
Abstract
Atherosclerosis, which is a vascular pathology characterized by inflammation and plaque build-up within arterial vessel walls, acts as the important cause of most cardiovascular diseases. Except for a lipid-depository and chronic inflammatory, increasing evidences propose that epigenetic modifications are increasingly associated with atherosclerosis and are of interest from both therapeutic and biomarker perspectives. The chronic progressive nature of atherosclerosis has highlighted atherosclerosis heterogeneity and the fact that specific cell types in the complex milieu of the plaque are, by far, not the only initiators and drivers of atherosclerosis. Instead, the ubiquitous effects of cell type are tightly controlled and directed by the epigenetic signature, which, in turn, is affected by many proatherogenic stimuli, including low-density lipoprotein, proinflammatory, and physical forces of blood circulation. In this review, we summarize the role of DNA methylation and histone post-translational modifications in atherosclerosis. The future research directions and potential therapy for the management of atherosclerosis are also discussed.
Video Abstract
Similar content being viewed by others

Background
Atherosclerosis is the important cause of most cardiovascular diseases, including stroke and coronary artery disease, which are the leading reasons for mortality worldwide [1]. Atherosclerosis is caused by vascular endothelial dysfunction, lipid deposition, huge phagocytosis, foam cell formation, abnormal migration, and the proliferation of vascular smooth muscle cells (VSMCs) and stromal cells under the action of promoting inflammatory factors [2, 3]. In response to pathological stimuli, such as low-density lipoprotein (LDL) and triglyceride levels, smoking, and obesity, vascular endothelial cells (ECs)—the innermost layer of blood vessels—become activated. Monocytes, a pro-inflammatory palette that can bind to adhesion molecules expressed by activated endothelial cells, are recruited into the intima through chemokine. Upon entering the intima, monocytes mature into macrophages expressing scavenger receptors and then bind lipoprotein and become differentiated into foam cells. In the progression of atherosclerotic lesions, VSMCs initially stabilize the plaque by producing extracellular matrix proteins, such as collagen and proteoglycans (reviewed in [4]). Over decades, the accumulation of foam cells, coupled with debris from dead and dying cells, results in the progression of atherosclerotic plaque. However, during the late stages of atherosclerosis, activated macrophages produce enzymes of the matrix metalloproteinases family and lead to plaque rupture, resulting in myocardial and cerebral infarctions, which significantly contribute to morbidity and mortality from atherosclerotic diseases (reviewed in [4]). Recent evidence reveals that VSMCs can also differentiate into macrophage- and mesenchymal stem cell-like cells that promote lesion expansion and instability.
Epigenetics, which was proposed by Conrad Waddington in 1942, is a phenotype involving heritability, passed on through either mitosis or meiosis [5]. Since then, epigenetics has been redefined multiple times [6]. Recently, an epigenetic trait was defined as “a stably heritable phenotype resulting from changes in a chromosome without alterations in the DNA sequence” [7]. Epigenetics has both positive and negative effects. On the one hand, epigenetic modulating agents may coordinately promote tumor immunogenicity by inducing de novo expression of transcriptionally repressed tumor-associated antigens, increasing expression of neoantigens and major histocompatibility complex (MHC) processing/presentation machinery, and activating tumor immunogenic cell death [8]. On the other hand, if exposed to environmental factors such as chemicals, drugs, stress, or infections, epigenetics is associated with the accumulation of senescent cells resulting from immune senescence [9].
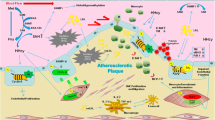
Recently, except for a lipid-depository and chronic inflammatory diseases, atherosclerosis is defined as an epigenetic disease [10,11,12,13]. Increasing evidences propose that epigenetic modifications are involved in the occurrence and development of atherosclerosis [14]. Microarray-based DNA methylation analysis reveales that patients with atherosclerosis have higher levels of DNA methylation than healthy controls [15]. Studies suggest that epigenetics not only regulates the expression of inflammatory cytokines but also controls epigenetic modifications through a reciprocal mechanism [16]. However, the chronic progressive nature of atherosclerosis has highlighted atherosclerosis heterogeneity and the fact that specific cell types in the complex milieu of the plaque are, by far, not the only initiators and drivers of atherosclerosis. Instead, the ubiquitous effects of cell type are tightly controlled and directed by the epigenetic signature, which, in turn, is affected by many proatherogenic stimuli, including LDL, proinflammatory cytokines, and physical forces of blood circulation. Besides, inflammatory molecular pathways (such as Toll-like receptors (TLR), NF-κB, and the JAK/STAT signaling pathway) are associated to epigenetic modifications (Fig. 1). Therefore, a better understanding of the role of epigenetics in the pathogenesis of atherosclerosis has enormous potential translational value.

Association with epigenetic modifications and the TLR, NF-κB, and JAK/STAT signaling pathway. TLRs can be regulated by DNA methylation, and histone modifications, which will eventually result in changed TLRs expression. DNA methylation that occurs on the promoter region of TLR genes, such as TLR1, 2, 3, 4, 5, 6, and 8, can reduce the expression of TLR on the membranes. Depending on the type of modification, the histone modifications that occurs on the nucleosome near the promoter of TLR genes, such as TLR2, 3, 4, and 5, can also positively or negatively regulate the expression of TLRs on the membranes; HATs interacts directly with NF-κB or induces its acetylation, and recruits NF-κB to the promoters of proinflammatory genes such as IL-6, IL-8 and cyclooxygenase-2 (COX-2) to regulate inflammatory signaling pathway activation; JAK kinases are activated in response to stimulation, which causes Stat3 activation, resulting in Stat3 nuclear transfer. Stat3 can also be activated by HDAC or DNMT. Activation of Stat3 leading to the DNA methylation of Stat3
In this Review, we discuss the latest major advances in our understanding of the functions of DNA methylation and histone post-translational modifications, as well as their establishment, maintenance, and erasure during atherosclerotic development. We also discuss new insights gained into the patterning of DNA methylation and histone post-translational modifications during atherosclerotic progression.
DNA methylation in atherosclerosis
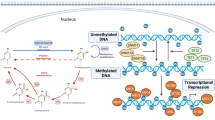
DNA methylation is one of the earliest discovered epigenetic modifications of genes. The activation or repression associated with DNA methylation goes hand in hand with the context and site of methylation [17]. DNA methylation is a dynamic and reversible modification process, which refers to the process of converting cytosine in the cytosine guanine (CG) into 5-methylcytosine (5mC) under the action of DNA methyltransferases (DNMTs) [12]. In this process, S-adenosylmethionine provides a methyl group, while DNA demethylase ten-eleven translocation protein (TET) converts 5mC into 5-hydroxymethylcytosine (5-hmC). The following are three phases of DNA methylation: establishment (de novo DNA methylation), maintenance, and demethylation. In mammals, de novo DNA methylation predominantly occurs at symmetric 5’-Cphosphate-G-3’ (CpG) sites under the action of DNMT3A/3B in an early stage of embryonal development [18]. Two major de novo DNA methylation enzymes, DNMT3A and DNMT3B [19, 20], contain a highly conserved DNMT domain (the MTase domain) in the carboxy terminus and two chromatin reading domains, ATRX-DNMT3-DNMT3L (ADD) and PWWP. Methylation maintenance enzyme DNMT1 in collaboration with another multidomain protein, E3 ubiquitin-protein ligase UHRF1, which specifically binds hemimethylated CpG dinucleotides at replication forks through its SET- and RING-associated (SRA) domain [21]. Additionally, TET methylcytosine dioxygenases (including TET1, TET2, and TET3) progressively oxidize 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC), result in activing DNA demethylation [22, 23].
Healthy individuals’ CpG islands in the promoter region of genes are normally hypomethylated, whereas the CpG islands in the non-promoter region are hypermethylated [24, 25]. Global DNA hypomethylation occurs in non-promoter regions, and it can cause the initiation of transcription at incorrect regions and high transcriptional activity in sites that are usually silent. Global DNA hypermethylation, on the other hand, typically suppresses expression, gene mutation and allelic loss. Under normal physiological conditions, DNMTs and TETs collaborate to maintain the balance of DNA methylation and demethylation. It has been demonstrated that aberrant DNA methylation is implicated in various diseases and plays an important role in atherosclerosis, including aberrant hypermethylation and hypomethylation [26]. Using whole-genome bisulfite sequencing, Zaina et al. [27] discovered that the atherosclerotic region of the aorta was hypermethylated across several genomic loci when compared to the corresponding healthy counterpart. However, there is genome-wide hypomethylation in atherosclerosis. Previous research has shown that hypomethylation of DNA predominates in atherosclerotic plaques [28]. In advanced human atherosclerotic lesions and ApoE knock-out mice lesions, genomic hypomethylation occurs with atherosclerosis [29]. These findings suggest that DNA methylation in atherosclerotic is a dynamic process, which is shown to be increased in early stages and decrease in late stages. The level of DNA methylation, on the other hand, is related not only to the stage of atherosclerosis, but also to the lesion grade of atherosclerosis. Using genome-wide DNA methylation sequencing, a positive correlation between DNA methylation and atherosclerotic lesion grade was discovered in atherosclerotic human aortas [30].
DNA hypermethylation and atherosclerosis
Increasing studies demonstrated that DNA methylation is regulated by inflammatory signaling pathways. For example, treatment of human umbilical vein endothelial cells (HUVECs) with pro-inflammatory stimuli, such as oxidized LDL (oxLDL), was shown to upregulate DNMT1 and lead to Kruppel-like factor 2 (KLF2) gene promoter methylation, resulting in KLF2 repression and endothelial inflammation accumulation [31]. Additionally, interleukin-6 (IL-6) can determine protein stabilization of DNMT1 and DNMT3B to induce changes in global and promoter-specific DNA methylation of genes [32]. Furthermore, inflammatory signaling pathways are regulated by DNA methylation. Inhibition of DNMT3b can increase the expression levels of forkhead box P3, transforming growth factor-β, and interleukin-10 and decrease the levels of interleukin-1β and interferon-gamma. In the process of regulating inflammatory factors, the generation mechanism of DNA methylation and the mechanism of DNA methylation in the three-dimensional structure of genome-related gene expression and regulation have not yet been elucidated. A recent survey of 542 human transcription factors found that 117 (22%) exhibited decreased binding to their motifs when methylated compared with unmethylated [33]. By preventing the binding of such transcription factors, DNA methylation can therefore impede transcription activation of CpG island promoters that contain their sequence recognition motifs.
In addition to inflammatory factors, DNA methylation is also affected by other stimuli. In vitro study revealed that ECs exposed to disturbed blood flow patterns have higher levels of DNMT1, resulting in DNA hypermethylation of their genome [34]. Obesity has been associated to an increased risk of serious cardiovascular or endocrinal diseases, such as atherosclerosis and stroke. In inguinal white adipose tissue of obese mice, Scara3 expression was reduced [35]. SCARA3 hypermethylation has been observed in patients with type 2 diabetes and atherosclerosis [35]. Moreover, findings from overweight/obese Korean subjects suggest an association between DNA hypermethylation at the TSPO-associated protein 1 antisense RNA 1 (TSPOAP1-AS1) promoter and overweight/obesity, as well as significant positive correlations between LDL cholesterol levels and TSPOAP1-AS1 DNA hypermethylation levels [36]. Whether hypermethylation in these promoter regions might be potential predictors of atherosclerosis is unclear; thus, this needs further investigation.
Atherosclerosis is a complex pathological process involving a variety of vascular wall cells and inflammatory cells. Endothelial dysfunction is the pathological basis of atherosclerosis, and it is accompanied by changes in vascular wall permeability. It leads to lipid accumulation, inflammatory cell infiltration, and smooth muscle cell migration and proliferation before developing into atherosclerosis. Gene expression changes caused by abnormal DNA methylation can lead to changes in cell phenotype and function. DNMT3b mediated hypermethylation of cellular repressor of E1A-stimulated genes (CREG) is observed in HUVECs treated with oxidized low-density lipoprotein (ox-LDL), a critical atherosclerogenic factor, result in CREG expression inhibition and endothelial dysfunction [37]. Homocysteine (Hcy), an independent risk factor for atherosclerosis, up-regulates phosphatase and tensin homologue on chromosome 10 (PTEN) methylation levels and enhances VSMCs proliferation, a primary pathological event in the development of atherosclerosis [38]. Hypermethylation of mitofusin-2 (MFN2), an important transmembrane GTPase in the mitochondrial outer membrane, can further facilitate VSMCs proliferation in Hcy-treated VSMCs. Increased c-Myc binding to DNMT1 promoter is a new and relevant molecular mechanism to contribute to MFN2 hypermethylation [39]. Hcy also plays a role in the inflammatory response and DNA methylation disorder in atherosclerosis, activating NF-κB-mediated vascular inflammatory response in human umbilical VSMCs via promoting SMAD7 promoter hypermethylation in a dose and time-dependent manner [40]. DNMT1 is a defining factor in macrophage inflammation both in vitro and in vivo. DNMT1 promotes macrophage M1 activation by suppressing the expression of Kruppel-like factor 4 (KLF4), and ApoE−/− mice deficient Dnmt1 had ameliorated atheroma formation and suppressed plaque inflammation [41]. In a study of swine aortic endothelium isolated from disturbed flow regions, researchers discovered that hemodynamics increased DNA methylation of CpG islands within the KLF4 promoter via DNMT3a enrichment, with regional consequences for atherosclerosis [42]. DNMT3b accelerates atherosclerosis and may be associated with forkhead box P3 hypermethylation status in human peripheral blood regulatory T cells. In ApoE−/− mice, Dnmt3b silencing attenuated atherosclerosis by decreasing lesion size and macrophage content while increasing collagen and smooth muscle cell content [43]. In both human atherosclerotic plaques and atherosclerosis patients, the SMAD7 promoter is hyper-methylated and it is positively related to homocysteine levels and carotid plaque scores [44]. In a sample of African-Americans, DNA methylation of AHRR, GFI1, and LRRC52 were associated with atherosclerosis after adjusting for cardiovascular diseases risk factors [45]. These findings suggest methylated SMAD7, AHRR, GFI1, and LRRC52 may be novel predicted biomarkers of atherosclerosis.
DNA hypomethylation and atherosclerosis
Atherosclerosis with DNA hypomethylation has been found in many studies in cell models, animals, and humans. Endothelial hypomethylation, which is induced by s-adenosylhomocysteine (SAH), the precursor of homocysteine [46, 47], led to augmented endothelial transmigration and decreased levels of aquaporin 1 and impaired water permeability, contributing to the development of atherosclerosis [48]. SAH levels by downregulating SAH hydrolase can also activate the expression of p66Shc, a key protein regulating oxidative stress, by reducing the expression of DNMT1 and the methylation of the p66Shc promoter. It then induces oxidative stress to damage endothelial function and contributes to the development of atherosclerosis in ApoE−/− mice, suggesting that SAH-associated endothelial injury may contribute to the development of atherosclerosis [49, 50]. Inhibition of SAH hydrolase in ApoE−/− mice epigenetically up-regulates Drp1 expression through repressing DNA methylation in endothelial cells, leading to vascular senescence and atherosclerosis [51]. Dynamin-related protein 1 (mdivi-1), a Drp1 specific inhibitor, alleviates atherosclerosis in ApoE−/− mice by suppressing mito-ROS/NLRP3-mediated M1 polarization [52]. Ribonuclease 6 expression was upregulated in the peripheral blood and plaque tissues of atherosclerosis patients. In vivo and vitro study showed that hypomethylation of Ribonuclease 6 promoter aggravates atherosclerosis in mice, enhances proliferation and migration of oxLDL treated murine aortic VSMCs, and upregulated ROS content and inflammatory factor secretion levels in the cells [53]. Additionally, hypomethylation of the IL-6 promoter region was found in coronary heart disease subjects as compared with controls, and DNA hypomethylation in the IL-6 gene is associated with increased IL-6 gene expression in atherosclerosis patients [54, 55]. The expression of signaling lymphocytic activation molecule 7 (SLAM7) was significantly higher in advanced plaque than early atherosclerotic tissue as well as in the unstable plaques than in the stable plaques in atherosclerosis patients. High expression of SLAM7 promoted the secretion of proinflammatory cytokines and inhibited proliferation of VSMCs, which is a key regulator and could be a target of potential therapeutic intervention in atherosclerosis [56]. A genome-wide analysis found that the CpG site (cg11874627) at the promoter region of lipoprotein-associated phospholipase A2 is hypomethylated in vulnerable atherosclerotic lesions when compared with the non-vulnerable lesions with an increased expression upon inflammation [57]. Overall, these results suggested that aberrant DNA demethylation modifications (ie. p66Shc, Drp1, Ribonuclease 6, IL-6, SLAM7, and lipoprotein-associated phospholipase A2) contribute to atherosclerosis progression and its potential role in atherosclerosis as a therapeutic target.
TET1s deficiency exacerbates oscillatory shear flow-induced atherosclerosis [58]. DNA demethylating enzyme TET2 represses the upregulation of pro-inflammatory cytokines, chemokine, and inflammasome activation, thus preventing atherosclerosis [59, 60]. In line with both reports, hematopoietic or myeloid cell-specific TET2 deletion also aggravates cardiac dysfunction in heart failure, with activation of the NLRP3 /IL-1β pathway [61]. Recently, it was demonstrated that TET2 ameliorated atherosclerosis progression in ApoE−/− mice via modulating Beclin1-dependent autophagic processes [62].
Histone modification in Atherosclerosis
Nucleosomes, which are made up of 147 base pairs of DNA and an octamer assembled by four core histones (H2A, H2B, H3, and H4), act as functional units of chromatin [12]. The N-terminal of histone extends beyond the nucleosome, and gene expression can be modified by acetylation, methylation, phosphorylation, ubiquitination, glycosylation, and ADP-ribosylation. This process is collectively defined as histone post‑translational modifications. Imbalances in histone modifications can lead to the development of cardiovascular diseases [63], and the loss of methylation and acetylation of histone H3 and H4 residues is a marker of atherosclerosis. Histone acetylation and methylation are the most studied modifications in inflammation and cardiovascular disease in these modifications [64].
Histone methylation
Histone methylation, which functions as maintenance and formation of heterochromatin structure, genomic imprinting, DNA repair, inactivation of X chromatin, and regulatory aspects of transcription, is a more stable epigenetic marker compared to histone acetylation. Histone methylation mainly occurs on lysine (k) or arginine (R) residues of histones H3 and H4. According to the methylation degree of each site, it can be divided into mono-methylation (me), di-methylation (me2), and tri-methylation (me3). Histone methylation is generally associated with transcriptional repression.
The histone methylation process is mainly catalyzed by two histone methyltransferases (HMT). Among them, histone lysine methyltransferase (HKMT) contains six histone lysine methyltransferase complexes (KMT1-6, listed in Table 1), whereas histone arginine methyltransferase (protein arginine methyltransferase, PRMT) contains PRMT1, 3, 5, 6, and CARM1. The histone demethylases are roughly divided into two families: LSD (Lysine-specific demethylase, contains LSD1 and LSD2) and JMJD (JmjC domain-containing family, contains KDM2, 3, 4, 5, 6). Generally, the methylation of different sites of histone H3 and H4 and the amount of methylation have great significance for the transcriptional regulation of genes. Among them, H3K9me3, H3K27me3, and H4K20me2/3 mediate transcriptional repression, whereas H3K4me1/2/3, H3K9me1, H3K27me1, H3K36me1/2/3, and H3k79me1/2/3 mediate transcriptional activation.
The development of atherosclerosis was significantly associated with inflammatory factors, which were secreted from the M1 pro-inflammatory phenotype. Transient receptor potential A1, a calcium-permeable non-selective cation channel, is overexpressed in atherosclerosis. It can change the H3K27 trimethylation level in macrophages and regulate the macrophages toward an inflammatory phenotype [65]. In monocytes, reduced methylation of H3K9 and H3K27 was shown in inflammatory cells [66]. The plasma concentrations of monocyte chemoattractant protein 1 (MCP1) in CD14+ monocytes from coronary heart disease patients were significantly upregulated, and the H3K9 tri-methylation of the MCP1 promoter was decreased [67]. MCP1 affects the chemotaxis of monocytes and is a key chemokine closely related to the development of atherosclerosis.
Upregulation of histone lysine methyltransferase SETDB2, a member of the KMT1 family, was observed in proinflammatory M1, whereas deficiency of SETDB2 in hematopoietic cells promoted vascular inflammation and accelerated atherosclerosis [68]. Histone H3K27 methyltransferase Ezh2 represses Socs3, the suppressor of cytokine signaling, to increase macrophage inflammatory responses. Myeloid-specific Ezh2 deficiency can reduce macrophage foam cell inflammatory response with reduced production of nitric oxide, IL-6, and IL-12, contributing to reduced atherosclerosis in mice [69]. In VSMC-specific disruptor of telomeric silencing 1-like (Dot1l) conditional knock-out mouse model, Dot1l and its uniquely induced H3K79me2 directly regulate the transcription of Nf-κB, result in increased expression of CCL5 and CXCL10 [70]. Dot1l deficiency in mice lowers atherosclerotic plaque stability and promotes inflammatory plaque macrophages activation by controlling lipid biosynthesis gene programs [71]. These findings suggest DOT1L as a potential therapeutic target for atherosclerosis.
On the other hand, lysine demethylase KDM4A/JMJD2A directly targets oxLDL-induced M1 polarization of macrophages independent of NF-κB and HIF activation, two signals critical for pro-inflammatory activation of macrophages [72]. LPS treatment promoted JMJD3 expression and enhanced JMJD3 nuclear accumulation in HUVECs. JMJD3 attenuated the methylation status in the promoter region of target genes, culminating in target gene expression [73].
Histone acetylation and deacetylation
The changes in histone acetyl groups have been widely considered an epigenetic marker of atherosclerosis. Acetylation results from the transfer of an acetyl group from acetyl-CoA to the ε-amino side chain of lysine by lysine acetyltransferases (KAT). This process can be reversed by KDAC. Adding acetyl groups to histone structure reduces its positive charge and affinity for negatively charged DNA, thus increasing the transcriptional accessibility of chromatin [74, 75]. Histone acetylation is the most widely studied form of histone modification. Histone acetyltransferases (HAT) and histone deacetylases (HDACs) were identified in the mid-1990s to late 1990s. HATs and HDACs were renamed KATs and lysine deacetylases (KDACs), respectively, to differentiate from non-histone acetylation. These two enzymes regulate histone acetylation level and gene transcription by reversible modification of histone. KATs are classified into three classes: GCN5 (GCN5-related N-acetyltransferases family; contains GCN5 and PCAF), MYST (MOZ, MORF, Ybf2/Sas3, Sas2, and Tip60), and P300 (CBP and P300). KDACs are classified into four classes: class 1 (histone deacetylase (HDAC) 1, 2, 3, 8), class 2 (HDAC 4, 5, 6, 7, 9, 10), class3 (SIRT 1–7), and class4 (HDAC11) [76]. Class1, 2, and 4 KDACs are classical Zn-dependent deacetylases, whereas the class 4 KDAC is NAD-dependent sirtuin deacetylases. In human macrophages, LPS induces the recruitment of p300 and enhances histone acetylation at the sites of active transcription within proximal promoter of NADPH oxidase 5 gene, suggesting that pharmacological targeting of epigenetic-based pathways that control NADPH oxidase 5 expression might be a noteworthy novel therapeutic strategy in atherosclerosis [74].
Class I KDACs
The increasing number of evidence has shown that HDACs are involved in the phenotype switch of VSMCs proliferation and migration. Recently study found that HDAC1, an important modulator, was critical for the migration and phenotypic switch of aortic VSMCs [77]. Regulatory factor X1 deficiency in CD14 + monocytes facilitated H3 and H4 acetylation and H3K9 tri-methylation in the MCP1 promoter region and contributed to MCP1 overexpression via reducing the recruitments of HDAC1 and suppressor of variegation 3–9 homolog 1 (SUV39H1) [78]. In endothelial cells, histone deacetylase 2 (HDAC2) protects against endothelial dysfunction and atherogenesis [79]. Endothelial-mesenchymal transition (EndMT) is a vital factor of plaque instability in atherosclerosis. HDACs are closely related to vascular endothelium stability and EndMT in atherosclerosis. HDAC3, an essential pro-survival molecule, is essential for differentiating endothelial progenitors. When HDAC3 is knocked down, ApoE−/− mice develop atherosclerosis, and their vessel ruptures [80]. HDAC3 also affects EndMT in atherosclerosis. In ApoE−/− mice and HUVECs, HDAC3 inhibitor suppresses EndMT via modulating inflammatory [81]. HDAC3 protects against atherosclerosis through inhibition of inflammation via the microRNA-19b/PPARγ/NF-κB axis in ox-LDL treated HUVECs and ApoE−/− mice [82].
Class II KDACs
HDAC4 is a key regulator and participates in proliferation and migration in various cell types [83,84,85]. HDAC4 can promote VSMC proliferation and migration and can be inhibited by interfering with HDAC4 [86]. Besides, HDAC4 was also involved in vascular calcification (VC). Recently, Abend et al. [87, 88] found that HDAC4 was upregulated early in VC and involved in vascular calcification and inflammatory response in VSMCs. VC is now widely known to be an active process occurring in VSMCs and is characterized by calcium deposition inside arteries. It is also associated with the morbidity and mortality of atherosclerosis [87, 89]. Whereas HDAC5 acts as a pro-inflammatory molecule in VSMCs, which is mediated by Nox4-dependent ROS production, and phosphatidylinositol 3-kinase (PI3K)/AKT pathways [90]. HDAC6, on the other hand, is upregulated by atherogenic stimuli via posttranslational modifications and is a critical regulator of CSEγ expression in vascular endothelium. In endothelial cells treated with oxLDL, the expression of CSEγ and H2S production are decreased, and it leads to endothelial dysfunction. The vascular endothelium is protected by cystathionine γ-lyase via inhibition of HDAC6 activity [91]. Targeting HDAC6 attenuates nicotine-induced macrophage pyroptosis via NF-κB/NLRP3 pathway in atherosclerosis [92].
Atherosclerosis-prone mice showed reduced EndMT and significantly reduced plaque area while endothelial-specific HDAC9 knockout while endothelial-specific HDAC9 controlled EndMT and the atherosclerotic plaque phenotype [93]. Malhotra et al. found that HDAC9 is associated with abdominal aortic calcification and affects VSMCs phenotype. In human aortic VSMCs, overexpression of HDAC9 promoted calcification and reduced contractility, whereas decreased expression of HDAC9 inhibited calcification and enhanced cell contractility [94]. HDAC9 promotes endothelial-mesenchymal transition and an unfavorable atherosclerotic plaque phenotype [93]. VSMCs calcification could be inhibited by HDAC inhibitors, such as apicidin, trichostatin, vorinostat, and tubacin [95,96,97].
Class III KDACs
SIRT6 (Sirtuin 6) is a nuclear deacetylase and plays a key role in regulating VSMCs senescence and atherosclerosis. In human and mouse plaque VSMCs, SIRT6 protein expression is reduced and regulated by CHIP. SIRT6 regulates telomere maintenance and VSMCs lifespan and then inhibits atherogenesis, which is dependent on its deacetylase activity. Endogenous SIRT6 deacetylase is an important inhibitor of VSMCs senescence and atherosclerosis [98].
Targeting DNA methylation and histone modification in the treatment of atherosclerosis
Extensive epigenetic modifications contribute to the development and progress of atherosclerotic plaque. Clinically, aspirin absorption leads to ABCB1 lower methylation in intracranial artery stenosis patients [99]. Folic acid (the deficiency of which increases homocysteine levels) is an atherosclerotic drug that induces endothelial dysfunction, accelerates atherosclerotic pathological processes, and increases DNA methylation of vascular peroxidase 1, MCP1, and vascular endothelial growth factor in high-fat diet-fed ApoE knockout mice [100, 101]. Therefore, epigenetic modification, including DNA methylation and histone post-translational modification, is considered to be a promising method for the treatment of many diseases, including atherosclerosis.
Treatment strategies targeting DNA methylation
DNMT inhibitors are among the first epigenetic drugs to be used in cancer therapeutics [102]. Drugs targeting DNMT include cytosine analogs, oligonucleotide drugs, DNA binder, and S-adenosylmethionine competitors. Cytosine analogs can be irreversibly incorporated into DNA during DNA synthesis. When DNMT tries to catalyze DNA methylation, these cytosine analogs DNMT can covalently bind to DNMT so that DNMT cannot be detached from chromatin, resulting in inhibition of activity. Presently, there are two FDA-approved cytosine analogs DNMT, namely 5-Azacytidine (5-Aza-C) and 5-Aza-CdR (trade name, decitabine). 5-Aza-C is used to treat myelodysplastic syndromes. It interferes not only with DNA methylation but also mRNA synthesis, thus having a relatively strong toxicity. Pharmacological upregulation of PTEN by 5-Aza-C reduces plaque area and preserves SMC contractile protein expression in vivo [103]. 5-Aza-CdR (trade name, decitabine), which is indicated for myelodysplastic syndromes and acute myeloid leukemia, only interferes with deoxyribonucleic acid without interfering with the nucleic acid. Inhibiting DNA methylation by 5-Aza-2'-deoxycytidine ameliorates atherosclerosis by suppressing macrophage inflammation [104]. Interestingly, increasing studies identified that 5-Aza-CdR effectively inhibits atherosclerosis development in several well-established animal models of atherosclerosis, including diet-induced atherosclerosis in ApoE−/− mice, LDLr−/− mice, and ApoE−/− mice undergoing carotid partial ligation [104,105,106].
However, pharmacological editing of global DNA methylation lacks specificity and may result in adverse reactions, such as autoimmune disorders [107]. In human atherosclerotic arteries, there was a negative correlation between DNMT3B and CREG expression levels, indicating blocking CREG methylation may represent a novel therapeutic approach to treat ox-LDL-induced atherosclerosis [37]. Recently, Ziltivekimab, a fully human monoclonal antibody directed against the IL-6 ligand, was shown to markedly reduced biomarkers of inflammation and thrombosis relevant to atherosclerosis in RESCUE trial [108, 109]. Furthermore, ziltivekimab-mediated IL-6 ligand inhibition is associated with a lower neutrophil–lymphocyte ratio, which independently predicts atherosclerotic events and is a potential biomarker for residual inflammatory risk, suggesting that it may disrupt multiple atherogenic inflammatory pathways [110]. On the other hand, Drp1 inhibition reduced macrophage burden, oxidative stress, and advanced calcified atherosclerotic plaque in aortic roots of diabetic ApoE−/− mice, as well as inflammatory cytokine production in human macrophages. Mdivi-1, a Drp1 specific inhibitor, was identified as a novel small molecule proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitor, which represents a cornerstone of cardiovascular prevention [111, 112].
Oligonucleotide drugs, such as MG98, target the active catalytic pocket of DNMT and prevent DNMT from binding to the promoter of a specific gene to inhibit its DNA methylation. DNA binders, such as SGI-1027, target cofactor-binding sites of DNMTs. Although the relationship between oligonucleotide drugs, DNA binder, and disease has not been studied, they may become an alternative approach, as they have low cytotoxicity without being incorporated into DNA. Additionally, several Chinese herbal medicines show a potential regulatory effect on DNA methylation in atherosclerosis [1]. Some herbs and herbal compounds, such as curcumin, geniposide, and resveratrol, have also shown promise in modulating epigenetic enzymes in vascular cells and atherosclerosis [26, 38, 113, 114]. Although the current literature has proven that some of the naturally occurring non-nucleoside DNMTi is capable of inhibiting atherosclerosis in mice, it is unknown how far their specific mechanism of action and the DNMTs inhibiting effects of these compounds contribute to their atheroprotective effects. Only a few drugs currently used to treat neuropsychiatric disorders have a direct effect on histone-modifying enzymes or on DNMTs. Importantly, global DNA methylation targeting cytidine analogues and non-intercalating methyltransferase inhibitors are FDA approved for the treatment of certain cancers [115]. DNA methylation is dynamic and reversible, with individual differences and specificity in time and space. Therefore, the design of specific drugs for DNA methylation markers of specific individuals and specific stages of the disease has become a challenge for future drug development.
Thus, targeting DNA methylation pathways may represent a promising avenue for therapy in atherosclerosis, similar to current clinical uses of DNA-hypomethylating agents in leukemia. However, further work is needed to decipher cell- and gene-specific DNA methylation changes (especially in humans) and to determine if treatment with DNMT inhibitors has disparate effects at distinct stages of the disease.
Treatment strategies targeting histone modification
Histone methylation is usually associated with transcriptional inhibition. Histone methylation inhibitors (HTMi), when compared to other epigenetic inhibitors, have not been extensively studied and remain an untapped resource. GSK126 is a potent histone methylation inhibitor, which is highly selective for the histone N-methyltransferase EZH2. Additionally, it can inhibit H3K27me3 and severely attenuate the expression of proinflammatory genes at both mRNA and protein levels [116, 117].
Regarding potential modulators of histone acetylation, garcinol and anacardic acid are natural compounds that show histone acetyltransferase inhibitors (HATi) activity. Garcinol, a polyisoprenylated benzophenone derived from the Garcinia indica fruit, was used to investigate the role of histone acetylation in the regulation of early growth response protein 1 (EGR1) gene [118, 119]. Recently, a new anacardic acid analogue, MG149, has been developed as an effective and selective inhibitor of the histone acetyltransferases (HATs) MYST family (Tip60, KAT5, and MOZ). MG149 can inhibit the NF-κB pathway, which is involved in the expression of various pro-inflammatory cytokines and plays a key role in inflammatory diseases, such as atherosclerosis [120, 121].
Histone acetylation is associated with open chromatin and gene transcription, which can be influenced by HDACs through counteracting HATs. Nowadays, HDAC inhibitors (HDACi), potential modifiers of histone acetylation, have been approved for the treatment of hematological malignancies, even though their application in atherosclerosis has not been investigated in clinical trials [122]. The pharmacological effects of HDACi are mediated through the reactivation of silenced genes by preventing histone deacetylation on target gene promoters. HDACi is mainly divided into four groups, including cyclic peptides, aliphatic acids, and benzamides [123]. Vorinostat (an HDAC inhibitor), also known as suberoylanilide hydroxamic acid (SAHA), is a compound derived from the hydroxamic acid class. It has been approved for T-cell cutaneous lymphoma therapy by the FDA, and it affects all classes of HDACs except class III. SAHA has been reported to decrease atherosclerotic lesion size in ApoE deficient mice in a KLF2-dependent manner [75, 124]. Trichostatin A (TSA), another specific HDACi, exacerbates atherosclerosis via increasing acetylation at the scavenger receptor CD36 promoter region, tumor necrosis factor (TNF)-alpha, and vascular cell adhesion molecule-1 (VCAM-1) and decreasing IL-6 and IL-1beta expressions in Ldlr−/− mice [125, 126]. Recently, TSA was found to target C/EBPα/PPARγ axis and induce acetylation of C/EBPα to alleviate atherosclerosis [127]. These varieties effect of TSA in atherosclerosis may attribute to its nonspecific, as TSA have an inhibitory effect toward HDAC I, IIA, and IIB. This phenomenon suggests that HDAC inhibition-dependent and -independent mechanisms must be explored when assessing the pharmacological effects of HDACi. HDACi are a promising class of anti-inflammatory drugs. Recently, an efficient drug delivery system carrying the class I/IIa selective HDACi, such as valproic acid (VPA), was developed to circumvent common disadvantages of free drug administration, e.g., short half-life and side effects. Additionally, it exhibits anti-inflammatory effects in primary human macrophages and is able to attenuate the lipopolysaccharide-induced inflammatory response [128]. Treatment with TMP195, a selective inhibitor of Class IIa HDAC, reduced critical inflammatory pathways and mitigated atherogenesis in advanced stage atherosclerosis, thereby offering a novel therapeutic strategy for reducing the consequence of vascular inflammation [129]. It is well known that the majority of existing or clinically available HDAC inhibitors are generic [130]. The development of selective HDACi could reduce the side effects of other target activities, such as the potential generic toxicity of HDAC6 [131]. Romidepsin (FK228) is a selective inhibitor against HDAC1/2 with anti-inflammatory properties and an effect on SMCs proliferation through regulating the deacetylation of several transcription factors (krüppel-like factor 5, CREB binding protein) [132, 133]. In ApoE−/− mice, HDAC3 specific inhibitor RGFP966 alleviated atherosclerotic lesions and inhibited EndMT of the atherosclerotic plaque [81]. Bossche et al. [134] found that inhibition of HDAC3 had the atherogenic protective effect of pan-HDAC inhibitors using specific HDAC inhibitor. Due to a partial reduction in M1 activation without an increase in foam cells, HDAC inhibition in macrophages, particularly HDAC3, displayed anti-atherogenic effects. In high-fat diet-fed ApoE−/− mice, Romidepsin-enhanced STAT3 acetylation epigenetically modulates VCAM-1 expression to suppress atherosclerosis [135]. As DNA methylation is accompanied by histone deacetylation, the combination of DNA methylation inhibitors and histone deacetylation inhibitors in the treatment of inflammatory diseases have become popular. Considering that existing chemotherapies, and additional drugs in development that modulate epigenetic silencing may increase risk of myocardial infarction, therapies that target specific cells may be an alternative to atherosclerosis treatment. From human genetics, ZEB2, a master regulator of EndMT, was found to be a coronary artery disease associated gene, and ZEB2 regulates SMC phenotypic transition through epigenetic inhibition of TGFβ and NOTCH signaling in atherosclerosis [136].
Nanomaterials for direct epigenetic therapy for atherosclerosis
Targeting DNA methylation or histone modification may represent a promising avenue for atherosclerosis therapy. However, as stated in the article, the main challenge of epigenetic therapy is the possibility of side-effects due to the fact that many targets of epigenetic-drugs are ubiquitously expressed, and that some epigenetic -drugs had a poor bioavailability, low stability and a short half-life [137]. Nanomaterials, which include organic nanoparticles (e.g. polymeric nanoparticle, liposomes, micelles, and high-density lipoprotein nanoparticle), and inorganic nanoparticles (e.g. gold nanoparticles, Fe3O4, mesoporous silica nanoparticles, and CuS) have been shown to be effective for therapy and diagnosis for atherosclerosis [138, 139]. Indeed, in recent years, a large number of nanoparticles have been developed with physical and chemical characteristics that allow them effectively deliver the epigenetic-drugs to the diseased cells and control their release [reviewed in [137]]. Nanoparticles comprising gelatinase with polyethylene glycol (PEG) and poly-ε-caprolactone) to specifically deliver DAC, for example, greatly inhibited tumor growth in mouse gastric cancer xenograft model [140]. Polymeric nanoparticles functionalized with histone deacetylase inhibitors (iHDACs) resulted in optimal iHDACs release in mesothelioma cancer, leading in a significant reduction in the weight of the tumor without toxicity [141]. Liposomes, on the other hand, have been used as nano-carriers of epigenetic-drugs: PEG-functionalized liposomes transport and release some anti-tumor drugs more efficiently, including the HDAC inhibitors SAHA, PXD101 and TSA [142].
Conclusion
The chronic progressive nature of atherosclerosis has highlighted atherosclerosis heterogeneity and the fact that specific cell types in the complex milieu of the plaque are, by far, not the only initiators and drivers of atherosclerosis. Instead, the ubiquitous effects of cell type are tightly controlled and directed by the epigenetic signature, which, in turn, is affected by many proatherogenic stimuli, including LDL, proinflammatory, and physical forces of blood circulation. Therefore, defining the role of epigenetics in vascular pathogenesis using human atherosclerotic tissue or animal atherosclerotic models is complicated by the dynamic nature of the disease and tissue heterogeneity. The ubiquitous effects of epigenetic changes on varieties of cell types limit clinical application in disease specificity or treatment. Therefore, systematic detection of epigenetic changes over different time periods allows us to develop comprehensive therapeutic strategies. Another problem faced is that when multiple post-translational modifications target the same amino acid residue (for example, lysine residues), there may be competitive antagonism between different modifications. How can atherosclerosis-related cells seek common ground while reserving difference? Further research is required to appropriately place the epigenetic modifiers in the treatment algorithm of atherosclerosis.
Availability of data and materials
Not applicable.
Abbreviations
- CpG:
-
5’-Cphosphate-G-3’
- CG:
-
Cytosine guanine
- DNMTs:
-
DNA methyltransferases
- ECs:
-
Endothelial cells
- EndMT:
-
Endothelial-mesenchymal transition
- HAT:
-
Histone acetyltransferases
- Hcy:
-
Homocysteine
- HDACs:
-
Histone deacetylases
- HKMT:
-
Histone lysine methyltransferase
- HMT:
-
Histone methyltransferases
- HUVECs:
-
Human umbilical vein endothelial cells
- IL-6:
-
Interleukin-6
- KLF4:
-
Kruppel-like factor 4
- LDL:
-
Low-density lipoprotein
- MCP1:
-
Monocyte chemoattractant protein 1
- oxLDL:
-
Oxidized LDL
- SAH:
-
S-adenosylhomocysteine
- SIRT6:
-
Sirtuin 6:
- TET:
-
Ten-eleven translocation protein
- VC:
-
Vascular calcification
- VSMCs:
-
Vascular smooth muscle cells
- 5-Aza-C:
-
5-Azacytidine
- 5-hmC:
-
5-Hydroxymethylcytosine
- 5mC:
-
5-Methylcytosine
References
Zhang Y, Mei J, Li J, Zhang Y, Zhou Q, Xu F. DNA methylation in atherosclerosis: a new perspective. Evid Based Complement Alternat Med. 2021;2021:6623657.
Aavik E, Babu M, Yla-Herttuala S. DNA methylation processes in atheosclerotic plaque. Atherosclerosis. 2019;281:168–79.
Lacey M, Baribault C, Ehrlich KC, Ehrlich M. Atherosclerosis-associated differentially methylated regions can reflect the disease phenotype and are often at enhancers. Atherosclerosis. 2019;280:183–91.
Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, Tokgözoğlu L, Lewis EF. Atherosclerosis Nat Rev Dis Primers. 2019;5(1):56.
Waddington CH. The epigenotype. Endeavour. 1942;1:18–20.
Nicoglou A, Merlin F. Epigenetics: a way to bridge the gap between biological fields. Stud Hist Philos Biol Biomed Sci. 2017;66:73–82.
Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23(7):781–3.
Dai E, Zhu Z, Wahed S, Qu Z, Storkus WJ, Guo ZS. Epigenetic modulation of antitumor immunity for improved cancer immunotherapy. Mol Cancer. 2021;20(1):171.
Saul D, Kosinsky RL. Epigenetics of aging and aging-associated diseases. Int J Mol Sci. 2021;22(1):401.
Wolf D, Ley K. Immunity and inflammation in atherosclerosis. Circ Res. 2019;124(2):315–27.
Xie M, Tang Q, Nie J, Zhang C, Zhou X, Yu S, Sun J, Cheng X, Dong N, Hu Y, Chen L. BMAL1-downregulation aggravates porphyromonas gingivalis-induced atherosclerosis by encouraging oxidative stress. Circ Res. 2020;126(6):e15–29.
Xu S, Pelisek J, Jin ZG. Atherosclerosis is an epigenetic disease. Trends Endocrinol Metab. 2018;29(11):739–42.
Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. 1999;340(2):115–26.
Khyzha N, Alizada A, Wilson MD, Fish JE. Epigenetics of atherosclerosis: emerging mechanisms and methods. Trends Mol Med. 2017;23(4):332–47.
Chen WD, Song T, Cao QH, Li R, Wang H, Chen XB, Chen ZT. Atherosclerosis prediction by microarray-based DNA methylation analysis. Exp Ther Med. 2020;20(3):2863–9.
Wierda RJ, Geutskens SB, Jukema JW, Quax PH, van den Elsen PJ. Epigenetics in atherosclerosis and inflammation. J Cell Mol Med. 2010;14(6a):1225–40.
de Mendoza A, Nguyen TV, Ford E, Poppe D, Buckberry S, Pflueger J, Grimmer MR, Stolzenburg S, Bogdanovic O, Oshlack A, et al. Large-scale manipulation of promoter DNA methylation reveals context-specific transcriptional responses and stability. Genome Biol. 2022;23(1):163.
Seisenberger S, Peat JR, Hore TA, Santos F, Dean W, Reik W. Reprogramming DNA methylation in the mammalian life cycle: building and breaking epigenetic barriers. Philos Trans R Soc Lond B Biol Sci. 2013;368(1609):20110330.
Okano M, Xie S, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet. 1998;19(3):219–20.
Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99(3):247–57.
Bostick M, Kim JK, Estève PO, Clark A, Pradhan S, Jacobsen SE. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 2007;317(5845):1760–4.
Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–5.
Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333(6047):1300–3.
Zhang Y, Zeng C. Role of DNA methylation in cardiovascular diseases. Clin Exp Hypertens. 2016;38(3):261–7.
Tabaei S, Tabaee SS. DNA methylation abnormalities in atherosclerosis. Artif Cells Nanomed Biotechnol. 2019;47(1):2031–41.
Chistiakov DA, Orekhov AN, Bobryshev YV. Treatment of cardiovascular pathology with epigenetically active agents: Focus on natural and synthetic inhibitors of DNA methylation and histone deacetylation. Int J Cardiol. 2017;227:66–82.
Zaina S, Heyn H, Carmona FJ, Varol N, Sayols S, Condom E, Ramírez-Ruz J, Gomez A, Gonçalves I, Moran S, Esteller M. DNA methylation map of human atherosclerosis. Circ Cardiovasc Genet. 2014;7(5):692–700.
Aavik E, Lumivuori H, Leppänen O, Wirth T, Häkkinen SK, Bräsen JH, Beschorner U, Zeller T, Braspenning M, van Criekinge W, et al. Global DNA methylation analysis of human atherosclerotic plaques reveals extensive genomic hypomethylation and reactivation at imprinted locus 14q32 involving induction of a miRNA cluster. Eur Heart J. 2015;36(16):993–1000.
Hiltunen MO, Turunen MP, Häkkinen TP, Rutanen J, Hedman M, Mäkinen K, Turunen AM, Aalto-Setälä K, Ylä-Herttuala S. DNA hypomethylation and methyltransferase expression in atherosclerotic lesions. Vasc Med. 2002;7(1):5–11.
Valencia-Morales Mdel P, Zaina S, Heyn H, Carmona FJ, Varol N, Sayols S, Condom E, Ramírez-Ruz J, Gomez A, Moran S, et al. The DNA methylation drift of the atherosclerotic aorta increases with lesion progression. BMC Med Genomics. 2015;8:7.
Kumar A, Kumar S, Vikram A, Hoffman TA, Naqvi A, Lewarchik CM, Kim YR, Irani K. Histone and DNA methylation-mediated epigenetic downregulation of endothelial Kruppel-like factor 2 by low-density lipoprotein cholesterol. Arterioscler Thromb Vasc Biol. 2013;33(8):1936–42.
Balakrishnan A, Guruprasad KP, Satyamoorthy K, Joshi MB. Interleukin-6 determines protein stabilization of DNA methyltransferases and alters DNA promoter methylation of genes associated with insulin signaling and angiogenesis. Lab Invest. 2018;98(9):1143–58.
Yin Y, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund-Sayeed S, Das PK, Kivioja T, Dave K, Zhong F, et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science. 2017;356(6337):eaaj2239.
Zhou J, Li YS, Wang KC, Chien S. Epigenetic mechanism in regulation of endothelial function by disturbed flow: induction of DNA Hypermethylation by DNMT1. Cell Mol Bioeng. 2014;7(2):218–24.
Peng H, Guo Q, Su T, Xiao Y, Li CJ, Huang Y, Luo XH. Identification of SCARA3 with potential roles in metabolic disorders. Aging (Albany NY). 2020;13(2):2149–67.
Yim NH, Cha MH, Kim MS. Hypermethylation of the TSPOAP1-AS1 promoter may be associated with obesity in overweight/obese Korean subjects. Int J Mol Sci. 2020;21(9):3307.
Liu Y, Tian X, Liu S, Liu D, Li Y, Liu M, Zhang X, Yan C, Han Y. DNA hypermethylation: a novel mechanism of CREG gene suppression and atherosclerogenic endothelial dysfunction. Redox Biol. 2020;32:101444.
Ma SC, Zhang HP, Jiao Y, Wang YH, Zhang H, Yang XL, Yang AN, Jiang YD. Homocysteine-induced proliferation of vascular smooth muscle cells occurs via PTEN hypermethylation and is mitigated by Resveratrol. Mol Med Rep. 2018;17(4):5312–9.
Xu L, Hao H, Hao Y, Wei G, Li G, Ma P, Xu L, Ding N, Ma S, Chen AF, Jiang Y. Aberrant MFN2 transcription facilitates homocysteine-induced VSMCs proliferation via the increased binding of c-Myc to DNMT1 in atherosclerosis. J Cell Mol Med. 2019;23(7):4611–26.
Wei LH, Chao NX, Gao S, Yu YT, Shi L, Ma XB, Liao N, Lan K, Luo Y, Xie ZY, Li YY. Homocysteine induces vascular inflammatory response via SMAD7 hypermethylation in human umbilical vein smooth muscle cells. Microvasc Res. 2018;120:8–12.
Tang RZ, Zhu JJ, Yang FF, Zhang YP, Xie SA, Liu YF, Yao WJ, Pang W, Han LL, Kong W, et al. DNA methyltransferase 1 and Krüppel-like factor 4 axis regulates macrophage inflammation and atherosclerosis. J Mol Cell Cardiol. 2019;128:11–24.
Jiang YZ, Jiménez JM, Ou K, McCormick ME, Zhang LD, Davies PF. Hemodynamic disturbed flow induces differential DNA methylation of endothelial Kruppel-Like Factor 4 promoter in vitro and in vivo. Circ Res. 2014;115(1):32–43.
Zhu L, Jia L, Liu N, Wu R, Guan G, Hui R, Xing Y, Zhang Y, Wang J. DNA Methyltransferase 3b Accelerates the Process of Atherosclerosis. Oxid Med Cell Longev. 2022;2022:5249367.
Wei L, Zhao S, Wang G, Zhang S, Luo W, Qin Z, Bi X, Tan Y, Meng M, Qin J, et al. SMAD7 methylation as a novel marker in atherosclerosis. Biochem Biophys Res Commun. 2018;496(2):700–5.
Ammous F, Zhao W, Lin L, Ratliff SM, Mosley TH, Bielak LF, Zhou X, Peyser PA, Kardia SLR, Smith JA. Epigenetics of single-site and multi-site atherosclerosis in African Americans from the Genetic Epidemiology Network of Arteriopathy (GENOA). Clin Epigenetics. 2022;14(1):10.
Esse R, Barroso M, Tavares de Almeida I, Castro R. The contribution of homocysteine metabolism disruption to endothelial dysfunction: state-of-the-art. Int J Mol Sci. 2019;20(4):867.
Huang X, Lv X, Song H, Yang Q, Sun Y, Zhang W, Yu X, Dong S, Yao W, Li Y, et al. The relationship between S-adenosylhomocysteine and coronary artery lesions: a case control study. Clin Chim Acta. 2017;471:314–20.
da Silva IV, Barroso M, Moura T, Castro R, Soveral G. Endothelial aquaporins and hypomethylation: potential implications for atherosclerosis and cardiovascular disease. Int J Mol Sci. 2018;19(1):130.
Kumar S. P66Shc and vascular endothelial function. Biosci Rep. 2019;39(4):BSR20182134.
Xiao Y, Xia J, Cheng J, Huang H, Zhou Y, Yang X, Su X, Ke Y, Ling W. Inhibition of S-Adenosylhomocysteine Hydrolase Induces Endothelial Dysfunction via Epigenetic Regulation of p66shc-Mediated Oxidative Stress Pathway. Circulation. 2019;139(19):2260–77.
You Y, Chen X, Chen Y, Pang J, Chen Q, Liu Q, Xue H, Zeng Y, Xiao J, Mi J, et al. Epigenetic modulation of Drp1-mediated mitochondrial fission by inhibition of S-adenosylhomocysteine hydrolase promotes vascular senescence and atherosclerosis. Redox Biol. 2023;65:102828.
Su ZD, Li CQ, Wang HW, Zheng MM, Chen QW. Inhibition of DRP1-dependent mitochondrial fission by Mdivi-1 alleviates atherosclerosis through the modulation of M1 polarization. J Transl Med. 2023;21(1):427.
Fang Y, Li J, Niu X, Ma N, Zhao J. Hypomethylation of Rnase6 promoter enhances proliferation and migration of murine aortic vascular smooth muscle cells and aggravates atherosclerosis in mice. Front Bioeng Biotechnol. 2021;9:695461.
Indumathi B, Katkam SK, Krishna LSR, Kutala VK. Dual effect of IL-6 -174 G/C polymorphism and promoter methylation in the risk of coronary artery disease among South Indians. Indian J Clin Biochem. 2019;34(2):180–7.
Mohammadpanah M, Heidari MM, Khatami M, Hadadzadeh M. Relationship of hypomethylation CpG islands in interleukin-6 gene promoter with IL-6 mRNA levels in patients with coronary atherosclerosis. J Cardiovasc Thorac Res. 2020;12(3):214–21.
Xia Z, Gu M, Jia X, Wang X, Wu C, Guo J, Zhang L, Du Y, Wang J. Integrated DNA methylation and gene expression analysis identifies SLAMF7 as a key regulator of atherosclerosis. Aging (Albany NY). 2018;10(6):1324–37.
Li J, Zhang X, Yang M, Yang H, Xu N, Fan X, Liu G, Jiang X, Fan J, Zhang L, et al. DNA methylome profiling reveals epigenetic regulation of lipoprotein-associated phospholipase A2 in human vulnerable atherosclerotic plaque. Clin Epigenetics. 2021;13(1):161.
Qu K, Wang C, Huang L, Qin X, Zhang K, Zhong Y, Ma Q, Yan W, Li T, Peng Q, et al. TET1s deficiency exacerbates oscillatory shear flow-induced atherosclerosis. Int J Biol Sci. 2022;18(5):2163–80.
Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377(2):111–21.
Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355(6327):842–7.
Sano S, Oshima K, Wang Y, MacLauchlan S, Katanasaka Y, Sano M, Zuriaga MA, Yoshiyama M, Goukassian D, Cooper MA, et al. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1β/NLRP3 inflammasome. J Am Coll Cardiol. 2018;71(8):875–86.
Peng J, Yang Q, Li AF, Li RQ, Wang Z, Liu LS, Ren Z, Zheng XL, Tang XQ, Li GH, et al. Tet methylcytosine dioxygenase 2 inhibits atherosclerosis via upregulation of autophagy in ApoE-/- mice. Oncotarget. 2016;7(47):76423–36.
Prasher D, Greenway SC, Singh RB. The impact of epigenetics on cardiovascular disease. Biochem Cell Biol. 2020;98(1):12–22.
Jiang W, Agrawal DK, Boosani CS. Cellspecific histone modifications in atherosclerosis (Review). Mol Med Rep. 2018;18(2):1215–24.
Wang Q, Chen K, Zhang F, Peng K, Wang Z, Yang D, Yang Y. TRPA1 regulates macrophages phenotype plasticity and atherosclerosis progression. Atherosclerosis. 2020;301:44–53.
Greissel A, Culmes M, Burgkart R, Zimmermann A, Eckstein HH, Zernecke A, Pelisek J. Histone acetylation and methylation significantly change with severity of atherosclerosis in human carotid plaques. Cardiovasc Pathol. 2016;25(2):79–86.
Xiao LI, Cao Y, Wang Y, Lai X, Gao KQ, Du P, Zhang BK, Jia SJ. Aberrant histone modifications of global histone and MCP-1 promoter in CD14(+) monocytes from patients with coronary artery disease. Pharmazie. 2018;73(4):202–6.
Zhang X, Sun J, Canfran-Duque A, Aryal B, Tellides G, Chang YJ, Suarez Y, Osborne TF, Fernandez-Hernando C. Deficiency of histone lysine methyltransferase SETDB2 in hematopoietic cells promotes vascular inflammation and accelerates atherosclerosis. JCI Insight. 2021;6(12):e147984.
Neele AE, Chen HJ, Gijbels MJJ, van der Velden S, Hoeksema MA, Boshuizen MCS, Van den Bossche J, Tool AT, Matlung HL, van den Berg TK, et al. Myeloid Ezh2 deficiency limits atherosclerosis development. Front Immunol. 2020;11:594603.
Farina FM, Serio S, Hall IF, Zani S, Cassanmagnago GA, Climent M, Civilini E, Condorelli G, Quintavalle M, Elia L. The epigenetic enzyme DOT1L orchestrates vascular smooth muscle cell-monocyte crosstalk and protects against atherosclerosis via the NF-κB pathway. Eur Heart J. 2022;43(43):4562–76.
Willemsen L, Prange KHM, Neele AE, van Roomen C, Gijbels M, Griffith GR, Toom MD, Beckers L, Siebeler R, Spann NJ, et al. DOT1L regulates lipid biosynthesis and inflammatory responses in macrophages and promotes atherosclerotic plaque stability. Cell Rep. 2022;41(8):111703.
Wang X, Wang S, Yao G, Yu D, Chen K, Tong Q, Ye L, Wu C, Sun Y, Li H, et al. Identification of the histone lysine demethylase KDM4A/JMJD2A as a novel epigenetic target in M1 macrophage polarization induced by oxidized LDL. Oncotarget. 2017;8(70):114442–56.
Yu S, Chen X, Xiu M, He F, Xing J, Min D, Guo F. The regulation of Jmjd3 upon the expression of NF-κB downstream inflammatory genes in LPS activated vascular endothelial cells. Biochem Biophys Res Commun. 2017;485(1):62–8.
Vlad ML, Manea SA, Lazar AG, Raicu M, Muresian H, Simionescu M, Manea A. Histone acetyltransferase-dependent pathways mediate upregulation of NADPH oxidase 5 in human macrophages under inflammatory conditions: a potential mechanism of reactive oxygen species overproduction in atherosclerosis. Oxid Med Cell Longev. 2019;2019:3201062.
Manea SA, Vlad ML, Fenyo IM, Lazar AG, Raicu M, Muresian H, Simionescu M, Manea A. Pharmacological inhibition of histone deacetylase reduces NADPH oxidase expression, oxidative stress and the progression of atherosclerotic lesions in hypercholesterolemic apolipoprotein E-deficient mice; potential implications for human atherosclerosis. Redox Biol. 2020;28: 101338.
Park SY, Kim JS. A short guide to histone deacetylases including recent progress on class II enzymes. Exp Mol Med. 2020;52(2):204–12.
Sun L, Wang C, Yuan Y, Guo Z, He Y, Ma W, Zhang J. Downregulation of HDAC1 suppresses media degeneration by inhibiting the migration and phenotypic switch of aortic vascular smooth muscle cells in aortic dissection. J Cell Physiol. 2020;235(11):8747–56.
Jia S, Yang S, Du P, Gao K, Cao Y, Yao B, Guo R, Zhao M. Regulatory factor X1 downregulation contributes to monocyte chemoattractant protein-1 overexpression in CD14+ monocytes via epigenetic mechanisms in coronary heart disease. Front Genet. 2019;10:1098.
Hori D, Nomura Y, Nakano M, Han M, Bhatta A, Chen K, Akiyoshi K, Pandey D. Endothelial-specific overexpression of histone deacetylase 2 protects mice against endothelial dysfunction and atherosclerosis. Cell Physiol Biochem. 2020;54(5):947–58.
Zampetaki A, Zeng L, Margariti A, Xiao Q, Li H, Zhang Z, Pepe AE, Wang G, Habi O, deFalco E, et al. Histone deacetylase 3 is critical in endothelial survival and atherosclerosis development in response to disturbed flow. Circulation. 2010;121(1):132–42.
Chen L, Shang C, Wang B, Wang G, Jin Z, Yao F, Yue Z, Bai L, Wang R, Zhao S, et al. HDAC3 inhibitor suppresses endothelial-to-mesenchymal transition via modulating inflammatory response in atherosclerosis. Biochem Pharmacol. 2021;192:114716.
Wang J, Xu X, Li P, Zhang B, Zhang J. HDAC3 protects against atherosclerosis through inhibition of inflammation via the microRNA-19b/PPARγ/NF-κB axis. Atherosclerosis. 2021;323:1–12.
Usui T, Morita T, Okada M, Yamawaki H. Histone deacetylase 4 controls neointimal hyperplasia via stimulating proliferation and migration of vascular smooth muscle cells. Hypertension. 2014;63(2):397–403.
Liu J, Zhou X, Li Q, Zhou SM, Hu B, Hu GW, Niu X, Guo SC, Wang Y, Deng ZF. Role of phosphorylated HDAC4 in stroke-induced angiogenesis. Biomed Res Int. 2017;2017:2957538.
Ren G, Zhang G, Dong Z, Liu Z, Li L, Feng Y, Su D, Zhang Y, Huang B, Lu J. Recruitment of HDAC4 by transcription factor YY1 represses HOXB13 to affect cell growth in AR-negative prostate cancers. Int J Biochem Cell Biol. 2009;41(5):1094–101.
Zheng X, Wu Z, Xu K, Qiu Y, Su X, Zhang Z, Zhou M. Interfering histone deacetylase 4 inhibits the proliferation of vascular smooth muscle cells via regulating MEG3/miR-125a-5p/IRF1. Cell Adh Migr. 2019;13(1):41–9.
Abend A, Shkedi O, Fertouk M, Caspi LH, Kehat I. Salt-inducible kinase induces cytoplasmic histone deacetylase 4 to promote vascular calcification. EMBO Rep. 2017;18(7):1166–85.
Lee CW, Lin CC, Luo SF, Lee HC, Lee IT, Aird WC, Hwang TL, Yang CM. Tumor necrosis factor-alpha enhances neutrophil adhesiveness: induction of vascular cell adhesion molecule-1 via activation of Akt and CaM kinase II and modifications of histone acetyltransferase and histone deacetylase 4 in human tracheal smooth muscle cells. Mol Pharmacol. 2008;73(5):1454–64.
Kwon DH, Ryu J, Kim YK, Kook H. Roles of histone acetylation modifiers and other epigenetic regulators in vascular calcification. Int J Mol Sci. 2020;21(9):3246.
Pietruczuk P, Jain A, Simo-Cheyou ER, Anand-Srivastava MB, Srivastava AK. Protein kinase B/AKT mediates insulin-like growth factor 1-induced phosphorylation and nuclear export of histone deacetylase 5 via NADPH oxidase 4 activation in vascular smooth muscle cells. J Cell Physiol. 2019;234(10):17337–50.
Leucker TM, Nomura Y, Kim JH, Bhatta A, Wang V, Wecker A, Jandu S, Santhanam L, Berkowitz D, Romer L, Pandey D. Cystathionine gamma-lyase protects vascular endothelium: a role for inhibition of histone deacetylase 6. Am J Physiol Heart Circ Physiol. 2017;312(4):H711–20.
Xu S, Chen H, Ni H, Dai Q. Targeting HDAC6 attenuates nicotine-induced macrophage pyroptosis via NF-κB/NLRP3 pathway. Atherosclerosis. 2021;317:1–9.
Lecce L, Xu Y, V’Gangula B, Chandel N, Pothula V, Caudrillier A, Santini MP, d’Escamard V, Ceholski DK, Gorski PA, et al. Histone deacetylase 9 promotes endothelial-mesenchymal transition and an unfavorable atherosclerotic plaque phenotype. J Clin Invest. 2021;131(15):e131178.
Malhotra R, Mauer AC, Lino Cardenas CL, Guo X, Yao J, Zhang X, Wunderer F, Smith AV, Wong Q, Pechlivanis S, et al. HDAC9 is implicated in atherosclerotic aortic calcification and affects vascular smooth muscle cell phenotype. Nat Genet. 2019;51(11):1580–7.
Kwon DH, Eom GH, Ko JH, Shin S, Joung H, Choe N, Nam YS, Min HK, Kook T, Yoon S, et al. MDM2 E3 ligase-mediated ubiquitination and degradation of HDAC1 in vascular calcification. Nat Commun. 2016;7:10492.
Gu J, Lu Y, Deng M, Qiu M, Tian Y, Ji Y, Zong P, Shao Y, Zheng R, Zhou B, et al. Inhibition of acetylation of histones 3 and 4 attenuates aortic valve calcification. Exp Mol Med. 2019;51(7):1–14.
Fu Z, Li F, Jia L, Su S, Wang Y, Cai Z, Xiang M. Histone deacetylase 6 reduction promotes aortic valve calcification via an endoplasmic reticulum stress-mediated osteogenic pathway. J Thorac Cardiovasc Surg. 2019;158(2):408-17 e2.
Grootaert MOJ, Finigan A, Figg NL, Uryga AK, Bennett MR. SIRT6 protects smooth muscle cells from senescence and reduces atherosclerosis. Circ Res. 2021;128(4):474–91.
Li X, Zhao K, Ma N, Sun S, Miao Z, Zhao Z. Association of ABCB1 promoter methylation with aspirin exposure, platelet function, and clinical outcomes in Chinese intracranial artery stenosis patients. Eur J Clin Pharmacol. 2017;73(10):1261–9.
Cui S, Lv X, Li W, Li Z, Liu H, Gao Y, Huang G. Folic acid modulates VPO1 DNA methylation levels and alleviates oxidative stress-induced apoptosis in vivo and in vitro. Redox Biol. 2018;19:81–91.
Hou H, Zhao H. Epigenetic factors in atherosclerosis: DNA methylation, folic acid metabolism, and intestinal microbiota. Clin Chim Acta. 2021;512:7–11.
Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31(1):27–36.
Strand KA, Lu S, Mutryn MF, Li L, Zhou Q, Enyart BT, Jolly AJ, Dubner AM, Moulton KS, Nemenoff RA, et al. High Throughput Screen Identifies the DNMT1 (DNA Methyltransferase-1) Inhibitor, 5-Azacytidine, as a Potent Inducer of PTEN (Phosphatase and Tensin Homolog): Central Role for PTEN in 5-Azacytidine Protection Against Pathological Vascular Remodeling. Arterioscler Thromb Vasc Biol. 2020;40(8):1854–69.
Cao Q, Wang X, Jia L, Mondal AK, Diallo A, Hawkins GA, Das SK, Parks JS, Yu L, Shi H, et al. Inhibiting DNA Methylation by 5-Aza-2’-deoxycytidine ameliorates atherosclerosis through suppressing macrophage inflammation. Endocrinology. 2014;155(12):4925–38.
Dunn J, Qiu H, Kim S, Jjingo D, Hoffman R, Kim CW, Jang I, Son DJ, Kim D, Pan C, et al. Flow-dependent epigenetic DNA methylation regulates endothelial gene expression and atherosclerosis. J Clin Invest. 2014;124(7):3187–99.
Zhuang J, Luan P, Li H, Wang K, Zhang P, Xu Y, Peng W. The Yin-Yang dynamics of DNA methylation is the key regulator for smooth muscle cell phenotype switch and vascular remodeling. Arterioscler Thromb Vasc Biol. 2017;37(1):84–97.
Chen SH, Lv QL, Hu L, Peng MJ, Wang GH, Sun B. DNA methylation alterations in the pathogenesis of lupus. Clin Exp Immunol. 2017;187(2):185–92.
Ridker PM, Devalaraja M, Baeres FMM, Engelmann MDM, Hovingh GK, Ivkovic M, Lo L, Kling D, Pergola P, Raj D, et al. IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet. 2021;397(10289):2060–9.
Wada Y, Jensen C, Meyer ASP, Zonoozi AAM, Honda H. Efficacy and safety of interleukin-6 inhibition with ziltivekimab in patients at high risk of atherosclerotic events in Japan (RESCUE-2): a randomized, double-blind, placebo-controlled, phase 2 trial. J Cardiol. 2023;82(4):279–85.
Adamstein NH, Cornel JH, Davidson M, Libby P, de Remigis A, Jensen C, Ekström K, Ridker PM. Association of interleukin 6 inhibition with Ziltivekimab and the neutrophil-lymphocyte ratio: a secondary analysis of the RESCUE clinical trial. JAMA Cardiol. 2023;8(2):177–81.
Rogers MA, Hutcheson JD, Okui T, Goettsch C, Singh SA, Halu A, Schlotter F, Higashi H, Wang L, Whelan MC, et al. Dynamin-related protein 1 inhibition reduces hepatic PCSK9 secretion. Cardiovasc Res. 2021;117(11):2340–53.
Bellino M, Galasso G, Silverio A, Tedeschi M, Formisano C, Romei S, Esposito L, Cancro FP, Vassallo MG, Accarino G, et al. Soluble PCSK9 inhibition: indications, clinical impact, new molecular insights and practical approach-where do we stand? J Clin Med. 2023;12(8):2922.
Lewinska A, Wnuk M, Grabowska W, Zabek T, Semik E, Sikora E, Bielak-Zmijewska A. Curcumin induces oxidation-dependent cell cycle arrest mediated by SIRT7 inhibition of rDNA transcription in human aortic smooth muscle cells. Toxicol Lett. 2015;233(3):227–38.
Li ZQ, Huang XY, Hu CY, Zhu ZS, Chen Y, Gong M. Geniposide protects against ox-LDL-induced foam cell formation through inhibition of MAPKs and NF-kB signaling pathways. Pharmazie. 2019;74(10):601–5.
Heerboth S, Lapinska K, Snyder N, Leary M, Rollinson S, Sarkar S. Use of epigenetic drugs in disease: an overview. Genet Epigenet. 2014;6:9–19.
McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A, 3rd, Diaz E, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492(7427):108-12.
Lv YC, Tang YY, Zhang P, Wan W, Yao F, He PP, Xie W, Mo ZC, Shi JF, Wu JF, et al. Histone methyltransferase enhancer of zeste homolog 2-mediated ABCA1 promoter DNA methylation contributes to the progression of atherosclerosis. PLoS One. 2016;11(6):e0157265.
Shin IS, Kim JM, Kim KL, Jang SY, Jeon ES, Choi SH, Kim DK, Suh W, Kim YW. Early growth response factor-1 is associated with intraluminal thrombus formation in human abdominal aortic aneurysm. J Am Coll Cardiol. 2009;53(9):792–9.
Abdel-Malak NA, Mofarrahi M, Mayaki D, Khachigian LM, Hussain SN. Early growth response-1 regulates angiopoietin-1-induced endothelial cell proliferation, migration, and differentiation. Arterioscler Thromb Vasc Biol. 2009;29(2):209–16.
Legartova S, Stixova L, Strnad H, Kozubek S, Martinet N, Dekker FJ, Franek M, Bartova E. Basic nuclear processes affected by histone acetyltransferases and histone deacetylase inhibitors. Epigenomics. 2013;5(4):379–96.
Rothgiesser KM, Fey M, Hottiger MO. Acetylation of p65 at lysine 314 is important for late NF-kappaB-dependent gene expression. BMC Genomics. 2010;11:22.
Petrella A, Fontanella B, Carratù A, Bizzarro V, Rodriquez M, Parente L. Histone deacetylase inhibitors in the treatment of hematological malignancies. Mini Rev Med Chem. 2011;11(6):519–27.
Zhao C, Dong H, Xu Q, Zhang Y. Histone deacetylase (HDAC) inhibitors in cancer: a patent review (2017-present). Expert Opin Ther Pat. 2020;30(4):263–74.
Xu Y, Xu S, Liu P, Koroleva M, Zhang S, Si S, Jin ZG. Suberanilohydroxamic acid as a pharmacological Kruppel-Like Factor 2 activator that represses vascular inflammation and atherosclerosis. J Am Heart Assoc. 2017;6(12):e007134.
Choi JH, Nam KH, Kim J, Baek MW, Park JE, Park HY, Kwon HJ, Kwon OS, Kim DY, Oh GT. Trichostatin A exacerbates atherosclerosis in low density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25(11):2404–9.
Song S, Kang SW, Choi C. Trichostatin A enhances proliferation and migration of vascular smooth muscle cells by downregulating thioredoxin 1. Cardiovasc Res. 2010;85(1):241–9.
Gao Q, Wei A, Chen F, Chen X, Ding W, Ding Z, Wu Z, Du R, Cao W. Enhancing PPARgamma by HDAC inhibition reduces foam cell formation and atherosclerosis in ApoE deficient mice. Pharmacol Res. 2020;160:105059.
Kühne M, Kretzer C, Lindemann H, Godmann M, Heinze T, Werz O, Heinzel T. Biocompatible valproic acid-coupled nanoparticles attenuate lipopolysaccharide-induced inflammation. Int J Pharm. 2021;601:120567.
Asare Y, Campbell-James TA, Bokov Y, Yu LL, Prestel M, El Bounkari O, Roth S, Megens RTA, Straub T, Thomas K, et al. Histone deacetylase 9 activates IKK to regulate atherosclerotic plaque vulnerability. Circ Res. 2020;127(6):811–23.
Bondarev AD, Attwood MM, Jonsson J, Chubarev VN, Tarasov VV, Schiöth HB. Recent developments of HDAC inhibitors: emerging indications and novel molecules. Br J Clin Pharmacol. 2021;87(12):4577–97.
Yang SS, Zhang R, Wang G, Zhang YF. The development prospection of HDAC inhibitors as a potential therapeutic direction in Alzheimer’s disease. Transl Neurodegener. 2017;6:19.
Nishida K, Komiyama T, Miyazawa S, Shen ZN, Furumatsu T, Doi H, Yoshida A, Yamana J, Yamamura M, Ninomiya Y, et al. Histone deacetylase inhibitor suppression of autoantibody-mediated arthritis in mice via regulation of p16INK4a and p21(WAF1/Cip1) expression. Arthritis Rheum. 2004;50(10):3365–76.
Zheng B, Han M, Shu YN, Li YJ, Miao SB, Zhang XH, Shi HJ, Zhang T, Wen JK. HDAC2 phosphorylation-dependent Klf5 deacetylation and RARalpha acetylation induced by RAR agonist switch the transcription regulatory programs of p21 in VSMCs. Cell Res. 2011;21(10):1487–508.
Van den Bossche J, Neele AE, Hoeksema MA, de Heij F, Boshuizen MC, van der Velden S, de Boer VC, Reedquist KA, de Winther MP. Inhibiting epigenetic enzymes to improve atherogenic macrophage functions. Biochem Biophys Res Commun. 2014;455(3–4):396–402.
Hu C, Peng K, Wu Q, Wang Y, Fan X, Zhang DM, Passerini AG, Sun C. HDAC1 and 2 regulate endothelial VCAM-1 expression and atherogenesis by suppressing methylation of the GATA6 promoter. Theranostics. 2021;11(11):5605–19.
Cheng P, Wirka RC, Shoa Clarke L, Zhao Q, Kundu R, Nguyen T, Nair S, Sharma D, Kim HJ, Shi H, et al. ZEB2 shapes the epigenetic landscape of atherosclerosis. Circulation. 2022;145(6):469–85.
Musolino E, Pagiatakis C, Serio S, Borgese M, Gamberoni F, Gornati R, Bernardini G, Papait R. The Yin and Yang of epigenetics in the field of nanoparticles. Nanoscale Adv. 2022;4(4):979–94.
Chen J, Zhang X, Millican R, Sherwood J, Martin S, Jo H, Yoon YS, Brott BC, Jun HW. Recent advances in nanomaterials for therapy and diagnosis for atherosclerosis. Adv Drug Deliv Rev. 2021;170:142–99.
Kiaie N, Gorabi AM, Penson PE, Watts G, Johnston TP, Banach M, Sahebkar A. A new approach to the diagnosis and treatment of atherosclerosis: the era of the liposome. Drug Discov Today. 2020;25(1):58–72.
Hong YD, Zhang J, Zhuang M, Li W, Wu PU, Li RT, Hu N, Bian BX, Song ZY, Wu FL. Efficacy of decitabine-loaded gelatinases-stimuli nanoparticles in overcoming cancer drug resistance is mediated via its enhanced demethylating activity to transcription factor AP-2 epsilon. Oncotarget. 2017;8(70):114495–505.
El Bahhaj F, Denis I, Pichavant L, Delatouche R, Collette F, Linot C, Pouliquen D, Grégoire M, Héroguez V, Blanquart C, Bertrand P. Histone deacetylase inhibitors delivery using nanoparticles with intrinsic passive tumor targeting properties for tumor therapy. Theranostics. 2016;6(6):795–807.
Urbinati G, Marsaud V, Plassat V, Fattal E, Lesieur S, Renoir JM. Liposomes loaded with histone deacetylase inhibitors for breast cancer therapy. Int J Pharm. 2010;397(1–2):184–93.
Funding
This work was supported by Shaanxi Province Social Development Research Project (No. 2021SF-324) and Air Force Military Medical University Research Project (No. 2022LGC2235).
Author information
Authors and Affiliations
Contributions
LZ and CHX: conceptualization, writing–original draft preparation. YJY and FFS have drafted the DNA methylation section or substantively revised it. YZ and HW have drafted the histone post-translational modifications section or substantively revised it. RL and MY: conceptualization, supervision. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, L., Xia, C., Yang, Y. et al. DNA methylation and histone post-translational modifications in atherosclerosis and a novel perspective for epigenetic therapy. Cell Commun Signal 21, 344 (2023). https://doi.org/10.1186/s12964-023-01298-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-023-01298-8