]. Activation of apelin signaling promotes brown adipocyte differentiation [160]. Apelin further promotes the browning of white fat [161]. Apelin also promotes angiogenesis [162]. In addition, apelin signaling stimulates nitric oxide release, which promotes vasodilation by relaxing the smooth muscle cells of the arterial walls [35]. In cancer, activation of the apelin-AJP pathway promotes the peritoneal dissemination of ovarian cancer cells [36]. Moreover, the loss of apelin blocks angiogenesis in lung cancer and melanoma cells in vivo [37]. Apelin activates the PI3K/AKT pathway and promotes the proliferation, migration, and glucose uptake of pancreatic cancer cell lines [38]. Apelin promotes tumor growth by facilitating endothelial cell migration, resulting in rapid angiogenesis [39]. Apelin KO mice bearing breast cancer tumors show prolonged survival, with or without anti-angiogenic treatment [40]. In melanoma, APJ-KO suppressed angiogenesis in vivo [163]. These results suggest that apelin strongly affects the vascular environment surrounding tumors and that it is a novel cancer treatment target.
Clinical trials using apelin and apelin inhibitors have not been reported on PubMed to date, but apelin receptor expression has been reported to correlate with prognosis in patients with gastric cancer who were treated with chemoradiotherapy [164]. Moreover, in vitro and in vivo experiments using colon and prostate cancer cell lines have reported that high apelin expression alters the vascular structure and immune environment, resulting in a reduction in tumor size [165]. In contrast, tumor growth, angiogenesis, and metastasis have been reported to be suppressed in vivo in tumors in mouse models in which apelin expression was suppressed [40]. Further, ML221, an antagonist of the apelin receptor, significantly suppressed liver metastasis in breast cancer when combined with dendritic cell vaccine therapy in vivo [166]. In summary, although the function of apelin in cancer is complex, it may serve as a potential therapeutic target.
Omentin
In 2001, omentin (also known as intelectin-1) was identified as a human lectin that binds to galactofuranosyl residues [72]. Omentin is composed of 295 amino acids and is expressed in the heart, intestine, thymus, and adipocytes [72, 73]. Omentin is expressed in large amounts in visceral white adipocytes and to some extent in subcutaneous white adipocytes [73]. Omentin is also widely expressed in organs other than adipose tissue, including the heart, intestinal tract, and kidneys [74]. Omentin levels are inversely associated with obesity and type 2 diabetes mellitus [75]. However, omentin receptors have not yet been identified.
The effect of omentin on cancer progression is controversial because the relationship between the omentin serum levels and cancer progression differs depending on the primary cancer sites and the patient’s health status [76, 77]. For example, serum omentin levels were positively correlated with colon cancer risk in study participants with a body mass index (BMI) < 30 kg/m2, but the correlation was not observed in participants with BMI ≥ 30 kg/m2 [167]. While omentin has been reported to promote apoptosis in hepatocellular carcinoma cells [168], it has also been reported to induce cell proliferation [169].
In patients with endometrial cancer, the blood levels of omentin correlate with lymph node metastasis [170], suggesting that it may be involved in the control of cancer malignancy. At present, no clinical trials targeting omentin have been conducted.
Taken together, these data suggest that omentin may have the potential to suppress tumors, but this effect is limited to certain conditions.
miRNAs, chemokines, extracellular vesicles, and other factors
Various types of miRNAs are secreted from adipose tissues [171,172,173]. Studies using the 3T3-L1 cell line reported that adipocytes secrete a large number of extracellular vesicles [174] and that a large number of miRNAs are contained within these extracellular vesicles [175]. In addition, specific miRNAs appear to be expressed in different adipocyte types [174].
Adipose tissue secretes various chemokines, such as CCL2, CCL20, and CXCL5, at regular intervals or during cancer therapy [176, 177]. Consequently, they induce inflammation and the reorganization of adipose tissue, which affects the progression of cancer.
Moreover, adipocytes also secrete various other factors, including free fatty acids [178, 179], lipokines [180, 181], vasoactive proteins [182, 183], and matrix metalloproteinases [184, 185], which are directly or indirectly involved in cancer nutrition, growth, and metastasis.
Conclusions
Aging and metabolic syndrome are both recognized in developed countries, and as a result, the total number of cancer patients with organs with adipocyte accumulation and adipocytes replacement is expected to increase [186,187,188,189]. Cancer therapy targeting adipocytes has the potential to be an innovative treatment not only for metastatic cancer, but also for a wide range of cancer patients across organs. In this review, we described the effects of various adipokines and other adipocyte-secreted factors on cancer. The molecular mechanisms by which the factors secreted by adipocytes affect cancer and the resulting effects on cancer survival, proliferation, invasion, metastasis, and resistance to therapy are diverse. Notably, while adipokines mainly promote tumor growth, certain adipokines, such as adiponectin and chemerin, have the potential to suppress tumor growth. Therefore, it is essential to consider the overall balance of adipocyte-derived factors to understand the role of adipocytes on tumors. We propose the establishment and expansion of “adipo-oncology” as a research field to enhance the comprehensive understanding of the role of adipocytes in metastatic cancers and the development of more robust metastatic cancer treatments. Integrating information of mechanisms regulating cancer by adipocyte-secreted factors, understanding the secretion status of each secreted factor, the type and distribution of adipocytes in patients with cancer, and carefully controlling fat secretion factors in each patient may lead to useful cancer treatments. Therefore, it is necessary to accumulate basic and clinical data for the future development of novel cancer therapies.
References
Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73:17–48.
Nagao M, Tsugane S. Cancer in Japan: Prevalence, prevention and the role of heterocyclic amines in human carcinogenesis. Genes Environ. 2016;38:16.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54.
Eggermont AMM, Blank CU, Mandala M, Long GV, Atkinson V, Dalle S, Haydon A, Lichinitser M, Khattak A, Carlino MS, et al. Adjuvant Pembrolizumab versus Placebo in Resected Stage III Melanoma. N Engl J Med. 2018;378:1789–801.
Horn L, Mansfield AS, Szczesna A, Havel L, Krzakowski M, Hochmair MJ, Huemer F, Losonczy G, Johnson ML, Nishio M, et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med. 2018;379:2220–9.
Cercek A, Lumish M, Sinopoli J, Weiss J, Shia J, Lamendola-Essel M, El Dika IH, Segal N, Shcherba M, Sugarman R, et al. PD-1 blockade in mismatch repair-deficient, locally advanced rectal cancer. N Engl J Med. 2022;386:2363–76.
Hurwitz HI, Fehrenbacher L, Hainsworth JD, Heim W, Berlin J, Holmgren E, Hambleton J, Novotny WF, Kabbinavar F. Bevacizumab in combination with fluorouracil and leucovorin: an active regimen for first-line metastatic colorectal cancer. J Clin Oncol. 2005;23:3502–8.
Ohtsu A, Shah MA, Van Cutsem E, Rha SY, Sawaki A, Park SR, Lim HY, Yamada Y, Wu J, Langer B, et al. Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: a randomized, double-blind, placebo-controlled phase III study. J Clin Oncol. 2011;29:3968–76.
Zalcman G, Mazieres J, Margery J, Greillier L, Audigier-Valette C, Moro-Sibilot D, Molinier O, Corre R, Monnet I, Gounant V, et al. Bevacizumab for newly diagnosed pleural mesothelioma in the Mesothelioma Avastin Cisplatin Pemetrexed Study (MAPS): a randomised, controlled, open-label, phase 3 trial. Lancet. 2016;387:1405–14.
Cazet AS, Hui MN, Elsworth BL, Wu SZ, Roden D, Chan CL, Skhinas JN, Collot R, Yang J, Harvey K, et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat Commun. 2018;9:2897.
Kahn CR, Wang G, Lee KY. Altered adipose tissue and adipocyte function in the pathogenesis of metabolic syndrome. J Clin Invest. 2019;129:3990–4000.
Rodriguez A, Becerril S, Hernandez-Pardos AW, Fruhbeck G. Adipose tissue depot differences in adipokines and effects on skeletal and cardiac muscle. Curr Opin Pharmacol. 2020;52:1–8.
He LY, Zhang SS, Peng DX, Guan LP, Wang SH. Synthesis and evaluations of selective COX-2 inhibitory effects: Benzo [d]thiazol analogs. Bioorg Med Chem Lett. 2020;30:127376.
Pestel J, Blangero F, Watson J, Pirola L, Eljaafari A. Adipokines in obesity and metabolic-related-diseases. Biochimie. 2023;212:48–59.
Renehan AG, Zwahlen M, Egger M. Adiposity and cancer risk: new mechanistic insights from epidemiology. Nat Rev Cancer. 2015;15:484–98.
Picon-Ruiz M, Morata-Tarifa C, Valle-Goffin JJ, Friedman ER, Slingerland JM. Obesity and adverse breast cancer risk and outcome: Mechanistic insights and strategies for intervention. CA Cancer J Clin. 2017;67:378–97.
Annett S, Moore G, Robson T. Obesity and Cancer Metastasis: Molecular and Translational Perspectives. Cancers (Basel). 2020;12(12):3798.
Sato S, Hiruma T, Koizumi M, Yoshihara M, Nakamura Y, Tadokoro H, Motomatsu S, Yamanaka T, Washimi K, Okubo Y, et al. Bone marrow adipocytes induce cancer-associated fibroblasts and immune evasion, enhancing invasion and drug resistance. Cancer Sci. 2023;114:2674–88.
Zhang Z, Du J, Shi H, Wang S, Yan Y, Xu Q, Zhou S, Zhao Z, Mu Y, Qian C, et al. Adiponectin suppresses tumor growth of nasopharyngeal carcinoma through activating AMPK signaling pathway. J Transl Med. 2022;20:89.
Ramzan AA, Bitler BG, Hicks D, Barner K, Qamar L, Behbakht K, Powell T, Jansson T, Wilson H. Adiponectin receptor agonist AdipoRon induces apoptotic cell death and suppresses proliferation in human ovarian cancer cells. Mol Cell Biochem. 2019;461:37–46.
Manieri E, Herrera-Melle L, Mora A, Tomas-Loba A, Leiva-Vega L, Fernandez DI, Rodriguez E, Moran L, Hernandez-Cosido L, Torres JL, et al. Adiponectin accounts for gender differences in hepatocellular carcinoma incidence. J Exp Med. 2019;216:1108–19.
Jiang J, Fan Y, Zhang W, Shen Y, Liu T, Yao M, Gu J, Tu H, Gan Y. Adiponectin suppresses human pancreatic cancer growth through attenuating the beta-catenin signaling pathway. Int J Biol Sci. 2019;15:253–64.
Mauro L, Pellegrino M, De Amicis F, Ricchio E, Giordano F, Rizza P, Catalano S, Bonofiglio D, Sisci D, Panno ML, Ando S. Evidences that estrogen receptor alpha interferes with adiponectin effects on breast cancer cell growth. Cell Cycle. 2014;13:553–64.
Kim AY, Lee YS, Kim KH, Lee JH, Lee HK, Jang SH, Kim SE, Lee GY, Lee JW, Jung SA, et al. Adiponectin represses colon cancer cell proliferation via AdipoR1- and -R2-mediated AMPK activation. Mol Endocrinol. 2010;24:1441–52.
Tan W, Wang L, Ma Q, Qi M, Lu N, Zhang L, Han B. Adiponectin as a potential tumor suppressor inhibiting epithelial-to-mesenchymal transition but frequently silenced in prostate cancer by promoter methylation. Prostate. 2015;75:1197–205.
Niu K, Asada M, Okazaki T, Yamanda S, Ebihara T, Guo H, Zhang D, Nagatomi R, Arai H, Kohzuki M, Ebihara S. Adiponectin pathway attenuates malignant mesothelioma cell growth. Am J Respir Cell Mol Biol. 2012;46:515–23.
Porcile C, Di Zazzo E, Monaco ML, D’Angelo G, Passarella D, Russo C, Di Costanzo A, Pattarozzi A, Gatti M, Bajetto A, et al. Adiponectin as novel regulator of cell proliferation in human glioblastoma. J Cell Physiol. 2014;229:1444–54.
Moon HS, Liu X, Nagel JM, Chamberland JP, Diakopoulos KN, Brinkoetter MT, Hatziapostolou M, Wu Y, Robson SC, Iliopoulos D, Mantzoros CS. Salutary effects of adiponectin on colon cancer: in vivo and in vitro studies in mice. Gut. 2013;62:561–70.
Lucas EA, Li W, Peterson SK, Brown A, Kuvibidila S, Perkins-Veazie P, Clarke SL, Smith BJ. Mango modulates body fat and plasma glucose and lipids in mice fed a high-fat diet. Br J Nutr. 2011;106:1495–505.
Sturgeon K, Digiovanni L, Good J, Salvatore D, Fenderson D, Domchek S, Stopfer J, Galantino ML, Bryan C, Hwang WT, Schmitz K. Exercise-induced dose-response alterations in adiponectin and leptin levels are dependent on body fat changes in women at risk for breast cancer. Cancer Epidemiol Biomarkers Prev. 2016;25:1195–200.
HooshmandMoghadam B, Golestani F, Bagheri R, Cheraghloo N, Eskandari M, Wong A, Nordvall M, Suzuki K, Pournemati P. The effects of high-intensity interval training vs. moderate-intensity continuous training on inflammatory markers, body composition, and physical fitness in overweight/obese survivors of breast cancer: a randomized controlled clinical trial. Cancers (Basel). 2021;13(17):4386.
Bahmannia M, Azizzade M, Heydari S, Nasrollahzadeh J, Rabiei S, Naja F, Sheikhi Mobarakeh Z, Hejazi J, Hejazi E. Effects of decaffeinated green coffee extract supplementation on anthropometric indices, blood glucose, leptin, adiponectin and neuropeptide Y (NPY) in breast cancer survivors: a randomized clinical trial. Food Funct. 2022;13:10347–56.
Sinicrope FA, Shi Q, Smyrk TC, Goldberg RM, Cohen SJ, Gill S, Kahlenberg MS, Nair S, Shield AF, Jahagirdar BN, et al. Association of adiponectin and vitamin D with tumor infiltrating lymphocytes and survival in stage III colon cancer. JNCI Cancer Spectr. 2021;5(5):pkab070.
Guercio BJ, Zhang S, Ou FS, Venook AP, Niedzwiecki D, Lenz HJ, Innocenti F, Pollak MN, Nixon AB, Mullen BC, et al. IGF-Binding Proteins, Adiponectin, and Survival in Metastatic Colorectal Cancer: Results From CALGB (Alliance)/SWOG 80405. JNCI Cancer Spectr. 2021;5(1):pkaa074.
Tatemoto K, Takayama K, Zou MX, Kumaki I, Zhang W, Kumano K, Fujimiya M. The novel peptide apelin lowers blood pressure via a nitric oxide-dependent mechanism. Regul Pept. 2001;99:87–92.
Dogra S, Neelakantan D, Patel MM, Griesel B, Olson A, Woo S. Adipokine Apelin/APJ pathway promotes peritoneal dissemination of ovarian cancer cells by regulating lipid metabolism. Mol Cancer Res. 2021;19:1534–45.
Azad AK, Campbell KR, Zhabyeyev P, Oudit GY, Moore RB, Murray AG. Loss of apelin blocks the emergence of sprouting angiogenesis in experimental tumors. FASEB J. 2022;36:e22560.
Chaves-Almagro C, Auriau J, Dortignac A, Clerc P, Lulka H, Deleruyelle S, Projetti F, Nakhle J, Frances A, Berta J, et al. Upregulated apelin signaling in pancreatic cancer activates oncogenic signaling pathways to promote tumor development. Int J Mol Sci. 2022;23(18):10600.
Bernier-Latmani J, Cisarovsky C, Mahfoud S, Ragusa S, Dupanloup I, Barras D, Renevey F, Nassiri S, Anderle P, Squadrito ML, et al. Apelin-driven endothelial cell migration sustains intestinal progenitor cells and tumor growth. Nat Cardiovasc Res. 2022;1:476–90.
Uribesalgo I, Hoffmann D, Zhang Y, Kavirayani A, Lazovic J, Berta J, Novatchkova M, Pai TP, Wimmer RA, Laszlo V, et al. Apelin inhibition prevents resistance and metastasis associated with anti-angiogenic therapy. EMBO Mol Med. 2019;11:e9266.
Muruganandan S, Roman AA, Sinal CJ. Role of chemerin/CMKLR1 signaling in adipogenesis and osteoblastogenesis of bone marrow stem cells. J Bone Miner Res. 2010;25:222–34.
Sell H, Laurencikiene J, Taube A, Eckardt K, Cramer A, Horrighs A, Arner P, Eckel J. Chemerin is a novel adipocyte-derived factor inducing insulin resistance in primary human skeletal muscle cells. Diabetes. 2009;58:2731–40.
Noppes K, Gross S, Hannemann A, Markus MRP, Bahls M, Volzke H, Dorr M, Nauck M, Friedrich N, Zylla S. Association of plasma chemerin with all-cause and disease-specific mortality - results from a population-based study. Int J Obes (Lond). 2023;47(10):956–62.
Song Y, Zhu X, Lin Z, Luo L, Wen D. The potential value of serum chemerin in patients with breast cancer. Sci Rep. 2021;11:6564.
Schmitt M, Gallistl J, Schuler-Toprak S, Fritsch J, Buechler C, Ortmann O, Treeck O. Anti-tumoral effect of chemerin on ovarian cancer cell lines mediated by activation of interferon alpha response. Cancers (Basel). 2022;14(17):4108.
Kim H, Lee JH, Lee SK, Song NY, Son SH, Kim KR, Chung WY. Chemerin treatment inhibits the growth and bone invasion of breast cancer cells. Int J Mol Sci. 2020;21(8):2871.
Friebus-Kardash J, Schulz P, Reinicke S, Karthaus C, Schefer Q, Bandholtz S, Grotzinger C. A chemerin peptide analog stimulates tumor growth in two xenograft mouse models of human colorectal carcinoma. Cancers (Basel). 2021;14(1):125.
Tan SK, Mahmud I, Fontanesi F, Puchowicz M, Neumann CKA, Griswold AJ, Patel R, Dispagna M, Ahmed HH, Gonzalgo ML, et al. Obesity-dependent adipokine chemerin suppresses fatty acid oxidation to confer ferroptosis resistance. Cancer Discov. 2021;11:2072–93.
Pachynski RK, Wang P, Salazar N, Zheng Y, Nease L, Rosalez J, Leong WI, Virdi G, Rennier K, Shin WJ, et al. Chemerin suppresses breast cancer growth by recruiting immune effector cells into the tumor microenvironment. Front Immunol. 2019;10:983.
Wei L, Chen Y, Zhang C, Liu M, Xiong H. Leptin induces IL-6 and IL-8 expression through leptin receptor Ob-Rb in human dental pulp fibroblasts. Acta Odontol Scand. 2019;77:205–12.
Li F, Zhao S, Guo T, Li J, Gu C. The Nutritional Cytokine Leptin Promotes NSCLC by Activating the PI3K/AKT and MAPK/ERK Pathways in NSCLC Cells in a Paracrine Manner. Biomed Res Int. 2019;2019:2585743.
Wang J, Zhou F, Li F, Wang B, Hu Y, Li X. Autocrined leptin promotes proliferation of non-small cell lung cancer (NSCLC) via PI3K/AKT and p53 pathways. Ann Transl Med. 2021;9:568.
Saxena NK, Sharma D, Ding X, Lin S, Marra F, Merlin D, Anania FA. Concomitant activation of the JAK/STAT, PI3K/AKT, and ERK signaling is involved in leptin-mediated promotion of invasion and migration of hepatocellular carcinoma cells. Cancer Res. 2007;67:2497–507.
Mauro L, Catalano S, Bossi G, Pellegrino M, Barone I, Morales S, Giordano C, Bartella V, Casaburi I, Ando S. Evidences that leptin up-regulates E-cadherin expression in breast cancer: effects on tumor growth and progression. Cancer Res. 2007;67:3412–21.
Shveid Gerson D, Gerson-Cwilich R, Lara Torres CO, Chousleb de Kalach A, Ventura Gallegos JL, Badillo-Garcia LE, Bargallo Rocha JE, Maffuz-Aziz A, Sanchez Forgach ER, Castorena Roji G, et al. Establishment of triple-negative breast cancer cells based on BMI: A novel model in the correlation between obesity and breast cancer. Front Oncol. 2022;12:988968.
Onuma M, Bub JD, Rummel TL, Iwamoto Y. Prostate cancer cell-adipocyte interaction: leptin mediates androgen-independent prostate cancer cell proliferation through c-Jun NH2-terminal kinase. J Biol Chem. 2003;278:42660–7.
Gorrab A, Pagano A, Ayed K, Chebil M, Derouiche A, Kovacic H, Gati A. Leptin promotes prostate cancer proliferation and migration by stimulating STAT3 pathway. Nutr Cancer. 2021;73:1217–27.
Xu Y, Tan M, Tian X, Zhang J, Zhang J, Chen J, Xu W, Sheng H. Leptin receptor mediates the proliferation and glucose metabolism of pancreatic cancer cells via AKT pathway activation. Mol Med Rep. 2020;21:945–52.
Ptak A, Kolaczkowska E, Gregoraszczuk EL. Leptin stimulation of cell cycle and inhibition of apoptosis gene and protein expression in OVCAR-3 ovarian cancer cells. Endocrine. 2013;43:394–403.
Chen C, Chang YC, Lan MS, Breslin M. Leptin stimulates ovarian cancer cell growth and inhibits apoptosis by increasing cyclin D1 and Mcl-1 expression via the activation of the MEK/ERK1/2 and PI3K/Akt signaling pathways. Int J Oncol. 2013;42:1113–9.
Han G, Wang L, Zhao R, Yue Z, Zhou X, Hu X, Cao Y, Dai D, Liu J. Leptin promotes human glioblastoma growth through activating Signal Transducers and Activators of Transcription 3 signaling. Brain Res Bull. 2012;87:274–9.
Bartucci M, Svensson S, Ricci-Vitiani L, Dattilo R, Biffoni M, Signore M, Ferla R, De Maria R, Surmacz E. Obesity hormone leptin induces growth and interferes with the cytotoxic effects of 5-fluorouracil in colorectal tumor stem cells. Endocr Relat Cancer. 2010;17:823–33.
Otvos L Jr, Kovalszky I, Riolfi M, Ferla R, Olah J, Sztodola A, Nama K, Molino A, Piubello Q, Wade JD, Surmacz E. Efficacy of a leptin receptor antagonist peptide in a mouse model of triple-negative breast cancer. Eur J Cancer. 2011;47:1578–84.
Garofalo C, Koda M, Cascio S, Sulkowska M, Kanczuga-Koda L, Golaszewska J, Russo A, Sulkowski S, Surmacz E. Increased expression of leptin and the leptin receptor as a marker of breast cancer progression: possible role of obesity-related stimuli. Clin Cancer Res. 2006;12:1447–53.
Uddin S, Bu R, Ahmed M, Abubaker J, Al-Dayel F, Bavi P, Al-Kuraya KS. Overexpression of leptin receptor predicts an unfavorable outcome in Middle Eastern ovarian cancer. Mol Cancer. 2009;8:74.
Garcia-Estevez L, Calvo I, Perez S, Gallegos I, Diaz E, Sampayo-Cordero M, Oltra SS, Moreno-Bueno G. Predictive role of leptin receptor (Ob-R) overexpression in patients with early breast cancer receiving neoadjuvant systemic treatment. Cancers (Basel). 2021;13(13):3269.
Rozenchan PB, Carraro DM, Brentani H, de CarvalhoMota LD, Bastos EP, Ferreira EN, Torres CH, Katayama ML, Roela RA, Lyra EC, et al. Reciprocal changes in gene expression profiles of cocultured breast epithelial cells and primary fibroblasts. Int J Cancer. 2009;125:2767–77.
Coradini D, Gambazza S, Oriana S, Ambrogi F. Adipokines expression and epithelial cell polarity in normal and cancerous breast tissue. Carcinogenesis. 2020;41:1402–8.
Linares RL, Benitez JGS, Reynoso MO, Romero CG, Sandoval-Cabrera A. Modulation of the leptin receptors expression in breast cancer cell lines exposed to leptin and tamoxifen. Sci Rep. 2019;9:19189.
Van Harmelen V, Reynisdottir S, Eriksson P, Thorne A, Hoffstedt J, Lonnqvist F, Arner P. Leptin secretion from subcutaneous and visceral adipose tissue in women. Diabetes. 1998;47:913–7.
Pommier AJ, Farren M, Patel B, Wappett M, Michopoulos F, Smith NR, Kendrew J, Frith J, Huby R, Eberlein C, et al. Leptin, BMI, and a metabolic gene expression signature associated with clinical outcome to VEGF inhibition in colorectal cancer. Cell Metab. 2016;23:77–93.
Tsuji S, Uehori J, Matsumoto M, Suzuki Y, Matsuhisa A, Toyoshima K, Seya T. Human intelectin is a novel soluble lectin that recognizes galactofuranose in carbohydrate chains of bacterial cell wall. J Biol Chem. 2001;276:23456–63.
Yang RZ, Lee MJ, Hu H, Pray J, Wu HB, Hansen BC, Shuldiner AR, Fried SK, McLenithan JC, Gong DW. Identification of omentin as a novel depot-specific adipokine in human adipose tissue: possible role in modulating insulin action. Am J Physiol Endocrinol Metab. 2006;290:E1253-1261.
Radzik-Zajac J, Wytrychowski K, Wisniewski A, Barg W. The role of the novel adipokines vaspin and omentin in chronic inflammatory diseases. Pediatr Endocrinol Diabetes Metab. 2023;29:48–52.
Eimal Latif AH, Anwar S, Gautham KS, Kadurei F, Ojo RO, Hafizyar F, Muhammad Haroon D, Rakesh F, Talpur AS. Association of plasma omentin-1 levels with diabetes and its complications. Cureus. 2021;13:e18203.
Feng Z, Sun H, Liu P, Shi W, Han W, Ma L. Analysis of the expression of plasma omentin-1 level in colorectal cancer and its correlation with prognosis. Transl Cancer Res. 2020;9:6479–86.
Nuszkiewicz J, Czuczejko J, Drozdz W, Wozniak A, Malkowski B, Szewczyk-Golec K. Concentration of selected adipokines and factors regulating carbohydrate metabolism in patients with head and neck cancer in respect to their body mass index. Int J Mol Sci. 2023;24(4):3283.
Viengchareun S, Zennaro MC, Pascual-Le Tallec L, Lombes M. Brown adipocytes are novel sites of expression and regulation of adiponectin and resistin. FEBS Lett. 2002;532:345–50.
Nogueiras R, Gallego R, Gualillo O, Caminos JE, Garcia-Caballero T, Casanueva FF, Dieguez C. Resistin is expressed in different rat tissues and is regulated in a tissue- and gender-specific manner. FEBS Lett. 2003;548:21–7.
Daquinag AC, Zhang Y, Amaya-Manzanares F, Simmons PJ, Kolonin MG. An isoform of decorin is a resistin receptor on the surface of adipose progenitor cells. Cell Stem Cell. 2011;9:74–86.
Sanchez-Solana B, Laborda J, Baladron V. Mouse resistin modulates adipogenesis and glucose uptake in 3T3-L1 preadipocytes through the ROR1 receptor. Mol Endocrinol. 2012;26:110–27.
Lee S, Lee HC, Kwon YW, Lee SE, Cho Y, Kim J, Lee S, Kim JY, Lee J, Yang HM, et al. Adenylyl cyclase-associated protein 1 is a receptor for human resistin and mediates inflammatory actions of human monocytes. Cell Metab. 2014;19:484–97.
Hasan R, Zhou GL. The Cytoskeletal Protein Cyclase-Associated Protein 1 (CAP1) in breast cancer: context-dependent roles in both the invasiveness and proliferation of cancer cells and underlying cell signals. Int J Mol Sci. 2019;20(11):2653.
Kim HJ, Lee YS, Won EH, Chang IH, Kim TH, Park ES, Kim MK, Kim W, Myung SC. Expression of resistin in the prostate and its stimulatory effect on prostate cancer cell proliferation. BJU Int. 2011;108:E77-83.
Deshmukh SK, Srivastava SK, Bhardwaj A, Singh AP, Tyagi N, Marimuthu S, Dyess DL, Dal Zotto V, Carter JE, Singh S. Resistin and interleukin-6 exhibit racially-disparate expression in breast cancer patients, display molecular association and promote growth and aggressiveness of tumor cells through STAT3 activation. Oncotarget. 2015;6:11231–41.
Pang L, Zhang Y, Yu Y, Zhang S. Resistin promotes the expression of vascular endothelial growth factor in ovary carcinoma cells. Int J Mol Sci. 2013;14:9751–66.
Tsai HC, Cheng SP, Han CK, Huang YL, Wang SW, Lee JJ, Lai CT, Fong YC, Tang CH. Resistin enhances angiogenesis in osteosarcoma via the MAPK signaling pathway. Aging (Albany NY). 2019;11:9767–77.
Qiu L, Zhang GF, Yu L, Wang HY, Jia XJ, Wang TJ. Novel oncogenic and chemoresistance-inducing functions of resistin in ovarian cancer cells require miRNAs-mediated induction of epithelial-to-mesenchymal transition. Sci Rep. 2018;8:12522.
Takebayashi K, Suetsugu M, Wakabayashi S, Aso Y, Inukai T. Association between plasma visfatin and vascular endothelial function in patients with type 2 diabetes mellitus. Metabolism. 2007;56:451–8.
Kumari B, Yadav UCS. Adipokine Visfatin’s Role in pathogenesis of diabesity and related metabolic derangements. Curr Mol Med. 2018;18:116–25.
Dalamaga M, Karmaniolas K, Papadavid E, Pelekanos N, Sotiropoulos G, Lekka A. Elevated serum visfatin/nicotinamide phosphoribosyl-transferase levels are associated with risk of postmenopausal breast cancer independently from adiponectin, leptin, and anthropometric and metabolic parameters. Menopause. 2011;18:1198–204.
Tian W, Zhu Y, Wang Y, Teng F, Zhang H, Liu G, Ma X, Sun D, Rohan T, Xue F. Visfatin, a potential biomarker and prognostic factor for endometrial cancer. Gynecol Oncol. 2013;129:505–12.
Chinapayan SM, Kuppusamy S, Yap NY, Perumal K, Gobe G, Rajandram R. Potential value of visfatin, omentin-1, nesfatin-1 and apelin in Renal Cell Carcinoma (RCC): a systematic review and meta-analysis. Diagnostics (Basel). 2022;12(12):3069.
Wang Y, Gao C, Zhang Y, Gao J, Teng F, Tian W, Yang W, Yan Y, Xue F. Visfatin stimulates endometrial cancer cell proliferation via activation of PI3K/Akt and MAPK/ERK1/2 signalling pathways. Gynecol Oncol. 2016;143:168–78.
Miethe C, Torres L, Zamora M, Price RS. Inhibition of PI3K/Akt and ERK signaling decreases visfatin-induced invasion in liver cancer cells. Horm Mol Biol Clin Investig. 2021;42:357–66.
Maeda K, Okubo K, Shimomura I, Funahashi T, Matsuzawa Y, Matsubara K. cDNA cloning and expression of a novel adipose specific collagen-like factor, apM1 (AdiPose Most abundant Gene transcript 1). Biochem Biophys Res Commun. 1996;221:286–9.
Martinez-Huenchullan SF, Tam CS, Ban LA, Ehrenfeld-Slater P, McLennan SV, Twigg SM. Skeletal muscle adiponectin induction in obesity and exercise. Metabolism. 2020;102:154008.
Ghoshal K, Bhattacharyya M. Adiponectin: Probe of the molecular paradigm associating diabetes and obesity. World J Diabetes. 2015;6:151–66.
Kita S, Maeda N, Shimomura I. Interorgan communication by exosomes, adipose tissue, and adiponectin in metabolic syndrome. J Clin Invest. 2019;129:4041–9.
Lautamaki R, Ronnemaa T, Huupponen R, Lehtimaki T, Iozzo P, Airaksinen KE, Knuuti J, Nuutila P. Low serum adiponectin is associated with high circulating oxidized low-density lipoprotein in patients with type 2 diabetes mellitus and coronary artery disease. Metabolism. 2007;56:881–6.
Takahashi N, Anan F, Nakagawa M, Yufu K, Shinohara T, Tsubone T, Goto K, Masaki T, Katsuragi I, Tanaka K, et al. Hypoadiponectinemia in type 2 diabetes mellitus in men is associated with sympathetic overactivity as evaluated by cardiac 123I-metaiodobenzylguanidine scintigraphy. Metabolism. 2007;56:919–24.
Wang X, You T, Murphy K, Lyles MF, Nicklas BJ. Addition of exercise increases plasma adiponectin and release from adipose tissue. Med Sci Sports Exerc. 2015;47:2450–5.
Nguyen TMD. Adiponectin: role in physiology and pathophysiology. Int J Prev Med. 2020;11:136.
Scheja L, Heeren J. The endocrine function of adipose tissues in health and cardiometabolic disease. Nat Rev Endocrinol. 2019;15:507–24.
Gantov M, Pagnotta P, Lotufo C, Rindone GM, Riera MF, Calvo JC, Toneatto J. Beige adipocytes contribute to breast cancer progression. Oncol Rep. 2021;45:317–28.
Than A, Xu S, Li R, Leow MK, Sun L, Chen P. Erratum: Author Correction: Angiotensin type 2 receptor activation promotes browning of white adipose tissue and brown adipogenesis. Signal Transduct Target Ther. 2018;3:10.
Shao M, Ishibashi J, Kusminski CM, Wang QA, Hepler C, Vishvanath L, MacPherson KA, Spurgin SB, Sun K, Holland WL, et al. Zfp423 maintains white adipocyte identity through suppression of the beige cell thermogenic gene program. Cell Metab. 2016;23:1167–84.
Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423:762–9.
Hug C, Wang J, Ahmad NS, Bogan JS, Tsao TS, Lodish HF. T-cadherin is a receptor for hexameric and high-molecular-weight forms of Acrp30/adiponectin. Proc Natl Acad Sci U S A. 2004;101:10308–13.
Nehme R, Diab-Assaf M, Decombat C, Delort L, Caldefie-Chezet F. Targeting Adiponectin in Breast Cancer. Biomedicines. 2022;10(11):2958.
Kadowaki T, Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev. 2005;26:439–51.
Denzel MS, Scimia MC, Zumstein PM, Walsh K, Ruiz-Lozano P, Ranscht B. T-cadherin is critical for adiponectin-mediated cardioprotection in mice. J Clin Invest. 2010;120:4342–52.
Lonnqvist F, Nordfors L, Jansson M, Thorne A, Schalling M, Arner P. Leptin secretion from adipose tissue in women. Relationship to plasma levels and gene expression. J Clin Invest. 1997;99:2398–404.
Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395:763–70.
Heymsfield SB, Greenberg AS, Fujioka K, Dixon RM, Kushner R, Hunt T, Lubina JA, Patane J, Self B, Hunt P, McCamish M. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. JAMA. 1999;282:1568–75.
Qing Y, Jamal MA, Shi D, Zhao S, Xu K, Jiao D, Zhao H, Li H, Jia B, Wang H, et al. Delayed body development with reduced triglycerides levels in leptin transgenic pigs. Transgenic Res. 2022;31:59–72.
Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, et al. Identification and expression cloning of a leptin receptor. OB-R Cell. 1995;83:1263–71.
Munzberg H, Bjornholm M, Bates SH, Myers MG Jr. Leptin receptor action and mechanisms of leptin resistance. Cell Mol Life Sci. 2005;62:642–52.
Myers MG, Cowley MA, Munzberg H. Mechanisms of leptin action and leptin resistance. Annu Rev Physiol. 2008;70:537–56.
Akasaka Y, Tsunoda M, Ogata T, Ide T, Murakami K. Direct evidence for leptin-induced lipid oxidation independent of long-form leptin receptor. Biochim Biophys Acta. 2010;1801:1115–22.
Chan JL, Bluher S, Yiannakouris N, Suchard MA, Kratzsch J, Mantzoros CS. Regulation of circulating soluble leptin receptor levels by gender, adiposity, sex steroids, and leptin: observational and interventional studies in humans. Diabetes. 2002;51:2105–12.
Sun Q, van Dam RM, Meigs JB, Franco OH, Mantzoros CS, Hu FB. Leptin and soluble leptin receptor levels in plasma and risk of type 2 diabetes in U.S. women: a prospective study. Diabetes. 2010;59:611–8.
Komori T, Morikawa Y, Nanjo K, Senba E. Induction of brain-derived neurotrophic factor by leptin in the ventromedial hypothalamus. Neuroscience. 2006;139:1107–15.
Belgardt BF, Bruning JC. CNS leptin and insulin action in the control of energy homeostasis. Ann N Y Acad Sci. 2010;1212:97–113.
Harvey J. Leptin: a multifaceted hormone in the central nervous system. Mol Neurobiol. 2003;28:245–58.
Mora-Munoz L, Guerrero-Naranjo A, Rodriguez-Jimenez EA, Mastronardi CA, Velez-van-Meerbeke A. Leptin: role over central nervous system in epilepsy. BMC Neurosci. 2018;19:51.
Ikejima K, Lang T, Zhang YJ, Yamashina S, Honda H, Yoshikawa M, Hirose M, Enomoto N, Kitamura T, Takei Y, Sato N. Expression of leptin receptors in hepatic sinusoidal cells. Comp Hepatol. 2004;3(Suppl 1):S12.
Ouyang S, Hsuchou H, Kastin AJ, Mishra PK, Wang Y, Pan W. Leukocyte infiltration into spinal cord of EAE mice is attenuated by removal of endothelial leptin signaling. Brain Behav Immun. 2014;40:61–73.
Philp LK, Rockstroh A, Sadowski MC, Taherian Fard A, Lehman M, Tevz G, Liberio MS, Bidgood CL, Gunter JH, McPherson S, et al. Leptin antagonism inhibits prostate cancer xenograft growth and progression. Endocr Relat Cancer. 2021;28:353–75.
Delort L, Bougaret L, Cholet J, Vermerie M, Billard H, Decombat C, Bourgne C, Berger M, Dumontet C, Caldefie-Chezet F. Hormonal therapy resistance and breast cancer: involvement of adipocytes and leptin. Nutrients. 2019;11(12):2839.
Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA. The hormone resistin links obesity to diabetes. Nature. 2001;409:307–12.
Huang X, Yang Z. Resistin’s, obesity and insulin resistance: the continuing disconnect between rodents and humans. J Endocrinol Invest. 2016;39:607–15.
Minn AH, Patterson NB, Pack S, Hoffmann SC, Gavrilova O, Vinson C, Harlan DM, Shalev A. Resistin is expressed in pancreatic islets. Biochem Biophys Res Commun. 2003;310:641–5.
Muse ED, Obici S, Bhanot S, Monia BP, McKay RA, Rajala MW, Scherer PE, Rossetti L. Role of resistin in diet-induced hepatic insulin resistance. J Clin Invest. 2004;114:232–9.
Gerber M, Boettner A, Seidel B, Lammert A, Bar J, Schuster E, Thiery J, Kiess W, Kratzsch J. Serum resistin levels of obese and lean children and adolescents: biochemical analysis and clinical relevance. J Clin Endocrinol Metab. 2005;90:4503–9.
Lobo TF, Torloni MR, Gueuvoghlanian-Silva BY, Mattar R, Daher S. Resistin concentration and gestational diabetes: a systematic review of the literature. J Reprod Immunol. 2013;97:120–7.
Jain SH, Massaro JM, Hoffmann U, Rosito GA, Vasan RS, Raji A, O’Donnell CJ, Meigs JB, Fox CS. Cross-sectional associations between abdominal and thoracic adipose tissue compartments and adiponectin and resistin in the Framingham Heart Study. Diabetes Care. 2009;32:903–8.
Xu JY, Sham PC, Xu A, Tso AW, Wat NM, Cheng KY, Fong CH, Janus ED, Lam KS. Resistin gene polymorphisms and progression of glycaemia in southern Chinese: a 5-year prospective study. Clin Endocrinol (Oxf). 2007;66:211–7.
Way JM, Gorgun CZ, Tong Q, Uysal KT, Brown KK, Harrington WW, Oliver WR Jr, Willson TM, Kliewer SA, Hotamisligil GS. Adipose tissue resistin expression is severely suppressed in obesity and stimulated by peroxisome proliferator-activated receptor gamma agonists. J Biol Chem. 2001;276:25651–3.
Zhang Z, Du J, Xu Q, Li Y, Zhou S, Zhao Z, Mu Y, Zhao AZ, Cao SM, Li F. Resistin promotes nasopharyngeal carcinoma metastasis through TLR4-Mediated Activation of p38 MAPK/NF-kappaB signaling pathway. Cancers (Basel). 2022;14(23):6003.
Santa-Maria CA, Coughlin JW, Sharma D, Armanios M, Blackford AL, Schreyer C, Dalcin A, Carpenter A, Jerome GJ, Armstrong DK, et al. The effects of a remote-based weight loss program on adipocytokines, metabolic markers, and telomere length in breast cancer survivors: the POWER-remote trial. Clin Cancer Res. 2020;26:3024–34.
Romacho T, Sanchez-Ferrer CF, Peiro C. Visfatin/Nampt: an adipokine with cardiovascular impact. Mediators Inflamm. 2013;2013:946427.
Samal B, Sun Y, Stearns G, Xie C, Suggs S, McNiece I. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol Cell Biol. 1994;14:1431–7.
Berndt J, Kloting N, Kralisch S, Kovacs P, Fasshauer M, Schon MR, Stumvoll M, Bluher M. Plasma visfatin concentrations and fat depot-specific mRNA expression in humans. Diabetes. 2005;54:2911–6.
Tan BK, Chen J, Digby JE, Keay SD, Kennedy CR, Randeva HS. Increased visfatin messenger ribonucleic acid and protein levels in adipose tissue and adipocytes in women with polycystic ovary syndrome: parallel increase in plasma visfatin. J Clin Endocrinol Metab. 2006;91:5022–8.
Pisani DF, Dumortier O, Beranger GE, Casamento V, Ghandour RA, Giroud M, Gautier N, Balaguer T, Chambard JC, Virtanen KA, et al. Visfatin expression analysis in association with recruitment and activation of human and rodent brown and brite adipocytes. Adipocyte. 2016;5:186–95.
Garcia-Miranda A, Garcia-Hernandez A, Castaneda-Saucedo E, Navarro-Tito N, Maycotte P. Adipokines as regulators of autophagy in obesity-linked cancer. Cells. 2022;11(20):3230.
Skoczen S, Tomasik PJ, Gozdzik J, Fijorek K, Krasowska-Kwiecien A, Wiecha O, Czogala W, Dluzniewska A, Sztefko K, Starzyk J, Siedlar M. Visfatin concentrations in children with leukemia before and after stem cell transplantation. Exp Hematol. 2014;42:252–60.
Lu Z, Fang Z, Guo Y, Liu X, Chen S. Cisplatin resistance of NSCLC cells involves upregulation of visfatin through activation of its transcription and stabilization of mRNA. Chem Biol Interact. 2022;351:109705.
Nagpal S, Patel S, Jacobe H, DiSepio D, Ghosn C, Malhotra M, Teng M, Duvic M, Chandraratna RA. Tazarotene-induced gene 2 (TIG2), a novel retinoid-responsive gene in skin. J Invest Dermatol. 1997;109:91–5.
Goralski KB, McCarthy TC, Hanniman EA, Zabel BA, Butcher EC, Parlee SD, Muruganandan S, Sinal CJ. Chemerin, a novel adipokine that regulates adipogenesis and adipocyte metabolism. J Biol Chem. 2007;282:28175–88.
Estienne A, Bongrani A, Reverchon M, Rame C, Ducluzeau PH, Froment P, Dupont J. Involvement of novel adipokines, chemerin, visfatin, resistin and apelin in reproductive functions in normal and pathological conditions in humans and animal models. Int J Mol Sci. 2019;20(18):4431.
Zhang Y, Shen WJ, Qiu S, Yang P, Dempsey G, Zhao L, Zhou Q, Hao X, Dong D, Stahl A, et al. Chemerin regulates formation and function of brown adipose tissue: Ablation results in increased insulin resistance with high fat challenge and aging. FASEB J. 2021;35:e21687.
Li JJ, Yin HK, Guan DX, Zhao JS, Feng YX, Deng YZ, Wang X, Li N, Wang XF, Cheng SQ, et al. Chemerin suppresses hepatocellular carcinoma metastasis through CMKLR1-PTEN-Akt axis. Br J Cancer. 2018;118:1337–48.
Farsam V, Basu A, Gatzka M, Treiber N, Schneider LA, Mulaw MA, Lucas T, Kochanek S, Dummer R, Levesque MP, et al. Senescent fibroblast-derived Chemerin promotes squamous cell carcinoma migration. Oncotarget. 2016;7:83554–69.
Klose R, Krzywinska E, Castells M, Gotthardt D, Putz EM, Kantari-Mimoun C, Chikdene N, Meinecke AK, Schrodter K, Helfrich I, et al. Targeting VEGF-A in myeloid cells enhances natural killer cell responses to chemotherapy and ameliorates cachexia. Nat Commun. 2016;7:12528.
Yu M, Yang Y, Zhao H, Li M, Chen J, Wang B, Xiao T, Huang C, Zhao H, Zhou W, Zhang JV. Targeting the chemerin/CMKLR1 axis by small molecule antagonist alpha-NETA mitigates endometriosis progression. Front Pharmacol. 2022;13:985618.
Tatemoto K, Hosoya M, Habata Y, Fujii R, Kakegawa T, Zou MX, Kawamata Y, Fukusumi S, Hinuma S, Kitada C, et al. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem Biophys Res Commun. 1998;251:471–6.
Boucher J, Masri B, Daviaud D, Gesta S, Guigne C, Mazzucotelli A, Castan-Laurell I, Tack I, Knibiehler B, Carpene C, et al. Apelin, a newly identified adipokine up-regulated by insulin and obesity. Endocrinology. 2005;146:1764–71.
Than A, He HL, Chua SH, Xu D, Sun L, Leow MK, Chen P. Apelin enhances brown adipogenesis and browning of white adipocytes. J Biol Chem. 2015;290:14679–91.
Kim DY, Choi MJ, Ko TK, Lee NH, Kim OH, Cheon HG. Angiotensin AT(1) receptor antagonism by losartan stimulates adipocyte browning via induction of apelin. J Biol Chem. 2020;295:14878–92.
Kasai A, Shintani N, Oda M, Kakuda M, Hashimoto H, Matsuda T, Hinuma S, Baba A. Apelin is a novel angiogenic factor in retinal endothelial cells. Biochem Biophys Res Commun. 2004;325:395–400.
Inukai K, Kise K, Hayashi Y, Jia W, Muramatsu F, Okamoto N, Konishi H, Akuta K, Kidoya H, Takakura N. Cancer apelin receptor suppresses vascular mimicry in malignant melanoma. Pathol Oncol Res. 2023;29:1610867.
Hao YZ, Li ML, Ning FL, Wang XW. APJ Is Associated with treatment response in gastric cancer patients receiving concurrent chemoradiotherapy and endostar therapy. Cancer Biother Radiopharm. 2017;32:133–8.
Kidoya H, Kunii N, Naito H, Muramatsu F, Okamoto Y, Nakayama T, Takakura N. The apelin/APJ system induces maturation of the tumor vasculature and improves the efficiency of immune therapy. Oncogene. 2012;31:3254–64.
Masoumi J, Zainodini N, Basirjafar P, Tavakoli T, Zandvakili R, Nemati M, Ramezani M, Rezayati MT, Ayoobi F, Khademalhosseini M, et al. Apelin receptor antagonist boosts dendritic cell vaccine efficacy in controlling angiogenic, metastatic and apoptotic-related factors in 4T1 breast tumor-bearing mice. Med Oncol. 2023;40:179.
Aleksandrova K, di Giuseppe R, Isermann B, Biemann R, Schulze M, Wittenbecher C, Fritsche A, Lehmann R, Menzel J, Weikert C, et al. Circulating omentin as a novel biomarker for colorectal cancer risk: data from the EPIC-potsdam cohort study. Cancer Res. 2016;76:3862–71.
Zhang YY, Zhou LM. Omentin-1, a new adipokine, promotes apoptosis through regulating Sirt1-dependent p53 deacetylation in hepatocellular carcinoma cells. Eur J Pharmacol. 2013;698:137–44.
Wu SS, Liang QH, Liu Y, Cui RR, Yuan LQ, Liao EY. Omentin-1 stimulates human osteoblast proliferation through PI3K/Akt signal pathway. Int J Endocrinol. 2013;2013:368970.
Cymbaluk-Ploska A, Chudecka-Glaz A, Jagodzinska A, Pius-Sadowska E, Sompolska-Rzechula A, Machalinski B, Menkiszak J. Evaluation of biologically active substances promoting the development of or protecting against endometrial cancer. Onco Targets Ther. 2018;11:1363–72.
Arner E, Mejhert N, Kulyte A, Balwierz PJ, Pachkov M, Cormont M, Lorente-Cebrian S, Ehrlund A, Laurencikiene J, Heden P, et al. Adipose tissue microRNAs as regulators of CCL2 production in human obesity. Diabetes. 2012;61:1986–93.
Arner P, Kulyte A. MicroRNA regulatory networks in human adipose tissue and obesity. Nat Rev Endocrinol. 2015;11:276–88.
Gesmundo I, Pardini B, Gargantini E, Gamba G, Birolo G, Fanciulli A, Banfi D, Congiusta N, Favaro E, Deregibus MC, et al. Adipocyte-derived extracellular vesicles regulate survival and function of pancreatic beta cells. JCI Insight. 2021;6(5):e141962.
Garcia-Martin R, Wang G, Brandao BB, Zanotto TM, Shah S, Kumar Patel S, Schilling B, Kahn CR. MicroRNA sequence codes for small extracellular vesicle release and cellular retention. Nature. 2022;601:446–51.
Sato S, Weaver AM. Extracellular vesicles: important collaborators in cancer progression. Essays Biochem. 2018;62:149–63.
Montecucco F, Lenglet S, Quercioli A, Burger F, Thomas A, Lauer E, da Silva AR, Mach F, Vuilleumier N, Bobbioni-Harsch E, et al. Gastric bypass in morbid obese patients is associated with reduction in adipose tissue inflammation via N-oleoylethanolamide (OEA)-mediated pathways. Thromb Haemost. 2015;113:838–50.
Kim EJ, Kim YK, Kim S, Kim JE, Tian YD, Doh EJ, Lee DH, Chung JH. Adipochemokines induced by ultraviolet irradiation contribute to impaired fat metabolism in subcutaneous fat cells. Br J Dermatol. 2018;178:492–501.
Bartelt A, Merkel M, Heeren J. A new, powerful player in lipoprotein metabolism: brown adipose tissue. J Mol Med (Berl). 2012;90:887–93.
Renovato-Martins M, Moreira-Nunes C, Atella GC, Barja-Fidalgo C, Moraes JA. Obese adipose tissue secretion induces inflammation in preadipocytes: role of toll-like receptor-4. Nutrients. 2020;12(9):2828.
Li VL, Kim JT, Long JZ. Adipose tissue lipokines: recent progress and future directions. Diabetes. 2020;69:2541–8.
Gao Y, Shabalina IG, Braz GRF, Cannon B, Yang G, Nedergaard J. Establishing the potency of N-acyl amino acids versus conventional fatty acids as thermogenic uncouplers in cells and mitochondria from different tissues. Biochim Biophys Acta Bioenerg. 2022;1863:148542.
DeClercq V, Taylor CG, Zahradka P. Isomer-specific effects of conjugated linoleic acid on blood pressure, adipocyte size and function. Br J Nutr. 2012;107:1413–21.
Pinheiro TA, Barcala-Jorge AS, Andrade JMO, Pinheiro TA, Ferreira ECN, Crespo TS, Batista-Jorge GC, Vieira CA, Lelis DF, Paraiso AF, et al. Obesity and malnutrition similarly alter the renin-angiotensin system and inflammation in mice and human adipose. J Nutr Biochem. 2017;48:74–82.
O’Hara A, Lim FL, Mazzatti DJ, Trayhurn P. Microarray analysis identifies matrix metalloproteinases (MMPs) as key genes whose expression is up-regulated in human adipocytes by macrophage-conditioned medium. Pflugers Arch. 2009;458:1103–14.
Kepczynska MA, Zaibi MS, Alomar SY, Trayhurn P. PCR arrays indicate that the expression of extracellular matrix and cell adhesion genes in human adipocytes is regulated by IL-1beta (interleukin-1beta). Arch Physiol Biochem. 2017;123:61–7.
James PT, Rigby N, Leach R. International obesity task F: The obesity epidemic, metabolic syndrome and future prevention strategies. Eur J Cardiovasc Prev Rehabil. 2004;11:3–8.
De Luca DE, Bonacci S, Giraldi G. Aging populations: the health and quality of life of the elderly. Clin Ter. 2011;162:e13-18.
Cohen SA, Greaney ML. Aging in rural communities. Curr Epidemiol Rep. 2023;10:1–16.
Madan K, Paliwal S, Sharma S, Kesar S, Chauhan N, Madan M. Metabolic syndrome: the constellation of co-morbidities, a global threat. Endocr Metab Immune Disord Drug Targets. 2023;23:1491–504.
Acknowledgements
This work was supported by the Japan Society for the Promotion of Science Grants-in-Aid for Scientific Research (KAKENHI; grant number 23K08708 and JP20K09422), Takeda Science Foundation, and Foundation for Promotion of Cancer Research.
Informed consent
Not applicable.
Funding
This work was supported by JSPS KAKENHI Grant Number 23K08708 and 20K09422, Takeda Science.
Foundation, and Foundation for Promotion of Cancer Research.
Author information
Authors and Affiliations
Contributions
S.S. wrote the main manuscript text, prepared figure and table, and reviewed the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sato, S. Adipo-oncology: adipocyte-derived factors govern engraftment, survival, and progression of metastatic cancers. Cell Commun Signal 22, 52 (2024). https://doi.org/10.1186/s12964-024-01474-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-024-01474-4