
Abstract
Melanoma is an aggressive kind of skin cancer; its rate has risen rapidly over the past few decades. Melanoma reports for only about 1% of skin cancers but leads to a high majority of skin cancer deaths. Thus, new useful therapeutic approaches are currently required, to state effective treatments to consistently enhance the overall survival rate of melanoma patients. Ferroptosis is a recently identified cell death process, which is different from autophagy, apoptosis, necrosis, and pyroptosis in terms of biochemistry, genetics, and morphology which plays an important role in cancer treatment. Ferroptosis happens mostly by accumulating iron and lipid peroxides in the cell. Recently, studies have revealed that ferroptosis has a key role in the tumor’s progression. Especially, inducing ferroptosis in cells can inhibit the tumor cells’ growth, leading to back warding tumorigenesis. Here, we outline the ferroptosis characteristics from its basic role in melanoma cancer and mention its possible applications in melanoma cancer treatment.
Video Abstract
Similar content being viewed by others

Introduction
Melanoma is the most invasive skin cancer, and metastatic melanoma has the highest risk of death with a median survival rate of nearly 6 months [1]. Melanoma prevalence is significantly rising all around the world [2, 3]. Specialized pigment cells are known as melanocytes, which are found in the basal epidermis, and lead to melanoma [4]. In a normal physiological condition, keratinocytes control melanocyte growth and activity [4]. Due to abnormalities in critical genes that control cell growth, melanocytes cannot adequately respond to regulatory cues from keratinocytes, which ultimately results in aberrant growth. Melanoma can develop without a precursor lesion, although in certain instances, the development of a nevus or mole marks the beginning of this aberrant growth [5]. Melanoma has also been identified from transformed stem cells. Stem cell markers such as CD20, and CD133, as well as OCT 4, NANOG, and pSTAT 3, have been recognized in melanoma [6, 7]. A challenge in treating melanoma is the variety of cell populations with stem cell characteristics since some of these cells are resistant to therapy [8]. Cancer stem cells are also known to secrete factors in response to hypoxia, increasing tumor angiogenesis, and thereby promoting disease progression [9]. In spite of the development mechanism of melanoma, neovessel formation precedes tumor progression.
Current cancer treatment methods include surgical resection, chemotherapy, photodynamic therapy, immunotherapy, and targeted therapy. Depending on the patient's health, tumor stage, and location, the therapeutic strategy may consist of single drugs or combined therapies. Due to the development of different resistance mechanisms, the efficacy of various treatments may be decreased. Studies of the genetic profile of melanocytes and the discovery of molecular factors involved in the pathogenesis of malignant transformation have provided new therapeutic targets [10]. Today, two main new therapeutic strategies are routinely used which are molecularly targeted therapy (using dabrafenib, vemurafenib, encorafenib, trametinib, cobimetinib, binimetinib) and immunotherapy (using pembrolizumab, nivolumab, ipilimumab). Additionally, in the case of the presence of mutations in genes other than BRAF (B-Raf proto-oncogene, serine/threonine kinase), alternative targeted therapy may be considered, e.g., with imatinib, when a mutation in the c-KIT gene is present. It is also possible to treat injectable melanoma with the genetically modified oncolytic virus (talimogene laherparepvec) [11]. Adjuvant therapy with kinase inhibitors (dabrafenib and trametinib) and immunotherapy (pembrolizumab, nivolumab, ipilimumab) for high-risk melanoma are also registered [12] Although many patients take advantage of these new therapies, some patients do not respond to both targeted and immunological tratment. The development of reliable markers of response would allow for better personalization of the treatment and consequently would lead to improved patient survival and lower costs of patient care [13, 14]. For patients with solitary melanoma metastasis, metastasectomy is the standard of care, and chemotherapy may be recommended in some metastatic melanoma instances [15]. Radiotherapy can be effective for the treatment of skin, bone, and brain metastases, despite being rarely advised for original tumor treatment. It was claimed that the combination of photodynamic therapy (PDT) with chemotherapy (dacarbazine) is an effective treatment for reducing resistance in pigmented and unpigmented metastatic melanomas [16].
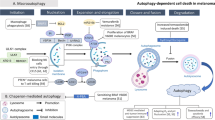
Cell death entities can be categorized into programmed or non-programmed cell death based on their signal dependency. Programmed cell death (PCD) is driven by tightly regulated intracellular signal transduction pathways. By contrast, accidental cell death is referred to as non-PCD as a result of unexpected cell injury. Given the morphological characteristics and molecular mechanisms, PCD can be further categorized into apoptotic cell death and non-apoptotic cell death. Apoptosis retains cell membrane integrity and occurs in a caspase-dependent manner. By contrast, non-apoptotic cell death is mostly characterized by membrane rupture and caspase independency (Fig. 1).

The main morphologic characteristics of cell death are apoptosis, pyroptosis, ferroptosis, and Necrosis. Apoptosis is characterized by DNA condensation and fragmentation, and the occurrence of apoptotic body. Cells with pyroptosis present DNA condensation and fragmentation, and the membrane is ruptured. Ferroptosis is defined as free iron accumulation, lipids peroxidation, ROS generation. Cells undergoing necrosis show DNA degradation and membrane rupture. Pyroptosis and necrosis are accompanied with cell membrane rupture and severe inflammatory reaction, while apoptosis and ferroptosis are devoid of these changes and there are no cell membrane alterations and no DNA fragmentation
Until ferroptosis identification as such in 2012, studies describing what is now known as ferroptotic cell death were attributed to alternative cell death mechanisms or not recognized as biologically significant [17]. In 2003, Dolma et al. identified erastin, a novel drug that had a selectively lethal effect on RAS-expressing cancer cells, although the mode of cell death was distinct from what had previously been observed. There were no nuclear morphological alterations, DNA fragmentation, or caspase activation, and caspase inhibitors had no effect on this process [18]. Subsequently, Yang [19] and Yagoda [20] found that This pattern of cell death is inhibited by iron chelating agents, whereas another substance, RSL3, can induce this pattern of cell death. The term ferroptosis was coined in 2012 to [21] describe an iron-dependent, non-apoptotic form of cell death triggered by erastin and RSL3. This discovery was followed by the development of ferrostatin-1, the first small molecule ferroptosis inhibitor, and the demonstration of glutamate-induced ferroptosis in organotypic rat brain slices, indicating the possible role of ferroptosis in neurodegeneration. Subsequently, numerous other researches began to demonstrate a similar ferroptotic process as the underlying mechanism for a variety of phenomena. In the presence of serum, amino acid starvation has been found to trigger non-apoptotic and non-necrotic cell death in multiple cell types [22, 23]. It was demonstrated that cystine deficiency was sufficient to cause the same serum-dependent cell death pathway and that transferrin, an iron carrier, was the essential serum component for cell death. Thus, cystine deficiency was similar to system Xc inhibition. These findings confirmed that ferroptosis is the cell death process triggered by amino acid starvation in the presence of an external iron source. Later, it was discovered that not all reactive oxygen species (ROS) function equally in ferroptosis, and that lipid peroxidation is the main factor of ferroptotic death [24], which has been supported by the identification of lipophilic antioxidants as inhibitors of ferroptotic death induced by erastin and other compounds [20].
In addition to revealing mechanisms of a fundamental biological process, cell death research has contributed to the development of novel cancer treatments over the past three decades. Knowledge of this mechanism has enabled the development of treatments that kill cancer cells by directly activating the cell death machinery and by synergizing with conventional chemotherapy and targeted therapies to improve cancer patients' outcomes [25]. The roles of autophagy-dependent cell death, necroptosis, ferroptosis, pyroptosis, and parthanatos have recently attracted considerable interest. This is despite the fact that melanoma cells are generally equipped with anti-apoptotic machinery and that recurrent genetic alterations in the RAS/RAF/MEK/ERK signaling significantly contribute to the pro-survival phenotype of melanoma [26]. In addition, the links between sensitivity to non-apoptotic cell death pathways and distinct cell morphologies have been identified, suggesting that the plasticity of melanoma cells can be exploited to alter their response to various cell death stimuli [26]. Increasing evidence indicates that the anti-tumor approach based on non-apoptotic cell death is a direction to solving existing problems in cancer treatment. On the one hand, numerous kinds of non-apoptotic cell death successfully bypass or overcome the resistance of tumor cells to apoptosis and provide alternative death pathways when the apoptosis pathway is deficient, so considerably enhancing the anti-cancer efficacy (Fig. 1) [27].
According to the studies ferroptosis can be introduced as a target for melanoma cancer treatment. In this review, we will describe ferroptosis and its mechanisms which are involved in different melanoma cancer therapies.
Ferroptosis definition and its morphological hallmarks
In 2012, Dixon has defined the concept of ferroptosis as an iron-dependent form of cell death described by the excessive accumulation of lipid peroxides and reactive oxygen species (ROS) [21]. Recent data has shown that ferroptosis has a role in the occurrence and progression of a variety of diseases, making it the central issue of controversy in current research about to the treatment and prognosis improvement of related disorders. Electron microscopy shows that morphologically, Ferroptosis is characterized by decreased mitochondrial volume, increased bilayer membrane density, and decreased or disappearance of mitochondrial cristae. However, there is no evidence of the cell membrane permeabilize, nucleus fragments, or chromatin condensation. Although oxidative damage in the DNA occurs by some activators of ferroptosis, the nucleus shows a normal size, without chromatin condensation [28]. In some cases, other features like detachment and rounding up of cells and an increased autophagosome is observed in ferroptosis cells [21, 29, 30]. In addition, ferroptosis is a defined form of inflammatory regulated cell death (RCD) that immune cell infiltration that could be observed in tissues with ferroptotic damage. For instance, acute pancreatitis is an inflammatory disorder characterized by an initial injury that results in acinar cell death. Ferroptotic acinar death contributes to experimental pancreatitis in mice, particularly when circadian rhythms are disrupted [31]. According to hematoxylin and eosin stain, the histological assessment revealed ferroptosis is associated with leukocyte infiltration and pancreatic damage [31].
Biochemical hallmarks of ferroptosis
Ferroptosis is a ROS-dependent form of controlled cell death characterized by two primary biochemical characteristics, iron accumulation and enhanced lipid peroxidation. Ferroptosis is mainly triggered by intracellular glutathione (GSH) depletion and a decrease in the activity of glutathione peroxidase 4 (GPX4). As a result, lipid peroxides are unable to be metabolized by the GPX4-catalyzed reduction mechanism, which results in an accumulation of lipid peroxides. Fe2+ triggers ferroptosis by oxidizing lipids in a Fenton-like way and by producing a large amount of reactive oxygen species [30].
Iron overload
Multiple iron metabolism regulators are involved in the process of ferroptosis. As they enhance intracellular iron accumulation, the common ferroptosis activators erastin and RSL3 block the antioxidant system. Iron can mediate the production of excessive ROS via the Fenton reaction and contribute to increased oxidative damage [21]. Heme and non-heme iron in excess can directly induce ferroptosis [32]. Arachidonate lipoxygenase (ALOX) and EGLN (also known as PHD) 2-oxoglutarate (2OG)-dependent dioxygenases, which are in control of lipid peroxidation and oxygen homeostasis, are two iron-containing enzymes that activity may be increased by iron. The sensitivity of ferroptosis is influenced by local and systemic cellular iron regulation [33]. Iron overload- or the usage of iron-chelating agents-related suppressor genes may successfully prevent ferroptosis cell death. It is not well understood why only iron (no other metals like zinc) also induces ROS generation via a Fenton reaction [34] to trigger ferroptosis [21]. This may be happening due to the iron overload and activate specific downstream effectors that participate in the performance of ferroptosis after the generation of lipid ROS.
Lipid peroxidation
Lipid peroxidation happens under conditions where ROS readily react with vulnerable lipids on cell membranes. Initial lipid hydroperoxides (LOOHs) and later reactive aldehydes, such as malondialdehyde (MDA) and 4-hydroxynonenal (4HNE), which rise during ferroptosis, are examples of lipid peroxidation's products. Saturated fatty acids (no double bond), monounsaturated fatty acids (MUFAs, 1 double bond), and polyunsaturated fatty acids (PUFAs, > 1 double bond) are the three different types of fatty acids. Only the peroxidation of PUFAs in phospholipids by ALOXs appears to be necessary for ferroptosis among all the cell membrane lipids that can be oxidized, including phosphatidylcholine, phosphatidylethanolamine (PE), and cardiolipin [35, 36]. Although there are extensive ultrastructural changes in mitochondria during ferroptosis, there is no evidence of cardiolipin peroxidation (Fig. 1) [37].
Genetic features
Genetically, multiple genes have been found to regulate ferroptosis [19, 20]. Ferroptosis particularly involves genetic changes in iron homeostasis and lipid peroxidation metabolism. However further study is needed to determine the specific regulatory mechanisms. Prostaglandin-endoperoxide synthase 2 (PTGS2/COX2), a crucial enzyme in prostaglandin production, is an example of how overexpression of a select few genes/proteins has been considered to be a genetic signature of ferroptosis [38]. The up-regulation of PTGS2 mRNA is used as a pharmacodynamic marker of ferroptotic tissues in mice exposed to erastin or RSL3 [38]. Although it is a widely used biomarker of ferroptosis in vitro or in vivo, PTGS2 inhibitor (e.g., indomethacin) fails to affect ferroptotic cell death indicating it is not a contributor of ferroptosis. In contradiction, MIR212-mediated downregulation of PTGS2 mRNA prevents ferroptotic neuronal death in a traumatic brain injury mouse model [39] suggesting a cell type-dependent role of PTGS2 in ferroptosis. Further mechanism studies suggest that the up-regulation of PTGS2 gene expression in ferroptosis requires lipid peroxidation because antioxidant vitamin E or toxic 4-HNE can inhibit or induce PTGS2 expression in cancer cells or macrophages, respectively [38].
A specific biomarker for ferroptosis is the enzyme acyl-CoA synthetase long-chain family member 4 (ACSL4), which is involved in fatty acid metabolism. When ACSL4 is overexpressed, it increases the quantity of polyunsaturated fatty acids (PUFAs) in phospholipids, which are sensitive to oxidation processes and cause ferroptosis [35, 40, 41]. Nevertheless, ferroptosis is not dependent on ACSL4 in all circumstances. Specific conditions can cause a cell to undergo ferroptosis when ACSL4 is low (Fig. 2) [42]. Therefore, in response to ferroptosis signals, cells "decide" whether to live or die based on the balance of injury and anti-injury conditions.

Ferroptosis cell death process. Ferroptosis is triggered by direct suppression of system Xc- or GPX4, which ultimately results in cell death. The ferroptosis process involves lipid ROS. On the one hand, PUFA peroxidation is thought to be a key element. In contrast, the execution of ferroptosis is based on iron overload
Selective ferroptosis inducers
Four classes of ferroptosis inducers can be classified:
Class1: inhibit system Xc- and prevent cystine import
Erastin is one of the ferroptosis inducers which is identified by antioxidant depletion generated by cystine glutamate antiporter inhibition (xCT). Another xCT inhibitor ferroptosis inducer is the clinical medicine sulfasalazine, used to treat inflammatory bowel disease [17]. Cysteine is the rate-limiting substrate for the important antioxidant glutathione, when system XC − is inhibited results in a reduction of cysteine, as a substrate for GSH synthesis, which will result in diminished levels of GSH [38, 43]. For GPX4 to catalyze the degradation of hydrogen peroxide and hydroperoxide and prevent the production of L-ROS, GSH is a crucial cofactor. As a result, erastin indirectly reduces the synthesis of GPX4 and further reduces the potential of cells to produce antioxidants by inhibiting the system XC [44]. It has recently been shown that GPX4 activity was decreased in a number of cancer cells treated with erastin. These drugs induce ferroptosis, which has an anticancer effect [21, 22]. Nonetheless, erastin has another physiological target, voltage-dependent anion channels (VDACs), which cause mitochondrial dysfunction. Additionally, it was recently shown that erastin's activation of ferroptosis is related to an increase in the lysosomal-associated membrane protein 2a, which in turn generates chaperone-mediated autophagy and, subsequently, increases the destruction of GPX4 [45].
A particular inhibitor of the xCT-mediated cystine transporter is sulfasalazine (SAS) [46, 47]. As an anti-inflammatory drug, SAS has the ability to scavenge ROS, reduce the production of IL-1 and IL-2, and inhibit nuclear factor κ B (NFκB), as well as leukocyte motility [48,49,50]. SAS has also been identified as a GSH depletion inducer (90%). It can arrest tumor growth, and improve sensitivity to chemotherapeutic agents in pancreatic, prostate, and mammary cancer [51,52,53]. SAS prevents a cystine-glutamate transporter and therefore plays important role in the induction of ferroptosis [21, 54]. The iron-dependent lethal accumulation of lipid ROS can make this process more sensitive when the cancer cells display high levels of Ras activity or p53 [21, 54]. Both ferroptosis and glutathione depletion can be driven by xCT inhibitors such sulfasalazine, glutamine, and sorafenib [55].
Class 2: inhibit GPX4:
RSL3 and DPI7, which directly inhibit GPX4 activity and induce ferroptosis, are classified as the second category. GPX4 is a key modulator of ferroptosis by inhibiting the formation of lipid peroxides. The enzyme GPX4 reduces the cytotoxic lipid peroxides (L-OOH) to the corresponding alcohols and converts GSH into glutathione disulfide (GSSG) (L-OH). GPX4 inhibition stops the conversion of lipid peroxides to lipid alcohols, which leads to the accumulation of lipid peroxides, which is a hallmark of Ferroptosis. RSL3, a ferroptosis inducer, directly interacts with GPX4 and suppresses its activity, reducing cells' capacity to defend against ROS and causing ferroptosis [38]. Additionally, the DPI7 and DPI10 compounds directly affect GPX4 and cause ferroptosis.
Class 3: degrade GPX4, bind to SQS and deplete antioxidant CoQ10
A member of the third category is FIN56, which has two ways to induce ferroptosis. First, FIN56 stimulates the degrading of GPX4. Second, FIN56 binds to the squalene synthase enzyme, which causes the endogenous antioxidant coenzyme Q10 to be depleted (COQ10). This procedure makes cells more sensitive to ferroptosis caused by FIN56 [56]. GPX4 protein levels are negatively regulated by FIN56, which also activates squalene synthase (SQS), a mevalonate pathway enzyme that functions downstream of HMG-CoA reductase and leads to ferroptosis. In one mechanism, ACC activity is required for FIN56 to enhance the degradation of GPX4 in a process. Tofa's inhibition of ACC prevents GPX4 from being degraded by FIN56, however, the link between FIN56, ACC, and GPX4 degradation is unclear. The second pathway involves the binding and activation of SQS, an enzyme that converts farnesyl pyrophosphate (FPP) into squalene. This decreases the amount of FPP available for protein prenylation and metabolite synthesis, which inevitably leads to the depletion of coenzyme Q10 (CoQ10). SQS inhibition increases the pool of FPP and its derivative products that are accessible, decreasing ferroptosis [57].
Class 4: oxidizing ferrous iron and directly inactivating GPX4 through lipidome
The final category includes FINO2 exerting ferroptosis by the dual-function effect of oxidation of labile iron and the inactivation of GPX4 [56]. The initiation of ferroptosis by FINO2 is highly dependent on the availability of iron and oxidizes a wide range of polyunsaturated lipids. FINO2 is able to initiate ferroptosis preferentially over other types of cell death, in contrast to other peroxide-containing compounds. Numerous elements can be considered for this remarkable selectivity, including the inactivation of GPX4, or the lipophilicity of FINO2. FINO2 can accumulate in lipid bilayers of cell membranes according to its high lipophilicity, causing oxidized PUFAs directly and triggering ferroptosis in the locations [58].
Ferroptosis-inducing nanoparticles in cancer
In recent years, researchers have tried to combine bio-nanotechnology with ferroptosis to develop candidates with a stronger antitumor effect [59, 60].
The delivery of nano-drugs is based on engineering technology. Nanoparticles are used to deliver and control drug release and adjust the intracellular chemical reaction to affect the ROS levels, thereby improving the pharmacokinetic properties of the drug [61, 62]. Nanomaterials can be used to supplement exogenous lipids in tumor cells to increase the accumulation of intracellular lipid peroxides, promote ferroptosis, and achieve the goal of curing cancer [63]. The current emerging nano therapies mainly focus on inhibiting the expression of GPX4 in tumor cells, increasing the accumulation of ferrous/iron ions in tumor cells, and regulating lipid peroxidation [64,65,66].
This process primarily involves triggering or promoting the Fenton response in tumor cells [64].
Some nanomaterials, such as sorafenib, are encapsulated into network-like nanostructures composed of Fe3+ and tannic acid (TA) [67]. Sorafenib is a typical small-molecule System Xc- inhibitor. It inhibits GPX4, leading to tumor-specific ferroptosis, and TA is used to chemically reduce Fe3+ to Fe2+ and continuously supply Fe2+ to maintain the iron redox cycle and maintain the Fenton reaction [67]. Shen et al. by using lactoferrin (LF) and RGD dimer (RGD2)-coupled cisplatin (CDDP) Fe3O4/Gd2O3 hybrid nanoparticles FeGd-HN@Pt@LF/RGD2 successfully combined and delivered Fe2+, Fe3+, and H2O2 (the reactants involved in the Fenton reaction) to the tumor sites. Their local concentration was increased to accelerate the Fenton reaction, significantly improving the efficacy of in situ brain tumor ferroptosis treatment [68]. In another study, Professor Song Yang and Associate Professor Zhu Xiaokang from Southwest University designed a poly-nanosystem Fe3O4-PLGA-Ce6 coated with PLGA, containing iron oxide (Fe3O4) and photosensitizer Ce6, and used it to synergize ferroptosis–photodynamics anticancer treatment. Fe3O4-PLGA-Ce6 nanosystem can dissociate in an acidic tumor microenvironment and release ferrous/iron ions and Ce6. Subsequently, the released ferrous/iron ions will react with excess hydrogen peroxide in the cell to produce a Fenton-like reaction generating hydroxyl free radicals (•OH), and induce ferroptosis of tumor cells [69].
Novel nanoparticles were reported as important in inhibiting tumor progression as presented in Table 1 [67, 70, 71].
Ferroptosis's role in melanoma
One of the most aggressive and challenging treatments among human cancers is skin melanoma whose annual incidence is rising. Over 60% of all fatal skin malignancies are cutaneous melanomas, the most dangerous form of skin cancer that results from melanocyte transformation. Melanoma has a substantial socioeconomic impact due to its high mortality rate in the metastatic form and its disproportionately high incidence in young adults [74]. It is significant to highlight that nevi, benign collections of melanocytes, are produced when melanocytes grow unevenly, but dysplastic nevus is thought to be a possible precursor to cutaneous melanoma since it displays a high level of cytologic and architectural atypia [75]. When tumor cells do not exhibit a significant proliferation capacity or metastasis, they are in the radial growth phase (RGP), which is the first observable malignant stage. Tumor cells can infiltrate the dermis as an increasing mass during the vertical growth phase (VGP), the main lesion, and subsequently move into the lymphatic and blood arteries, causing systemic dissemination. The progression to the invasive stage is accelerated by the accumulation of the initial genetic alterations that occur during the precursor stage. The final stage of tumor development is metastasis (metastatic melanoma) [76].
Various factors have been considered to involve in melanoma progression [4], namely genetic alteration in multiple genes (oncogenic and tumor suppressor genes) such as cyclin-dependent kinase inhibitor 2A (CDKN2A), melanocortin receptor (MC1R), cyclin-dependent kinase 4 (CDK4), Ras, and BRAF (v-raf murine sarcoma viral oncogene homolog B1) genes. A list of onco-suppressor and oncogenic factors involved in melanoma is presented in Table 2.
Indeed, despite significant advancements in the therapeutic management of human cancers in recent years, patients with metastatic melanoma still have not greatly benefited from these medical developments. To establish and define successful treatments to consistently improve the overall survival rate of patients affected by this malignancy, new worthwhile therapeutic techniques are urgently required [92]. Recently, it has also been demonstrated that ferroptosis is related to resistance to cancer therapy. Additionally, a number of studies have suggested that controlling ferroptosis may affect the effectiveness of cancer treatment and perhaps overcome resistance [93,94,95]. Here, we give a thorough explanation of the mechanics behind ferroptosis and discuss how controlling it can treat melanoma cancer. Ferroptosis can initiate glutamate-induced cytotoxicity. Therefore, iron chelators and other ferroptosis inhibitors can suppress glutamate-induced cytotoxicity. Ferroptosis can also be regulated by glutaminolysis and glutamine metabolism in various ways.
For instance, glutamine is taken in and converted into glutamate and -ketoglutarate (-KG) by the glutamate importer (SLC1A5/SLC28A1), glutaminase (GLS), and glutamic-oxaloacetic transaminase-1 (GOT1). Inactivation of any of these genes may cause resistance of cells to ferroptosis [23]. Reduced SLC1A5 expression has been linked to increased ferroptosis, decreased glutamine synthesis, and decreased glutamine accumulation in melanoma [96]. Additionally, the reduction in glutamic-oxaloacetic transaminase prevented the depletion of Glu, consequently leads anti-ferroptosis action on melanoma cells [97].
According to Sato et al. study on melanoma pathogenesis and metastasis, ferroptosis initiate by inducing cysteine-glutamate antiporter (System Xc−) deficient B16F10 melanoma cells. Deficiency in System Xc− resulting in a reduction of cysteine uptake, cellular glutathione, cell cycle progression, and proliferation in vitro, tumor spheroid formation ex vivo, and subcutaneous tumor formation in vivo. Notably, the ferroptosis inhibitor liproxstatin-1 was unable to reverse any of these alterations. Additionally, by using the tail vein, intrasplenic, IP, and footpad injections, loss of System Xc- generally have fewer metastases in vivo and is attached poorly to the lung vascular endothelium in vitro as well as reduced migration. The summary of the study isassessing the link between ferroptosis susceptibility and metastatic potential in melanoma [98]. Melanoma that metastasizes through the blood rather than the lymphatic system became dependent on the ferroptosis inhibitor GPX4. Cells with chemical ferroptosis inhibitors treatment metastases than were those that did not treat after intravenous, but not intra-lymphatic, injection. In this study, they observed differences between lymph fluid and blood plasma that may involve in the reduction of oxidative stress and ferroptosis in lymph, such as higher levels of glutathione and oleic acid and less free iron in the lymph. Oleic acid improved the ability of melanoma cells to generate metastatic tumors and prevented ferroptosis in an Acsl3-dependent manner. Melanoma cells in lymph nodes have shown resistance to ferroptosis. When intravenous injection was followed by metastases, these cells were more dominant than melanoma cells from subcutaneous tumors. Melanoma cells protected from ferroptosis which increase their capacity for survival during following metastasis through the blood [99].
Ferroptosis in melanoma cancer-associated signaling pathways
Ferroptosis as programmed cell death is very important in the development and progression of cancer. Cell susceptibility to ferroptosis has been observed at different stages of melanoma progression. Ferroptosis was initially thought to occur only in RAS-mutant cancer cells, but it was later found that induction of ferroptosis could be independent of the mutated state of the RAS [100]. Given that the BRAF-activating mutations have been identified as the most common genetic variation in melanoma, BRAF inhibitors can increase the susceptibility of melanoma cells to ferroptosis. In fact, BRAF inhibition can activate an oxidative phosphorylation system in cells, induce ROS generation, and by altering the metabolism in the cell can increase ferroptosis [101,102,103]. It has been suggested that DNA damage can initiate ferroptosis in melanoma cells as well as several oncogenic pathways have been identified in melanoma, which predisposes cells to ferroptosis by affecting essential cell regulators [104]. Various regulators of ferroptosis have been identified in melanoma. TP53, which encodes P53, is mutated in many cancer cells. But its mutation in melanoma has been found to be very rare [105]. It is suggested that P53 function regulates ferroptosis by regulating cellular redox and metabolism. Researchers have observed that P53 suppresses SLC7A11 activity. Downregulation of SLC7A11 has been proposed as a marker of induction of ferroptosis in melanoma metastatic cells [100, 103]. It has been reported that inhibiting SLC7A11 activity, increases the efficacy of ferroptosis-promoting drugs in melanoma cells [92]. P53 also reduces the uptake of cysteine and acts as a rheostat in the cell due to the stimuli present. In the case of low oxidative stress, P53 reduces ferroptosis, while in the case of high ROS content, it increases ferroptosis. In fact, the expression of several ferroptosis-regulating proteins and redox homeostasis is regulated by P53 [100].
Iron metabolism also plays a key role in inducing ferroptosis in cancer cells, including melanoma. YAO et al., Showed that iron regulatory protein 1(IRP1) induced ferroptosis in melanoma cell lines A375 and G-361. It was observed that the expression of IRP1 and IRP2 were upregulated in the melanoma cells through the inducer of ferropotosis such as erastin and RSL3. IRP1 played a major role in regulating iron homeostasis and thus promoted ferroptosis, and IRP2 increased function IRP1. IRP1 regulated the expression of proteins involved in iron metabolism, such as transferrin receptor, ferroportin, and ferritin heavy chain 1 which increased ferroptosis by increasing the amount of intracellular iron [106].
High nuclear factor erythroid 2-related factor 2 (Nrf2) has also been observed in malignant melanoma cells, leading to the intrinsic resistance of cells to anticancer therapies [107]. Gagliardi et al. investigated the role of Nrf2 in ferroptosis-resistant melanoma cells. Their studies showed that Nrf2 expression was increased in ferroptosis-resistant cells that lead to the expression of glutathione-specific gamma-glutamylcyclotransferase 1 (CHAC1) and the aldo–keto reductase (AKRs). Expression of these markers reduced 12/15-LOX-generated lipid peroxides and inhibited ferroptosis. Inhibition of Nrf2 activity re-induced ferroptosis in the cells [103]. Zhu et al., also showed that Nrf2 regulated the expression of UV-induced programmed cell death ligand 1 (PD-L1) in melanoma cells. Targeted therapy of Nrf2 induced tumor infiltration via CD8 + and CD4 + T cells and inhibited tumor progression. They showed simultaneous inhibition of Nrf2 and anti-programmed cell death protein-1 (a checkpoint protein on T cells) increased melanoma cell death [108]. AKRs such as AKR1C1, AKR1C2, and AKR1C3 play an important role in ferroptosis cell death in melanoma cells. It has been observed that the activity of these genes inhibits the cell death of ferroptosis in melanoma cells by reducing the amount of lipid peroxide. Inhibition of AKRs led to lipid ROS production and the induction of ferroptosis in resistant melanoma cells [92].
MicroRNAs (miRNAs) also play an important role in regulating ferroptosis in melanoma. It has been observed that they regulate the process of ferroptosis in cells by regulating glutamate metabolism. For example, overexpression of miR-137 in melanoma cells downregulated the expression of glutamine transporter SLC1A5, reduced the process of lipid peroxidation, and the accumulation of iron, which reduced ferroptosis. Thus up-regulation of the miR-137 gene increased tumor growth, tumor volume, and drug resistance in melanoma cells [96]. Overexpression of miR-9, also down- regulated the expression of glutamic-oxaloacetic transaminase 1 (GOT1) in melanoma, decreased lipid peroxidation, and iron accumulation, causing cells to escape from ferroptosis. Conversely, inhibition of miR-9 function increased the susceptibility of melanoma cells to inducers of ferroptosis [97].
Ferroptosis in melanoma cancer therapy
The research showed that certain subtypes of melanoma cells could be successfully treated using multiple therapies including chemotherapy, radiotherapy, and immunotherapy in combination with ferroptosis-inducing drugs [109]. Mechanisms of several ferroptosis inducers and their combination therapy in melanoma are listed in Table 3.
Chemotherapy
Despite the spread of chemotherapy drugs, their function has been limited due to drug resistance. Therefore, identifying new treatment goals seems necessary. In the meantime, ferroptosis has been considered by researchers as a new type of programmed cell death. Various studies have shown the importance of ferroptosis in the treatment of cancer cells. Also, the combination of chemotherapy and ferroptosis inducers has shown a significant synergistic effect on cancer cells [44, 94, 129]. On the other hand, dysregulation and ineffective ferroptosis lead to resistance of cancer cells to chemotherapy. Many chemotherapeutic drugs have been shown to induce ferroptosis in cancer cells by pharmacologically regulating or genetic pathways and eliminate the treatment resistance by targeting lipid metabolism, iron metabolism, and canonical GPX4-regulated pathways (Fig. 1) [130]. Tang et al., showed sorafenib enhanced the function of vemurafenib in vemurafenib-resistant melanoma A375 and SK-Mel-28 cells by inducing ferroptosis. The combination of sorafenib and vemurafenib reduced the concentration of GSH and increased the production of ROS, MDA (an end product of lipid peroxidation), and iron which led to ferroptosis [110]. Viswanathan et al. Reported that fluvastatin downregulated the expression of GPX4 in various cancer cells, including melanoma, thereby promoting ferroptosis [111]. Osrodek et al., showed that Vemurafenib and trametinib downregulated the expression of SLC7A11 in melanoma cells [112]. Vemurafenib, along with erastin or RSL3, also increased ferroptosis in resistance melanoma cells by targeting GPX4 and System Xc− transporter [113]. The researchers showed that dioscin induced ferroptosis in melanoma cells by producing ROS and regulating the expression of transferrin and ferroportin, which caused an increase in intracellular iron. Dioscin in combination with chemotherapy drugs such as cisplatin, vemurafenib, rapamycin, and dacrbazine, also had synergistic effects in the melanoma cells [114]. Zeng et al., have shown that paclitaxel, nelarabine, dolastatin 10, actinomycin D, eribulin mesylate, vinorelbine, vinblastine, chelerythrine, docetaxel, and homoharringtonine are closely linked to ferroptosis in melanoma cells, the activation of ferroptosis showed good results in the patient survival. Therefore, they suggested that these drugs could be used as supplements or in combination with other drugs in the treatment of melanoma [131]. Generally, researchers showed the inducers of ferroptosis increased the therapeutic effects of chemotherapy in the melanoma cells (Fig. 2).
Radiotherapy
Radiotherapy often causes cell death by causing breaks in DNA structure. It has also been shown that radiotherapy indirectly reduces GSH and increases ROS production by inducing cell water radiolysis and increasing oxidase activity (Fig. 2). The effectiveness of radiotherapy increases with the reduction of GSH [109]. Lang et al., reported the radiotherapy-induced ferroptosis in cancer cells, such as melanoma. They also showed that immunotherapy synergistically increased the sensitivity of tumors to radiotherapy by reducing SLC7A11 and inducing ferroptosis (Fig. 2) [115]. Another study reported ferroptosis inducers (FINs) such as sorafenib, RSL3, sulfasalazine, and erastin, synergistically increased the effect of radiotherapy in various cancers, including melanoma, by reducing SLC7A11 expression or inhibiting GPX4 [116]. The therapeutic effects of cyst(e)inase, a recombinant human enzyme, which causes the breakdown of extracellular cysteine and cystine, have been studied in tumor cells. Cyst(e)inase increased ROS production and cell death by decreasing intracellular GSH levels. Researchers reported cyst(e)inase combined with radiotherapy enhanced lipid oxidation and had a synergistic effect on B16F10 melanoma cells [117]. Khorsandi et al., reported that pre-irradiation increased the anti-cancer function of gallic acid in melanoma cells by producing ROS, reducing GPX activity, and inducing lipid peroxidation [118]. Nagane et al., reported sulfasalazine, an inhibitor of the cystine-glutamate antiporter, reduced repair of damaged DNA, and intratumorally GSH concentration in B16F10 melanoma cells and synergistically increased the effect of radiotherapy in the cells [119].
Photodynamic therapy
Photodynamic therapy (PDT), a non-invasive and highly selective cancer treatment modality, has been studied in melanoma treatment. It involves the use of a photosensitizer activated by light to generate ROS, leading to localized cytotoxicity in tumor cells [132]. However, the efficacy of PDT in advanced melanoma still faces challenges that need to be addressed. It is worth exploring whether combining ferroptosis-targeted strategies with PDT can overcome the limitations.
The photodynamic treatment (PDT) and ferroptosis combined therapy were successful by loading methylene blue (MB) into SFT through the deposition of tannic acid (TA) and Fe3 + onto SRF nanocrystal [38]. Ferroptosis-induced nanomaterials can also happen through GSH metabolism. Based on the high surface area to volume ratio, the arginine-capped manganese silicate nanobubbles (AMSNs) were created with a high efficiency of GSH depletion [56]. According to an in-vivo investigation, AMSNs could suppress the formation of Huh7 xenograft tumors by downregulating GPX4. Liproxstatin-1, a ferroptosis inhibitor, might prevent this [57]. Researchers have synthesized a potent mitochondria-localized photosensitizer called cyclometalated Ir(III) complexes Ir-pbt-Bpa, which exhibits a strong antitumor impact on melanoma cells by inducing ferroptosis and restraining tumor growth in murine models [133].
Another study constructed a nanoparticle-based material named protoporphyrin IX-based polysilsesquioxane platform (PpIX-PSilQ NPs), which synergizes with PDT to mainly induce ferroptotic cell death by upregulating lipid peroxides and inactivation of GpX enzymes [134].
Hence, the use of combined ferroptosis-targeted strategies may provide alternative approaches in designing PDT to improve treatment outcomes Fig. 3.

Mechanisms governing ferroptosis by radiotherapy, chemotherapy, and immunotherapy. A The canonical GPX4-regulated pathway, the iron metabolism pathway, and the lipid metabolism pathway are the three pathways that start the process of ferroptosis and chemotherapy resistance reversal. The canonical GPX4-regulated pathway is regulated as follows: Directly inhibit GPX4 via increasing miR-324-3p, decreasing AR and KIF20A, inhibiting GSH production with ent-kaurane diterpenoids, and blocking cystine absorption with erastin and sorafenib, miR-375, and ATF3. The iron metabolism pathway is regulated as follows: DHA increases cellular LIP while repressing DMT1 and LCN2. The lipid metabolism pathway is regulated as follows: Target LOX by decreasing miR-522 and ACSL4 by decreasing ARF6. B T cells that have been stimulated by immunotherapy treatments release interferon (IFN), which causes ferroptosis. IFN- may reduce tumor cells' ability to take up cystine, which reduces the effectiveness of intracellular GPX4. C There were four phases in the mechanism of radiotherapy-induced ferroptosis. First, radiotherapy impairs system XC transport via ATM, which in turn impacts GSH production. Second step: By increasing ACSL4 expression, radiotherapy encourages lipid production. Third step: By generating DNA damage, radiotherapy triggers autophagy-dependent ferroptosis. Fourth step: Radiation therapy makes it easier for RT-MPs to be made, which leads to lipid peroxidation in nearby cells
Immunotherapy
Today, despite many advances in the treatment of melanoma, most patients experience resistance mechanisms of treatment and patient survival is limited due to progression, invasion, and metastasis. Immunotherapy using immune checkpoint inhibitors has dramatically improved the treatment of melanoma the deadliest type of skin cancer [135]. Immunotherapy is a relatively new method of cancer treatment that helps the immune system removes cancer cells and despite its specific benefits and performance, creating resistance to it is a major treatment challenge. Mechanisms of resistance to immunotherapy consist of two parts: 1- Tumor cell-intrinsic factors and 2- tumor cell-extrinsic factors [136]. Intrinsic factors are related to changes in the tumor cells themselves, such as the up-regulation or down-regulation of specific genes and pathways that prevent the penetration or function of immune cells in the microenvironment of the tumor. Tumor cell-extrinsic factors include factors separate from the tumor cells in the tumor microenvironment, such as regulatory T cells, and inhibitory immune checkpoints, which lead to the inhibition of immunity against tumor cells and the development of primary and/or adaptive resistance [136]. Inhibition of immune checkpoints by activating CD + 8 T cells induced ferroptosis in tumor cells, including melanoma. The researchers reported overexpression of TYRO3-suppressed ferroptosis and increased resistance to α-PD-1/PD-L1 immune checkpoint inhibitors. Upregulation of TYRO3 has been suggested as one of the pathways of ferroptosis resistance in tumors. It was observed that up-regulation of TYRo3 was associated with lower survival of treated melanoma patients with α-PD- 1checkpoint inhibitors. In cells with TYRO3 overexpression, the expression of ferroptosis-inhibiting genes such as SLC40A1, SLC7A11, SLC3A2, and GPX4 increased, while the genes that promoted ferroptosis such as SLC5A1 and TFRC decreased (Fig. 2) [135].
Another study has shown that cytokines secreted by T cells such as TNF-α and IFNγ in melanoma cell culture medium induced dedifferentiation and increased ferroptosis in the cells through activating of NF-κB or STAT1 signaling pathways [113]. It has also been shown that immunotherapy by activating CD8 + cells increase lipid peroxidation in melanoma cells and activates ferroptosis as a cytotoxic pathway in melanoma cells. Thus, the induction of ferroptosis in cells increased the effectiveness of immunotherapy. IFNγ secreted by T cells reduced SLC3A2 and SLC7A11 expression, thereby reducing the uptake of cyctine, which affects intracellular GHS levels and lipid peroxidation. Transcriptome analysis in nivolumab-treated melanoma patients showed the benefits of increasing IFNγ and decreasing SLC3A2 expression, which improved patient survival [120].
Combining a ferroptosis inducer with immunotherapy can also enhance the anti-tumor capacity. A study demonstrated that the joint treatment of erastin with an oncolytic virus (OV)-mediated cancer therapy resulted in a synergistic effect [126].
Erastin induced cytotoxicity on melanoma cells via ferroptosis but failed to generate productive and active antitumor immunity. However, co-treatment with OV and erastin improved the efficacy of OV and increased the infiltration of immune cells.
Furthermore, targeting ferroptosis-related signaling pathways can further enhance the performance of immunotherapy. Wnt/β-catenin signaling was also proven to regulate melanoma ferroptosis by increasing lipid peroxidation production [127].
ICG001 is a Wnt inhibitor that can enhance the effectiveness of anti-PD-1 immunotherapy by facilitating ferroptosis [127]. The introduction of ferroptosis improved the response to immunotherapy as well [137].
Conclusions and future outlooks
Melanoma cancer treatment is still a crucial challenge for humans. So far, various effective treatment approaches have been explored which most focus on apoptotic cancer cell death. Meanwhile, ferroptosis has defined which is different from apoptosis in biochemistry and morphology. Due to the fact that ferroptosis has shown good anticancer efficacy since its discovery, it can unveil a novel treatment horizon for defeating apoptosis resistance in multidrug-resistant cancers.
FDA-approved drugs altretamine, SAS, sorafenib, and nanoparticles as ferroptosis inducers in cancer build high chances for treatment of resistant cancer like melanoma. Taking into consideration these positive observations, ferroptosis is promised to be a bright melanoma treatment strategy soon, either alone or in combination therapy. However, there are still many concerns that more research is needed to address them.
Availability of data and materials
The data presented in this study are available in this manuscript.
References
Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445(7130):851–7.
Tod BM, Kellett PE, Singh E, Visser WI, Lombard CJ, Wright CY. The incidence of melanoma in South Africa: an exploratory analysis of National Cancer Registry data from 2005 to 2013 with a specific focus on melanoma in black Africans. S Afr Med J. 2019;109(4):246.
Godar DE, Subramanian M, Merrill SJ. Cutaneous malignant melanoma incidences analyzed worldwide by sex, age, and skin type over personal Ultraviolet-B dose shows no role for sunburn but implies one for Vitamin D 3. Dermatoendocrinol. 2017;9(1):e1267077.
Amaro A, Gangemi R, Piaggio F, Angelini G, Barisione G, Ferrini S, et al. The biology of uveal melanoma. Cancer Metastasis Rev. 2017;36(1):109–40.
Damsky WE, Bosenberg M. Melanocytic nevi and melanoma: unraveling a complex relationship. Oncogene. 2017;36(42):5771–92.
Turdo A, Veschi V, Gaggianesi M, Chinnici A, Bianca P, Todaro M, et al. Meeting the challenge of targeting cancer stem cells. Front Cell Dev Biol. 2019;18:7.
Wickremesekera AC, Brasch HD, Lee VM, Davis PF, Woon K, Johnson R, et al. Expression of cancer stem cell markers in metastatic melanoma to the brain. J Clin Neurosci. 2019;60:112–6.
Rambow F, Marine JC, Goding CR. Melanoma plasticity and phenotypic diversity: therapeutic barriers and opportunities. Genes Dev. 2019;33(19–20):1295–318.
López de Andrés J, Griñán-Lisón C, Jiménez G, Marchal JA. Cancer stem cell secretome in the tumor microenvironment: a key point for an effective personalized cancer treatment. J Hematol Oncol. 2020;13(1):136.
Domingues B, Lopes J, Soares P, Populo H. Melanoma treatment in review. Immunotargets Ther. 2018;7:35–49.
Ferrucci PF, Pala L, Conforti F, Cocorocchio E. Talimogene Laherparepvec (T-VEC): an intralesional cancer immunotherapy for advanced melanoma. Cancers (Basel). 2021;13(6):1383.
Olbryt M. Molecular background of skin melanoma development and progression: therapeutic implications. Adv Dermatol Allergol. 2019;36(2):129–38.
van Zeijl MCT, van den Eertwegh AJ, Haanen JB, Wouters MWJM. (Neo)adjuvant systemic therapy for melanoma. Eur J Surg Oncol. 2017;43(3):534–43.
Austin E, Mamalis A, Ho D, Jagdeo J. Laser and light-based therapy for cutaneous and soft-tissue metastases of malignant melanoma: a systematic review. Arch Dermatol Res. 2017;309(4):229–42.
Batus M, Waheed S, Ruby C, Petersen L, Bines SD, Kaufman HL. Optimal management of metastatic melanoma: current strategies and future directions. Am J Clin Dermatol. 2013;14(3):179–94.
Biteghe FN, Davids L. A combination of photodynamic therapy and chemotherapy displays a differential cytotoxic effect on human metastatic melanoma cells. J Photochem Photobiol B. 2017;166:18–27.
Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266–82.
Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3(3):285–96.
Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15(3):234–45.
Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. RAS–RAF–MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447(7146):865–9.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–72.
Gao M, Monian P, Jiang X. Metabolism and iron signaling in ferroptotic cell death. Oncotarget. 2015;6(34):35145–6.
Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59(2):298–308.
Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26(3):165–76.
Strasser A, Vaux DL. Cell death in the origin and treatment of cancer. Mol Cell. 2020;78(6):1045–54.
Hartman ML. Non-Apoptotic cell death signaling pathways in melanoma. Int J Mol Sci. 2020;21(8):2980.
Wang X, Hua P, He C, Chen M. Non-apoptotic cell death-based cancer therapy: Molecular mechanism, pharmacological modulators, and nanomedicine. Acta Pharm Sin B. 2022;12(9):3567–93.
Chen X, Comish PB, Tang D, Kang R. Characteristics and biomarkers of ferroptosis. Front Cell Dev Biol. 2021;21:9.
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25(3):486–541.
Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16(12):1180–91.
Liu Y, Wang Y, Liu J, Kang R, Tang D. The circadian clock protects against ferroptosis-induced sterile inflammation. Biochem Biophys Res Commun. 2020;525(3):620–5.
Li Q, Han X, Lan X, Gao Y, Wan J, Durham F, et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight. 2017;2(7):e90777. https://doi.org/10.1172/jci.insight.90777.
Chen X, Yu C, Kang R, Tang D. Iron metabolism in ferroptosis. Front Cell Dev Biol. 2020;7:8.
Ayala A, Muñoz MF, Argüelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev. 2014;2014:1–31.
Kagan VE, Mao G, Qu F, Angeli JPF, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81–90.
Wenzel SE, Tyurina YY, Zhao J, St. Croix CM, Dar HH, Mao G, et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell. 2017;171(3):628–641.e26.
Kapralov AA, Yang Q, Dar HH, Tyurina YY, Anthonymuthu TS, Kim R, et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol. 2020;16(3):278–90.
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317–31.
Xiao X, Jiang Y, Liang W, Wang Y, Cao S, Yan H, et al. miR-212-5p attenuates ferroptotic neuronal death after traumatic brain injury by targeting Ptgs2. Mol Brain. 2019;12(1):78.
Yuan H, Li X, Zhang X, Kang R, Tang D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun. 2016;478(3):1338–43.
Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91–8.
Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. 2019;21(5):579–91.
Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc. 2014;136(12):4551–6.
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23(3):369–79.
Wu Z, Geng Y, Lu X, Shi Y, Wu G, Zhang M, et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci. 2019;116(8):2996–3005.
Lo M, Wang YZ, Gout PW. The xc− cystine/glutamate antiporter: a potential target for therapy of cancer and other diseases. J Cell Physiol. 2008;215(3):593–602.
Zhang W, Trachootham D, Liu J, Chen G, Pelicano H, Garcia-Prieto C, et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat Cell Biol. 2012;14(3):276–86.
Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347(6):417–29.
Nielsen OH, Verspaget HW, Elmgreen J. Inhibition of intestinal macrophage chemotaxis to leukotriene B4 by sulphasalazine, olsalazine, and 5-aminosalicylic acid. Aliment Pharmacol Ther. 2007;2(3):203–11.
Wahl C, Liptay S, Adler G, Schmid RM. Sulfasalazine: a potent and specific inhibitor of nuclear factor kappa B. J Clin Investig. 1998;101(5):1163–74.
Lo M, Ling V, Low C, Wang YZ, Gout PW. Potential use of the anti-inflammatory drug, sulfasalazine, for targeted therapy of pancreatic cancer. Curr Oncol. 2010;17(3):9–16.
Doxsee DW, Gout PW, Kurita T, Lo M, Buckley AR, Wang Y, et al. Sulfasalazine-induced cystine starvation: potential use for prostate cancer therapy. Prostate. 2007;67(2):162–71. https://doi.org/10.1002/pros.20508.
Narang VS, Pauletti GM, Gout PW, Buckley DJ, Buckley AR. Sulfasalazine-induced reduction of glutathione levels in breast cancer cells: enhancement of growth-inhibitory activity of doxorubicin. Chemotherapy. 2007;53(3):210–7.
Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci. 2016;73(11–12):2195–209.
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–85.
Liang C, Zhang X, Yang M, Dong X. Recent progress in ferroptosis inducers for cancer therapy. Adv Mater. 2019;31(51):1904197.
Cotto-Rios XM, Gavathiotis E. Unraveling cell death mysteries. Nat Chem Biol. 2016;12(7):470–1.
Gaschler MM, Andia AA, Liu H, Csuka JM, Hurlocker B, Vaiana CA, et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat Chem Biol. 2018;14(5):507–15.
Lin H, Chen Y, Shi J. Nanoparticle-triggered in situ catalytic chemical reactions for tumour-specific therapy. Chem Soc Rev. 2018;47(6):1938–58.
Shen Z, Song J, Yung BC, Zhou Z, Wu A, Chen X. Emerging strategies of cancer therapy based on ferroptosis. Adv Mater. 2018;30(12):1704007.
Wang M, Chen Y, Cai W, Feng H, Du T, Liu W, et al. In situ self-assembling Au-DNA complexes for targeted cancer bioimaging and inhibition. Proc Natl Acad Sci. 2020;117(1):308–16.
Wang M, Zhao J, Jiang H, Wang X. Tumor-targeted nano-delivery system of therapeutic RNA. Mater Horiz. 2022;9(4):1111–40.
Shan X, Li S, Sun B, Chen Q, Sun J, He Z, et al. Ferroptosis-driven nanotherapeutics for cancer treatment. J Control Release. 2020;319:322–32.
Qian X, Zhang J, Gu Z, Chen Y. Nanocatalysts-augmented Fenton chemical reaction for nanocatalytic tumor therapy. Biomaterials. 2019;211:1–13.
Wang L, Huo M, Chen Y, Shi J. Iron-engineered mesoporous silica nanocatalyst with biodegradable and catalytic framework for tumor-specific therapy. Biomaterials. 2018;163:1–13.
Huo M, Wang L, Chen Y, Shi J. Tumor-selective catalytic nanomedicine by nanocatalyst delivery. Nat Commun. 2017;8(1):357.
Liu T, Liu W, Zhang M, Yu W, Gao F, Li C, et al. Ferrous-supply-regeneration nanoengineering for cancer-cell-specific ferroptosis in combination with imaging-guided photodynamic therapy. ACS Nano. 2018;12(12):12181–92.
Shen Z, Liu T, Li Y, Lau J, Yang Z, Fan W, et al. Fenton-reaction-acceleratable magnetic nanoparticles for ferroptosis therapy of orthotopic brain tumors. ACS Nano. 2018;12(11):11355–65.
Chen Q, Ma X, Xie L, Chen W, Xu Z, Song E, et al. Iron-based nanoparticles for MR imaging-guided ferroptosis in combination with photodynamic therapy to enhance cancer treatment. Nanoscale. 2021;13(9):4855–70.
Wang S, Li F, Qiao R, Hu X, Liao H, Chen L, et al. Arginine-rich manganese silicate nanobubbles as a ferroptosis-inducing agent for tumor-targeted theranostics. ACS Nano. 2018;12(12):12380–92.
Jiang M, Qiao M, Zhao C, Deng J, Li X, Zhou C. Targeting ferroptosis for cancer therapy: exploring novel strategies from its mechanisms and role in cancers. Transl Lung Cancer Res. 2020;9(4):1569–84.
He S, Jiang Y, Li J, Pu K. Semiconducting polycomplex nanoparticles for photothermal ferrotherapy of cancer. Angew Chem Int Ed. 2020;59(26):10633–8.
Ou W, Mulik RS, Anwar A, McDonald JG, He X, Corbin IR. Low-density lipoprotein docosahexaenoic acid nanoparticles induce ferroptotic cell death in hepatocellular carcinoma. Free Radic Biol Med. 2017;112:597–607.
Tsao H, Atkins MB, Sober AJ. Management of cutaneous melanoma. N Engl J Med. 2004;351(10):998–1012.
Kraemer KH, Greene MH. Dysplastic nevus syndrome. Familial and sporadic precursors of cutaneous melanoma. Dermatol Clin. 1985;3(2):225–37.
Letter to Editor, Cell Biol Res Ther Vol: 1 Issue: 2 Harnessing Autophagy for Melanoma Benefit Marco Corazzari1,2* and Penny E Lovat3.
Tang DYL, Ellis RA, Lovat PE. Prognostic impact of autophagy biomarkers for cutaneous melanoma. Front Oncol. 2016;9:6.
Karami Fath M, Azargoonjahromi A, Kiani A, Jalalifar F, Osati P, Akbari Oryani M, et al. The role of epigenetic modifications in drug resistance and treatment of breast cancer. Cell Mol Biol Lett. 2022;27(1):52.
Rosenkranz AA, Slastnikova TA, Durymanov MO, Sobolev AS. Malignant melanoma and melanocortin 1 receptor. Biochem Mosc. 2013;78(11):1228–37.
Cheng L, Lopez-Beltran A, Massari F, MacLennan GT, Montironi R. Molecular testing for BRAF mutations to inform melanoma treatment decisions: a move toward precision medicine. Mod Pathol. 2018;31(1):24–38.
González-Ruiz L, González-Moles MÁ, González-Ruiz I, Ruiz-Ávila I, Ayén Á, Ramos-García P. An update on the implications of cyclin D1 in melanomas. Pigment Cell Melanoma Res. 2020;33(6):788–805.
Muñoz-Couselo E, Zamora Adelantado E, Ortiz Vélez C, Soberino-García J, Perez-Garcia JM. <em>NRAS</em>-mutant melanoma: current challenges and future prospect. Onco Targets Ther. 2017;10:3941–7.
Ponti G, Manfredini M, Greco S, Pellacani G, Depenni R, Tomasi A, et al. BRAF, NRAS and C-KIT Advanced Melanoma: Clinico-pathological Features, Targeted-Therapy Strategies and Survival. Anticancer Res. 2017;37(12):7043–8. https://doi.org/10.21873/anticanres.12175.
Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, et al. Mutations in GNA11 in Uveal Melanoma. N Engl J Med. 2010;363(23):2191–9.
Xu D, Yuan R, Gu H, Liu T, Tu Y, Yang Z, et al. The effect of ultraviolet radiation on the transforming growth factor beta 1/Smads pathway and p53 in actinic keratosis and normal skin. Arch Dermatol Res. 2013;305(9):777–86.
Hajkova N, Hojny J, Nemejcova K, Dundr P, Ulrych J, Jirsova K, et al. Germline mutation in the TP53 gene in uveal melanoma. Sci Rep. 2018;8(1):7618.
Zhang H, Rosdahl I. Deletion in p16INK4a and loss of p16 expression in human skin primary and metastatic melanoma cells. Int J Oncol. 2004;24(2):331–5.
Mologni L, Costanza M, Sharma GG, Viltadi M, Massimino L, Citterio S, et al. Concomitant BCORL1 and BRAF mutations in vemurafenib-resistant melanoma cells. Neoplasia. 2018;20(5):467–77.
O’Connor CM, Perl A, Leonard D, Sangodkar J, Narla G. Therapeutic targeting of PP2A. Int J Biochem Cell Biol. 2018;96:182–93.
Arafeh R, Qutob N, Emmanuel R, Keren-Paz A, Madore J, Elkahloun A, et al. Recurrent inactivating RASA2 mutations in melanoma. Nat Genet. 2015;47(12):1408–10.
Stark M, Hayward N. Genome-wide loss of heterozygosity and copy number analysis in melanoma using high-density single-nucleotide polymorphism arrays. Cancer Res. 2007;67(6):2632–42.
Gagliardi M, Saverio V, Monzani R, Ferrari E, Piacentini M, Corazzari M. Ferroptosis: a new unexpected chance to treat metastatic melanoma? Cell Cycle. 2020;19(19):2411–25.
Friedmann Angeli JP, Krysko DV, Conrad M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer. 2019;19(7):405–14.
Li B, Yang L, Peng X, Fan Q, Wei S, Yang S, et al. Emerging mechanisms and applications of ferroptosis in the treatment of resistant cancers. Biomed Pharmacother. 2020;130:110710. https://doi.org/10.1016/j.biopha.2020.110710.
Wu Y, Yu C, Luo M, Cen C, Qiu J, Zhang S, et al. Ferroptosis in cancer treatment: another way to rome. Front Oncol. 2020;25:10.
Luo M, Wu L, Zhang K, Wang H, Zhang T, Gutierrez L, et al. miR-137 regulates ferroptosis by targeting glutamine transporter SLC1A5 in melanoma. Cell Death Differ. 2018;25(8):1457–72.
Zhang K, Wu L, Zhang P, Luo M, Du J, Gao T, et al. miR-9 regulates ferroptosis by targeting glutamic-oxaloacetic transaminase GOT1 in melanoma. Mol Carcinog. 2018;57(11):1566–76.
Sato M, Onuma K, Domon M, Hasegawa S, Suzuki A, Kusumi R, et al. Loss of the cystine/glutamate antiporter in melanoma abrogates tumor metastasis and markedly increases survival rates of mice. Int J Cancer. 2020;147(11):3224–35.
Ubellacker JM, Tasdogan A, Ramesh V, Shen B, Mitchell EC, Martin-Sandoval MS, et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature. 2020;585(7823):113–8.
Hartman ML. Non-Apoptotic Cell Death Signaling Pathways in Melanoma. Int J Mol Sci. 2020;21(8):2980. https://doi.org/10.3390/ijms21082980.
Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell. 2013;23(3):302–15. https://doi.org/10.1016/j.ccr.2013.02.003.
Schöckel L, Glasauer A, Basit F, Bitschar K, Truong H, Erdmann G, et al. Targeting mitochondrial complex I using BAY 87–2243 reduces melanoma tumor growth. Cancer Metab. 2015;3(1):1–16.
Gagliardi M, Cotella D, Santoro C, Corà D, Barlev NA, Piacentini M, et al. Aldo-keto reductases protect metastatic melanoma from ER stress-independent ferroptosis. Cell Death Dis. 2019;10(12):902. https://doi.org/10.1038/s41419-019-2143-7.
Wang Z, Jin D, Ma D, Ji C, Wu W, Xu L, et al. Ferroptosis suppressed the growth of melanoma that may be related to DNA damage. Dermatol Ther. 2019;32(4):e12921. https://doi.org/10.1111/dth.12921.
Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell. 2015;161(7):1681–96. https://doi.org/10.1016/j.cell.2015.05.044.
Yao F, Cui X, Zhang Y, Bei Z, Wang H, Zhao D, et al. Iron regulatory protein 1 promotes ferroptosis by sustaining cellular iron homeostasis in melanoma. Oncol Lett. 2021;22(3):657. https://doi.org/10.3892/ol.2021.12918.
Miura S, Shibazaki M, Kasai S, Yasuhira S, Watanabe A, Inoue T, et al. A somatic mutation of the KEAP1 gene in malignant melanoma is involved in aberrant NRF2 activation and an increase in intrinsic drug resistance. J Investig Dermatol. 2014;134(2):553–6.
Zhu B, Tang L, Chen S, Yin C, Peng S, Li X, et al. Targeting the upstream transcriptional activator of PD-L1 as an alternative strategy in melanoma therapy. Oncogene. 2018;37(36):4941–54.
Wu Y, Yu C, Luo M, Cen C, Qiu J, Zhang S, Hu K. Ferroptosis in Cancer Treatment: Another Way to Rome. Front Oncol. 2020;10:571127. https://doi.org/10.3389/fonc.2020.571127.
Tang F, Li S, Liu D, Chen J, Han C. Sorafenib sensitizes melanoma cells to vemurafenib through ferroptosis. Transl Cancer Res. 2020;9(3):1584–93.
Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547(7664):453–7. https://doi.org/10.1038/nature23007.
Osrodek M, Hartman ML, Czyz M. Physiologically Relevant Oxygen Concentration (6% O2) as an Important Component of the Microenvironment Impacting Melanoma Phenotype and Melanoma Response to Targeted Therapeutics In Vitro. Int J Mol Sci. 2019;20(17):4203. https://doi.org/10.3390/ijms20174203.
Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, et al. Multi-stage differentiation defines melanoma subtypes with differential vulnerability to drug-induced iron-dependent oxidative stress. Cancer Cell. 2018;33(5):890–904.e5.
Xie Y, Chen G. Biochemical and Biophysical Research Communications Dioscin induces ferroptosis and synergistic cytotoxicity with chemotherapeutics in melanoma cells. Biochem Biophys Res Commun. 2021;557:213–20.
Jiang L, Liao P, Zhou J, Zhang Q, Dow A, Saripalli AL, et al. Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of SLC7A11. Cancer Discov. 2019;9(12):1673–85.
Nie Q, Hu Y, Yu X, Li X, Fang X. Induction and application of ferroptosis in cancer therapy. Cancer Cell Int. 2022;22(1):1–19.
Cramer SL, Saha A, Liu J, Tadi S, Tiziani S, Yan W, et al. Systemic depletion of serum L-Cyst(e)ine with an engineered human enzyme induces production of reactive oxygen species and suppresses tumor growth in mice. Nat Med. 2017;23(1):120–7.
Khorsandi K, Kianmehr Z, Hosseinmardi Z, Hosseinzadeh R. Anti-cancer effect of gallic acid in presence of low level laser irradiation: ROS production and induction of apoptosis and ferroptosis. Cancer Cell Int. 2020;20(1):1–14.
Nagane M, Kanai E, Shibata Y, Shimizu T, Yoshioka C, Maruo T, et al. Sulfasalazine, an inhibitor of the cystine-glutamate antiporter, reduces DNA damage repair and enhances radiosensitivity in murine B16F10 melanoma. PLoS One. 2018;13(4):1–19.
Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569(7755):270–4. https://doi.org/10.1038/s41586-019-1170-y.
Mbah NE, Lyssiotis CA. Metabolic regulation of ferroptosis in the tumor microenvironment. J Biol Chem. 2022;298(3):101617.
Zhang F, Li F, Lu G hong, Nie W, Zhang L, Lv Y, et al. Engineering Magnetosomes for Ferroptosis/Immunomodulation Synergism in Cancer. ACS Nano. 2019;13:5662–73.
Chang MT, Tsai LC, Nakagawa-Goto K, Lee KH, Shyur LF. Phyto-sesquiterpene lactones DET and DETD-35 induce ferroptosis in vemurafenib sensitive and resistant melanoma via GPX4 inhibition and metabolic reprogramming. Pharmacol Res. 2022;178:106148.
Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547(7664):453–7.
Zeng F, Ye L, Zhou Q, He Y, Zhang Y, Deng G, et al. Inhibiting SCD expression by IGF1R during lorlatinib therapy sensitizes melanoma to ferroptosis. Redox Biol. 2023;61: 102653.
Liu W, Chen H, Zhu Z, Liu Z, Ma C, Lee YJ, et al. Ferroptosis Inducer Improves the Efficacy of Oncolytic Virus-Mediated Cancer Immunotherapy. Biomedicines. 2022;10(6):1425.
Wang H, Zhang H, Chen Y, Wang H, Tian Y, Yi X, et al. Targeting Wnt/β-catenin signaling exacerbates ferroptosis and increases the efficacy of melanoma immunotherapy via the regulation of MITF. Cells. 2022;11(22):3580.
Ke L, Wei F, Xie L, Karges J, Chen Y, Ji L, et al. A Biodegradable Iridium(III) Coordination Polymer for Enhanced Two-Photon Photodynamic Therapy Using an Apoptosis-Ferroptosis Hybrid Pathway. Angew Chem Int Ed Engl. 2022;61(28):e202205429. https://doi.org/10.1002/anie.202205429.
Lu B, Chen XB, Ying MD, He QJ, Cao J, Yang B. The role of ferroptosis in cancer development and treatment response. Front Pharmacol. 2018;8(JAN):1–8.
Zhang C, Liu X, Jin S, Chen Y, Guo R. Ferroptosis in cancer therapy: a novel approach to reversing drug resistance. Mol Cancer. 2022;21(1):1–12.
Zeng H, You C, Zhao L, Wang J, Ye X, Yang T, et al. Ferroptosis-Associated Classifier and Indicator for Prognostic Prediction in Cutaneous Melanoma. J Oncol. 2021;2021:3658196. https://doi.org/10.1155/2021/3658196.
Khorsandi K, Hosseinzadeh R, Esfahani H, et al. Accelerating skin regeneration and wound healing by controlled ROS from photodynamic treatment. Inflamm Regen. 2022;42:40. https://doi.org/10.1186/s41232-022-00226-6.
Wang L, Karges J, Wei F, Xie L, Chen Z, Gasser G, et al. A mitochondria-localized iridium( <scp>iii</scp> ) photosensitizer for two-photon photodynamic immunotherapy against melanoma. Chem Sci. 2023;14(6):1461–71.
Vadarevu H, Juneja R, Lyles Z, Vivero-Escoto JL. Light-Activated Protoporphyrin IX-Based Polysilsesquioxane Nanoparticles Induce Ferroptosis in Melanoma Cells. Nanomaterials. 2021;11(9):2324.
Talty R, Bosenberg M. The role of ferroptosis in melanoma. Pigment Cell Melanoma Res. 2022;35(1):18–25.
Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168(4):707–23.
Wang G, Xie L, Li B, Sang W, Yan J, Li J, et al. A nanounit strategy reverses immune suppression of exosomal PD-L1 and is associated with enhanced ferroptosis. Nat Commun. 2021;12(1):5733.
Acknowledgements
All figures were created in BioRender.com.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
KK (Khatereh Khorsandi) contributed to the design and supervise the review. KK, HSE (HomaSadat Esfahani), SKG (Saeedeh Keyvani- Ghamsari), and PL (Parisa lakhshehei) contributed to the data collection and draft of the manuscript. KK read and edited the final version. All authors gave final approval.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Khorsandi, K., Esfahani, H., Ghamsari, S.K. et al. Targeting ferroptosis in melanoma: cancer therapeutics. Cell Commun Signal 21, 337 (2023). https://doi.org/10.1186/s12964-023-01296-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-023-01296-w