
Abstract
Background
Metabolic reprogramming is a critical event for cell fate and function, making it an attractive target for clinical therapy. The function of metabolic reprogramming in Helicobacter pylori (H. pylori)-infected gastric intestinal metaplasia remained to be identified.
Methods
Xanthurenic acid (XA) was measured in gastric cancer cells treated with H. pylori or H. pylori virulence factor, respectively, and qPCR and WB were performed to detect CDX2 and key metabolic enzymes expression. A subcellular fractionation approach, luciferase and ChIP combined with immunofluorescence were applied to reveal the mechanism underlying H. pylori mediated kynurenine pathway in intestinal metaplasia in vivo and in vitro.
Results
Herein, we, for the first time, demonstrated that H. pylori contributed to gastric intestinal metaplasia characterized by enhanced Caudal-related homeobox transcription factor-2 (CDX2) and mucin2 (MUC2) expression, which was attributed to activation of kynurenine pathway. H. pylori promoted kynurenine aminotransferase II (KAT2)-mediated kynurenine pathway of tryptophan metabolism, leading to XA production, which further induced CDX2 expression in gastric epithelial cells. Mechanically, H. pylori activated cyclic guanylate adenylate synthase (cGAS)-interferon regulatory factor 3 (IRF3) pathway in gastric epithelial cells, leading to enhance IRF3 nuclear translocation and the binding of IRF3 to KAT2 promoter. Inhibition of KAT2 could significantly reverse the effect of H. pylori on CDX2 expression. Also, the rescue phenomenon was observed in gastric epithelial cells treated with H. pylori after IRF3 inhibition in vitro and in vivo. Most importantly, phospho-IRF3 was confirmed to be a clinical positive relationship with CDX2.
Conclusion
These finding suggested H. pylori contributed to gastric intestinal metaplasia through KAT2-mediated kynurenine pathway of tryptophan metabolism via cGAS-IRF3 signaling, targeting the kynurenine pathway could be a promising strategy to prevent gastric intestinal metaplasia caused by H. pylori infection.
Video Abstract
Similar content being viewed by others


Background
Helicobacter pylori (H. pylori), classified as a class I carcinogen by World Health Organization, is a critical factor for gastritis and gastric intestinal metaplasia (IM) [1, 2]. IM has been regarded as a precancerous lesion in the H.pylori-induced metaplasia-dysplasia-carcinoma sequence, which was characterized by increased CDX2 and/or MUC2 expression [3, 4], an intestinal specific homeobox gene that regulated the development and maintenance of the intestinal mucosa phenotype [5]. The previous work has revealed that H. pylori virulence factor CagA contributed to gastric carcinoma cells invasiveness through upregulating CDX2-mediated Claudin-2 expression in AGS gastric cancer cells, thereby disrupting tight junction [6], and H. pylori promoted CDX2-mediated IM through NF-κB [7], SOX2 [8] and Activin A receptor type I (ACVR1) [3]. However, the mechanisms that regulate CDX2 expression during H. pylori-induced IM have not been fully defined.
Alteration of metabolic reprogramming plays a crucial role in the development of H. pylori-related gastric disease [9, 10]. For instance, in H.pylori-infected macrophages, cystathionine γ-lyase (CTH, also known as CSE) triggered the mammalian reverse transsulfuration pathway (RTP)-mediated SAM metabolism and induced macrophages activation through enhancing mitochondrial function and glycolysis [11], and H.pylori aggravated the severity of gastric inflammation and the development of premalignant lesions in the setting of iron deficiency through enhanced bile acid production [12, 13]. What’s more, the study showed that H. pylori virulence factor CagA contributed to 5-Fu resistance of gastric cancer cells through upregulating Hexokinase 2 (HK2) and LDHA-mediated glycolysis [14]. In addition, our previous work have demonstrated that fatty acid synthase (FASN) and ATP citrate lyase (ACLY)-mediated De novo lipogenesis (DNL) was increased in gastric mucosa with H.pylori infection [15], and HK2-mediated glycolysis was enhanced in gastric mucosa of patients with H.pylori infection [16]. Interestingly, bile acids (BAs) was reported to induce CDX2-mediated gastric intestinal metaplasia through HNF4α [17] and a major RNA N6-adenosine demethylase, named alkylation repair homolog protein 5 (ALKBH5) [18] as well as FXR [19, 20]. However, the further work was required to elucidate the role of metabolism reprogramming during H. pylori infection.
Kynurenine pathway is the main route of tryptophan metabolism and produces several intermediate metabolites with various biologic properties [21, 22]. Tryptophan(Trp) could be converted into kynurenine(KYN) by the first and rate-limiting enzymes indoleamine 2,3-dioxygenase (IDO) and/or tryptophan 2,3-dioxygenase (TDO) [23]. KYN could be catalyzed into 3-Hydroxy kynurenine (3HK) by kynurenine 3-monoxygenase (KMO), which can be further metabolized into 3-Hydroxy anthranilic acid (3HAA) and xanthurenic acid (XA) by kynureninase (KYNU) and kynurenine aminotransferase II (KAT2), respectively [24, 25]. What’s more, 3HAA could be further converted into quinolinic acid (QUIN) and picolinic acid (PIC) by 3-hydroxyanthranilate-3,4-dioxygenase (QPRT) and 7, 2-amino-3-carboxymuconic acid semialdehyde decarboxylase (ACMSD), respectively. In addition, kynurenine can be converted to anthranilic acid (QA) [26]. Recently, Trp depletion and KYN metabolites generation were identified in plasma of GC through disease progression within the Correa’s cascade [27], and enhanced KYN level in serum has been showed to facilitate Treg induction in vitro to promote chemotherapy resistance in gastric cancer cells through IL-10/STAT3/BCL2 [28]. What’s more, KYN interacted with AHR to suppress immune and subsequently, susceptibility to gastric cancer [29]. In addition, L-Kyn was confirmed to promote COL12A1 expression to induce gastric cancer cell growth and migration [30]. Importantly, the clinical data further showed significant increased kyn/Trp ratio, observed in H. pylori seropositive cancer patients, could be identified as potential gastric cancer biomarkers (<10.1515_pteridines.2010.21.1.110.pdf>) [31]. Herein, in this work, we further demonstrated that H. pylori triggered IRF3-mediated kynurenine pathway, leading to XA production, which induced CDX2 expression in gastric epithelial cells. Inhibition of KAT2 expression by PF-04859989 hydrochloride could reverse the promotion of CagA and VacA on CDX2 expression. Mechanically, H. pylori triggered cGAS/STING/IRF3 pathway to enhance IRF3 nuclear translocation and increase the binding of IRF3 to KAT2 promoter confirmed by luciferase and ChIP assay in BGC823 cells. Inhibition of IRF3 in gastric epithelial cells could rescue KAT2 expression induced by H. pylori in vivo and in vitro. Taken together, we identified a previously unreported mechanism that H. pylori-mediated gastric IM through activation of kynurenine pathway.
Methods and materials
Reagents and antibodies
Reagents
Dulbecco’s modified Eagle’s medium (DMEM, C11995500BT), and fetal bovine serum (FBS, 10099141C) were purchased from Life Technologies (Carlsbad, CA, USA); Recombinant helicobacter pylori CagA (ab224836), VacA (ab225655), Phenylmethanesulfonyl fluoride (PMSF, P0100) and protease inhibitor cocktail (PIC, P6730) were from Solarbio (Beijing, China); Ratio-immunoprecipitation assay (RIPA) lysis buffer(P0013B), Nuclear and Cytoplasmic Protein Extraction Kit (P0028), bicinchoninic acid (BCA) protein assay kit (P0012) and Beyozol RNA Isolation Kit (R0011) were from Beyotime Biotechnology (Shanghai, China); All-in-One First-Strand cDNA Synthesis Kit (QP006) and All-in-One qPCR Mix (QP005) were obtained from GeneCopoeia (Rockville, MD, USA). Dual-Luciferase Reporter Assay was purchased from Promega. SimpleChIP® Plus Enzymatic Chromatin IP Kit (Magnetic Beads) was purchased from CST. PF-04859989 HCI(T28368), MRT67307 HCI(1190378–57-4(free base))( T5162) and Xanthurenic Acid(S4774) was from Topscience and Selleck, respectively. All ultrapure reagents were from Sigma (St Louis, MO, USA).
Antibodies
Antibody against CDX2(60243–1-Ig, Proteintech1:2000 for WB, 1:800 for IF of human sample), CDX2 Monoclonal Antibody(14H6) (YM3057, immunoway, 1:800 for IF and IHC of mice gastric mucosa), MUC2(ab272692, Abcam,1:2000 for WB, 1:800 for IF), TDO2(15880–1-AP, Proteintech, 1:1500 for WB), KMO(YT6625, immunoway, 1:2000 for WB), AADAT (A13089, Abclonal, 1:1000 for WB), AADAT polyclonal antibody(13031–1-AP, Proteintech,1:800 for IHC/IF), α-tubulin(RM2007, Ray, 1:4000 for WB), Histone 3 (RM2005, Ray, 1:4000 for WB), phospho-cGAS-S291 Rabbit pAb(Abclonal, AP1176, 1:1000 for WB), cGAS Rabbit pAb (Abclonal, A8335, 1:1000 for WB), phospho-STING-S365 Rabbit pAb (Abclonal, AP1199, 1:1000 for WB), STING/TMEM173 Rabbit pAb (Abclonal, AP1176, 1:1000 for WB); IRF3(Immunoway, YT2398, 1:1000 for WB), Phospho-IRF3(s396)(Immunoway,YP0326, 1:1000 for WB), phospho-IRF3 (Ser396) Antibody (AF2436, affinity, 1:800 for IF), alexa-488- and 594-conjugated secondary antibodies were from Immunoway (Beijing, China).
Cell culture, treatment, and transfection
As described in our pervious study [32], GES-1 and BGC823 cells were from American Type Culture Collection and were cultured in DMEM supplemented with 10% fetal bovine serum (FBS) according to the manufacturer’s recommendations. For treatment, gastric epithelial cells were used in the whole study and treated with CagA(1ug/ml), VacA(1ug/ml) or CagA combined with vacA for 48 h. For transfection, plasmid or siRNA was transfected into cell using lipofectamine 3000 according to the manufacturer’s protocol. siRNA targeted IRF3(sc-35710) was purchased from Santacruz. KAT2 plasmid was from Youbio (Changshang, China).
H. pylori bacterial strains culture and infection
According to the work from Soutto et al. [33], The CagA+ H. pylori strains “J166”, a clinical isolate of human-derived H. pylori, was cultured on columbia agar plates containing 5% (v/v) sheep blood at 37 °C under microaerobic conditions. H. pylori was collected and resuspended in PBS. For infection, BGC823 (a human gastric carcinoma undifferentiated cell line) was cultured and washed with PBS three times and then infected with H. pylori at an MOI of 100 for 48 h after analysis of the quantity of bacteria by spectrophotometry.
RNA isolation and quantitative real-time PCR
As described in our pervious study [34], Total RNA was extracted and reverse transcripted into cDNA according to Beyozol RNA Isolation Kit and the All-in-one™ first-strand cDNA synthesis kit (Genecopoeia™, FulenGen), respectively. Quantitative PCR (qPCR) were carried out using the All-in-one™ qPCR mix (Genecopoeia™, FulenGen) according to the manufacturer’s instructions. Primer sequences used in this study were as followed: CDX2: forward:5′-CTCGGCAGCCAAGTGAAAACCA-3′, and reverse: 5′-GCTTTCCTCCGGATGGTGATGTA-3′ [35]; Villin: forward:5′-TCGGCCTCCAGTATGTAG-3′, and reverse: 5′- CGTCTTCGGGGTAGAACT-3′ [36]; MUC2: forward:5’-CAGGATGGCGCCTTCTGCTA-3’, and reverse:5’-ATGCTGCTCCAAGCTGAGGT-3’ [37]; TDO2: forward:5’-TCCTCAGGCTATCACTACCTGC-3’ and reverse: 5’-ATCTTCGGTATCCAGTGTCGG-3’; KMO: forward:5’-TGCCATCCCTCTAATTGGAGA-3’ and reverse: 5’-GCCCGCATTCATTCCTTGC-3’; KAT2: forward:5’- CACTTCAGTATTCTCCGAGTGC-3’ and reverse: 5’- AGCAGGTTCATCTAGGAGGAC-3’; UBC (internal control): forward:5’-ATTTGGGTCGCGGTTCTTG-3’ and reverse: 5’-TGCCTTGACATTCTCGATGGT-3’.
Western blotting (WB) analysis
As described in our work [5], after treatment, cells were collected and lysed in RIPA buffer containing 1% protease inhibitors, SDS-PAGE was performed to separate after quantitation. Proteins were transferred nitrocellulose (NC) membranes (PALL) and incubated primary antibodies for overnight at 4 °C. the further incubation with the secondary antibodies for 1 h at room temperature was required after washing with PBS. The protein level was detected using an enhanced chemiluminescence (Perkin Elmer).
Immunofluorescence (IF) analysis
As described in pervious study [38], after treatment, fixed and permeabilized gastric epithelial cell or gastric mucosa tissue slides were incubated with 2% BSA in PBS/0.05% Triton X-100 for 30 min. the slips were incubated with the primary antibody overnight at 4 °C, followed by incubation with Alexa-488- or Alexa 594- conjugated secondary antibodies for 1 h at room temperature. The coverslips were mounted onto glass slides with Prolong Gold Antifade reagent (after staining the nuclei with DAPI), and stained cells were imaged with a Leica TCS SP8 Inverted Fluorescence Microscope (Leica Microsystems). Post-acquisition processing (brightness, opacity, contrast, and color balance) was applied to the entire image and accurately reflected the results of the original image. For statistical analysis, as described in our previous work [39], 5 areas of each single gastric mucosa section were randomly selected for quantification. The number per square millimeter was counted and analyzed using Leica X image analysis software (Leica, Hamburg, Germany) and ImageJ software (National Institutes of Health, MD, USA).
Immunohistochemical staining
Briefly, the paraffin-embedded tissues were deparaffinized in xylene and gradient ethanol. Three percent hydrogen peroxide (H2O2) solution was used to block endogenous peroxidase activity after antigen retrieval with sodium citrate buffer. The slide was incubated overnight at 4 °C with the primary antibody. After washing with PBS, the sections were incubated with secondary antibody using PV-6001/6002 kit (Zhongshan Biotech, Beijing, China). The slides were stained with diaminobenzidine (DAB) chromogen and counterstained with hematoxylin. All images were acquired under a microscope (Leica, Hamburg, Germany). The immunoreactivity of the samples was scored for intensity (scaled 0–4) and frequency (scaled 0–4). For the statistical analysis, the intensity and frequency of targets were transformed into a composite expression score using the following formula intensity × frequency. The score ranges from 0 to 16.
Xanthurenic acid (XA) detection assay
After serum starved for 24 h, cells were treated with CagA and VacA or CagA combined with VacA or H. pylori, respectively, for 48 h, the level of XA in indicated supernatant were collected and detected according to the manufacturer’s protocol (MEIMIAN, MM-9263001).
Luciferase reporter assay
Cells were co‐transfected with the promoter reporter plasmid (KAT2‐Luc) or internal control plasmid (pGL4.74[hRluc/TK], Promega) with siRNA targeted IRF3. Twenty‐four hours after transfection, the cells were treated for further 24 h, The Firefly and Renilla luciferase value in cells were measured using the Dual‐Luciferase Reporter Assay System (Promega).
Chromatin immunoprecipitation (ChIP)
As described in previous studies [34, 40], gastric epithelial cells were grown up to 80% confluence, the cell was treated with serum-free medium for 24 h, and stimulated for another 1 h, the ChIP was performed using the SimpleChIP® Plus Enzymatic Chromatin IP Kit (Magnetic Beads) according to the manufacturer’s protocol. Quantitative PCR of co-immunoprecipitated genomic DNA fragments was performed with specific primers was synthesized from thermo life. Primers to amplify the proximal region of the KAT2 promoter containing a IRF3‐binding site was forward:5’- TGCAGAAAAGTTGAGGGAGGT-3’ and reverse: 5’- TTTTTGGGTCTAGCACCTTGCC-3’.
Animals infection model
As described in Soutto et al. [33], for in vivo, 6–8 weeks of age were orally gavaged with rodent-adapted CagA+ H. pylori strain pre-murine Sydney Strain 1 (PMSS1, 2 × 109 CFU)/mouse) every other day or challenged with Brucella Broth (BB) as an uninfected control group, After two weeks, one group of PMSS1 infected mice received PF-04859989(50 mg/kg) or MRT67307 (50 mg/kg) through intraperitoneal injection once every 2 days. Uninfected and H. pylori infected mice were used as control groups receiving injection with 1xPBS. Mice were euthanized a day after the last injection. All mice were euthanized to collect gastric mucosa for experiment.
Ethical approval of clinical sample studies
This study was conducted in a cohort of 14 Hp-infected patients diagnosed with gastric intestinal metaplasia, 12 healthy control upon the declaration of Helsinki as reflected in a prior approval by the institution's human research committee from 2016 to 2022 approved by the affiliated hospital of Putian university. Written informed consent was given by the caregiver of the patient for his clinical records used, which are not publicly available since the database is currently not anonymous and contains all patient's name; however, it could be available upon request.
Statistics analysis
GraphPad Prism V software was used to conduct all analysis. A P value less than 0.05 was considered to be statistical significant. Statistical differences among groups were determined by Student's t test, one sample t tests, one‐way ANOVA or two‐way ANOVA were used to determine the significance.
Results
KAT2/XA promoted gastric intestinal metaplasia
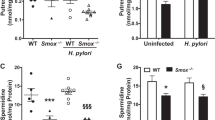
Metabolic reprogramming is critical for determination of cell fate. Specifically, a total of 37 differentially expressed metabolites (DEMs) metabolites, including xanthurenic acid (XA), were identified in BGC823 cells treated with H. pylori virulence factor CagA and VacA compared with Control group by liquid chromatography-tandem mass spectrometry (LC–MS/MS) (data unpublished). Consistently, enhanced xanthurenic acid (XA), a production of kynurenine pathway, was found in BGC823 cells in response to CagA, VacA, and CagA + VacA treatment comparing with Control group confirmed by Elisa (Fig. 1A). In line with this, the increased XA level, enhanced protein and mRNA level of CDX2, MUC2 and villin expression were significantly observed in BGC823 cells in response to H. pylori infection (Fig. 1B and C). these results implied H. pylori infection induced gastric intestinal metaplasia accompanying with XA production.

Enhanced XA/KAT2 in H. pylori infection induced gastric epithelial. Elisa was employed to detect the level of xanthurenic acid (XA) in BGC823 cells in response to CagA, VacA and CagA + VacA (A) or H. pylori infection (B) for 48 h, data presented as the means ± s.d. of three independent experiments and were analyzed by one-way ANOVA with multiple comparisons, followed by Dunnett post hoc test for significance, **p < 0.01, ***p < 0.001.****p < 0.0001. C After serum starvation for 24 h, BGC823 cells were incubated with H. pylori for additional 48 h. The total lysate was collected to separate by SDS to detect indicated protein expression. The band intensity was measured and quantified by Image J. Data represent the mean ± s.d. of three independent experiments and were analyzed by t test for significance. ****p < 0.0001. D the total RNA was isolated and quantitative real-time PCR was performed to analyzed indicated gene expression in BGC823 cells treated with XA at 5uM and 10uM. Data presented as the means ± s.d. of three independent experiments and were analyzed by one-way ANOVA with multiple comparisons, followed by Dunnett post hoc test for significance, ****p < 0.0001. E immunoblotting was used to detect CDX2 expression in BGC823 and GES-1 cells treated with 10uM XA for 48 h, respectively. α-tubulin was determined to ensure equal loading. Data presented as the means ± s.d. of three independent experiments and were analyzed by t test for significance, ****p < 0.0001. F BGC823 cells were transfected with pCDNA3.0 and KAT2 plasmid for 48 h, and WB was used to test indicated protein expression, the band intensity was quantified by Image J. Data represent the mean ± s.d. of three independent experiments and were analyzed by t test for significance. ****p < 0.0001. G-H HE, IHC and IF assay was used to detect KAT2 expression in gastric mucosa of patients with IM and in animal model. The KAT2 expression level was quantified and analyzed by t test for significance, ***p < 0.001, ****p < 0.0001
To explore whether XA caused by H. pylori infection involved in gastric intestinal metaplasia, real-time PCR and WB analysis were utilized to examine. The results showed that XA treatment in BGC823 cells led to a significant upregulation of CDX2, MUC2 and Villin mRNA expression in a dose-dependent manner (Fig. 1D), and WB and quantified results showed that 10uM XA could induce CDX2 expression in BGC823 and GES-1 cells, respectively (Fig. 1E). Because of XA synthesis is dependent on the activity of mitochondrial KAT2, which focus us to determine the role of KAT2 in gastric intestinal metaplasia. As shown in Fig. 1F, ectopic expression of KAT2 in BGC823 cells led to enhance CDX2 and MUC2 expression. Interestingly, immunohistochemical staining and fluorescence displayed KAT2 expression was drastically increased in patients diagnosed with gastric intestinal metaplasia and positive H. pylori infection (IM) in comparsion with healthy control (HC) (Fig. 1G). What’s more, histological examination of the gastric mucosa revealed that mice infected with the H. pylori PMSS1 strain showed KAT2 expression was notably enhanced in gastric mucosa of mice with H. pylori PMSS1 strain infection (Fig. 1H). These findings suggested KAT2/XA was increased in response to H. pylori infection and promoted gastric intestinal metaplasia.
H. pylori contributed to gastric intestinal metaplasia through KAT2-mediated kynurenine pathway
The above results implied that kynurenine pathway of tryptophan metabolism might play a vital role in H. pylori-induced gastric intestinal metaplasia, which focused us to explore the effect of H. pylori on the rate-limiting enzymes expression of 3-HK branch of tryptophan metabolism, including TDO2, KMO and KAT2. Our results demonstrated that treatment with H. pylori virulence factor CagA/VacA in BGC823 cells led to a significant upregulation of KAT2 expression at the mRNA level compared with control group; no significant difference in the expression of KMO and TDO2 was observed (Fig. 2A). In line with this, the result from WB and quantified analysis showed that H. pylori virulence factors, including CagA and VacA, or H. pylori infection resulted in enhancing CDX2, MUC2 and KAT2 expression in BGC823 cells, while failed to alter KMO and TDO2 expression (Fig. 2B and C). Taken together, these results suggested H. pylori promoted kynurenine pathway by triggering KAT2 expression, leading to XA production.

H.pylori triggered KAT2-mediated kynurenine pathway to induce CDX2 expression in gastric epithelial cells. A Real-time PCR was used to analyze the indicated genes expression in response to CagA, VacA treatment for 48 h in BGC823 and GES-1 cells, respectively. Data presented as the means ± s.d. of three independent experiments and were analyzed by one-way ANOVA with multiple comparisons, followed by Dunnett post hoc test for significance, ***p < 0.001, NS:no significant. B WB was performed to detect the indicated proteins expression in BGC823 cells stimulated by CagA, VacA and CagA + VacA for 48 h, α-tubulin was taken as internal control. Data presented as the means ± s.d. of three independent experiments and were quantified by two-way ANOVA with multiple comparisons, followed by Dunnett post hoc test for significance, ****p < 0.001, NS: no significant. C WB was employed to detect the indicated proteins expression in BGC823 cells treated with or without H.pylori for 48 h, α-tubulin was taken as internal control. Data presented as the means ± s.d. of three independent experiments, the band intensity was quantified and quantified by t test for significance, ****p < 0.001, NS: no significant, n = 3. After serum starvation for 24 h, BGC823 cells were pre-treated for 1 h with KAT2 inhibitor PF-04859989 (10uM) following by treatment as indicated (D-F) for additional 48 h. The total lysate was harvested to detect indicated proteins, (D right panel, G-H) Data represent the mean ± s.d. of three independent experiments and were analyzed by One-way ANOVA test for significance for D and two-way ANOVA with multiple comparisons, followed by Dunnett post hoc test for significance for E–F. ***p < 0.001, ****p < 0.0001. I representative histological image of gastric mucosa of C57BL/6 mice in different groups (scale bars: 75 μm), IHC staining showing CDX2 expression in gastric tissue from the indicated groups of mice (scale bars: 50 μm), Data represented the mean ± s.d. and was analyzed by One-way ANOVA test for significance. ****P < 0.0001. J Immunofluorescence showing CDX2 and MUC2 expression in gastric mucosa tissue in indicated group, respectively. Data represented the mean ± s.d. and was analyzed by One-way ANOVA test for significance. ****P < 0.0001
To further confirm whether KAT2 was required for in the H. pylori-mediated gastric intestinal metaplasia, the rescue experiment was performed through KAT2 inhibitor PF-04859989 [41, 42]. As shown in Fig. 2D, KAT2 inhibition in BGC823 cells drastically overcame the effect of H. pylori on gastric intestinal metaplasia characterized by enhanced CDX2 and MUC2 expression. What’s more, inhibition of KAT2 by PF-04859989 in BGC823 and GES-1 cells could largely abrogate the upregulation of CDX2 and MUC2 expression caused by H. pylori virulence factors (Fig. 2E-H). Most importantly, In vivo results also further confirmed that PF-04859989 alleviated the gastric intestinal metaplasia caused by H. pylori PMSS1 infection as confirmed by CDX2 expression through IHC and IF (Fig. 2I), in addition to IHC, IF also showed reduced CDX2 and MUC2 expression level was observed in H. pylori PMSS1 infection receiving KAT2 inhibitor treatment (Fig. 2J). Taken together, these findings suggested that H. pylori induced gastric intestinal metaplasia through KAT2-mediated kynurenine.
IRF3 was required for H. pylori-mediated KAT2 expression
Activation of Interferon Regulatory Factor 3 (IRF3) signaling has been shown to regulate type I interferon β (IFN- β) expression [43], which is belong to tryptophan cluster factors [44]. To explore whether H. pylori-dependent expression of KAT2 could be attributed to activation of IRF3 signaling. As shown in Fig. 3A, overexpression of IRF3 in BGC823 cells led to a significant increased relative luciferase unit (RLU) of KAT. While depletion of IRF3 expression in BGC823 cells drastically inhibited KAT2 transactivity, which further reversed the enhancement of KAT2 transactivity caused by CagA and/or VacA (Fig. 3B). In line with this, H. pylori infection in BGC823 cells led to increase KAT2 transactivation, while knockdown of IRF3 expression could reverse the enhanced KAT2 transactivation caused by H. pylori infection (Fig. 3C), these finding suggested that IRF3 is vital for H. pylori-mediated KAT2 expression.

IRF3 was crucial for H. pylori-mediated KAT2 transactivation. A BGC823 cells were co-transfected with indicated plasmids combined with KAT2-Luc plasmid and a control Renilla luciferase expression vector for 48 h, the relative luciferase unit (RLU) was presented as the fold activation relative to Renilla luciferase activity. Data presented as the means ± s.d. of three independent experiments and was analyzed by t test, ****p < 0.0001. B After transfection with si-CTL and si-IRF3 combined with KAT2-Luc and Renilla plasmids, respectively, for 24 h, BGC823 cells were starved for 12 h following by stimulation with CagA, VacA and CagA + VacA for further 48 h, the RLU was detected using dual‐luciferase reporter assay system (Promega). Data represented as the mean ± s.d. of three independent experiments and were analyzed by two-way ANOVA with multiple comparisons, followed by Bonferroni post hoc test for significance. **p < 0.01, ****p < 0.0001 versus si-CTL, ####p < 0.0001 versus si-IRF3. C The BGC823 cells were transfected as described in B and starved for 12 h following by stimulation with H. pylori for further 48 h, the RLU was detected using dual‐luciferase reporter assay system (Promega). Data represented as the mean ± s.d. of three independent experiments and were analyzed by two-way ANOVA with multiple comparisons, followed by Bonferroni post hoc test for significance. ****p < 0.0001 versus si-CTL. D left panel: BGC823 cells were serum starved and treated as indicated for 1 h. The whole cell lysates were immunoprecipitated with an anti-IRF3 antibody to collect the chromosome segment that binding of IRF3 to KAT2 promoter in vivo, the fragments were amplified and quantified by real-time PCR. right panel: real-time PCR of a nonspecific region corresponding to CDS of the KAT2 gene enriched by IRF3 (negative control). Data represent the mean ± s.d. of three independent experiments and were analyzed by two-way ANOVA with multiple comparisons, followed by Bonferroni post hoc test for significance. ***p < 0.001. E BGC823 cells were starved for 12 h following by stimulation with or without H. pylori for 1 h, the ChIP was performed with anti-IRF3 antibody as described in D. Data represent the mean ± s.d. of three independent experiments and were analyzed by two-way ANOVA with multiple comparisons, followed by Bonferroni post hoc test for significance. ****p < 0.0001
To further confirm the possibility that upregulation of KAT2 transactivation resulting from CagA/VacA stimulation or H. pylori infection was attributed to the increased binding of IRF3 to the KAT2 promoter sequence, we utilized chromatin immunoprecipitation (ChIP) to confirm this hypothesis. As shown in Fig. 3D, the baseline of the binding of IRF3 to the KAT2 promoter region were dramatically increased in response to CagA/VacA treatment in BGC823 cells. As a negative control, no nonspecific amplifying was obtained using the primer targeted CDS of KAT2, indicating that the IP and real-time PCR-based amplification of the KAT2 promoter sequence was specific. In addition, H. pylori infection led to a significant binding of IRF3 to KAT2 promoter in BGC823 cells (Fig. 3E). Taken together, these results showed that IRF3 is critical for H. pylori -mediated KAT2 expression.
H. pylori triggered cGAS/STING/IRF3 cascade signaling
The above results suggested IRF3 was required for H. pylori-mediated KAT2 expression, the exact mechanism through which H. pylori regulated IRF3 signaling remained to be identified. The stimulator of interferon genes (STING)-IRF3 pathway has recently been shown to play an important role in immune and metabolic diseases. IRF3,a transcription factor, was activated by phosphorylation and translocated into the nucleus, initiating transcription of IFN-β and other antiviral genes [45]. As shown in Fig. 4A, a subcellular fractionation approach was utilized to analyze the influence of H. pylori on the level of nuclear IRF3 in BGC823 cells. The results showed that H. pylori infection led to a significant IRF3 nuclear translocation in the time course experiment (Fig. 4A). In line with this, immunofluorescence staining further confirmed that a remarkable nuclear translocation of IRF3 was observed in response to H. pylori infection and CagA/VacA stimulation in comparison with control group (Fig. 4B-C).

H. pylori mediated the cGAS-STING-IRF3 cascade pathway (A) BGC823 cells were serum starved for 24 h and then left untreated or treated with H. pylori for another 1 h, respectively. nuclear (Nuc) and cytosolic (Cyt) IRF3 were tested by immunoblotting. α-tubulin and Histone 3 were taken as internal controls for the cytosolic and nuclear fractions, respectively. B Immunofluorescence of IRF3 localization in BGC823 cells treated with or without H. pylori infection for 1 h after serum starved for 24 h. C After serum starvation for 24 h, BGC823 cells were treated with CagA, VacA and CagA + VacA for 1 h, respectively. Immunofluorescence was performed to display IRF3 nuclear translocation. After serum starvation for 24 h, BGC823 cells were treated with CagA, VacA and CagA + VacA (D) for 1 h or H. pylori (E) for indicated time. The total protein was collected to detect indicated protein with corresponding antibodies. Total cGAS/STING/IRF3 was determined to ensure equal loading. Data represent the mean ± s.d. of three independent experiments and were analyzed by one-way ANOVA with multiple comparisons, followed by Dunnett post hoc test for significance versus Control. **p < 0.01, ***p < 0.001, ****p < 0.0001. After serum starvation for 24 h, BGC823 cells were pre-treated for 1 h with IRF3 inhibitor MRT67307(5uM) and stimulated with H. pylori (F) or CagA, VacA and CagA + VacA (G) for additional 48 h. The total lysate was harvested to detect indicated proteins, Data represent the mean ± s.d. of three independent experiments and were analyzed by One-way ANOVA test for significance for F and two-way ANOVA with multiple comparisons, followed by Dunnett post hoc test for significance for G. *p < 0.01, ***p < 0.001, ****p < 0.0001
The above results indicated that H. pylori have an pivotal role in IRF3 nuclear translocation. Here, we sought to further reveal the possible mechanisms underlying IRF3 nuclear translocation caused by H. pylori. Because cGAS was activated in response to double-stranded DNA in a sequence-independent manner, and activation of cGAS-STING/IRF3 signaling is the classical pathway that mediates that mediated IRF3 nuclear translocation, which focused us to explore the effect of H. pylori on cGAS-STING/IRF3 signaling. WB analysis showed that in BGC823 cells, a significant upregulation of phosphorylation of cGAS/STING/IRF3 was observed in response to CagA and VacA stimulation for 1 h (Fig. 4D) and H. pylori infection for time course experiment (Fig. 4E). Moreover, inhibition of cGAS/STING/IRF3 signaling by MRT67307 in BGC823 cells could rescue the effect of H. pylori and its virulence factor on KAT2, even CDX2 and MUC2 expression (Fig. 4F-G). Taken together, these finding suggested H. pylori triggered cGAS/STING/IRF3 activation to induce IRF3 nuclear translocation, initiating KAT2 expression.
IRF3 Inhibition protected against H. pylori-mediated gastric intestinal metaplasia In vivo
To further provide insights into the effect of IRF3 inhibition on KAT2 expression in H. pylori-induced gastric intestinal metaplasia model. Mice were inoculated by oral gavage with H. pylori (PMSS1 strain) strain combined with IRF3 signal inhibitor MRT67307. The gastric tissues from uninfected or H. pylori-infected mice combined with or without MRT67307 treatment were subjected to immunohistochemical staining of CDX2 and MUC2. As shown in Fig. 5A, histological analysis of the gastric mucosa revealed that mice infected with the H. pylori PMSS1 strain showed significant inflammation, and both CDX2 and KAT2 staining were significantly observed in the H. pylori-infected mice, of note, these phenomena were alleviated after receiving MRT67307 treatment. What’s more, the analysis of IF staining displayed that gastric intestinal metaplasia characterized by enhanced CDX2 and MUC2 expression was drastically suppressed in the mucosa in H. pylori-infected mice after MRT67307 treatment, which was attributed to the decreased KAT2 expression caused by IRF3 inhibitor MRT67307 (Fig. 5B). This in vivo experiment demonstrated that IRF3 is a critical regulator of H. pylori-induced KAT2-mediated kynurenine pathway activation to trigger intestinal metaplasia. The strategy that targeting IRF3 could protect against the progression of H. pylori-induced gastric intestinal metaplasia.

IRF3 inhibition alleviated H. pylori-infected gastric intestinal metaplasia. A HE staining of representative histological features of gastric mucosa of C57BL/6 mice in different groups (scale bars: 75 μm), IHC experiment staining showing representative images of CDX2 and KAT2 expression in gastric tissue from the indicated groups of mice (scale bars: 50 μm), Data represented the mean ± s.d. and was analyzed by One-way ANOVA test for significance. ****P < 0.0001; **P < 0.01. B Immunofluorescence was performed to detect KAT2, CDX2 and MUC2 expression in gastric mucosa tissue in indicated group, respectively. Data represented the mean ± s.d. and was analyzed by One-way ANOVA test for significance. ****P < 0.0001. C Immunofluorescence was performed to examine phosphor-IRF3 and CDX2 expression in gastric mucosa tissue in indicated group, respectively. The relationship of the indicated protein expression was analyzed using simple linear regression, ****P < 0.0001 (D) Schematic model of H. pylori triggered XA production to induced CDX2 in gastric epithelial cells. H. pylori triggered cGAS/STING/IRF3 pathway to promote IRF3 nuclear translocation in gastric epithelial cells, leading to increase the binding of IRF3 to KAT2 promoter and induce KAT2 transactivation, which further induced XA production and CDX2-mediated gastric intestinal metaplasia
The above results suggested the critical role of KAT2 in H. pylori-mediated gastric intestinal metaplasia, which focused us to further explore the clinical relationship between KAT2 and CDX2. As shown in Table 1, we have recruited 26 subjects, including 14 H. pylori-infected patients diagnosed with gastric intestinal metaplasia, 12 healthy controls with aged between 30 and 70 years. Detailed clinical characteristics of the subjects, which are not public, could be available upon reasonable request. IF was performed in a set slide tissue to analyze phosphorylated IRF3 and CDX2 expression in gastric intestinal metaplasia with H. pylori infection. The results showed that phosphorylation of IRF3 was significantly increased in the gastric epithelial cells in patients with H. pylori-mediated gastric intestinal metaplasia characterized by enhanced CDX2 expression in IM group in comparsion to HC group (Fig. 5C), further linear regression analysis showed that phosphorylation of IRF3 is positively correlation with CDX2 (p < 0.0001). Overall, these results suggested that activation of IRF3 pathway is critical role for H. pylori-infected gastric intestinal metaplasia.
Discussion
Gastric intestinal metaplasia (IM), a precancerous or para-cancerous lesion, is strongly associated with chronic H. pylori infection. In this study, as shown in Fig. 5D, we revealed that H. pylori contributed to IM by triggering KAT2-mediated kynurenine pathway through IRF3 pathway. KAT2 inhibition in gastric epithelial cells could reverse the promotion of H. pylori on CDX2 expression. Mechanically, H. pylori activated cGAS/STING/TBK1/IRF3 signaling, resulting in nuclear translocation of IRF3 and initiating KAT2 transactivation, which further induced XA production to induce IM characterized by enhanced CDX2 expression. Inhibition of IRF3 could rescue the expression of KAT2 caused by H. pylori. Most importantly phosphorylation IRF3 was positively correlation with CDX2 expression in clinical sample with H. pylori infection diagnosed with IM, suggesting IRF3-induced kynurenine pathway is critical for H. pylori-mediated IM. Taken together, these finding greatly extended our insight into the metabolic reprogramming of kynurenine pathway in H. pylori-induced IM.
Up to date, there is limit available reports about the metabolic reprogramming during H. pylori-induced IM. Cellular metabolism regulates normal cell functions as well as the pathophysiology of multiple disease conditions. Recently, 11 metabolites involved in glycolysis, tricarboxylic acid (TCA) cycle, and amino acid metabolism, such as asparagine, serine, glycine, citric acid, malic acid and isocitric acid, were found to be significantly altered in AGS cells infected with H.pylori [46]. In this work, we have identified XA was largely enhanced in gastric epithelial cells in response to CagA and VacA or H. pylori treatment, and further analysis showed that XA treatment in gastric epithelial cells could induce CDX2 expression, leading to gastric IM, which was attributed to enhanced KAT2 expression confirmed by qPCR, WB. Interestingly, the study demonstrated that XA-stimulated cGMP synthesis through GEP1 (gametogenesis essential protein 1) and the subsequent signaling and cellular events, such as Ca2+ mobilization, gamete formation, and gametes egress out of erythrocytes [47, 48]. However, the further work is required to discuss the work that how XA induced CDX2 expression and the possible function of XA in H. pylori-induced pathology, including H.pylori replication and colonization, immunity, and drug resistance. In addition to XA, the investigation is required to explore that there is any possible metabolites in kynurenine pathway involved in H. pylori-induced IM, such as kynurenine, 3-hydroxy kynurenine.
In line with the Song et al. study showed that infection with H. pylori is well recognized as a kind of genotoxic DNA pathogens to trigger STING signaling, including IRF3 phosphorylation [49], we found that phosphorylation and nuclear translocation of IRF3 was increased in gastric epithelial cells in response to H. pylori stimulation, which was attributed to activation of cGAS-STING pathway, including phosphorylation of TBK and STING. Further analysis suggested that H. pylori infection in gastric epithelial cells led to the binding of IRF3 to KAT2 promoter confirmed by ChIP and luciferase assay, identifying KAT2 is a direct target of IRF3. What’s more, inhibition of IRF3 in gastric epithelial cells could reverse the enhanced KAT2, even CDX2 and MUC2 expression caused by H. pylori, while KAT2 suppression by could rescue the inducible of CDX2 expression in gastric epithelial cells in response to H. pylori stimulation. However, in addition to KAT2, the critical molecular through which H. pyloritriggered IM could be required to address, and the work is also needed to explore the novel pathway involved in H. pylori-mediated kynurenine pathway. This unsolved issue would be addressed in our future work.
In summary, this work is the first to unravel the role and mechanism that H. pylori induced gastric intestinal metaplasia by activation of kynurenine pathway in gastric epithelial cells. H. pylori promoted kynurenine pathway and XA production through activation of IRF3, which further led to CDX2 and MUC2 expression in gastric epithelial cells. We have identified KAT2 is a direct target of IRF3, and most importantly, H. pylori regulated IRF3 phosphorylation, leading to IRF3 nuclear translocation and increase the binding of IRF3 to KAT2 promoter, which further enhanced KAT2 transcription. Despite how XA induced CDX2 expression remained unknown, the current study provided insights into the novel metabolic reprogramming mechanism of H. pylori in gastric intestinal metaplasia.
Conclusions
This work has revealed H. pylori triggered cGAS/STING/IRF3-mediated kynurenine pathway of tryptophan metabolism to promote XA production, which further induced gastric intestinal metaplasia.
Availability of data and materials
The datasets generated during and/or analyses during the current study are available from the corresponding author on reasonable request.
Abbreviations
- 3HK:
-
3-Hydroxy kynurenine
- 3HAA:
-
3-Hydroxy anthranilic acid
- ACVR1:
-
Activin A receptor type I
- ACMSD:
-
7, 2-Amino-3-carboxymuconic acid semialdehyde decarboxylase
- ALKBH5:
-
Alkylation repair homolog protein 5
- BCL2:
-
B-cell lymphoma-2
- BA:
-
Bile acids
- CagA:
-
Cytotoxin associated gene A
- CDX2:
-
Caudal-related homeobox transcription factor-2
- CRC:
-
Colorectal cancer
- ChIP:
-
Chromatin Immunoprecipitation
- COL12A1:
-
Collagen Type XII
- DMEM:
-
Dulbecco's Modified Eagle Medium
- DNL:
-
De novo lipogenesis
- FBS:
-
Fetal bovine serum
- FXR:
-
Farnesoid X Receptor
- FASN:
-
Fatty acid synthase
- GC:
-
Gastric cancer
- GECs:
-
Gastric epithelial cells
- HK2:
-
Hexokinase 2
- H. pylori:
-
Helicobacter pylori
- HNF4α:
-
Hepatocyte nuclear factor 4 alpha
- LC–MS:
-
Liquid chromatography-mass spectrometry
- IRF3:
-
Interferon regulatory factor 3
- IF:
-
Immunofluorescence
- IM:
-
Intestinal metaplasia
- IL:
-
Interleukin
- KYNA:
-
Kynurenine to Kynurenic acid
- KYNU:
-
Kynureninase
- KAT:
-
Kynurenine aminotransferase
- KMO:
-
Kynurenine 3-monoxygenase
- KYN:
-
Kynurenine
- IDO1:
-
Indoleamine-2,3-dioxygenase-1
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- Trp:
-
Tryptophan
- TDO2:
-
Tryptamin 2,3 dioxygenase
- PBS:
-
Phosphate Buffer solution
- PIC:
-
Picolinic acid
- QA:
-
Anthranilic acid
- QUIN:
-
Quinolinic acid
- QPRT:
-
3-Hydroxyanthranilate-3,4-dioxygenase
- QPCR:
-
Quantitative polymerase chain reaction
- RLU:
-
Relative luciferase unit
- SHMT2:
-
Serine hydroxymethyltransferase-2
- STAT3:
-
Signal transducer and activator of transcription 3
- SOX2:
-
SRY (sex determining region Y)-box 2
- SDS-PAGE:
-
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- TBST:
-
Tris Buffered Saline Tween-20
- VacA:
-
Vacuolating cytotoxin A
- WB:
-
Western blotting
- XA:
-
Xanthurenic acid
References
Scida S, Russo M, Miraglia C, Leandro G, Franzoni L, Meschi T, et al. Relationship between Helicobacter pylori infection and GERD. Acta Biomed. 2018;89(8-S):40–3.
Zheng J, Zhang G, Gao C, Xu G, Lin W, Jiang C, et al. Linked color imaging-based endoscopic grading of gastric intestinal metaplasia and histological gastritis staging in the assessment of gastric cancer risk. Scand J Gastroenterol. 2022;57(11):1–7.
Chen HY, Hu Y, Xu XB, Zhou YA, Li NS, He C, et al. Upregulation of oncogene Activin A receptor type I by Helicobacter pylori infection promotes gastric intestinal metaplasia via regulating CDX2. Helicobacter. 2021;26(6):e12849.
Chen HY, Hu Y, Lu NH, Zhu Y. Caudal type homeoboxes as a driving force in Helicobacter pylori infection-induced gastric intestinal metaplasia. Gut Microbes. 2020;12(1):1–12.
Huang L, Tang X, Yang F, Pan W, Liang X, Xu Z, et al. Shikonin contributes to intestinal epithelial cell differentiation through PKM2/NRF2-mediated Polyol pathway. Pharmacological Res - Modern Chin Med. 2021;1:100004.
Song X, Chen HX, Wang XY, Deng XY, Xi YX, He Q, et al. H. pylori-encoded CagA disrupts tight junctions and induces invasiveness of AGS gastric carcinoma cells via Cdx2-dependent targeting of Claudin-2. Cell Immunol. 2013;286(1–2):22–30.
Asano N, Imatani A, Watanabe T, Fushiya J, Kondo Y, Jin X, et al. Cdx2 Expression and Intestinal Metaplasia Induced by H. pylori Infection of Gastric Cells Is Regulated by NOD1-Mediated Innate Immune Responses. Cancer Res. 2016;76(5):1135–45.
Asonuma S, Imatani A, Asano N, Oikawa T, Konishi H, Iijima K, et al. Helicobacter pylori induces gastric mucosal intestinal metaplasia through the inhibition of interleukin-4-mediated HMG box protein Sox2 expression. Am J Physiol Gastrointest Liver Physiol. 2009;297(2):G312–22.
Li Y, Zhao J, Chen R, Chen S, Xu Y, Cai W. Integration of clinical and transcriptomics reveals programming of the lipid metabolism in gastric cancer. BMC Cancer. 2022;22(1):955.
Huo J, Guan J, Li Y. Metabolism reprogramming signature associated with stromal cells abundance in tumor microenvironment improve prognostic risk classification for gastric cancer. BMC Gastroenterol. 2022;22(1):364.
Latour YL, Sierra JC, Finley JL, Asim M, Barry DP, Allaman MM, et al. CTH exacerbates Helicobacter pylori immunopathogenesis by promoting macrophage metabolic remodeling and activation. JCI Insight. 2022;7(12):e155338.
Noto JM, Piazuelo MB, Shah SC, Romero-Gallo J, Hart JL, Di C, et al. Iron deficiency linked to altered bile acid metabolism promotes Helicobacter pylori-induced inflammation-driven gastric carcinogenesis. J Clin Invest. 2022;132(10):e147822.
Alizadeh M, Raufman JP. Gastrointestinal neoplasia: carcinogenic interaction between bile acids and Helicobacter pylori in the stomach. J Clin Invest. 2022;132(10):e160194.
Gao S, Song D, Liu Y, Yan H, Chen X. Helicobacter pylori CagA Protein Attenuates 5-Fu Sensitivity of Gastric Cancer Cells Through Upregulating Cellular Glucose Metabolism. Onco Targets Ther. 2020;13:6339–49.
Chen P, Li L, Wang H, Zhao J, Cheng Y, Xie J, et al. Omeprazole, an inhibitor of proton pump, suppresses De novo lipogenesis in gastric epithelial cells. Biomed Pharmacother. 2020;130:110472.
Zhou Y, Chen S, Yang F, Zhang Y, Xiong L, Zhao J, et al. Rabeprazole suppresses cell proliferation in gastric epithelial cells by targeting STAT3-mediated glycolysis. Biochem Pharmacol. 2021;188:114525.
Ni Z, Min Y, Han C, Yuan T, Lu W, Ashktorab H, et al. TGR5-HNF4alpha axis contributes to bile acid-induced gastric intestinal metaplasia markers expression. Cell Death Discov. 2020;6:56.
Yue B, Cui R, Zheng R, Jin W, Song C, Bao T, et al. Essential role of ALKBH5-mediated RNA demethylation modification in bile acid-induced gastric intestinal metaplasia. Mol Ther Nucleic Acids. 2021;26:458–72.
Li S, Chen X, Zhou L, Wang BM. Farnesoid X receptor signal is involved in deoxycholic acid-induced intestinal metaplasia of normal human gastric epithelial cells. Oncol Rep. 2015;34(5):2674–82.
Yu JH, Zheng JB, Qi J, Yang K, Wu YH, Wang K, et al. Bile acids promote gastric intestinal metaplasia by upregulating CDX2 and MUC2 expression via the FXR/NF-kappaB signalling pathway. Int J Oncol. 2019;54(3):879–92.
Oxenkrug G. Interferon-gamma - Inducible Inflammation: Contribution to Aging and Aging-Associated Psychiatric Disorders. Aging Dis. 2011;2(6):474–86.
Ala M, Eftekhar SP. The Footprint of Kynurenine Pathway in Cardiovascular Diseases. Int J Tryptophan Res. 2022;15:11786469221096644.
Liang H, Li T, Fang X, Xing Z, Zhang S, Shi L, et al. IDO1/TDO dual inhibitor RY103 targets Kyn-AhR pathway and exhibits preclinical efficacy on pancreatic cancer. Cancer Lett. 2021;522:32–43.
Beaupre BA, Reabe KR, Roman JV, Moran GR. Hydrogen movements in the oxidative half-reaction of kynurenine 3-monooxygenase from Pseudomonas fluorescens reveal the mechanism of hydroxylation. Arch Biochem Biophys. 2020;690:108474.
Liu JJ, Raynal S, Bailbe D, Gausseres B, Carbonne C, Autier V, et al. Expression of the kynurenine pathway enzymes in the pancreatic islet cells. Activation by cytokines and glucolipotoxicity. Biochim Biophys Acta. 2015;1852(5):980–91.
Fukuoka S, Ishiguro K, Yanagihara K, Tanabe A, Egashira Y, Sanada H, et al. Identification and expression of a cDNA encoding human alpha-amino-beta-carboxymuconate-epsilon-semialdehyde decarboxylase (ACMSD). A key enzyme for the tryptophan-niacine pathway and “quinolinate hypothesis.” J Biol Chem. 2002;277(38):35162–7.
Kuligowski J, Sanjuan-Herraez D, Vazquez-Sanchez MA, Brunet-Vega A, Pericay C, Ramirez-Lazaro MJ, et al. Metabolomic Analysis of Gastric Cancer Progression within the Correa’s Cascade Using Ultraperformance Liquid Chromatography-Mass Spectrometry. J Proteome Res. 2016;15(8):2729–38.
Wu D, Wang Z. Gastric Cancer Cell-Derived Kynurenines Hyperactive Regulatory T Cells to Promote Chemoresistance via the IL-10/STAT3/BCL2 Signaling Pathway. DNA Cell Biol. 2022;41(4):447–55.
Pirzadeh M, Khalili N, Rezaei N. The interplay between aryl hydrocarbon receptor, H. pylori, tryptophan, and arginine in the pathogenesis of gastric cancer. Int Rev Immunol. 2022;41(3):299–312.
Xiang Z, Li J, Song S, Wang J, Cai W, Hu W, et al. A positive feedback between IDO1 metabolite and COL12A1 via MAPK pathway to promote gastric cancer metastasis. J Exp Clin Cancer Res. 2019;38(1):314.
Sadok I, Jedruchniewicz K, Rawicz-Pruszynski K, Staniszewska M. UHPLC-ESI-MS/MS Quantification of Relevant Substrates and Metabolites of the Kynurenine Pathway Present in Serum and Peritoneal Fluid from Gastric Cancer Patients-Method Development and Validation. Int J Mol Sci. 2021;22(13):6972.
Zhou L, Liu T, Huang B, Luo M, Chen Z, Zhao Z, et al. Excessive deubiquitination of NLRP3-R779C variant contributes to very-early-onset inflammatory bowel disease development. J Allergy Clin Immunol. 2021;147(1):267–79.
Soutto M, Romero-Gallo J, Krishna U, Piazuelo MB, Washington MK, Belkhiri A, et al. Loss of TFF1 promotes Helicobacter pylori-induced beta-catenin activation and gastric tumorigenesis. Oncotarget. 2015;6(20):17911–22.
Zhang S, Xu W, Wang H, Cao M, Li M, Zhao J, et al. Inhibition of CREB-mediated ZO-1 and activation of NF-kappaB-induced IL-6 by colonic epithelial MCT4 destroys intestinal barrier function. Cell Prolif. 2019;52(6):e12673.
Sun X, Yang Q, Rogers CJ, Du M, Zhu MJ. AMPK improves gut epithelial differentiation and barrier function via regulating Cdx2 expression. Cell Death Differ. 2017;24(5):819–31.
Gao T, Wang Z, Cao J, Dong Y, Chen Y. The Role of Aeromonas-Goblet Cell Interactions in Melatonin-Mediated Improvements in Sleep Deprivation-Induced Colitis. Oxid Med Cell Longev. 2022;2022:8133310.
Li YY, Hsieh LL, Tang RP, Liao SK, Yeh KY. Macrophage-derived interleukin-6 up-regulates MUC1, but down-regulates MUC2 expression in the human colon cancer HT-29 cell line. Cell Immunol. 2009;256(1–2):19–26.
Wang Y, Zhou X, Zou K, Chen G, Huang L, Yang F, et al. Monocarboxylate Transporter 4 Triggered Cell Pyroptosis to Aggravate Intestinal Inflammation in Inflammatory Bowel Disease. Front Immunol. 2021;12:644862.
Huang B, Chen Z, Geng L, Wang J, Liang H, Cao Y, et al. Mucosal Profiling of Pediatric-Onset Colitis and IBD Reveals Common Pathogenics and Therapeutic Pathways. Cell. 2019;179(5):1160-76 e24.
Xu W, Zhang Z, Zou K, Cheng Y, Yang M, Chen H, et al. MiR-1 suppresses tumor cell proliferation in colorectal cancer by inhibition of Smad3-mediated tumor glycolysis. Cell Death Dis. 2017;8(5):e2761.
Dounay AB, Anderson M, Bechle BM, Campbell BM, Claffey MM, Evdokimov A, et al. Discovery of Brain-Penetrant, Irreversible Kynurenine Aminotransferase II Inhibitors for Schizophrenia. ACS Med Chem Lett. 2012;3(3):187–92.
Linderholm KR, Alm MT, Larsson MK, Olsson SK, Goiny M, Hajos M, et al. Inhibition of kynurenine aminotransferase II reduces activity of midbrain dopamine neurons. Neuropharmacology. 2016;102:42–7.
Cheng Q, Yuan L, Guo J, Guo D, Liu X, Li S. Phosphorylation of Ser82 on IRF3 acts as negative-feedback regulation of IRF3-dependent innate immunity. Int J Biochem Cell Biol. 2022;150:106275.
Holland JW, Bird S, Williamson B, Woudstra C, Mustafa A, Wang T, et al. Molecular characterization of IRF3 and IRF7 in rainbow trout, Oncorhynchus mykiss: functional analysis and transcriptional modulation. Mol Immunol. 2008;46(2):269–85.
Popli S, Chakravarty S, Fan S, Glanz A, Aras S, Nagy LE, et al. IRF3 inhibits nuclear translocation of NF-kappaB to prevent viral inflammation. Proc Natl Acad Sci U S A. 2022;119(37):e2121385119.
Matsunaga S, Nishiumi S, Tagawa R, Yoshida M. Alterations in metabolic pathways in gastric epithelial cells infected with Helicobacter pylori. Microb Pathog. 2018;124:122–9.
Muhia DK, Swales CA, Deng W, Kelly JM, Baker DA. The gametocyte-activating factor xanthurenic acid stimulates an increase in membrane-associated guanylyl cyclase activity in the human malaria parasite Plasmodium falciparum. Mol Microbiol. 2001;42(2):553–60.
Jiang Y, Wei J, Cui H, Liu C, Zhi Y, Jiang Z, et al. An intracellular membrane protein GEP1 regulates xanthurenic acid induced gametogenesis of malaria parasites. Nat Commun. 2020;11(1):1764.
Song S, Peng P, Tang Z, Zhao J, Wu W, Li H, et al. Decreased expression of STING predicts poor prognosis in patients with gastric cancer. Sci Rep. 2017;7:39858.
Acknowledgements
Not applicable.
Funding
This work was supported by National Natural Science Foundation of China (No.82200607), Guangdong Basic and Applied Basic Research Foundation (No.2020A1515110109, No.2021A1515012194, No.2023A1515030064), Basic and applied research project of Guangzhou Municipal Science and Technology Project (No.202201020631), Guangzhou Medical Key Disciplines and Specialties (No.011006003). Guangzhou key laboratory of Pediatric Inflammatory Bowel Disease (2023A03J0866), National Health Commission Key Laboratory of Tropical Disease Prevention and Control (2022NHCTDCKFKT21001), Fujian Province young and middle-aged teachers Education Research Project (JAT210416).
Author information
Authors and Affiliations
Contributions
WFX, LLG and STG conceived and designed the experiments, XHL, WJD, LH and FFZ performed experiments, LX, WXP, YWX and FYY analyzed data, LLG, STG and WFX wrote the manuscript and revised manuscript. All authors read and approved the final revised manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Upon the declaration of Helsinki, the study was reviewed and approved by The Medical Ethics Committee for Clinical Ethical Review of The affiliated hospital of Putian University. The patients gave written informed consent for his clinical records used, which are not publicly available; however, it could be available upon request.
Consent for publication
All authors have agreed to publish this manuscript.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Thedetailed information of subjects enrolled in the study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liang, X., Du, W., Huang, L. et al. Helicobacter pylori promotes gastric intestinal metaplasia through activation of IRF3-mediated kynurenine pathway. Cell Commun Signal 21, 141 (2023). https://doi.org/10.1186/s12964-023-01162-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-023-01162-9