
Abstract
The accumulation of unfolded or misfolded proteins in the endoplasmic reticulum (ER) causes ER stress and activates the unfolded protein response (UPR). As an adaptive cellular response to hostile microenvironments, such as hypoxia, nutrient deprivation, oxidative stress, and chemotherapeutic drugs, the UPR is activated in diverse cancer types and functions as a dynamic tumour promoter in cancer development; this role of the UPR indicates that regulation of the UPR can be utilized as a target for tumour treatment. T-cell exhaustion mainly refers to effector T cells losing their effector functions and expressing inhibitory receptors, leading to tumour immune evasion and the loss of tumour control. Emerging evidence suggests that the UPR plays a crucial role in T-cell exhaustion, immune evasion, and resistance to immunotherapy. In this review, we summarize the molecular basis of UPR activation, the effect of the UPR on immune evasion, the emerging mechanisms of the UPR in chemotherapy and immunotherapy resistance, and agents that target the UPR for tumour therapeutics. An understanding of the role of the UPR in immune evasion and therapeutic resistance will be helpful to identify new therapeutic modalities for cancer treatment.
Video Abstract
Similar content being viewed by others
Introduction
The endoplasmic reticulum (ER) is an extensive membrane-bound organelle in eukaryotic cells that plays an important role in many cellular processes, such as the storage and release of calcium [1]. Ample evidence suggests that nutrient fluctuations, hypoxia and pathological insults result in the accumulation of unfolded or misfolded proteins in the ER lumen and disruption of protein-folding homeostasis, a condition referred to as ER stress [1,2,3]. In response to ER stress, cytoprotective signalling pathways are triggered to restore protein-folding homeostasis in the ER in a process termed the unfolded protein response (UPR). When the UPR is triggered, global protein synthesis is transiently attenuated, and protein degradation pathways, including ER-associated degradation (ERAD) and autophagy, are activated, thus restoring ER homeostasis. If the protein-folding defect is persistent, cells undergo apoptosis. Thus, chronic or persistent ER stress results in cellular injury which is associated with various human diseases, including neurodegeneration, diabetes and cancers [2, 4, 5]. Chemotherapy is an additional extrinsic challenge that leads to the activation of UPR signalling. Previous studies have shown that the UPR plays an important role in chemotherapy resistance [6,7,8], suggesting that targeting the UPR is a promising strategy for tumour treatment. Understanding the mechanism of UPR-mediated resistance to antitumour drugs could provide a reference for developing a new therapeutic approach to overcome chemotherapy resistance.
Immunosuppression is crucial for tumorigenesis and tumour development, metastasis and recurrence. Establishing an immunosuppressive network derived from interactions between cancer cells and host stromal cells (e.g., T cells, macrophages, B cells, fibroblasts, dendritic cells) in the tumour microenvironment could promote tumour growth, protect the tumour from immune attack, and attenuate the efficacy of immunotherapeutic approaches [9,10,11]. Tumour-infiltrating T cells in the tumour microenvironment often lose their ability to kill tumour cells and are in a state of exhaustion. Exhausted T cells have the following two characteristics: (1) high expression of immune checkpoint molecules under persistent infection or antigen stimulation, such as programmed cell death protein 1 (PD1) [12,13,14], lymphocyte activating 3 (LAG3) [15], cytotoxic T-lymphocyte associated protein 4 (CTLA4) [16], T-cell immunoreceptor with Ig and ITIM domains (TIGIT) [17], CD160 [18], TNF receptor superfamily member 9 (TNFRSF9) [19], and hepatitis A virus cellular receptor 2 (HAVCR2) [20]; and (2) decreased proliferation, viability, and the loss of cytotoxic activity against tumours in vitro [21]. Although T-cell exhaustion does not effectively control tumour cells, this process is reversible. The clinical application of immune checkpoint therapies (ICTs), including anti-PD1/PD-L1 (PD1 ligand 1) antibody therapy, can restore T-cell immune function to a certain extent and increased the likelihood of tumour patient survival [22]. Overwhelming evidence indicates that the combination of PD1 with HAVCR2, CTLA4 or TIGIT inhibitors can more effectively improve T-cell function and eliminate tumour cells [23]. An increasing number of studies have shown that the UPR plays an important role in resistance to immune checkpoint inhibitors (ICIs). Thus, targeting the UPR may be a promising therapeutic strategy for overcoming resistance to ICIs.
In this article, we reviewed the molecular basis of UPR activation, the effect of the UPR on T-cell exhaustion and immune evasion, mechanisms of the UPR in resistance to chemotherapy and immunotherapy, and agents that target the UPR for tumour therapeutics. Finally, we discuss the applications and limitations of targeting the UPR in cancer treatment.
Key sensors in the UPR
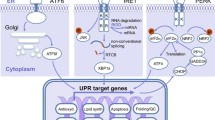
In mammals, UPR signalling is mainly transduced by the following three sensors: protein kinase R-like endoplasmic reticulum kinase (PERK) [24], activating transcription factor 6 alpha (ATF6α) [25], and inositol requiring enzyme 1 alpha (IRE1α) [26]. PERK and IRE1α are type I transmembrane proteins with cytosolic kinase domains. Under nonstress conditions, binding-immunoglobulin protein (BIP; also known as GRP78) binds to the luminal domain of PERK, IRE1α and ATF6α to prevent their activation. When unfolded and misfolded proteins accumulate in the ER lumen, BIP dissociates from PERK, IRE1α and ATF6α and binds to unfolded proteins, which allows PERK and IRE1α homodimerization and autophosphorylation [27, 28] and ATF6α translocation to the Golgi apparatus [29] (Fig. 1). A recent study showed that the above sensors could also be activated without disrupting their binding with BIP [30], which is indicative of the complexity of UPR signalling.

Unfolded protein response. Three ER stress sensors (PERK, IRE1α and ATF6α) are present in the ER membrane. Accumulation of misfolded protein in the ER lumen causes ER stress and dissociation of BiP from UPR sensors. Subsequently, IRE1α and PERK auto-transphosphorylated and are activated, and ATF6α exposes an ER export motif and translocates to the nucleus. Activated PERK phosphorylates eIF2α and leads to attenuation of global translation, but increases the expression of transcription factor ATF4. ATF4 induces transcription of genes that restore proteostasis and promote survival. Once ER stress is resolved, the dephosphorylation of eIF2α is triggered by growth arrest and DNA damage-inducible protein 34 (GADD34), and translation is reinitiated. Activated IRE1α has endoribonuclease activity and can splice an intron from unspliced X-box binding protein 1 (XBP1u), which leads to the production of XBP1s mRNA. Subsequently, XBP1s mRNA increases transcription of a set of genes associated with protein folding and degradation of misfolded proteins. Meanwhile, ATF6α is translocated from ER to Golgi, in which it is processed by S1P and S2P, producing a cytosolic p50 fragment (pATF6α) that functions as a transcription factor. Collectively, UPR activation aims to restore ER homeostasis, otherwise cells enter death
Under ER stress, PERK phosphorylates eIF2α, a translation initiation factor 2 subunit, leading to inactivation of eIF2α and inhibition of global protein translation due to the limited amount of the eIF2α–GTP–tRNA met ternary complex [31,32,33]. Although the translation of most mRNAs is suppressed in response to ER stress, the translation of some species of mRNA is increased, such as transcription factor ATF4 and C/EBP homologous protein (CHOP; also known as DDIT3). ATF4 is a transcription factor that regulates the expression of genes associated with the ER stress response, the antioxidant response, amino acid biosynthesis, autophagy and apoptosis [34,35,36] (Fig. 1). ATF4 also upregulates the transcription of CHOP and the growth of arrest and DNA damage-inducible protein 34 (GADD34; also known as PPP1R15A) [37, 38] (Fig. 1). CHOP is a pro-apoptotic factor that upregulates the expression of apoptosis-related genes, such as DR5 and PUMA [39,40,41]. In addition, PERK also phosphorylates and activates nuclear factor erythroid 2-related factor 2 (NRF2) to upregulate the expression of antioxidant genes, such as glutathione synthetase and haem oxygenase-1 (HO-1), thereby limiting the accumulation of cellular reactive oxygen species (ROS) [42,43,44].
IRE1α, a type I transmembrane protein, contains a kinase domain and an endoribonuclease (RNase) domain at its cytoplasmic tail [45]. In response to ER stress, IRE1α dimerizes and transautophosphorylates, inducing conformational alteration of its RNase domain and thereby activating its RNase activity. Afterwards, IRE1α splices a 26-nucleotide intron from the XBP1 mRNA and produces XBP1s mRNA, leading to a shift of the reading frame to translate a stable and active transcription factor known as XBP1s [1, 46,47,48,49] (Fig. 1). XBP1s upregulates a subset of target genes associated with lipid synthesis, protein folding, secretion, protein degradation, redox homeostasis, and amino acid metabolism [50, 51] (Fig. 1), which helps cells restore ER homeostasis. In addition, IRE1α increases the expression of several genes associated with cell death in response to ER stress. For instance, IRE1α utilizes its RNase activity to selectively cleave microRNA that normally suppresses the expression of pro-apoptosis targets, such as pro-oxidant protein TXNIP (thioredoxin-interacting protein) and caspase-2, resulting in increased expression of these pro-apoptosis genes [52,53,54]. IRE1α also interacts with the adaptor protein tumour necrosis factor (TNF) receptor-associated factor 2 (TRAF2) to recruit apoptosis signal-regulating kinase 1 (ASK1) and JUN N-terminal kinase (JNK), leading to activation of the apoptosis pathway [55,56,57] (Fig. 1).
ATF6α is a type II ER transmembrane protein with a cytosolic cAMP-responsive element-binding protein (CREB)/ATF basic leucine zipper (bZIP) domain. Upon ER stress, ATF6α translocates to the Golgi apparatus and is cleaved by serine protease site-1 (S1P) and metalloprotease site-2 (S2P), leading to the production of an active transcription factor (pATF6α) [58,59,60] (Fig. 1). Then, pATF6α translocates to the nucleus, where it activates the expression of genes associated with protein folding and protein degradation [25, 61]. In addition, in response to ER stress, several cAMP-responsive element-binding proteins, including cAMP-responsive element-binding protein H (CREBH or CREB3L3), CREB3, CREB3L1, CREB3L2, and CREB, are activated similarly to ATF6α [62,63,64].
In addition to the canonical sensors mentioned above, Ca2+ is also an important player that transduces UPR signalling. Canonically, initiation of ER stress/the UPR is associated with a reduction in ER Ca2+ levels [65]. The reduction in ER Ca2+ enhances the UPR by the following mechanisms: (1) inhibition of the activity of several protein folding enzymes [66] and (2) impairment of the folding capacity by binding chaperones [66]. For example, Ca2+ binds to the chaperone calreticulin and activates it, leading to a reduction in misfolded proteins in the ER and thereby alleviating the UPR [67]. Mechanically, the Ca2+-binding chaperone calreticulin interacts with protein disulfide isomerase (PDI) and ERp57, two important protein folding enzymes that promote the proper folding of synthesized proteins, and activates them, thereby reducing the accumulation of misfolded proteins in the ER [68]. These studies demonstrated that Ca2+ plays an important role in the quality control and proper folding of newly synthesized proteins. Therefore, ER Ca2+ imbalance can greatly impact folding capacity and induce ER stress-mediated apoptosis. In multiple myeloma (MM), inhibition of TRPV1, which is involved in the regulation of calcium signalling, induces the accumulation of mitochondrial calcium and decreases the level of HSP70 induced by bortezomib, thereby impairing protein folding capacity and sensitizing MM cells to bortezomib [69].
The source of the UPR
Due to cell-intrinsic factors, including the hyperactivation of oncogenes and inactivation of tumour suppressors, as well as the conditions of the tumour microenvironment, such as hypoxia and nutrient deprivation, cancer cells exhibit higher ER stress than normal cells [70]. Cell-intrinsic factors that trigger the UPR have been extensively reviewed elsewhere [70, 71]. In this section, we mainly focus on the impact of hypoxia on the UPR.
When tumour cells encounter a hostile environment, protein-folding homeostasis is disrupted, leading to activation of the UPR. All three UPR branches are activated by hypoxia, and the multiple mechanisms by which hypoxia activates the UPR have been identified. First, the formation of disulfide bonds during posttranslational folding or isomerization in the ER is oxygen-dependent [72]. Thus, hypoxia inhibits disulfide bond formation, leading to the accumulation of misfolded proteins in the ER [72]. Second, hypoxia promotes the expression of many components of the UPR, such as BIP [73, 74] and CHOP [75], through hypoxia-inducible factors. Third, hypoxia reduces the content of desaturated lipids and thereby limits the expansion of the ER, thus disrupting ER homeostasis [76, 77]. However, hypoxia can also increase the expression of ERO1α, an oxidoreductase involved in disulfide bond formation and protein folding in the endoplasmic reticulum, to restore ER homeostasis [78]. This phenomenon creates a feedback mechanism that contributes to the restoration of ER homeostasis. While writing this review, a detailed summary of hypoxia in relation to the unfolded protein response was published elsewhere [79, 80]. These findings contribute to our understanding of the well-established role of hypoxia in UPR activation.
Role of the UPR in cancer
A moderate increase in UPR signalling can promote cell survival, whereas an excessive and prolonged UPR drives cells into apoptosis. This phenomenon applies to both normal and cancer cells because UPR activation-induced cell death pathways are integrated in at least some tumour cells. Thus, it is not surprising that the UPR could induce either the survival or apoptosis of cancer cells.
The PERK-eIF2α pathway in cancer
The PERK-mediated UPR induces either survival or apoptosis in response to ER stress. Thus, the UPR may promote or inhibit malignant transformation [81], depending on the context. PERK deficiency has been shown to inhibit tumour progression [82], while activation of PERK signalling contributes to Myc-mediated malignant transformation by inducing autophagy [8, 83, 84]. In contrast, certain studies substantiate the fact that inhibition of PERK signalling promotes cell transformation [85]. For example, deletion of CHOP, a gene downstream of PERK signalling, has been reported to promote tumorigenesis in a KrasG12V-induced mouse model of lung cancer, indicating that CHOP has a tumour-suppressive role [86]. Moreover, PERK has been reported to be a haploinsufficient tumour suppressor in melanoma [85]. Thus, PERK is thought to be a tumour suppressor or proadaptive tumour promoter based on gene dose.
The IRE1α-XBP1 pathway in cancer
IRE1α mutations have been found to be frequent in human cancers [87, 88]. Mounting evidence suggests that the IRE1α-mediated UPR branch could promote tumour development in glioblastoma by increasing the expression of genes associated with inflammation and angiogenesis, while loss-of-function mutations in the IRE1α signalling pathway increased the expression of matrix proteins, leading to inhibition of tumour invasion [89,90,91,92,93]. In addition, XBP1 inhibits lipid metabolism and antigen presentation in dendritic cells, leading to the suppression of antitumour immunity [94]. Studies have also shown that an increase in XBP1 expression can promote triple-negative breast cancer development by upregulating the expression of HIF-1α [95]. In contrast, in mouse models of intestinal cancer, loss of XBP1 function contributes to tumour incidence [89]. In multiple myeloma, loss-of-function mutations in IRE1α and XBP1 play pro-oncogenic roles [96], suggesting that the IRE1α-XBP1 pathway acts as a tumour suppressor in some tumours.
The ATF6α signalling pathway in cancer
The role of ATF6α in cancer is poorly understood. Current evidence suggests that ATF6α is associated with hepatocellular carcinoma, bladder cancer, non-small cell lung cancer (NSCLC) and prostate cancer [97,98,99,100]. ATF6α functions as a transcription factor, which may be associated with the regulation of tumour cell dormancy [101]. Collectively, existing evidence indicates that ATF6α may help dormant cells adapt to chemotherapy and nutritional stress, although the underlying mechanism remains unclear.
The UPR and chemotherapy resistance
Chemotherapy is an additional extrinsic challenge. In response to chemotherapeutic drugs, tumour cells have evolved a variety of mechanisms of drug resistance to adapt to this challenge. Multiple mechanisms associated with chemotherapy resistance have been identified, such as (i) a reduction in drug accumulation; (ii) enhancement of DNA damage repair (DDR); (iii) insensitivity of drug-induced cell death; (iv) induction of drug inactivation; and (v) induction of autophagy. Emerging evidence shows that the UPR is closely associated with the above mechanisms.
The UPR and the reduction of drug accumulation
Drugs can enter tumour cells through diffusion, active transport, and endocytosis [102,103,104]. However, tumour cells limit this entry by increasing drug efflux or reducing drug uptake. The proteins responsible for drug efflux are named ATP-binding cassette (ABC) transporters; these transporters include multidrug resistance protein 1 (MDR1), MDR-associated protein 1 (MRP1), and breast cancer resistance protein (BCRP) [105,106,107,108]. In addition, downregulated expression or loss-of-function mutation of cell surface uptake transporters can reduce drug uptake. For example, downregulated expression or mutation of folate carriers reduced their drug affinity in osteosarcoma [109, 110].
Many studies have demonstrated that the UPR promotes drug efflux by upregulating the expression of ABC transporters. A recent study showed that the ATF4-mediated stress response–like transcriptional program is activated by daunorubicin, induces the expression of MDR1, and increases drug efflux [111], which leads to resistance to chemotherapeutic drugs (Fig. 2). Similarly, studies have also demonstrated that the 5-fluorouracil-activated IRE1α-XBP1 pathway upregulates the expression of ABC transporters and increases drug efflux in colon cancer cells [112]. In addition to the IRE1α-XBP1 pathway, the upregulation of GRP78 expression can also increase the efflux activity of ABC transporters, thereby conferring resistance to chemotherapeutic drugs in pancreatic cancer cells [113]. Additionally, in colorectal cancer, ATF6 is activated by TAM-secreted C–C motif chemokine ligand 17 (CCL17) and CCL22 via membrane receptor CCR4, leading to the upregulation of GRP78 expression [114]. Then, GRP78 interacts with MRP1 and promotes its translocation to the cell membrane, causing TAM-induced 5-FU efflux [114] (Fig. 2).

The UPR and chemotherapy resistance. UPR promotes chemotherapy resistance of tumor cells by increasing the expression of antioxidant genes, efflux pumps genes and DNA damage response (DDR) genes. In response to chemotherapeutic drugs, PERK directly phosphorylates NRF2, which increases the expression of antioxidant genes. The upregulation of antioxidant genes eliminates cellular ROS induced by chemotherapeutic drugs to promote cell survival. Certain antioxidant genes can also catalyze the conjugation of glutathione with chemotherapeutic drugs and lead to inactivation of chemotherapeutic drugs. In addition, activation of PERK can increase the expression of ATF4. ATF4 is closely associated with the expression of DDR and efflux pump genes. In response to chemotherapeutic drugs, the IRE1α-XBP1 pathway is also involved in the upregulation of a number of DDR genes. The third branch of UPR, ATF6, is associated with efflux pump translocation, anti-apoptosis and activation of mTOR signalling. Thus, activation of UPR is thought to be crucial for chemotherapy resistance
The UPR and the increase in DNA damage repair
The antitumour activity of most drugs depends on the induction of DNA damage. For example, platinum-based drugs interact with DNA and form DNA inter- and intrastrand crosslinks, which leads to cancer cell apoptosis [115]. Anthracyclines, including adriamycin and daunorubicin, induce DNA damage in the following ways: (1) suppression of DNA replication and RNA synthesis by inhibiting DNA topoisomerase [116, 117]; (2) embedding of the DNA double strand and formation of DNA intrastrand crosslinks [118]; and (3) induction of ROS by Fe3+ chelation [119].
Many studies have demonstrated that DDR is associated with chemotherapy resistance. For instance, decreased expression of DNA topoisomerase I enhances the resistance of cancer cells to camptothecin [120]. Similarly, loss-of-function mutation of DNA topoisomerase-II, a target of doxorubicin and etoposide, has been reported in resistant cancer cell lines [121]. Furthermore, high expression of ERCC1, an important member of the nucleotide excision repair pathway, implies a poor response to chemotherapeutic drugs in many tumours, including NSCLC, gastric cancer and ovarian cancer [122, 123]. In particular, testicular cancer tissues have a very low ERCC1 level and are very sensitive to cisplatin [124]. A previous study also showed that overexpression of chromosomal instability (CIN) genes, which are involved in the maintenance of genomic integrity, is correlated with a poor prognosis for tumour patients [125].
An increasing body of evidence suggests that UPR sensors or downstream transcription factors are closely associated with DDR. For example, mammalian IRE1α-XBP1 has been identified to play an important role in the nonhomologous end-joining (NHEJ) repair pathway through the regulation of H4 acetylation [126]. Similar studies have shown that XBP1s in human hepatic cells directly upregulates the transcription of multiple DDR genes [127] (Fig. 2). Therefore, knockdown of XBP1s results in the formation and increase in γH2AX foci, downregulation of MRE11–RAD50–NBS1 (MRN) expression, and a decrease in ATM phosphorylation [128].
PERK-eIF2α-ATF4 is another UPR branch involved in DDR. Recent studies have shown that decreased expression of PERK in human breast cancer cells is associated with increased global phosphorylation of ATM and increased phosphorylation of its downstream effector CHK2, a cell cycle checkpoint kinase associated with DNA damage repair [82, 129] (Fig. 2). PERK-deficient tumour cells exhibit increased oxidative DNA damage, which leads to G2/M cell cycle checkpoint activation [82]. Moreover, PERK, IRE1α and ATF6α interact with DNA damage proteins (e.g., ATM, ATR, p53, p21, CHK1 and CHK2) to promote DDR in response to genotoxic stress, which enhances resistance to chemotherapeutic drugs [82, 128, 130,131,132,133,134,135]. Similarly, ATF4 increases the expression of cleavable topoisomerase complexes, leading to drug resistance to a variety of DNA-interactive agents [136,137,138].
The UPR and insensitivity to drug-induced cell death
Mounting evidence indicates that cancer cells are usually "addicted" to a few antiapoptotic proteins and decrease the expression of proapoptotic proteins for their survival, thereby leading to resistance to chemotherapeutic drugs. These studies provide a strong theoretical basis for using these antiapoptotic and proapoptotic proteins as therapeutic targets. Mutations, amplifications, chromosomal translocations and overexpression of these antiapoptotic genes, including BCL‑2 family members, inhibitor of apoptosis proteins (IAPs), and the cellular FLICE-inhibitory protein (FLIP), are usually associated with chemotherapy resistance [139]. Antiapoptotic Bcl-2 family inhibitors have made substantial progress as anticancer therapies. UPR sensors, such as ATF4, upregulate the expression of the antiapoptotic genes BCL-2 and FLIP and protect tumour cells against apoptosis in response to drug-induced stress, leading to tumour cell resistance to chemotherapeutic drugs [139]. UPR sensors can also mediate the inactivation or downregulated expression of pro-apoptosis proteins and thus prevent tumour cells from undergoing chemotherapeutic drug-induced apoptosis. For instance, studies have demonstrated that GRP78 inhibits the pro-apoptosis proteins Bax (Bcl2-associated X protein), BIK (Bcl2-interacting killer) and caspase-7 activation [140,141,142,143,144,145], thereby reducing chemotherapeutic drug-induced apoptosis. In relapsed/refractory osteosarcoma (OS), ATF6α cleavage and activity were enhanced in OS cells compared to normal osteoblasts, and knockdown of ATF6α expression sensitized OS cells to chemotherapeutic drugs and induced cell death by activating Bax [146] (Fig. 2). In addition, UPR sensors can couple with other UPR sensors to promote tumour resistance to chemotherapeutic drugs. For example, functional coupling of GRP78 overexpression and PERK activation enhances dormant tumour cell resistance to chemotherapeutic drug-induced apoptosis [147]. Interestingly, GRP78 can interact with the tumour suppressor BRCA1 and inhibit apoptosis of ovarian and breast cancer cells [148]. Accordingly, silencing GRP78 increases ovarian and breast cancer cell sensitivity to chemotherapeutic drugs [148].
Moreover, UPR-mediated upregulation of antioxidant gene expression and the elimination of ROS may, at least in part, account for tumour cell resistance to chemotherapeutic drug-induced apoptosis. In response to chemotherapeutic drugs, PERK directly phosphorylates the transcription factor NRF2, a transcription factor that regulates the expression of antioxidant genes, leading to the elimination of drug-induced ROS and inhibition of cancer cell apoptosis [6, 149].
The UPR and the induction of drug inactivation
Platinum drugs, including cisplatin, carboplatin, and oxaliplatin, can be covalently linked with thiol glutathione, which leads to their inactivation, decreases the availability of the native drug to bind its target, and promotes drug efflux by ABC transporters [130, 150]. Accordingly, high levels of glutathione have been reported to play a critical role in resistance to platinum drugs.
In response to chemotherapeutic drugs, the expression of ATF4 and NRF2, two downstream transcription factors of PERK, is upregulated, leading to increased expression of antioxidant genes, such as glutathione S transferase (GST) and glutamate-cysteine ligase catalytic subunit (GCLC) [2, 151, 152] (Fig. 2). Subsequently, GST conjugates hydrophobic electrophiles of drugs with glutathione, leading to inactivation of multiple chemotherapeutic drugs [130, 153]. Accordingly, targeting GST may be an effective therapeutic strategy against tumours.
The UPR and the induction of autophagy
Autophagy is a cellular degradation process in which long-lived cellular proteins and organelles are encapsulated in double-membrane vesicles termed autophagosomes and degraded by lysosomal hydrolases [154]. Under normal conditions, autophagy occurs at basal levels and promotes the upkeep of cytoplasmic components. Under nutrient deprivation, oxidative stress, hypoxia and ER stress, autophagy can be significantly induced [155]. For example, under ER stress, the assembly of pre-autophagosomal structures is promoted and increases ATG1 kinase activity, leading to the induction of autophagy [156]. Many studies have demonstrated that PERK-regulated autophagy promotes tumour growth and contributes to the chemotherapeutic resistance of tumour cells. For example, PERK-mediated autophagy can enhance c-Myc-induced transformation and tumour growth [83]. When breast cancer cells are exposed to bortezomib, ATF4 is induced and triggers autophagy, which promotes breast cancer cell survival [7]. In response to chemotherapeutic drugs, the PERK-eIF2α pathway is activated and induces autophagy, which promotes drug resistance in human osteosarcoma [157]. In melanoma, PERK-mediated autophagy enhanced resistance to BRAF and MEK inhibitors through the PERK-ERK-ATF4 axis [158]. In this study, PERK phosphorylated ERK. Subsequently, ERK phosphorylated ATF4, which induced cytoprotective autophagy, leading to melanoma cell survival. Therefore, targeting PERK or ERK is a promising therapeutic strategy for overcoming resistance to BRAF and MEK inhibitors [158]. Additionally, the ATF6-mediated UPR was also found to promote chemotherapy resistance by inducing autophagy [159]. In high-grade serous ovarian cancer, ATF6 is activated by STAT3 and in turn induces the UPR to promote autophagy, thereby leading to cancer cell resistance to both cisplatin and paclitaxel treatment [159].
The UPR and immunotherapy resistance
Cancer immunotherapies manipulate the immune system to recognize and attack cancer cells, and these therapies have revolutionized cancer therapy [160, 161]. However, immunotherapy resistance prevents most patients from benefiting from cancer immunotherapy. Recently, multiple mechanisms associated with immunotherapy resistance have been identified, including (1) impairment of T-cell recruitment; (2) deterioration of T-cell function; (3) suppression of antigen presentation; and (4) cancer cell stemness. Impairment of T-cell recruitment and deterioration of T-cell function in solid tumours are frequently associated with an immunosuppressive tumour microenvironment. An immunosuppressive tumour microenvironment is composed of all immune cells involved in the body’s immune response, such as CD4 + T cells, CD8 + T cells, dendritic cells, macrophages, and B cells, but the function of these immune cells is usually suppressed, which allows tumour cells to escape immune surveillance. Emerging evidence suggests that the UPR frequently participates in shaping an immunosuppressive tumour microenvironment and is closely associated with immunotherapy resistance.
The UPR and the impairment of T-cell recruitment
Many tumours exhibit few T cells and a high number of immunosuppressive cells, including myeloid-derived suppressor cells, tumour-associated macrophages, and regulatory T cells, which exhibit immune-cold characteristics and lead to resistance to anti-PD1 therapy [162, 163]. In ovarian cancer, IRE1α-XBP1 signalling has been found to inhibit T-cell infiltration and IFNγ expression, thereby promoting tumour progression [164]. T-cell-specific XBP1 deletion increases T-cell infiltration, exhibits superior antitumour immunity and leads to inhibition of malignant progression and an increase in overall survival [164]. In patients with vitiligo, activation of IRE1α-XBP1 signalling increases CD8+ T-cell recruitment by upregulating CXCL16 expression, thereby leading to melanocyte-specific autoimmune responses and depigmentation of the skin [165]. Targeting the IRE1α-XBP1-CXCL16 axis could inhibit CD8+ T-cell infiltration and is thought to be a promising therapeutic strategy for treating vitiligo. In primary brain tumours, glioblastoma (GBM) is remarkably resistant to immunotherapy. Studies have demonstrated that XBP1-mediated expression of T-cell-suppressive checkpoints in myeloid cells, such as PD1, prevents the activation of tumour infiltrating T cells in GBM, thereby leading to resistance to anti-PD1 therapy [166]. These results suggest that XBP1 may suppress the activation of antitumour T cells in solid cancer by upregulating PD1 expression in myeloid cells. Therefore, targeting IRE1α-XBP1 signalling could help increase T-cell infiltration and enhance immunotherapy efficacy.
PERK-eIF2α signalling is also involved in the inhibition of CD8+ T-cell antitumour immunity. A previous study showed that activation of PERK-eIF2α signalling could inhibit CD8+ T-cell infiltration and promote tumour growth, while inhibition of PERK-eIF2α signalling decreased the number of infiltrating CD8+ T cells, increased tumour clearance, and enhanced the efficacy of anti-PD-1 therapy in sarcoma [167] (Fig. 3). Similarly, CHOP, a downstream sensor of PERK-eIF2α signalling, is widely thought to be a negative regulator of CD8+ T-cell antitumour immunity. An increase in CHOP expression has been shown in tumour-infiltrating CD8+ T cells and implies poor clinical outcomes in ovarian cancer patients [168]. CHOP deletion in T cells promotes spontaneous CD8+ T-cell antitumour immunity and increases the effectiveness of T-cell-based immunotherapy [169]. Thus, targeting the PERK-eIF2α pathway may be a promising strategy for overcoming immunotherapy tolerance and increasing the effectiveness of immunotherapy.

Effects of UPR signals on antitumor immunity. Nutrient starvation/excess or accumulation of ROS in tumor microenvironment leads to induction of ER stress in T cells and thus causes T cell exhaustion. High cholesterol levels in TME can activate PERK-eIF2α and IRE1α–XBP1 signaling in intratumoral T cells and induce the expression of PD1, which leads to the inhibition of CD8+ T cell antitumor immunity. Cancer cells undergoing ER stress activate PERK and IRE1α signals, which leads to upregulation of ATF4 and XBP1s. ATF4 and XBP1s directly upregulate the expression of PD-L1, triggering T cell exhaustion. Under hostile environmental condition, ER stress activates IRE1α and ATF6α and induces the transformation of HPSC into MDSCs. PERK can also induce the transformation of PMN-MDSCs into MDSCs, leading to immunosuppression. Among three UPR branches, activated PERK directly phosphorylates NRF2 and increases the expression of antioxidant genes, leading to limitation of ROS and maintenance of mitochondrial genome stability in MDSCs. This increase of mitochondrial genome stability inhibits the expression of IFN α/β and thereby suppresses T cell infiltration. Activated PERK also increases the expression of CHOP, leading to upregulation of IL-6 and thus promotes CD8+ T cell exhaustion. DCs are responsible for antigen processing and presentation and promote the transformation of T cells into CD8+T cells. ROS accumulation in DCs causes IRE1α-XBP1 activation, which drives uncontrolled lipid droplet formation and leads to the inhibition of antigen presentation of DCs. The hostile microenvironment can also activate IRE1α–XBP1 signaling and upregulate the expression of miR-23a in macrophages, leading to the increase of PD-L1 expression. Additionally, CD4+ T cells exploit IRE1α–XBP1 signaling to control Ca2+ mobilization and expression of IL4, which is necessary for the activation of CD8+ T cells
The UPR and the deterioration of T-cell function
Deterioration of T-cell function refers to the loss of effector function and the expression of various immune checkpoint molecules in T cells, leading to a loss of tumour control. Multiple mechanisms associated with the deterioration of T-cell function have been identified, including (1) the expression of immune checkpoint molecules and their ligands; (2) the suppression of T-cell development and differentiation; and (3) the maintenance of the immunosuppressive function of myeloid-derived suppressor cells (MDSCs). An increasing number of studies show that the UPR is associated with the above mechanisms and causes tumour resistance to immunotherapy.
The UPR promotes the expression of immune checkpoint molecules and their ligands
Exposure to persistent antigens or inflammatory stimulation leads to the deterioration of T-cell function. Such deterioration is referred to as T-cell exhaustion, which is often associated with a loss of tumour control [21, 170,171,172]. Exhausted CD8+ T cells were characterized by high expression of immune checkpoint molecules, such as PD-1, LAG-3, TIM-3, 2B4, and CTLA-4 [173]. Many studies have shown that ER stress and chronic activation of the UPR lead to T-cell exhaustion [174,175,176,177]. In melanoma, high levels of cholesterol induce the expression of XBP1s, leading to upregulated expression of immune checkpoint molecules and inhibition of CD8+ T-cell antitumour immunity [178] (Fig. 3), while inhibition of XBP1 or a decrease in cholesterol could restore CD8+ T-cell antitumour immunity and reduce tumour progression [178]. Accordingly, reducing cholesterol or ER stress to restore T-cell function represents a new strategy to enhance the effectiveness of T-cell-based immunotherapy.
In addition to promoting the expression of immune checkpoint molecules on T cells, the UPR also promotes the expression of their ligands. Tumour-associated macrophages (TAMs) are abnormal myeloid cells that exist in the tumour microenvironment [179]. The relationship between TAMs and immune evasion has been well established. For example, TAM PD1 expression inhibits the phagocytic potency of macrophages against tumour cells. Accordingly, the PD1 inhibitor increases macrophage phagocytosis, reduces tumour growth, and prolongs the survival of mice in models of cancer in a macrophage-dependent fashion, which demonstrates that macrophages play a role in tumour evasion [180]. Recently, an increasing number of reports have demonstrated that the UPR is implicated in macrophage-associated immune evasion. In KSHV infection, the IRE1α-XBP1 pathway is activated and increases the expression of PD-L1 in KSHV-infected macrophages [181], leading to the loss of viral control (Fig. 3). In melanoma, activation of the IRE1α-XBP1 pathway upregulates the expression of macrophage PD-L1 and promotes tumour immune evasion [182] (Fig. 3). In addition, ER stress has been shown to upregulate PD-L1 expression in macrophages through the miR-23a–PTEN–AKT pathway, thereby promoting tumour progression [183] (Fig. 3). These studies show that ER stress-mediated PD-L1 expression in macrophages can inhibit T-cell function and promote tumour cell escape from antitumour immunity.
Although exhausted T cells lose their ability to kill tumour cells, this phenotype is reversible. The immune checkpoint blockade (PD1/PD-L1 immunotherapy) currently used in clinical practice can restore the antitumour function of T cells to a certain extent and improve the survival of tumour patients. Additionally, anti-PD1 combined with anti-HAVCR2, CTLA4, or TIGIT has been recently found to more effectively improve T-cell function, overcome resistance to anti-PD1 therapy, and eliminate tumour cells. These results show that high expression of multiple immune checkpoint molecules on T cells may be an important reason for immunotherapy resistance. Interestingly, XBP1 can simultaneously regulate the expression of multiple immune checkpoint molecules, such as PD1, LAG3, and HAVCR2, in some types of tumours [176], suggesting that XBP1 is a potential target for overcoming immunotherapy resistance.
The UPR regulates T-cell development and differentiation
In addition to CD8+ T cells, CD4+ T cells are also a part of the cancer immune cycle. CD4+ T cells play auxiliary roles for other cells of the immune system, especially antigen-presenting cells (APCs), such as macrophages, dendritic cells and B cells, and participate in the activation and maturation of these cells. CD4+ T cells have different subsets, such as Th1, Th2, Th17 and regulatory T cells. Growing evidence suggests that the UPR plays an important role in CD4+ T-cell differentiation and development and antitumour immunity. For example, in IRE1α knockout CD4+ T cells, CD4+ T cells cannot be activated and differentiated, leading to a decrease in IL4 production [184]. Additionally, a study showed that CD4+ T cells that are deficient in the ER stress chaperone GRP94 cannot be activated due to defective Ca2+ mobilization [185], leading to inhibition of tumour progression (Fig. 3). These studies suggest that UPR signalling is important for CD4+ T-cell differentiation and antitumour immunity. In another study, ATF4, a transcription factor of the PERK-eIF2α pathway, was shown to be essential for the CD4+ T-cell-mediated immune response [186]. Additionally, Yang et al. found that loss of ATF4 diminished Th1 effector function in high- and low-oxidizing environments. In this study, a moderate reduction in IL-17 production caused by loss of ATF4 was also observed, suggesting that ATF4 is involved in regulating Th17 cell development. In addition to ATF4, CHOP, another transcription factor of the PERK-eIF2α pathway, has also been implicated in the regulation of IL17 expression and Th17 cell differentiation. However, XBP1, a transcription factor of IRE1α-XBP1 signalling, inhibits the CD4+ T-cell-mediated immune response and promotes tumour progression [164]. Thus, moderate activation of PERK-eIF2α signalling combined with inhibition of IRE1α-XBP1 signalling in CD4+ T cells may promote the CD4+ T-cell-mediated immune response, allow cells to overcome immunotherapy resistance, and increase immunotherapy efficacy.
In addition to participating in CD4+ T-cell differentiation and development, the UPR also promotes CD8+ T-cell differentiation and the gain of effector function. Kamimura, D et al. found that XBP1 contributes to CD8+ T-cell differentiation during acute infection [187]. The ER stress chaperone GRP78 seems to be implicated in the regulation of CD8+ T-cell function. Studies have shown that GRP78 promotes the expression of granzyme B, an effector molecule, in CD8+ T cells [188], suggesting that the expression of GRP78 contributes to enhancing CD8+ T-cell cytotoxicity.
The UPR maintains the immunosuppressive function of MDSCs
MDSCs are marrow-derived heterogeneous cells that suppresses T-cell function [189,190,191]. PERK signalling has been demonstrated to be involved in the immunosuppressive function of MDSCs. For example, MDSCs expressing the ER stress sensor CHOP inhibited T-cell function and promoted tumour growth by upregulating IL-6 expression [192]. Accordingly, MDSCs lacking CHOP exhibited a decrease in immune regulatory functions and induction of antitumour responses, while IL-6 overexpression in CHOP-deficient MDSCs restored their immunosuppressive activity [192] (Fig. 3). These studies showed that IL-6 secreted by MDSCs was induced by CHOP and exerted immunosuppressive activity by suppressing T-cell function.
PERK signalling is also important for the maintenance of MDSCs. For example, activation of PERK signalling in MDSCs was found to be necessary for maintaining the MDSC population and immunosuppressive function [193] (Fig. 3). PERK deletion in MDSCs increased cytosolic mitochondrial DNA and activated the cGAS-Sting pathway, leading to the transformation of MDSCs into myeloid cells that activated CD8+ T-cell-mediated antitumour immunity [193] (Fig. 3). PERK signalling is also associated with the development of MDSCs [194] (Fig. 3). In mouse and human haematopoietic stem/progenitor cells (HSPCs), pharmacological and genetic inhibition of PERK signalling has been demonstrated to suppress the transformation of myeloid progeny cells into MDSCs [194]. These studies showed that PERK signalling plays an important role in the development, maintenance and regulation of MDSCs, suggesting that PERK signalling is a potential target of ICT.
Moreover, the IRE1α and ATF6α pathways were also involved in the maintenance of MDSCs. Tumour-bearing mouse models have shown that activation of the IRE1α and ATF6α pathways induces the development of polymorphonuclear MDSCs (PMNMDSCs) and promotes the immunosuppressive activity of PMNMDSCs (Fig. 3), thus leading to inhibition of the tumour-specific immune response and an increase in tumour progression [195]. These studies showed that the IRE1α and ATF6α pathways are important for MDSC-mediated immunosuppression, suggesting that targeting the IRE1α and ATF6α pathways is a promising therapeutic strategy for increasing the efficacy of ICT.
The UPR and the impairment of antigen presentation
Antigen presentation is mediated by major histocompatibility complex (MHC) class I and MHC class II molecules and is essential for T-cell-dependent immune responses. Almost all cells express endogenous antigenic peptide-loaded MHC class I molecules on the surface, which are presented to cytotoxic CD8+ T cells and lead to CD8+ T-cell recognition of tumour cells and induction of the T-cell immune response. However, low expression of MHC class I molecules in tumour cells leads to the loss of efficient MHC class I-mediated antigen presentation and thereby promotes tumour cell evasion of immune surveillance and inhibits CD8+ T-cell infiltration [196,197,198]. Professional antigen-presenting cells (APCs), including dendritic cells, macrophages and thymic epithelial cells, constitutively express antigenic peptide-loaded MHC class II molecules (peptide-MHC class II) on the surface, which are presented to antigen-specific CD4+ T cells and lead to the activation of CD4+ T cells and the enhancement of T-cell-dependent anticancer immunity [197]. In tumour tissue, due to the immunosuppressive microenvironment, APCs cannot be effectively activated and effectively present antigens. Recent studies have shown that the UPR inhibits antigen presentation, leading to the suppression of T-cell-dependent anticancer immunity and tumour resistance to immunotherapy [199, 200]. The UPR inhibits antigen presentation by the following mechanisms: (1) the inhibition of antigen-presenting cell function and (2) the low expression of antigen processing and presentation genes.
The UPR inhibits the function of APCs
Conventional dendritic cells (cDCs) represent a diverse group of specialized antigen-presenting cells (APCs) that promote antitumour adaptive immunity by presenting antigens to T cells. Dendritic cells (DCs) are needed to initiate and sustain T-cell-dependent anticancer immunity [201]. Tumours often evade immune control by reducing normal DC function. A previous study showed that the UPR was involved in the inhibition of DC function. For example, constitutive activation of XBP1 in tumour-associated DCs (tDCs), driven by lipid peroxidation byproducts, has been found to induce a triglyceride biosynthetic program, which leads to abnormal lipid accumulation and subsequent inhibition of the capacity of tDCs to support antitumour T cells [94] (Fig. 3). DC-specific XBP1 deletion or selective nanoparticle-mediated XBP1 silencing in tDCs led to the restoration of immunostimulatory activity, which evoked protective type 1 antitumour responses and extended survival [94]. This study showed that targeting the XBP1-mediated ER stress response could significantly inhibit tumour growth and enhance anticancer immunity, thus providing a unique approach to cancer immunotherapy.
In addition to dendritic cells, the UPR also inhibits the antigen processing and presentation of macrophages by promoting a shift in M1–M2 polarization [202]. In melanoma, IREIα-XBP1 signalling is observed to promote M2 macrophage polarization, including the upregulation of interleukin 6 (IL-6), IL-23, and arginase 1 expression, leading to tumour immunosuppression [182]. Similarly, another group found that lipid-induced IRE1-XBP1 signalling increases the expression of Arginase1 and MRC1-associated M2 macrophages and facilitates M2 polarization [203]. In addition, in a mouse lung cancer model, XBP1 was found to upregulate the expression of IL10, TGFβ, and Arginase1 associated with M2 macrophages, downregulate the expression of IL-12, TNF-α, and iNOS associated with M1 macrophages, facilitate M2 polarization, decrease M2 macrophages in the tumour region, and enhance T-cell infiltration, thereby improving the efficacy of anti-PD1 therapy [204]. In addition to IRE1-XBP1 signalling, PERK-eif2α signalling has also been found to be involved in M2 macrophage polarization. CHOP, a transcription factor of the PERK-eif2α pathway, increased M2 macrophage production in a mouse model of bleomycin-induced pulmonary fibrosis, suggesting that CHOP inhibits antigen processing and presentation [205]. In addition, induction of PERK signalling in macrophages promotes immunosuppressive M2 activation and proliferation and inhibits the efficacy of anti-PD1 therapy in melanoma [206]. Although these studies reveal that the PERK-eif2α and IRE1-XBP1 branches of the UPR are promising therapeutic targets for sustaining host antitumour immunity, the overall picture is far from complete. In the future, we need to confirm which UPR branch in tumours is crucial for antigen presentation to guide targeted therapy combinations with immunotherapy.
The UPR inhibits the expression of antigen processing and presentation genes
The presentation of intracellular antigens by MHC-I is a complex process. First, antigens are primarily processed by the proteasome, which generates sources of peptides for MHC-I loading [207]. Then, these peptides are imported into the ER by the transporter associated with antigen processing (TAP). Subsequently, the peptide-loading complex (PLC), which is composed of MHC-I heavy chain and b2 microglobulin, TAP, tapasin, calreticulin and ERp57, promotes the binding of newly synthesized MHC-I molecules to these peptides and thereby forms peptide–MHC-I complexes [207]. Finally, peptide–MHC-I complexes are released from the ER and transported via the Golgi to the plasma membrane for antigen presentation to CD8+ T cells [207].
Notably, IRE1α is involved in the regulation of several members of the MHC-I antigen presentation pathway. Guttman et al. found that DCs process internalized protein antigens into antigen-derived peptides, enter the ER and masquerade as unfolded proteins, thereby leading to the activation of IRE1α [199]. IRE1α activation promotes MHC-I heavy-chain mRNA degradation by regulating IRE1α-dependent decay (RIDD) and attenuating antigen cross-presentation [199]. In tumour-bearing mice, IRE1α inhibition enhanced MHC-I expression on tumour-infiltrating DCs and increased CD8+ T-cell infiltration and activation [199]. IRE1α inhibition synergized with anti-PD-L1 therapy to inhibit tumour progression and enhance the efficacy of immunotherapy [199]. At the cellular level, activation of IRE1α upregulates the expression of miR-346. The increase in miR-346 expression downregulates the expression of its target genes, including MHC I and TAP, leading to inhibition of MHC I-mediated antigen presentation [189]. In mice infected with Toxoplasma gondii, infection led to specific activation of the IRE1α pathway of the cDC1 subset, while IRE1α promoted MHC I antigen presentation of secreted parasite antigens [190]. This evidence indicates that IRE1α is needed to inhibit the gene expression of key members of the MHC-I antigen presentation pathway, suggesting that targeting IRE1 is a promising strategy for cancer immunotherapy.
The UPR and cancer cell stemness
Cancer stem cells (CSCs) refer to a small subpopulation of cancer cells that can self-renew, recapitulate the heterogeneity of original tumours, and differentiate into the whole bulk of a new tumour. Cancer stem cells have been shown to increase resistance to cancer immunotherapy in many studies [191, 208]. For example, cancer stem cells suppress CD8 + T-cell infiltration and promote the recruitment of type 2 macrophages (M2), leading to systemic immunosuppression and subsequent immunotherapy resistance [209]. In a skin cancer model for squamous cell carcinoma (SCC), transforming growth factor β (TGF-β)-responsive tumour-initiating stem cells promoted skin cancer resistance to adoptive cytotoxic T-cell transfer (ACT)-based immunotherapy [191]. Notably, the UPR is implicated in the regulation of cancer stem cells. In ovarian cancer, FOXK2, as a highly expressed stemness-specific transcription factor, increased stemness features and tumour initiation capacity by directly activating the IRE1α-XBP1 pathway, while genetic or pharmacological blockade of this pathway inhibited ovarian CSCs [210]. In breast cancer, ER-associated protein p97 inhibition increased the expression of multiple stemness and pluripotency regulators, including C/EBPδ, c-MYC, SOX2, and SKP2, by activating the PERK-eif2α pathway, leading to an increase in CSCs [211]. In GBM, PERK signalling was also found to promote the expression of the stemness regulator SOX2 and increase GBM cell stemness [212]. Moreover, the epithelial-to-mesenchymal transition (EMT), a phenotype of CSCs, is thought to promote both tumour progression and drug resistance. A study found that EMT gene expression correlates strongly with that of PERK-eIF2α genes, suggesting that the PERK-eif2α pathway is involved in the regulation of CSCs [213]. These studies revealed that the UPR plays an important role in CSCs and is a promising therapeutic target for cancer treatment. Many other components of the UPR that are involved in the regulation of CSCs have been described and reviewed elsewhere. These findings contribute to our understanding of the relationship between the UPR and CSCs.
Targeting the UPR to overcome chemotherapy resistance
Given that cancer cells exhibit elevated levels of ER stress, these malignant cells could be dependent on resistance to ER stress for cell survival. Thus, targeting the UPR may be a promising strategy for cancer treatment [214, 215].
Targeting the IRE1α-XBP1 pathway
As acute ER stress promotes tumour cell apoptosis, agents that elevate ER stress promote apoptosis in cancer cells (Table 1). The ER stress-generating agent bortezomib, the first proteasome inhibitor approved by the US Food and Drug Administration (FDA) for cancer treatment, functions as an XBP1s activator to treat multiple myeloma [216]. Studies have shown that XBP1 (or XBP1s) levels are positively correlated with bortezomib efficacy in patients with multiple myeloma [217], suggesting that XBP1s can act as a predictive marker of treatment outcomes.
Inhibition of IRE1α-XBP1 signalling can attenuate cancer cell adaptation to ER stress and augment ER stress, which promotes tumour cell apoptosis; this method could be used as an anticancer strategy (Fig. 4). Recently, some new drugs have been developed to inhibit the IRE1α-XBP1 pathway and augment ER stress. IRE1α inhibitors targeting the catalytic core of the RNase domain or ATP-binding pocket of the kinase domain, such as sunitinib, APY29, toyocamycin, STF-083010, 4μ8C, MKC-3946 and B-I09 [218,219,220,221, 223,224,225, 238, 239], were identified by high-throughput screens (Table 1). Among these inhibitors, sunitinib and APY29 inhibit IRE1α kinase activity by interacting with the ATP-binding pocket of the kinase domain [238], while toyocamycin, STF-083010, 4μ8C, MKC-3946 and B-I09 inhibit IRE1α RNase activity by targeting the catalytic core of the RNase domain. MKC-3946 and STF-083010 have been demonstrated to inhibit tumour formation in multiple myeloma xenograft models [219, 226], while toyocamycin produced by an Actinomycete strain synergistically potentiated the therapeutic effectiveness of bortezomib by inducing the apoptosis of multiple myeloma cells at nanomolar concentrations [222]. Notably, although sunitinib and APY29 are thought to inhibit IRE1α kinase activity, some studies have demonstrated that they also activate IRE1α RNase activity in vitro [221, 224].

Therapeutic strategies to target UPR in tumors. Compounds, such as 4μ8c, MKC-3946 B-I09, and STF-083010 can directly inhibit IRE1α ribonuclease domain and prevent XBP1 RNA splicing. PERK inhibitors, such as GSK2606414 and GSK2656157, can directly inhibit activation of PERK and overcome chemotherapy resistance. ATF6 inhibitors, such as AEBSF and Ceapins, can directly inhibit the transactivation of ATF6. Treatment with these IRE1α, PERK, or ATF6 inhibitors can effectively reduce the hypoxia tolerance, angiogenesis, drug resistance and tumor metastasis
Targeting the PERK-eIF2α pathway
Since the PERK-eIF2α pathway plays an important role in the drug resistance of cancer cells [81, 240], targeting the PERK-eIF2α pathway with chemotherapy is thought to be effective for cancer treatment and contributes to overcoming chemotherapy resistance (Fig. 4). At present, PERK-eIF2α signalling inhibitors have been designed and developed for cancer treatment. For example, GSK2606414, the first reported PERK inhibitor, inhibits the PERK-mediated UPR and increases the amount of misfolded proteins in the ER [227], resulting in cancer cell apoptosis (Table 1). GSK2606414 has demonstrated oral activity and decreased tumour growth in a xenograft model of pancreatic cancer [227]. GSK2656157, another PERK inhibitor, has been reported to have better inhibitory properties (Table 1) [228]. Studies have shown that GSK2656157 inhibits ER stress-induced PERK autophosphorylation and eIF2α phosphorylation in multiple cell lines [228]. The twice-daily dosing of GSK2656157 results in dose-dependent inhibition of multiple human tumour xenograft growth in mice. The antitumour activity of GSK2656157 may be attributed to alterations in amino acid metabolism and decreases in blood vessel density and vascular perfusion, which led to dose-dependent inhibition of the growth of multiple human tumour xenografts in mice [228].
Targeting the ATF6α pathway
Although ATF6α is proposed to promote the dormancy of tumour cells and resistance to chemotherapeutic drugs, ATF6α inhibitors have rarely been developed. 4-(2-Aminoethyl) benzenesulfonyl fluoride (AEBSF), a serine protease inhibitor, has been found to prevent ER stress-induced cleavage of ATF6α and ATF6β (Fig. 4), resulting in the inhibition of transcriptional induction of ATF6 target genes [241]. Mechanically, AEBSF seems to directly prevent the cleavage of ATF6α and ATF6β by inhibiting Site-1 protease [241]. Using ER stress response (ERSE)-luciferase assays, Gallagher et al. discovered and developed Ceapins, a class of pyrazole amides that block ATF6α signalling in response to ER stress [242] (Fig. 4). Further study found that Ceapins are highly specific inhibitors of ATF6α signalling and do not affect signalling through the other branches of the UPR. Genome-wide CRISPR interference and proteomics studies revealed that the ABCD3 peroxisomal transporter interacts with ER-resident ATF6α in a Ceapin-dependent manner, leading to the inhibition of ATF6α cleavage [243].
Targeting other components of the UPR
Other agents that inhibit UPR signalling include chaperone and ERAD pathway inhibitors. For example, chaperone HSP90 and GRP78 inhibitors include 17-AAG [229, 230], AUY-922 [231], IPI-504 [232], radicicol [229], docosahexaenoic acid (DHA) [233], PAT-SM6 [234] and arctigenin [235] (Table 1), which bind to the amino-terminal ATP-binding domain of their targets and lead to cell apoptosis. ERAD pathway inhibitors include ML240 [236] and Eeyarestatin [237] (Table 1), which are involved in the suppression of valosin-containing protein (VCP) ATPase.
Targeting UPR to enhance the efficacy of immunotherapy
The UPR is closely associated with the antitumour activity of immune cells [244]. Many studies have shown that targeting the UPR in combination with ICT is an effective therapeutic strategy for tumour treatment.
Targeting PERK-eif2α signalling
PERK-eif2α signalling plays an important role in T-cell exhaustion, the development and maintenance of MDSCs, and PD-L1 expression in tumour cells, suggesting that targeting PERK-eif2α signalling is a promising strategy for the treatment of cancers. GSK2606414, a PERK inhibitor, significantly increased CD8+ T-cell infiltration, inhibited tumour growth, and enhanced the efficacy of anti-PD1 immunotherapy [167]. Similar to GSK2606414, an ERO1α (a PERK axis target gene) inhibitor also promoted CD8+ T-cell immunity, controlled tumour progression, and improved the immunotherapy response in mouse tumour models [167]. In addition, GSK2606414 and AMG-44 (a potent and selective PERK inhibitor) have been reported to activate cGAS-sting signalling in MDSCs by inhibiting the NRF2-mediated antioxidant pathway, thereby impairing the immunosuppressive activity of tumour MDSCs. This impairment increases the infiltration of CD8+ T cells and delays tumour growth in B16 tumour-bearing mice [193]. Surface-localized GRP78 is also considered to be a potential target of immunotherapy [245, 246]. These studies suggest that targeting PERK-eif2α signalling enhances the efficacy of ICIs and could provide benefits for cancer patients.
Targeting IRE1α-XBP1 signalling
An increasing number of studies have demonstrated that IRE1α-XBP1 signalling is associated with immunosuppression, suggesting that IRE1α-XBP1 signalling may be a potential target for immunotherapy. STF083010, an IRE1α RNase inhibitor, could impair PD1 expression in CD8+ T cells by inhibiting the transcriptional activity of XBP1 and enhancing the antitumour immunity of CD8+ T cells in mouse models of melanoma [176]. In a CARM1-expressing ovarian cancer model, the IRE1α inhibitor B-I09 increased the effectiveness of anti-PD1 therapy [225]. However, the underlying mechanism is still unclear. A reasonable explanation is that XBP1s may have promoted PD1 expression in a CARM1-expressing ovarian cancer model and thus increased the efficacy of anti-PD1 therapy. In addition, IRE1α-XBP1 signalling increased PD-L1 expression in TAMs and thus promoted melanoma growth [182], suggesting that targeting IRE1α-XBP1 signalling to TAMs combined with anti-PD-L1 therapy may be an effective strategy for the treatment of melanoma. These studies demonstrate that targeting IRE1α-XBP1 signalling may be a potential strategy to enhance the effectiveness of immunotherapy.
Conclusions
When tumour cells encounter a hostile microenvironment, such as nutrient deprivation, oxygen limitation, oxidative stress or chemotherapeutic drugs, the protein-folding capacity of the ER is disturbed, leading to activation of the UPR. Over the past decade, although we have made great progress in understanding the role of the UPR in cancer, many questions remain. For example, UPR-induced cell death pathways are integrated into some tumour cells. How do cancer cells avoid cell death in response to chronic UPR activation? Preclinical studies have suggested that a therapeutic threshold exists for UPR inhibitors. How can the optimal dose of UPR inhibitors be determined in individual patients? Chemotherapy represents an additional extrinsic challenge that cancer cells have to face and to which they adapt in the case of resistance. The main problem is how to determine the window of opportunity to target the UPR, that is, when UPR-targeting agents should be given after chemotherapy.
ICT has revolutionized cancer management, but resistance to ICT is emerging as an urgent problem to be solved. Increasing evidence suggests that targeting UPR sensors or UPR-associated components could sensitize aggressive tumours to immunotherapy, but the availability of UPR targets is limited. Thus, larger prospective and preclinical studies and retrospective clinical trial analyses are needed to uncover potent UPR targets to overcome resistance to chemotherapeutic drugs and ICIs and prevent cancer progression and/or recurrence.
Availability of data and materials
Not applicable.
Abbreviations
- ER:
-
Endoplasmic reticulum
- UPR:
-
Unfolded protein response
- ICT:
-
Immune checkpoint therapy
- PERK:
-
Protein kinase RNA-like endoplasmic reticulum kinase
- IRE1:
-
Inositol-requiring enzyme 1
- ATF6:
-
Activating transcription factor 6
- ATF4:
-
Activating transcription factor 4
- BiP:
-
Binding immunoglobulin protein
- GADD34:
-
Growth arrest and DNA damage-inducible protein 34
- DC:
-
Dendritic cell
- XBP1:
-
X-box binding protein 1
- ERAD:
-
ER-associated protein degradation
- ROS:
-
Respective oxygen species
- TME:
-
Tumor microenvironment
- PD1:
-
Programmed cell death protein 1
- MDSC:
-
Myeloid-derived suppressor cells
References
Hetz C, Papa FR. The Unfolded Protein Response and Cell Fate Control. Mol Cell. 2018;69(2):169–81.
Hetz C, Chevet E, Oakes SA. Proteostasis control by the unfolded protein response. Nat Cell Biol. 2015;17(7):829–38.
Karagoz GE, Acosta-Alvear D, Walter P. The Unfolded Protein Response: Detecting and Responding to Fluctuations in the Protein-Folding Capacity of the Endoplasmic Reticulum. Cold Spring Harb Perspect Biol. 2019;11(9):a033886.
Villalobos-Labra R, Subiabre M, Toledo F, Pardo F, Sobrevia L. Endoplasmic reticulum stress and development of insulin resistance in adipose, skeletal, liver, and foetoplacental tissue in diabesity. Mol Aspects Med. 2019;66:49–61.
Ochoa CD, Wu RF, Terada LS. ROS signaling and ER stress in cardiovascular disease. Mol Aspects Med. 2018;63:18–29.
Del Vecchio CA, Feng Y, Sokol ES, Tillman EJ, Sanduja S, Reinhardt F, Gupta PB. De-differentiation confers multidrug resistance via noncanonical PERK-Nrf2 signaling. PLoS Biol. 2014;12(9):e1001945.
Milani M, Rzymski T, Mellor HR, Pike L, Bottini A, Generali D, Harris AL. The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with Bortezomib. Can Res. 2009;69(10):4415–23.
Chaurasia M, Gupta S, Das A, Dwarakanath BS, Simonsen A, Sharma K. Radiation induces EIF2AK3/PERK and ERN1/IRE1 mediated pro-survival autophagy. Autophagy. 2019;15(8):1391–406.
Allegrezza MJ, Conejo-Garcia JR. Targeted therapy and immunosuppression in the tumor microenvironment. Trends Cancer. 2017;3(1):19–27.
Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, Sirven P, Magagna I, Fuhrmann L, Bernard C, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell. 2018;33(3):463-479 e410.
Baldominos P, Barbera-Mourelle A, Barreiro O, Huang Y, Wight A, Cho JW, Zhao X, Estivill G, Adam I, Sanchez X, et al. Quiescent cancer cells resist T cell attack by forming an immunosuppressive niche. Cell. 2022;185(10):1694-1708 e1619.
Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, Bordbar D, Shan D, Samanamud J, Mahajan A, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med. 2019;25(3):462–9.
Patsoukis N, Wang Q, Strauss L, Boussiotis VA. Revisiting the PD-1 pathway. Sci Adv. 2020;6(38):eabd2712.
Cunha LD, Yang M, Carter R, Guy C, Harris L, Crawford JC, Quarato G, Boada-Romero E, Kalkavan H, Johnson MDL, et al. LC3-associated phagocytosis in myeloid cells promotes tumor immune tolerance. Cell. 2018;175(2):429-441 e416.
Klumper N, Ralser DJ, Bawden EG, Landsberg J, Zarbl R, Kristiansen G, Toma M, Ritter M, Holzel M, Ellinger J, et al. LAG3 (LAG-3, CD223) DNA methylation correlates with LAG3 expression by tumor and immune cells, immune cell infiltration, and overall survival in clear cell renal cell carcinoma. J Immunother Cancer. 2020;8(1):e000552.
Pai CS, Simons DM, Lu X, Evans M, Wei J, Wang YH, Chen M, Huang J, Park C, Chang A, et al. Tumor-conditional anti-CTLA4 uncouples antitumor efficacy from immunotherapy-related toxicity. J Clin Investig. 2019;129(1):349–63.
Freed-Pastor WA, Lambert LJ, Ely ZA, Pattada NB, Bhutkar A, Eng G, Mercer KL, Garcia AP, Lin L, Rideout WM 3rd, et al. The CD155/TIGIT axis promotes and maintains immune evasion in neoantigen-expressing pancreatic cancer. Cancer cell. 2021;39(10):1342–13601314.
Liu S, Zhang W, Liu K, Wang Y. CD160 expression on CD8(+) T cells is associated with active effector responses but limited activation potential in pancreatic cancer. Cancer Immunol Immunother. 2020;69(5):789–97.
Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, Kang B, Liu Z, Jin L, Xing R, et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med. 2018;24(7):978–85.
Bassez A, Vos H, Van Dyck L, Floris G, Arijs I, Desmedt C, Boeckx B, Vanden Bempt M, Nevelsteen I, Lambein K, et al. A single-cell map of intratumoral changes during anti-PD1 treatment of patients with breast cancer. Nat Med. 2021;27(5):820–32.
Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, Lynn RC, Philip M, Rao A, Restifo NP, et al. Defining “T cell exhaustion”. Nat Rev Immunol. 2019;19(11):665–74.
Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16(5):275–87.
Rotte A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J Exp Clin Cancer Res. 2019;38(1):255.
Fan P, Jordan VC. PERK, Beyond an Unfolded Protein Response Sensor in Estrogen-Induced Apoptosis in Endocrine-Resistant Breast Cancer. Mol Cancer Res. 2022;20(2):193–201.
Glembotski CC, Rosarda JD, Wiseman RL. Proteostasis and beyond: ATF6 in Ischemic disease. Trends Mol Med. 2019;25(6):538–50.
Raymundo DP, Doultsinos D, Guillory X, Carlesso A, Eriksson LA, Chevet E. Pharmacological targeting of IRE1 in cancer. Trends Cancer. 2020;6(12):1018–30.
Gonzalez-Quiroz M, Blondel A, Sagredo A, Hetz C, Chevet E, Pedeux R. When endoplasmic reticulum proteostasis meets the DNA damage response. Trends Cell Biol. 2020;30(11):881–91.
Hughes D, Mallucci GR. The unfolded protein response in neurodegenerative disorders - therapeutic modulation of the PERK pathway. FEBS J. 2019;286(2):342–55.
Jin JK, Blackwood EA, Azizi K, Thuerauf DJ, Fahem AG, Hofmann C, Kaufman RJ, Doroudgar S, Glembotski CC. ATF6 decreases myocardial ischemia/reperfusion damage and links ER stress and oxidative stress signaling pathways in the heart. Circ Res. 2017;120(5):862–75.
Liang W, Qi W, Geng Y, Wang L, Zhao J, Zhu K, Wu G, Zhang Z, Pan H, Qian L, et al. Necroptosis activates UPR sensors without disrupting their binding with GRP78. Proc Natl Acad Sci U S A. 2021;118(39):e2110476118.
Dahal B, Lehman CW, Akhrymuk I, Bracci NR, Panny L, Barrera MD, Bhalla N, Jacobs JL, Dinman JD, Kehn-Hall K. PERK is critical for alphavirus nonstructural protein translation. Viruses. 2021;13(5):892.
Chen S, Henderson A, Petriello MC, Romano KA, Gearing M, Miao J, Schell M, Sandoval-Espinola WJ, Tao J, Sha B, et al. Trimethylamine N-oxide binds and activates PERK to promote metabolic dysfunction. Cell Metabol. 2019;30(6):1141-1151 e1145.
Wang YC, Li X, Shen Y, Lyu J, Sheng H, Paschen W, Yang W. PERK (Protein Kinase RNA-Like ER Kinase) branch of the unfolded protein response confers neuroprotection in ischemic stroke by suppressing protein synthesis. Stroke. 2020;51(5):1570–7.
Wortel IMN, van der Meer LT, Kilberg MS, van Leeuwen FN. Surviving stress: modulation of ATF4-mediated stress responses in normal and malignant cells. Trends Endocrinol Metab. 2017;28(11):794–806.
Han S, Zhu L, Zhu Y, Meng Y, Li J, Song P, Yousafzai NA, Feng L, Chen M, Wang Y, et al. Targeting ATF4-dependent pro-survival autophagy to synergize glutaminolysis inhibition. Theranostics. 2021;11(17):8464–79.
Chen D, Fan Z, Rauh M, Buchfelder M, Eyupoglu IY, Savaskan N. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene. 2017;36(40):5593–608.
Kaspar S, Oertlin C, Szczepanowska K, Kukat A, Senft K, Lucas C, Brodesser S, Hatzoglou M, Larsson O, Topisirovic I, et al. Adaptation to mitochondrial stress requires CHOP-directed tuning of ISR. Sci Adv. 2021;7(22):eabf0971.
Kasetti RB, Patel PD, Maddineni P, Patil S, Kiehlbauch C, Millar JC, Searby CC, Raghunathan V, Sheffield VC, Zode GS. ATF4 leads to glaucoma by promoting protein synthesis and ER client protein load. Nat Commun. 2020;11(1):5594.
Lee YS, Lee DH, Choudry HA, Bartlett DL, Lee YJ. Ferroptosis-induced endoplasmic reticulum stress: cross-talk between ferroptosis and apoptosis. Mol Cancer Res. 2018;16(7):1073–6.
Kim BR, Park SH, Jeong YA, Na YJ, Kim JL, Jo MJ, Jeong S, Yun HK, Oh SC, Lee DH. RUNX3 enhances TRAIL-induced apoptosis by upregulating DR5 in colorectal cancer. Oncogene. 2019;38(20):3903–18.
Galehdar Z, Swan P, Fuerth B, Callaghan SM, Park DS, Cregan SP. Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J Neurosci. 2010;30(50):16938–48.
Li AL, Shen T, Wang T, Zhou MX, Wang B, Song JT, Zhang PL, Wang XL, Ren DM, Lou HX, et al. Novel diterpenoid-type activators of the Keap1/Nrf2/ARE signaling pathway and their regulation of redox homeostasis. Free Radical Biol Med. 2019;141:21–33.
Shin D, Kim EH, Lee J, Roh JL. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radical Biol Med. 2018;129:454–62.
Wei R, Zhao Y, Wang J, Yang X, Li S, Wang Y, Yang X, Fei J, Hao X, Zhao Y, et al. Tagitinin C induces ferroptosis through PERK-Nrf2-HO-1 signaling pathway in colorectal cancer cells. Int J Biol Sci. 2021;17(11):2703–17.
Diego Acosta-Alvear GEK. Florian Frohlich, Han Li, Tobias C Walther PW: The unfolded protein response and endoplasmic reticulum protein targeting machineries converge on the stress sensor IRE1. Elife. 2018;7:e43036.
Madden E, Logue SE, Healy SJ, Manie S, Samali A. The role of the unfolded protein response in cancer progression: From oncogenesis to chemoresistance. Biol Cell. 2019;111(1):1–17.
Dong H, Adams NM, Xu Y, Cao J, Allan DSJ, Carlyle JR, Chen X, Sun JC, Glimcher LH. The IRE1 endoplasmic reticulum stress sensor activates natural killer cell immunity in part by regulating c-Myc. Nat Immunol. 2019;20(7):865–78.
Zhao N, Cao J, Xu L, Tang Q, Dobrolecki LE, Lv X, Talukdar M, Lu Y, Wang X, Hu DZ, et al. Pharmacological targeting of MYC-regulated IRE1/XBP1 pathway suppresses MYC-driven breast cancer. J Clin Investig. 2018;128(4):1283–99.
Barua D, Gupta A, Gupta S. Targeting the IRE1-XBP1 axis to overcome endocrine resistance in breast cancer: Opportunities and challenges. Cancer Lett. 2020;486:29–37.
Waldherr SM, Strovas TJ, Vadset TA, Liachko NF, Kraemer BC. Constitutive XBP-1s-mediated activation of the endoplasmic reticulum unfolded protein response protects against pathological tau. Nat Commun. 2019;10(1):4443.
Luizet JB, Raymond J, Lacerda TLS, Barbieux E, Kambarev S, Bonici M, Lembo F, Willemart K, Borg JP, Celli J, et al. The Brucella effector BspL targets the ER-associated degradation (ERAD) pathway and delays bacterial egress from infected cells. Proc Natl Acad Sci U S A. 2021;118(32):e2105324118.
Upton JP, Wang L, Han D, Wang ES, Huskey NE, Lim L, Truitt M, McManus MT, Ruggero D, Goga A, et al. IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science. 2012;338(6108):818–22.
Lerner AG, Upton JP, Praveen PV, Ghosh R, Nakagawa Y, Igbaria A, Shen S, Nguyen V, Backes BJ, Heiman M, et al. IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012;16(2):250–64.
Wang B, Zhang J, Liu X, Chai Q, Lu X, Yao X, et al. Protein disulfide isomerases (PDIs) negatively regulate ebolavirus structural glycoprotein expression in the endoplasmic reticulum (ER) via the autophagy-lysosomal pathway. Autophagy. 2022;18(10):2350–67.
Quan JH, Gao FF, Lee M, Yuk JM, Cha GH, Chu JQ, Wang H, Lee YH. Involvement of endoplasmic reticulum stress response and IRE1-mediated ASK1/JNK/Mcl-1 pathways in silver nanoparticle-induced apoptosis of human retinal pigment epithelial cells. Toxicology. 2020;442:152540.
Corazzari M, Rapino F, Ciccosanti F, Giglio P, Antonioli M, Conti B, Fimia GM, Lovat PE, Piacentini M. Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differ. 2015;22(6):946–58.
Yi S, Chen K, Zhang L, Shi W, Zhang Y, Niu S, Jia M, Cong B, Li Y. Endoplasmic Reticulum Stress Is Involved in Stress-Induced Hypothalamic Neuronal Injury in Rats via the PERK-ATF4-CHOP and IRE1-ASK1-JNK Pathways. Front Cell Neurosci. 2019;13:190.
Papaioannou A, Higa A, Jegou G, Jouan F, Pineau R, Saas L, Avril T, Pluquet O, Chevet E. Alterations of EDEM1 functions enhance ATF6 pro-survival signaling. FEBS J. 2018;285(22):4146–64.
Zheng Z, Zhang X, Huang B, Liu J, Wei X, Shan Z, Wu H, Feng Z, Chen Y, Fan S, et al. Site-1 protease controls osteoclastogenesis by mediating LC3 transcription. Cell Death Differ. 2021;28(6):2001–18.
Zielke S, Kardo S, Zein L, Mari M, Covarrubias-Pinto A, Kinzler MN, Meyer N, Stolz A, Fulda S, Reggiori F, et al. ATF4 links ER stress with reticulophagy in glioblastoma cells. Autophagy. 2021;17(9):2432–48.
Larabi A, Barnich N, Nguyen HTT. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy. 2020;16(1):38–51.
Sampieri L, Di Giusto P, Alvarez C. CREB3 Transcription Factors: ER-Golgi Stress Transducers as Hubs for Cellular Homeostasis. Front Cell Dev Biol. 2019;7:123.
Hillary RF, FitzGerald U. A lifetime of stress: ATF6 in development and homeostasis. J Biomed Sci. 2018;25(1):48.
Bailey D, O’Hare P. Transmembrane bZIP transcription factors in ER stress signaling and the unfolded protein response. Antioxid Redox Signal. 2007;9(12):2305–21.
Carreras-Sureda A, Pihán P, Hetz C. Calcium signaling at the endoplasmic reticulum: fine-tuning stress responses. Cell Calcium. 2018;70:24–31.
Pihán P, Carreras-Sureda A, Hetz C. BCL-2 family: integrating stress responses at the ER to control cell demise. Cell Death Differ. 2017;24(9):1478–87.
Michalak M, Groenendyk J, Szabo E, Gold Leslie I, Opas M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem J. 2009;417(3):651–66.
Jessop CE, Tavender TJ, Watkins RH, Chambers JE, Bulleid NJ. Substrate specificity of the oxidoreductase ERp57 is determined primarily by its interaction with calnexin and calreticulin. J Biol Chem. 2009;284(4):2194–202.
Beider K, Rosenberg E, Dimenshtein-Voevoda V, Sirovsky Y, Vladimirsky J, Magen H, Ostrovsky O, Shimoni A, Bromberg Z, Weiss L, et al. Blocking of Transient Receptor Potential Vanilloid 1 (TRPV1) promotes terminal mitophagy in multiple myeloma, disturbing calcium homeostasis and targeting ubiquitin pathway and bortezomib-induced unfolded protein response. J Hematol Oncol. 2020;13(1):158.
Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer. 2014;14(9):581–97.
Cubillos-Ruiz JR, Bettigole SE, Glimcher LH. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell. 2017;168(4):692–706.
Koritzinsky M, Levitin F, van den Beucken T, Rumantir RA, Harding NJ, Chu KC, Boutros PC, Braakman I, Wouters BG. Two phases of disulfide bond formation have differing requirements for oxygen. J Cell Biol. 2013;203(4):615–27.
Yan Y, He M, Zhao L, Wu H, Zhao Y, Han L, Wei B, Ye D, Lv X, Wang Y, et al. A novel HIF-2α targeted inhibitor suppresses hypoxia-induced breast cancer stemness via SOD2-mtROS-PDI/GPR78-UPRER axis. Cell Death Differ. 2022;29(9):1769–89.
Binet F, Sapieha P. ER Stress and Angiogenesis. Cell Metab. 2015;22(4):560.
Xie P, Duan Y, Guo X, Hu L, Yu M. SalA attenuates hypoxia-induced endothelial endoplasmic reticulum stress and apoptosis via down-regulation of VLDL receptor expression. Cell Physiol Biochem. 2015;35(1):17–28.
Han J, Kaufman RJ. The role of ER stress in lipid metabolism and lipotoxicity. J Lipid Res. 2016;57(8):1329–38.
Volmer R, Ron D. Lipid-dependent regulation of the unfolded protein response. Curr Opin Cell Biol. 2015;33:67–73.
Takei N, Yoneda A, Kosaka M, Sakai-Sawada K, Tamura Y. ERO1alpha is a novel endogenous marker of hypoxia in human cancer cell lines. BMC Cancer. 2019;19(1):510.
Bartoszewska S, Collawn JF. Unfolded protein response (UPR) integrated signaling networks determine cell fate during hypoxia. Cell Mol Biol Lett. 2020;25(1):18.
Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. 2008;8(11):851–64.
He J, Liu L, Tang F, Zhou Y, Liu H, Lu C, Feng D, Zhu H, Mao Y, Li Z, Zhang L, Duan Y, Xiao Z, Zeng M, Weng L, Sun LQ. Paradoxical effects of DNA tumor virus oncogenes on epithelium-derived tumor cell fate during tumor progression and chemotherapy response. Signal Transduct Targeted Ther. 2021;6:408.
Bobrovnikova-Marjon E, Grigoriadou C, Pytel D, Zhang F, Ye J, Koumenis C, Cavener D, Diehl JA. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene. 2010;29(27):3881–95.
Hart LS, Cunningham JT, Datta T, Dey S, Tameire F, Lehman SL, Qiu B, Zhang H, Cerniglia G, Bi M, et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J Clin Investig. 2012;122(12):4621–34.
Dey S, Tameire F, Koumenis C. PERK-ing up autophagy during MYC-induced tumorigenesis. Autophagy. 2013;9(4):612–4.
Pytel D, Gao Y, Mackiewicz K, Katlinskaya YV, Staschke KA, Paredes MC, Yoshida A, Qie S, Zhang G, Chajewski OS, et al. PERK Is a Haploinsufficient Tumor Suppressor: Gene Dose Determines Tumor-Suppressive Versus Tumor Promoting Properties of PERK in Melanoma. PLoS Genet. 2016;12(12):e1006518.
Huber AL, Lebeau J, Guillaumot P, Petrilli V, Malek M, Chilloux J, Fauvet F, Payen L, Kfoury A, Renno T, et al. p58(IPK)-mediated attenuation of the proapoptotic PERK-CHOP pathway allows malignant progression upon low glucose. Mol Cell. 2013;49(6):1049–59.
Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446(7132):153–8.
Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, Yue P, Haverty PM, Bourgon R, Zheng J, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466(7308):869–73.
Niederreiter L, Fritz TM, Adolph TE, Krismer AM, Offner FA, Tschurtschenthaler M, Flak MB, Hosomi S, Tomczak MF, Kaneider NC, et al. ER stress transcription factor Xbp1 suppresses intestinal tumorigenesis and directs intestinal stem cells. J Exp Med. 2013;210(10):2041–56.
Ghosh R, Wang L, Wang ES, Perera BG, Igbaria A, Morita S, Prado K, Thamsen M, Caswell D, Macias H, et al. Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell. 2014;158(3):534–48.
Drogat B, Auguste P, Nguyen DT, Bouchecareilh M, Pineau R, Nalbantoglu J, Kaufman RJ, Chevet E, Bikfalvi A, Moenner M. IRE1 signaling is essential for ischemia-induced vascular endothelial growth factor-A expression and contributes to angiogenesis and tumor growth in vivo. Can Res. 2007;67(14):6700–7.
Auf G, Jabouille A, Guerit S, Pineau R, Delugin M, Bouchecareilh M, Magnin N, Favereaux A, Maitre M, Gaiser T, et al. Inositol-requiring enzyme 1alpha is a key regulator of angiogenesis and invasion in malignant glioma. Proc Natl Acad Sci USA. 2010;107(35):15553–8.
Dejeans N, Pluquet O, Lhomond S, Grise F, Bouchecareilh M, Juin A, Meynard-Cadars M, Bidaud-Meynard A, Gentil C, Moreau V, et al. Autocrine control of glioma cells adhesion and migration through IRE1alpha-mediated cleavage of SPARC mRNA. J Cell Sci. 2012;125(Pt 18):4278–87.
Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, Zhang S, Bettigole SE, Gupta D, Holcomb K, et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell. 2015;161(7):1527–38.
Leung-Hagesteijn C, Erdmann N, Cheung G, Keats JJ, Stewart AK, Reece DE, Chung KC, Tiedemann RE. Xbp1s-negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell. 2013;24(3):289–304.
Perini T, Materozzi M, Milan E. The immunity-malignancy equilibrium in multiple myeloma: lessons from oncogenic events in plasma cells. FEBS J. 2022;289(15):4383–97.
Arai M, Kondoh N, Imazeki N, Hada A, Hatsuse K, Kimura F, Matsubara O, Mori K, Wakatsuki T, Yamamoto M. Transformation-associated gene regulation by ATF6alpha during hepatocarcinogenesis. FEBS Lett. 2006;580(1):184–90.
Zhang HH, Li C, Ren JW, Liu L, Du XH, Gao J, Liu T, Li SZ. OTUB1 facilitates bladder cancer progression by stabilizing ATF6 in response to endoplasmic reticulum stress. Cancer Sci. 2021;112(6):2199–209.
Jeon YJ, Kim T, Park D, Nuovo GJ, Rhee S, Joshi P, Lee BK, Jeong J, Suh SS, Grotzke JE, et al. miRNA-mediated TUSC3 deficiency enhances UPR and ERAD to promote metastatic potential of NSCLC. Nat Commun. 2018;9(1):5110.
Zhao R, Lv Y, Feng T, Zhang R, Ge L, Pan J, Han B, Song G, Wang L. ATF6alpha promotes prostate cancer progression by enhancing PLA2G4A-mediated arachidonic acid metabolism and protecting tumor cells against ferroptosis. Prostate. 2022;82(5):617–29.
Garcia-Carbonero N, Li W, Cabeza-Morales M, Martinez-Useros J, Garcia-Foncillas J. New hope for pancreatic ductal adenocarcinoma treatment targeting endoplasmic reticulum stress response: a systematic review. Int J Mol Sci. 2018;19(9):2468.
Tzanova MM, Randelov E, Stein PC, Hiorth M, di Cagno MP. Towards a better mechanistic comprehension of drug permeation and absorption: Introducing the diffusion-partitioning interplay. Int J Pharm. 2021;608:121116.
Ju Y, Guo H, Edman M, Hamm-Alvarez SF. Application of advances in endocytosis and membrane trafficking to drug delivery. Adv Drug Deliv Rev. 2020;157:118–41.
Taskar KS, Harada I, Alluri RV. Physiologically-based Pharmacokinetic (PBPK) modelling of transporter mediated drug absorption, clearance and drug-drug interactions. Curr Drug Metab. 2021;22(7):523–31.
Nobili S, Lapucci A, Landini I, Coronnello M, Roviello G, Mini E. Role of ATP-binding cassette transporters in cancer initiation and progression. Semin Cancer Biol. 2020;60:72–95.
Liu X. ABC family transporters. Adv Exp Med Biol. 2019;1141:13–100.
Robey RW, Pluchino KM, Hall MD, Fojo AT, Bates SE, Gottesman MM. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat Rev Cancer. 2018;18(7):452–64.
Thomas C, Tampe R. Structural and Mechanistic Principles of ABC Transporters. Annu Rev Biochem. 2020;89:605–36.
Carron PM, Crowley A, O’Shea D, McCann M, Howe O, Hunt M, Devereux M. Targeting the folate receptor: improving efficacy in inorganic medicinal chemistry. Curr Med Chem. 2018;25(23):2675–708.
Benjamin RS. Adjuvant and neoadjuvant chemotherapy for osteosarcoma: a historical perspective. Adv Exp Med Biol. 2020;1257:1–10.
Williams MS, Amaral FM, Simeoni F, Somervaille TC. A stress-responsive enhancer induces dynamic drug resistance in acute myeloid leukemia. J Clin Investig. 2020;130(3):1217–32.
Gao Q, Li XX, Xu YM, Zhang JZ, Rong SD, Qin YQ, Fang J. IRE1alpha-targeting downregulates ABC transporters and overcomes drug resistance of colon cancer cells. Cancer Lett. 2020;476:67–74.
Dauer P, Sharma NS, Gupta VK, Nomura A, Dudeja V, Saluja A, Banerjee S. GRP78-mediated antioxidant response and ABC transporter activity confers chemoresistance to pancreatic cancer cells. Mol Oncol. 2018;12(9):1498–512.
Zhang L, Lu X, Xu Y, La X, Tian J, Li A, Li H, Wu C, Xi Y, Song G, et al. Tumor-associated macrophages confer colorectal cancer 5-fluorouracil resistance by promoting MRP1 membrane translocation via an intercellular CXCL17/CXCL22–CCR4–ATF6–GRP78 axis. Cell Death Dis. 2023;14(9):582.
Stefanou DT, Souliotis VL, Zakopoulou R, Liontos M, Bamias A. DNA damage repair: predictor of platinum efficacy in ovarian cancer? Biomedicines. 2021;10(1):82.
Schaaf L, Schwab M, Ulmer C, Heine S, Murdter TE, Schmid JO, Sauer G, Aulitzky WE, van der Kuip H. Hyperthermia synergizes with chemotherapy by inhibiting PARP1-dependent DNA replication arrest. Can Res. 2016;76(10):2868–75.
Martins-Teixeira MB, Carvalho I. Antitumour anthracyclines: progress and perspectives. ChemMedChem. 2020;15(11):933–48.
Mahata T, Chakraborty J, Kanungo A, Patra D, Basu G, Dutta S. Intercalator-induced DNA superstructure formation: doxorubicin and a synthetic quinoxaline derivative. Biochemistry. 2018;57(38):5557–63.
Songbo M, Lang H, Xinyong C, Bin X, Ping Z, Liang S. Oxidative stress injury in doxorubicin-induced cardiotoxicity. Toxicol Lett. 2019;307:41–8.
Sugimoto Y, Tsukahara S, Oh-hara T, Isoe T, Tsuruo T. Decreased expression of DNA topoisomerase I in camptothecin-resistant tumor cell lines as determined by a monoclonal antibody. Can Res. 1990;50(21):6925–30.
Sugimoto Y, Tsukahara S, Oh-hara T, Liu LF, Tsuruo T. Elevated expression of DNA topoisomerase II in camptothecin-resistant human tumor cell lines. Can Res. 1990;50(24):7962–5.
Lord RV, Brabender J, Gandara D, Alberola V, Camps C, Domine M, Cardenal F, Sanchez JM, Gumerlock PH, Taron M, et al. Low ERCC1 expression correlates with prolonged survival after cisplatin plus gemcitabine chemotherapy in non-small cell lung cancer. Clin Cancer Res. 2002;8(7):2286–91.
Deng Q, Yang H, Lin Y, Qiu Y, Gu X, He P, Zhao M, Wang H, Xu Y, Lin Y, et al. Prognostic value of ERCC1 mRNA expression in non-small cell lung cancer, breast cancer, and gastric cancer in patients from Southern China. Int J Clin Exp Pathol. 2014;7(12):8312–21.
Usanova S, Piee-Staffa A, Sied U, Thomale J, Schneider A, Kaina B, Koberle B. Cisplatin sensitivity of testis tumour cells is due to deficiency in interstrand-crosslink repair and low ERCC1-XPF expression. Mol Cancer. 2010;9:248.
Swanton C, Nicke B, Schuett M, Eklund AC, Ng C, Li Q, Hardcastle T, Lee A, Roy R, East P, et al. Chromosomal instability determines taxane response. Proc Natl Acad Sci USA. 2009;106(21):8671–6.
Tao R, Chen H, Gao C, Xue P, Yang F, Han JD, Zhou B, Chen YG. Xbp1-mediated histone H4 deacetylation contributes to DNA double-strand break repair in yeast. Cell Res. 2011;21(11):1619–33.
Argemi J, Kress TR, Chang HCY, Ferrero R, Bertolo C, Moreno H, Gonzalez-Aparicio M, Uriarte I, Guembe L, Segura V, et al. X-box binding protein 1 regulates unfolded protein, acute-phase, and DNA damage responses during regeneration of mouse liver. Gastroenterology. 2017;152(5):1203–12161215.
Lyu X, Zhang M, Li G, Cai Y, Li G, Qiao Q. Interleukin-6 production mediated by the IRE1-XBP1 pathway confers radioresistance in human papillomavirus-negative oropharyngeal carcinoma. Cancer Sci. 2019;110(8):2471–84.
Rozpedek W, Pytel D, Nowak-Zdunczyk A, Lewko D, Wojtczak R, Diehl JA, Majsterek I. Breaking the DNA damage response via serine/threonine kinase inhibitors to improve cancer treatment. Curr Med Chem. 2019;26(8):1425–45.
Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13(10):714–26.
Fusee LTS, Marin M, Fahraeus R, Lopez I. Alternative mechanisms of p53 action during the unfolded protein response. Cancers. 2020;12(2):401.
Kim HS, Kim Y, Lim MJ, Park YG, Park SI, Sohn J. The p38-activated ER stress-ATF6alpha axis mediates cellular senescence. FASEB J. 2019;33(2):2422–34.
Lopez I, Tournillon AS, Prado Martins R, Karakostis K, Malbert-Colas L, Nylander K, Fahraeus R. p53-mediated suppression of BiP triggers BIK-induced apoptosis during prolonged endoplasmic reticulum stress. Cell Death Differ. 2017;24(10):1717–29.
Mlynarczyk C, Fahraeus R. Endoplasmic reticulum stress sensitizes cells to DNA damage-induced apoptosis through p53-dependent suppression of p21(CDKN1A). Nat Commun. 2014;5:5067.
Bu Y, Diehl JA. PERK integrates oncogenic signaling and cell survival during cancer development. J Cell Physiol. 2016;231(10):2088–96.
Leu JI, Murphy ME, George DL. Functional interplay among thiol-based redox signaling, metabolism, and ferroptosis unveiled by a genetic variant of TP53. Proc Natl Acad Sci USA. 2020;117(43):26804–11.
Szewczyk MM, Luciani GM, Vu V, Murison A, Dilworth D, Barghout SH, Lupien M, Arrowsmith CH, Minden MD, Barsyte-Lovejoy D. PRMT5 regulates ATF4 transcript splicing and oxidative stress response. Redox Biol. 2022;51:102282.
Mokarram P, Albokashy M, Zarghooni M, Moosavi MA, Sepehri Z, Chen QM, Hudecki A, Sargazi A, Alizadeh J, Moghadam AR, et al. New frontiers in the treatment of colorectal cancer: Autophagy and the unfolded protein response as promising targets. Autophagy. 2017;13(5):781–819.
Wu MZ, Fu T, Chen JX, Lin YY, Yang JE, Zhuang SM. LncRNA GOLGA2P10 is induced by PERK/ATF4/CHOP signaling and protects tumor cells from ER stress-induced apoptosis by regulating Bcl-2 family members. Cell Death Dis. 2020;11(4):276.
Khaled J, Kopsida M, Lennernas H, Heindryckx F. Drug resistance and endoplasmic reticulum stress in hepatocellular carcinoma. Cells. 2022;11(4):632.
Mhaidat NM, Alzoubi KH, Almomani N, Khabour OF. Expression of glucose regulated protein 78 (GRP78) determines colorectal cancer response to chemotherapy. Cancer Biomark. 2015;15(2):197–203.
Liu G, Yu J, Wu R, Shi L, Zhang X, Zhang W, Zhong X, Wang Y, Li H, Shen Y, et al. GRP78 determines glioblastoma sensitivity to UBA1 inhibition-induced UPR signaling and cell death. Cell Death Dis. 2021;12(8):733.
Pyrko P, Schonthal AH, Hofman FM, Chen TC, Lee AS. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Can Res. 2007;67(20):9809–16.
Mingming Yan JN. Deye Song, Muliang Ding, Jun Huang: Activation of unfolded protein response protects osteosarcoma cells from cisplatin-induced apoptosis through NF-κB pathway. Int J Clin Exp Pathol. 2015;8(9):10204–15.
Fu Y, Li J, Lee AS. GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Can Res. 2007;67(8):3734–40.
Yarapureddy S, Abril J, Foote J, Kumar S, Asad O, Sharath V, Faraj J, Daniel D, Dickman P, White-Collins A, et al. ATF6α activation enhances survival against chemotherapy and serves as a prognostic indicator in osteosarcoma. Neoplasia. 2019;21(6):516–32.
Ranganathan AC, Zhang L, Adam AP, Aguirre-Ghiso JA. Functional coupling of p38-induced up-regulation of BiP and activation of RNA-dependent protein kinase-like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Can Res. 2006;66(3):1702–11.
Yeung BH, Kwan BW, He QY, Lee AS, Liu J, Wong AS. Glucose-regulated protein 78 as a novel effector of BRCA1 for inhibiting stress-induced apoptosis. Oncogene. 2008;27(53):6782–9.
Moon EJ, Giaccia A. Dual roles of NRF2 in tumor prevention and progression: possible implications in cancer treatment. Free Radical Biol Med. 2015;79:292–9.
Giddings EL, Champagne DP, Wu MH, Laffin JM, Thornton TM, Valenca-Pereira F, Culp-Hill R, Fortner KA, Romero N, East J, et al. Mitochondrial ATP fuels ABC transporter-mediated drug efflux in cancer chemoresistance. Nat Commun. 2021;12(1):2804.
Gabryel B, Bontor K, Jarzabek K, Plato M, Pudelko A, Machnik G, Urbanek T. Sulodexide up-regulates glutathione S-transferase P1 by enhancing Nrf2 expression and translocation in human umbilical vein endothelial cells injured by oxygen glucose deprivation. Arch Med Sci. 2020;16(4):957–63.
Brown B, Mitra S, Roach FD, Vasudevan D, Ryoo HD. The transcription factor Xrp1 is required for PERK-mediated antioxidant gene induction in Drosophila. Elife. 2021;10:e74047.
Cheng SY, Chen NF, Wen ZH, Yao ZK, Tsui KH, Kuo HM, Chen WF. Glutathione S-transferase M3 is associated with glycolysis in intrinsic temozolomide-resistant glioblastoma multiforme cells. Int J Mol Sci. 2021;22(13):7080.
Yao RQ, Ren C, Xia ZF, Yao YM. Organelle-specific autophagy in inflammatory diseases: a potential therapeutic target underlying the quality control of multiple organelles. Autophagy. 2021;17(2):385–401.
Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306(5698):990–5.
Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J Biol Chem. 2006;281(40):30299–304.
Tang Z, Wei X, Li T, Wang W, Wu H, Dong H, Liu Y, Wei F, Shi L, Li X, et al. Sestrin2-mediated autophagy contributes to drug resistance via endoplasmic reticulum stress in human osteosarcoma. Front Cell Dev Biol. 2021;9:722960.
Ojha R, Leli NM, Onorati A, Piao S, Verginadis II, Tameire F, Rebecca VW, Chude CI, Murugan S, Fennelly C, et al. ER translocation of the MAPK pathway drives therapy resistance in BRAF-mutant melanoma. Cancer Discov. 2019;9(3):396–415.
McMellen A, Yamamoto TM, Qamar L, Sanders BE, Nguyen LL, Ortiz Chavez D, Bapat J, Berning A, Post MD, Johnson J, et al. ATF6-mediated signaling contributes to PARP inhibitor resistance in ovarian cancer. Mol Cancer Res. 2023;21(1):3–13.
Wang Y, Li C, Wang Z, Wang Z, Wu R, Wu Y, Song Y, Liu H. Comparison between immunotherapy efficacy in early non-small cell lung cancer and advanced non-small cell lung cancer: a systematic review. BMC Medicine. 2022;20(1):426.
Naimi A, Mohammed RN, Raji A, Chupradit S, Yumashev AV, Suksatan W, Shalaby MN, Thangavelu L, Kamrava S, Shomali N, et al. Tumor immunotherapies by immune checkpoint inhibitors (ICIs); the pros and cons. Cell Commun Signal. 2022;20(1):44.
Mączyńska J, Raes F, Da Pieve C, Turnock S, Boult JKR, Hoebart J, Niedbala M, Robinson SP, Harrington KJ, Kaspera W, et al. Triggering anti-GBM immune response with EGFR-mediated photoimmunotherapy. BMC Med. 2022;20(1):16.
Wang S, Sun J, Chen K, Ma P, Lei Q, Xing S, Cao Z, Sun S, Yu Z, Liu Y, et al. Perspectives of tumor-infiltrating lymphocyte treatment in solid tumors. BMC Med. 2021;19(1):140.
Song M, Sandoval TA, Chae CS, Chopra S, Tan C, Rutkowski MR, Raundhal M, Chaurio RA, Payne KK, Konrad C, et al. IRE1alpha-XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature. 2018;562(7727):423–8.
Li S, Zhu G, Yang Y, Jian Z, Guo S, Dai W, Shi Q, Ge R, Ma J, Liu L, et al. Oxidative stress drives CD8 + T-cell skin trafficking in patients with vitiligo through CXCL16 upregulation by activating the unfolded protein response in keratinocytes. J Allergy Clin Immunol. 2017;140(1):177-189.e179.
Lee AH, Sun L, Mochizuki AY, Reynoso JG, Orpilla J, Chow F, Kienzler JC, Everson RG, Nathanson DA, Bensinger SJ, et al. Neoadjuvant PD-1 blockade induces T cell and cDC1 activation but fails to overcome the immunosuppressive tumor associated macrophages in recurrent glioblastoma. Nat Commun. 2021;12(1):6938.
Hurst KE, Lawrence KA, Essman MT, Walton ZJ, Leddy LR, Thaxton JE. Endoplasmic Reticulum Stress Contributes to Mitochondrial Exhaustion of CD8(+) T Cells. Cancer Immunol Res. 2019;7(3):476–86.
Chen X, Cubillos-Ruiz JR. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat Rev Cancer. 2021;21(2):71–88.
Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, Lim E, Tam WL, Ni M, Chen Y, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature. 2014;508(7494):103–7.
Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol. 2018;18(3):153–67.
Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–99.
Wang Z, Wang X, Xu Y, Li J, Zhang X, Peng Z, Hu Y, Zhao X, Dong K, Zhang B, et al. Mutations of PI3K-AKT-mTOR pathway as predictors for immune cell infiltration and immunotherapy efficacy in dMMR/MSI-H gastric adenocarcinoma. BMC Med. 2022;20(1):133.
Xia ZA, Lu C, Pan C, Li J, Li J, Mao Y, Sun L, He J. The expression profiles of signature genes from CD103+LAG3+ tumour-infiltrating lymphocyte subsets predict breast cancer survival. BMC Med. 2023;21(1):268.
Moloney JN, Cotter TG. ROS signalling in the biology of cancer. Semin Cell Dev Biol. 2018;80:50–64.
Chen X, Song M, Zhang B, Zhang Y. Reactive oxygen species regulate T cell immune response in the tumor microenvironment. Oxid Med Cell Longev. 2016;2016:1580967.
Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, Wang Q, Yang M, Kalady MF, Qian J, et al. Cholesterol induces CD8+ T cell exhaustion in the tumor microenvironment. Cell Metab. 2019;30(1):143-156.e145.
Cao SS, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Signal. 2014;21(3):396–413.
Ma XH, Piao SF, Dey S, McAfee Q, Karakousis G, Villanueva J, Hart LS, Levi S, Hu J, Zhang G, et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J Clin Investig. 2014;124(3):1406–17.
Chen Y, Zhang X. Pivotal regulators of tissue homeostasis and cancer: macrophages. Exp Hematol Oncol. 2017;6:23.
Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, Gupta R, Tsai JM, Sinha R, Corey D, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545(7655):495–9.
Gilardini Montani MS, Falcinelli L, Santarelli R, Granato M, Romeo MA, Cecere N, Gonnella R, D’Orazi G, Faggioni A, Cirone M. KSHV infection skews macrophage polarisation towards M2-like/TAM and activates Ire1 alpha-XBP1 axis up-regulating pro-tumorigenic cytokine release and PD-L1 expression. Br J Cancer. 2020;123(2):298–306.
Batista A, Rodvold JJ, Xian S, Searles SC, Lew A, Iwawaki T, Almanza G, Waller TC, Lin J, Jepsen K, et al. IRE1alpha regulates macrophage polarization, PD-L1 expression, and tumor survival. PLoS Biol. 2020;18(6):e3000687.
Liu J, Fan L, Yu H, Zhang J, He Y, Feng D, Wang F, Li X, Liu Q, Li Y, Guo Z, Gao B, Wei W, Wang H, Sun G. Endoplasmic reticulum stress causes liver cancer cells to release exosomal miR-23a-3p and up-regulate programmed death ligand 1 expression in macrophages. Hepatology. 2019;70(1):241–58.
Kemp KL, Lin Z, Zhao F, Gao B, Song J, Zhang K, Fang D. The serine-threonine kinase inositol-requiring enzyme 1alpha (IRE1alpha) promotes IL-4 production in T helper cells. J Biol Chem. 2013;288(46):33272–82.
Thaxton JE, Wallace C, Riesenberg B, Zhang Y, Paulos CM, Beeson CC, Liu B, Li Z. Modulation of Endoplasmic Reticulum Stress Controls CD4(+) T-cell Activation and Antitumor Function. Cancer Immunol Res. 2017;5(8):666–75.
Yang X, Xia R, Yue C, Zhai W, Du W, Yang Q, Cao H, Chen X, Obando D, Zhu Y, et al. ATF4 regulates CD4(+) T cell immune responses through metabolic reprogramming. Cell Rep. 2018;23(6):1754–66.
Kamimura D, Bevan MJ. Endoplasmic reticulum stress regulator XBP-1 contributes to effector CD8+ T cell differentiation during acute infection. J Immunol. 2008;181(8):5433–41.
Chang J-S, Ocvirk S, Berger E, Kisling S, Binder U, Skerra A, Lee AS, Haller D. Endoplasmic reticulum stress response promotes cytotoxic phenotype of CD8αβ+ intraepithelial lymphocytes in a mouse model for Crohn’s disease-like Ileitis. J Immunol. 2012;189(3):1510–20.
Bartoszewski R, Brewer JW, Rab A, Crossman DK, Bartoszewska S, Kapoor N, Fuller C, Collawn JF, Bebok Z. The unfolded protein response (UPR)-activated transcription factor X-box-binding protein 1 (XBP1) induces MicroRNA-346 expression that targets the human antigen peptide transporter 1 (TAP1) mRNA and governs immune regulatory genes. J Biol Chem. 2011;286(48):41862–70.
Poncet AF, Bosteels V, Hoffmann E, Chehade S, Rennen S, Huot L, Peucelle V, Maréchal S, Khalife J, Blanchard N, et al. The UPR sensor IRE1α promotes dendritic cell responses to control Toxoplasma gondii infection. EMBO Rep. 2021;22(3):e49617.
Miao Y, Yang H, Levorse J, Yuan S, Polak L, Sribour M, Singh B, Rosenblum MD, Fuchs E. Adaptive immune resistance emerges from tumor-initiating stem cells. Cell. 2019;177(5):1172-1186.e1114.
Thevenot PT, Sierra RA, Raber PL, Al-Khami AA, Trillo-Tinoco J, Zarreii P, Ochoa AC, Cui Y, Del Valle L, Rodriguez PC. The stress-response sensor chop regulates the function and accumulation of myeloid-derived suppressor cells in tumors. Immunity. 2014;41(3):389–401.
Mohamed E, Sierra RA, Trillo-Tinoco J, Cao Y, Innamarato P, Payne KK, de Mingo Pulido A, Mandula J, Zhang S, Thevenot P, et al. The unfolded protein response mediator PERK governs myeloid cell-driven immunosuppression in tumors through inhibition of STING signaling. Immunity. 2020;52(4):668-682 e667.
Liu M, Wu C, Luo S, Hua Q, Chen HT, Weng Y, Xu J, Lin H, Wang L, Li J, et al. PERK reprograms hematopoietic progenitor cells to direct tumor-promoting myelopoiesis in the spleen. J ExpMed. 2022;219(4):e20211498.
Tcyganov EN, Hanabuchi S, Hashimoto A, Campbell D, Kar G, Slidel TW, Cayatte C, Landry A, Pilataxi F, Hayes S, et al. Distinct mechanisms govern populations of myeloid-derived suppressor cells in chronic viral infection and cancer. J Clin Invest. 2021;131(16):e145971.
Dersh D, Hollý J, Yewdell JW. A few good peptides: MHC class I-based cancer immunosurveillance and immunoevasion. Nat Rev Immunol. 2020;21(2):116–28.
Neefjes J, Jongsma MLM, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol. 2011;11(12):823–36.
Cheng Y, Wang C, Wang H, Zhang Z, Yang X, Dong Y, Ma L, Luo J. Combination of an autophagy inhibitor with immunoadjuvants and an anti-PD-L1 antibody in multifunctional nanoparticles for enhanced breast cancer immunotherapy. BMC Med. 2022;20(1):411.
Guttman O, Le Thomas A, Marsters S, Lawrence DA, Gutgesell L, Zuazo-Gaztelu I, Harnoss JM, Haag SM, Murthy A, Strasser G, et al. Antigen-derived peptides engage the ER stress sensor IRE1α to curb dendritic cell cross-presentation. J Cell Biol. 2022;221(6):e202111068.
Logue SE, Gorman AM, Samali A. New insights into IRE1α activation and function in anti-tumor immunity. J Cell Biol. 2022;221(6):e202205019.
Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20(1):7–24.
Yuan Y, Jiao P, Wang Z, Chen M, Du H, Xu L, Xu J, Dai Y, Wu FG, Zhang Y, et al. Endoplasmic reticulum stress promotes the release of exosomal PD-L1 from head and neck cancer cells and facilitates M2 macrophage polarization. Cell Commun Signal. 2022;20(1):12.
Di Conza G, Tsai C-H, Gallart-Ayala H, Yu Y-R, Franco F, Zaffalon L, Xie X, Li X, Xiao Z, Raines LN, et al. Tumor-induced reshuffling of lipid composition on the endoplasmic reticulum membrane sustains macrophage survival and pro-tumorigenic activity. Nat Immunol. 2021;22(11):1403–15.
Jiang M, Li X, Zhang J, Lu Y, Shi Y, Zhu C, Liu Y, Qin B, Luo Z, Du Y, et al. Dual inhibition of endoplasmic reticulum stress and oxidation stress manipulates the polarization of macrophages under hypoxia to sensitize immunotherapy. ACS Nano. 2021;15(9):14522–34.
Yao Y, Wang Y, Zhang Z, He L, Zhu J, Zhang M, He X, Cheng Z, Ao Q, Cao Y, et al. Chop deficiency protects mice against bleomycin-induced pulmonary fibrosis by attenuating M2 macrophage production. Mol Ther. 2016;24(5):915–25.
Raines LN, Zhao H, Wang Y, Chen H-Y, Gallart-Ayala H, Hsueh P-C, Cao W, Koh Y, Alamonte-Loya A, Liu P-S, et al. PERK is a critical metabolic hub for immunosuppressive function in macrophages. Nat Immunol. 2022;23(3):431–45.
Pishesha N, Harmand TJ, Ploegh HL. A guide to antigen processing and presentation. Nat Rev Immunol. 2022;22(12):751–64.
Dutta I, Dieters-Castator D, Papatzimas JW, Medina A, Schueler J, Derksen DJ, Lajoie G, Postovit L-M, Siegers GM. ADAM protease inhibition overcomes resistance of breast cancer stem-like cells to γδ T cell immunotherapy. Cancer Lett. 2021;496:156–68.
Saha D, Martuza RL, Rabkin SD. Macrophage polarization contributes to glioblastoma eradication by combination immunovirotherapy and immune checkpoint blockade. Cancer Cell. 2017;32(2):253-267.e255.
Zhang Y, Wang Y, Zhao G, Tanner EJ, Adli M, Matei D. FOXK2 promotes ovarian cancer stemness by regulating the unfolded protein response pathway. J Clin Invest. 2022;132(10):e151591.
Li C, Huang Y, Fan Q, Quan H, Dong Y, Nie M, Wang J, Xie F, Ji J, Zhou L, et al. p97/VCP is highly expressed in the stem-like cells of breast cancer and controls cancer stemness partly through the unfolded protein response. Cell Death Dis. 2021;12(4):286.
Peñaranda-Fajardo NM, Meijer C, Liang Y, Dijkstra BM, Aguirre-Gamboa R, den Dunnen WFA, Kruyt FAE. ER stress and UPR activation in glioblastoma: identification of a noncanonical PERK mechanism regulating GBM stem cells through SOX2 modulation. Cell Death Dis. 2019;10(10):690.
Feng YX, Sokol ES, Del Vecchio CA, Sanduja S, Claessen JHL, Proia TA, Jin DX, Reinhardt F, Ploegh HL, Wang Q, et al. Epithelial-to-mesenchymal transition activates PERK–eIF2α and sensitizes cells to endoplasmic reticulum stress. Cancer Discov. 2014;4(6):702–15.
Khateb A, Ronai ZA. Unfolded protein response in leukemia: from basic understanding to therapeutic opportunities. Trends Cancer. 2020;6(11):960–73.
Xu D, Liu Z, Liang MX, Fei YJ, Zhang W, Wu Y, Tang J-H. Endoplasmic reticulum stress targeted therapy for breast cancer. Cell Commun Signal. 2022;20(1):174.
Fels DR, Ye J, Segan AT, Kridel SJ, Spiotto M, Olson M, Koong AC, Koumenis C. Preferential cytotoxicity of bortezomib toward hypoxic tumor cells via overactivation of endoplasmic reticulum stress pathways. Can Res. 2008;68:9223–30.
Ling SC, Lau EK, Al-Shabeeb A, Nikolic A, Catalano A, Iland H, Horvath N, Ho PJ, Harrison S, Fleming S, et al. Response of myeloma to the proteasome inhibitor bortezomib is correlated with the unfolded protein response regulator XBP-1. Haematologica. 2012;97(1):64–72.
Mimura N, Fulciniti M, Gorgun G, Tai YT, Cirstea D, Santo L, Hu Y, Fabre C, Minami J, Ohguchi H, et al. Blockade of XBP1 splicing by inhibition of IRE1alpha is a promising therapeutic option in multiple myeloma. Blood. 2012;119(24):5772–81.
Cross BC, Bond PJ, Sadowski PG, Jha BK, Zak J, Goodman JM, Silverman RH, Neubert TA, Baxendale IR, Ron D, Harding HP. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc Natl Acad Sci USA. 2012;109:e869–78.
Papandreou I, Denko NC, Olson M, Van Melckebeke H, Lust S, Tam A, Solow-Cordero DE, Bouley DM, Offner F, Niwa M, et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood. 2011;117(4):1311–4.
Yuan J, Yao C, Tang J, Liu Y, Huang C, Yu S, Wei H, Han Y, Chen G. Enhanced GRP78 protein expression via the IRE1α/ASK1/p38 MAPK pathway during As2O3-induced endoplasmic reticulum stress in BEAS-2B cells. Toxicology. 2021;462:152962.
Ri M, Tashiro E, Oikawa D, Shinjo S, Tokuda M, Yokouchi Y, Narita T, Masaki A, Ito A, Ding J, et al. Identification of Toyocamycin, an agent cytotoxic for multiple myeloma cells, as a potent inhibitor of ER stress-induced XBP1 mRNA splicing. Blood Cancer J. 2012;2(7):e79.
Bagheri-Yarmand R, Sinha KM, Li L, Lu Y, Cote GJ, Sherman SI, Gagel RF. Combinations of tyrosine kinase inhibitor and ERAD inhibitor promote oxidative stress-induced apoptosis through ATF4 and KLF9 in medullary thyroid cancer. Mol Cancer Res. 2019;3(17):751–60.
Ali MM, Bagratuni T, Davenport EL, Nowak PR, Silva-Santisteban MC, Hardcastle A, McAndrews C, Rowlands MG, Morgan GJ, Aherne W, et al. Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. EMBO J. 2011;30(5):894–905.
Lin J, Liu H, Fukumoto T, Zundell J, Yan Q, Tang CA, Wu S, Zhou W, Guo D, Karakashev S, et al. Targeting the IRE1alpha/XBP1s pathway suppresses CARM1-expressing ovarian cancer. Nat Commun. 2021;12(1):5321.
Suh DH, Kim MK, Kim HS, Chung HH, Song YS. Unfolded protein response to autophagy as a promising druggable target for anticancer therapy. Ann N Y Acad Sci. 2012;1271:20–32.
Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, Li WH, Heerding DA, Minthorn E, Mencken T, Atkins C, Liu Q, Rabindran S, Kumar R, Hong X, Goetz A, Stanley T, Taylor JD, Sigethy SD, Tomberlin GH, Hassell AM, Kahler KM. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-p yrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J Med Chem. 2012;55(16):7193–207.
Atkins C, Liu Q, Minthorn E, Zhang SY, Figueroa DJ, Moss K, Stanley TB, Sanders B, Goetz A, Gaul N, et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Can Res. 2013;73(6):1993–2002.
Zhou Z, Li X, Qian Y, Liu C, Huang X, Fu M. Heat shock protein 90 inhibitors suppress pyroptosis in THP-1 cells. Biochem J. 2020;477(20):3923–34.
Li J, Xue W, Wang X, Huang W, Wang XX, Li H, Cui X, Li M, Mu H, Ren Y, et al. HSP90 as a novel therapeutic target for posterior capsule opacification. Exp Eye Res. 2019;189:107821.
Kubra KT, Uddin MA, Akhter MS, Barabutis N. Hsp90 inhibitors induce the unfolded protein response in bovine and mice lung cells. Cell Signal. 2020;67:109500.
Zheng ZG, Zhang X, Liu XX, Jin XX, Dai L, Cheng HM, Jing D, Thu PM, Zhang M, Li H, et al. Inhibition of HSP90beta improves lipid disorders by promoting mature SREBPs degradation via the ubiquitin-proteasome system. Theranostics. 2019;9(20):5769–83.
Chen Y, Mi Y, Zhang X, Ma Q, Song Y, Zhang L, Wang D, Xing J, Hou B, Li H, et al. Dihydroartemisinin-induced unfolded protein response feedback attenuates ferroptosis via PERK/ATF4/HSPA5 pathway in glioma cells. J Exp Clin Cancer Res. 2019;38(1):402.
Rasche L, Menoret E, Dubljevic V, Menu E, Vanderkerken K, Lapa C, Steinbrunn T, Chatterjee M, Knop S, Dull J, et al. A GRP78-Directed Monoclonal Antibody Recaptures Response in Refractory Multiple Myeloma with Extramedullary Involvement. Clin Cancer Res. 2016;22(17):4341–9.
Baba Y, Shigemi Z, Hara N, Moriguchi M, Ikeda M, Watanabe T, Fujimuro M. Arctigenin induces the apoptosis of primary effusion lymphoma cells under conditions of glucose deprivation. Int J Oncol. 2018;52(2):505–17.
Sen M, Al-Amin M, Kickova E, Sadeghi A, Puranen J, Urtti A, Caliceti P, Salmaso S, Arango-Gonzalez B, Ueffing M. Retinal neuroprotection by controlled release of a VCP inhibitor from self-assembled nanoparticles. J Control Release. 2021;339:307–20.
Nadeau ME, Rico C, Tsoi M, Vivancos M, Filimon S, Paquet M, Boerboom D. Pharmacological targeting of valosin containing protein (VCP) induces DNA damage and selectively kills canine lymphoma cells. BMC Cancer. 2015;15:479.
Volkmann K, Lucas JL, Vuga D, Wang X, Brumm D, Stiles C, Kriebel D, Der-Sarkissian A, Krishnan K, Schweitzer C, et al. Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. J Biol Chem. 2011;286(14):12743–55.
Zundell JA, Fukumoto T, Lin J, Fatkhudinov N, Nacarelli T, Kossenkov AV, Liu Q, Cassel J, Hu CA, Wu S, et al. Targeting the IRE1alpha/XBP1 endoplasmic reticulum stress response pathway in ARID1A-mutant ovarian cancers. Can Res. 2021;81(20):5325–35.
Jin Y, Saatcioglu F. Targeting the Unfolded Protein Response in Hormone-Regulated Cancers. Trends Cancer. 2020;6(2):160–71.
Okada T, Haze K, Nadanaka S, Yoshida H, Seidah NG, Hirano Y, Sato R, Negishi M, Mori K. A serine protease inhibitor prevents endoplasmic reticulum stress-induced cleavage but not transport of the membrane-bound transcription factor ATF6. J Biol Chem. 2003;278(33):31024–32.
Gallagher CM, Garri C, Cain EL, Ang KK, Wilson CG, Chen S, Hearn BR, Jaishankar P, Aranda-Diaz A, Arkin MR, Renslo AR, Walter P. Ceapins are a new class of unfolded protein response inhibitors, selectively targeting the ATF6α branch. Elife. 2016;20(5):e11878.
Torres SE, Gallagher CM, Plate L, Gupta M, Liem CR, Guo X, Tian R, Stroud RM, Kampmann M, Weissman JS, Walter P. Ceapins block the unfolded protein response sensor ATF6α by inducing a neomorphic inter-organelle tether. Elife. 2019;31(8):e46595.
Parish IA, Marshall HD, Staron MM, Lang PA, Brustle A, Chen JH, Cui W, Tsui YC, Perry C, Laidlaw BJ, et al. Chronic viral infection promotes sustained Th1-derived immunoregulatory IL-10 via BLIMP-1. J Clin Investig. 2014;124(8):3455–68.
Rauschert N, Brandlein S, Holzinger E, Hensel F, Muller-Hermelink HK, Vollmers HP. A new tumor-specific variant of GRP78 as target for antibody-based therapy. Lab Invest. 2008;88(4):375–86.
Hebbar N, Epperly R, Vaidya A, Thanekar U, Moore SE, Umeda M, Ma J, Patil SL, Langfitt D, Huang S, et al. CAR T cells redirected to cell surface GRP78 display robust anti-acute myeloid leukemia activity and do not target hematopoietic progenitor cells. Nat Commun. 2022;13(1):587.
Acknowledgements
Not applicable.
Funding
This work was funded by the following grants and associations: National Natural Science Foundations of China (81530084 and 81702721), Hunan province natural science funds (2023JJ30880).
Author information
Authors and Affiliations
Contributions
LQS supported this work. JH wrote the draft of manuscript. YZ managed the literature searches. All authors revised the manuscript before submission. All authors approved the submission.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
He, J., Zhou, Y. & Sun, L. Emerging mechanisms of the unfolded protein response in therapeutic resistance: from chemotherapy to Immunotherapy. Cell Commun Signal 22, 89 (2024). https://doi.org/10.1186/s12964-023-01438-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-023-01438-0