
Abstract
Metabolic reprogramming and immune escape play a major role in tumorigenesis. Increasing number of studies have shown that reprogramming of glutamine metabolism is a putative determinant of the anti-tumor immune response in the tumor microenvironment (TME). Usually, the predatory uptake of glutamine by tumor cells in the TME results in the limited utilization of glutamine by immune cells and affects the anti-tumor immune response. The cell-programmed glutamine partitioning also affects the anti-tumor immune response. However, the reprogramming of glutamine metabolism in tumors modulates immune escape by regulating tumor PD-L1 expression. Likewise, the reprogramming of glutamine metabolism in the immune cells also affects their immune function. Additionally, different types of glutamine metabolism inhibitors extensively regulate the immune cells in the TME while suppressing tumor cell proliferation. Herein, we discuss how metabolic reprogramming of tumor and immune cells regulates anti-tumor immune responses, as well as functional changes in different immune cells in the context of targeting tumor glutamine metabolism, which can better explain the potential of targeting glutamine metabolism in combination with immunotherapy for cancer.
Video abstract
Similar content being viewed by others
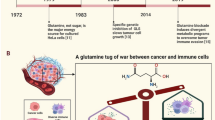

Introduction
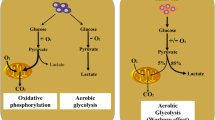
The onset and development of tumors are dependent on the impairment in the ecological balance of the body’s microenvironment, wherein metabolic reprogramming and the body’s immune response play important roles. Metabolic reprogramming and immune escape are considered to be the main characteristics of malignant tumors, which can be used to diagnose, detect and treat tumors [1, 2].In the past few decades, the Warburg effect has been considered as a typical example of metabolic reprogramming, wherein the tumor cells still rely on the conversion of glucose to lactate for energy needs under aerobic conditions, which is crucial for tumor initiation and progression [3, 4]. In addition to the key role of glucose, glutamine is also known to play an important role in tumorigenesis. As an important part of metabolic reprogramming in the tumors, reprogramming of glutamine metabolism is considered to have pleiotropic effects on cellular functions, such as macromolecule synthesis, energy generation, mTOR activation, maintenance of reactive oxygen species (ROS) balance and anti-tumor acidic microenvironment. Meanwhile, glutamine transporter mutants have been shown to promote metabolic reprogramming of tumors [5,6,7]. Recent studies have shown that glutamine is also an important raw material for the immune system, and plays a major role in regulating lymphocyte functions such as, the release of secretory factors; proliferation; and their general maintenance [8].
The body’s immune response involves a complex and dynamic biological network, including the innate and adaptive immune response, which play an important role in the onset and development of tumors. The innate immune response consists of many immune and non-immune cells, mainly including monocytes, macrophages, neutrophils, eosinophils, basophils, natural killer cells (NK cells), dendritic cells (DCs), platelets, epithelial cells, and fibroblasts. These cells together constitute an important barrier to prevent pathogens from infecting the body and help maintain the body’s homeostasis [9]. Adaptive immunity involves tightly regulated interactions between T and B lymphocytes and antigen-presenting cells, which promote the activation of pathogen-specific immune responses, the generation of immune memory, and the regulation of body’s homeostasis [10]. In fact, immune cells can sense various signal changes in the TME and turn on specific immune functions in response to those stimuli. Changes in nutrients and metabolites such as lactic acid, glucose and glutamine, in the TME, can affect the function of immune cells. Competition between tumor cells and immune cells in the TME can also affect the function of immune cells [11,12,13,14,15,16,17,18,19]. There is increasing number of studies reporting that glutamine affects the function of immune cells in the TME through multiple pathways, suggesting that intervention of glutamine metabolism may improve the effectiveness of anti-tumor immunotherapy.
In this review, we discuss the reprogramming of glutamine metabolism in tumor cells and immune cells within the TME, and the crosstalk between these cells. We also discuss how glutamine metabolism affects the biological changes in the tumor and immune cells, and affects the immune response. Finally, we also discuss the impact of glutamine metabolism inhibitors on the immune response.
Overview of glutamine metabolism in tumor and immune cells
Glutamine metabolism in tumor cells
Glutamine is the most abundant and widely used amino acid in the human body, which is an important source of nitrogen, and the respiratory fuel for tumor cells. It is an indispensable source of energy for maintaining tumor survival and progression [20]. It is known that some tumor cells consume a large amount of glutamine to meet their own metabolic needs. Tumor cells transport glutamine into cells through specific transporters (such as solute carrier family 1 neutral amino acid transporter member 5, SLC1A5; also known as alanine, serine, cysteine-preferring transporter 2, ASCT2), and then convert it into glutamate under the action of glutaminase (GLS), and further convert it into α-ketoglutarate (α-KG), which enters the Tricarboxylic Acid cycle (TCA) and participates in the onset, development and dissemination of tumors [21, 22]. For example, glutamine metabolites in tumor cells provides energy for tumor progression after entering the TCA cycle [6]. Glutaminolysis generates raw materials for the synthesis of macromolecular substances such as amino acids, nucleotides, fatty acids and hexosamines required by the tumor cells [23]. Glutamine contributes to the synthesis of uridine diphosphate-N-acetylglucosamine (UDP-GlcNAc), which is part of the hexosamine biosynthesis pathway (HBP) and is required for protein glycosylation and endoplasmic reticulum stress response in tumor cells [6]. Glutathione (GSH) and nicotinamide adenine dinucleotide phosphate (NADPH) synthesized by the glutamine metabolic pathway regulate the level of ROS in tumor cells to stabilize their redox homeostasis and ensure the survival of tumor cells [24, 25]. In addition, a complex relationship exists between tumor glutamine metabolism and autophagy. For example, ammonia produced by glutamine metabolism promotes autophagy in tumors [26, 27], and glutamine metabolism inhibits autophagy in tumor cells by activating the mTOR pathway [28].
The alterations in glutamine metabolism in tumor cells are an important outcome of the changes in the energy metabolism of tumor cells. Compared to the normal cells, the tumor cells express an abnormal level of regulatory molecules involved in glutamine metabolism. These regulatory molecules are often oncogenes or tumor suppressors that abnormally expressed in the tumors, and involved in tumor initiation and progression, such as Myc, p53, Ras, Hypoxia-inducible factor (HIF), Rho GTPase, etc. The above oncogenes or proteins may play a role in abnormal glutamine metabolism in the TME. For example, the amplification of Myc causes cellular addiction to glutamine, which may be related to the combined effects of Myc and glutamine transporter (such as SLC7A5 and SLC1A5) promoter elements, leading to enhanced glutamine uptake, which induces the activation of Lactate Dehydrogenase A (LDHA) and transports glucose-derived citrate out of the mitochondria, thereby increasing the requirement of glutamine [29,30,31]. As a suppressor gene, TP53 is mutated or deleted in most tumors. On the one hand, p53 promotes glutamine metabolism in tumor cells and makes them tolerant to the lack of glutamine by up-regulating GLS2, contributing to the survival of cancer cells. However, on the other hand, p53 also increases the level of glutathione (GSH) in tumor cells, reduces ROS, and inhibits tumorigenesis [32,33,34]. The Ras oncogene promotes autophagy and glycolysis and regulates energy metabolism. K-Ras is known to make tumor cells more sensitive to glutamine deficiency, inhibit the expression of LDHA, and increase the expression of aspartate aminotransferase. Glutamine is the main carbon source for the TCA cycle when Ras is activated [21, 35,36,37,38]. HIF-1α and HIF-2α are highly expressed in most tumors [39]. In the human non-small cell lung cancer cell line A549, it was found that silencing HIF-1α expression reduced glutamine consumption in the tumor cells [40]. Furthermore, HIF-2α has been reported to enhance the activity of c-MYC, which in turn drives glutamine catabolism by regulating numerous genes including glutaminase [30, 41]. Rho GTPase regulates glutamine metabolism in a nuclear factor-kappa B (NF-κB)-dependent manner. For example, in human breast and lymphoid cancer cells, cancer cells that are dependent on Rho GTPase signaling have a higher GLS1 activity, which promotes tumor cell proliferation [42]. It can be appreciated that there is usually a reprogramming of glutamine metabolism in tumors, and tumor cells eventually choose the best mode of glutamine metabolism to adapt to their own survival and metabolic needs by regulating the mutation and expression of related genes.
Glutamine metabolism in immune cells
Metabolic reprogramming plays a major role in: (a) the activation of immune cells; (b) the regulation of immune cell phenotype and function; (c) mounting a robust anti-tumor immune response [43,44,45]. Since immune cells play a key role in the host’s defense against infection and tumorigenesis, the metabolic changes in the immune cells have a major influence in regulating their pro-tumor or anti-tumor functions. These unique metabolic characteristics of immune cells are mainly reflected in the different metabolic patterns of immune cells during different cellular states, such as, quiescence, infection, or tumorigenesis [44, 46].For example, T cells exhibit completely different metabolic patterns depending on their activation state, for example, naive T cells have a minimum glycolytic rate and a minimum glutamine metabolism to maintain biosynthetic pathways for survival, whereas Teff cells have an increased glycolytic rate, and elevated glutamine metabolism, which enable the synthesis of proteins and nucleotides to meet the needs of rapidly proliferating tumor cells [47, 48]. For DCs, oxidative phosphorylation (OXPHOS) is the main source of energy at the resting state, while glycolysis is mainly used in the activated state [49].
Generally, the ratio of glutamine intake by immune cells is similar to or greater than that of glucose [50], and glutamine is converted to glutamate, alanine, and aspartate by partial oxidation to CO2 in immune cells. This unique transformation plays an important role in the functioning of immune cells. At the same time, the availability of glutamine largely determines the expression of certain genes in immune cells [51]. For example, during immune cell proliferation, glutamine induces the transcription of cell proliferation-related genes and promotes the proliferation of immune cells by activating proteins, such as ERK and JNK kinases and then acts on transcription factors, such as JNK and AP-1 [52]. Appropriate concentrations of glutamine promotes the expression of lymphocyte surface markers such as CD71, CD25, and CD45RO, and the production of cytokines such as IL-6, γ-interferon (IFN-γ), and TNF-α [53,54,55,56].Glutamine metabolism plays a major role in the activation of lymphocytes and is necessary for the differentiation of B lymphocytes into plasma cells and lymphoblasts. At the same time, glutamine is also necessary for T and B lymphocytes, for their proliferation, protein and antibody synthesis, and IL-2 production [57].Glutamine metabolism plays a key role in regulating macrophage activation, and the synthesis and secretion of pro-inflammatory cytokines, such as IL-1, TNF-α and IL-6. In addition, α-KG produced by glutamine metabolism promotes the differentiation of M2 macrophages [52, 58].Therefore, exploring the reprogramming of glutamine metabolism in immune cells and its impact on immune function will enable a better understanding of the mechanism by which glutamine metabolism regulates the TME and the body's immune response to tumors.
Glutamine metabolic association between tumor cells and immune cells
Glutamine competition
Like cancer cells, immune cells in the TME also undergo metabolic reprogramming [44]. Reprogramming of glucose metabolism is the most common phenomenon affecting energy metabolism in both tumor cells and immune cells. Both of these cells require glucose as an energy source, leading to a competition between them for glucose uptake in the TME [59,60,61]. Similarly, reprogramming of glutamine metabolism is also critical for the survival of tumor and immune cells, and competition for glutamine uptake also exists between these cells in the TME. For example, in glutamine-addicted clear cell renal cell carcinomas, the competitive consumption of glutamine by tumor cells results in local deprivation of extracellular glutamine, which activates HIF-1α and induces tumor-infiltrating macrophages to secrete IL-23. IL-23 further promotes the proliferation and activation of Treg cells, thereby suppressing the anti-tumor activity of Teff cells [62]. In triple-negative breast cancer (TNBC), studies have demonstrated that tumor cells competitively prey on glutamine in the TME, resulting in the limited availability of glutamine for tumor-infiltrating T lymphocytes, which affects their anti-tumor immune responses. Consistently, in the GLS-deficient mouse tumor model, the increased concentration of glutamine in the TME due to restricted glutamine utilization by tumor cells leads to elevated levels of glutamine available to tumor-infiltrating T lymphocytes, thereby enhancing its anti-tumor activity [17]. Mechanistically, the activation of the MAPK/ERK pathway plays a major role in promoting competition between tumor cells and T cells for glutamine uptake. Activation of the MAPK/ERK pathway not only up-regulates glutamine uptake by T cells, but also up-regulates glutamine uptake by tumor cells. Therefore, the differential expression of MAPK/ERK pathway-related proteins in the tumor cells and T cells may determine the cellular fate of competition between them for glutamine [63, 64]. In fact, a “glutamine steal” hypothesis has been proposed, which suggests that the selective blocking of glutamine metabolism in tumor cells could eliminate the metabolic competition for glutamine in the TME, while releasing glutamine for use by immune cells, so as to enhance anti-tumor immune response [17, 65] (Fig. 1A).

Association of glutamine metabolism with tumor cells and immune cells. The competition for glutamine between tumor cells and immune cells in the TME causes glutamine deficiency, which affects the function of immune cells, including macrophages, DCs, Treg cells, neutrophils, B cells and so on (A). Cell-programmed glutamine partitioning results in the highest consumption of glutamine by tumor cells in the TME. CAFs can up-regulate their glutamine synthesis, and complement glutamine depletion in the TME by secreting glutamine into the TME (B)
Cell-programmed glutamine partitioning
Despite the growing evidence related to the competition for nutrients between tumor cells and immune cells in the TME, it is still unclear whether the dysregulation of immune cells metabolism and function in the TME arises due to cell-intrinsic programming or competition with cancer cells for the limited nutrients. Recent research has revealed significant differences in the uptake of glucose and glutamine by different cell subsets in the TME. Cancer cells are known to consume the highest amount of glutamine, while immune cells consume the most amount of glucose. This unique nutrient distribution is mainly regulated by the mTORC1 signaling pathway and the expression of genes related to the metabolism of glucose and glutamine. Thus, cell-intrinsic programs drive immune cells and cancer cells to preferentially consume glucose and glutamine, respectively [66].Similarly, there are cell-intrinsic programs in stromal cells in the TME, which regulate the local sources of glutamine to compensate for the glutamine depletion in the TME. For example, cancer-associated fibroblasts (CAFs) are usually in a state of metabolic symbiosis with cancer cells, and compared to the normal fibroblasts, glutamine synthesis is up-regulated in CAFs, and is accompanied by glutamine secretion to supplement the concentration of glutamine in the TME. Thus, co-culture with CAFs rescues cancer cell growth in glutamine-deficient TME as compared to co-culture with normal fibroblasts. At the same time, selective abrogation of glutamine anabolism in vivo in the CAFs has been shown to inhibit ovarian tumor growth in mice [67] (Fig. 1B).
Effects of glutamine metabolism on immune response
Metabolic reprogramming of glutamine metabolism in tumor cells and its impact on immune response
Glutamine metabolism maintains tumor survival and progression, and is very important for multiple biological processes such as nucleotide synthesis, amino acid production, protein glycosylation modification, extracellular matrix production, epigenetic modifications, maintenance of cellular redox balance, and autophagy [68]. In addition to the direct effects of altered glutamine consumption on the function of immune cells and glutamine metabolism in the TME, the functional changes in the tumor cells themselves also directly affects the anti-tumor response. For example, after glutamine deprivation in the culture medium, the renal cancer cell lines and bladder cancer cell lines up-regulated the expression of PD-L1 in tumor cells by activating the EGFR/ERK/C-Jun pathway, which is an important immunosuppressive molecule that binds to the PD-1 receptor on the surface of immune cells, inhibiting the anti-tumor immune response. Therefore, co-culture with peripheral blood T lymphocytes (PBTLs) may inhibit the production of IFN-γ from T cells, thereby inhibiting the anti-tumor immune response [69, 70].Interestingly, in another study, researchers found that restricting glutamine consumption by tumors up-regulated the expression of tumor PD-L1, by reducing the expression of GSH in the tumor cells. Mechanistically, glutamine is a major precursor for glutathione synthesis, therefore, limiting the utilization of glutamine by tumor cells leads to the reduction in GSH, inhibits the activity of sarcoplasmic reticulum Ca2+-ATPase (SERCA), and activates the NF-κB signaling pathway, thereby promoting the expression of tumor PD-L1 and inactivating the co-cultured T cells. In mouse models of tumor, targeting glutamine metabolism combined with monoclonal antibody against PD-L1 may further improve the anti-tumor immune response [71] (Fig. 2).

Reprogramming of glutamine metabolism in tumor cells and T cells and its impact on immune response. Inhibition of the glutamine transporter inhibits the differentiation of Teff cells while simultaneously promotes the differentiation of Treg cells (①). Inhibition of the GLS promotes the differentiation and effector function of Teff cells (②). Glutamine deprivation affects the differentiation of naive T cells, and up-regulates the expression of PD-L1 in tumor cells by activating the EGFR/ERK/C-Jun signaling pathway or reducing GSH levels, inhibiting SERCA activity, and then activating the NF-κB signaling pathway (③)
The unique metabolic properties of tumor cells are essential features that distinguish them from normal cells. However, the molecular mechanisms regulating glutamine metabolism in the tumor is not yet fully understood. Also, the molecular mechanisms regulating immune checkpoints such as PD-L1 and CD47 are still unclear. Moreover, little is known about the regulatory relationship between glutamine metabolism and immune checkpoint regulation. Some recent studies have shown that there were same regulatory molecules enabling the crosstalk between glutamine metabolism and the expression of immune checkpoint proteins. As previously highlighted, the proto-oncogene Myc has been shown to be critical for glutamine metabolism. MYC specifically activates the expression of glutamine transporter and glutaminase in tumors, thereby regulating the reprogramming of glutamine metabolism in tumors [30, 72, 73].At the same time, MYC also regulates the expression of PD-L1 and CD47 in the tumor cells. MYC expressed by the tumor cells not only regulates the tumor immune microenvironment by acting on innate and acquired immune cells and the secretion of cytokines, but also by direct action on the promoters of the genes encoding CD47 and PD-L1, which in turn regulates their mRNA and protein expression, eventually causing immunosuppression and tumor growth [74].The Ras oncogene promotes the reprogramming of glutamine metabolism in tumor cells by up-regulating the expression of glutaminase [75]. Mutation of the K-Ras gene activates the downstream signaling pathway involved in stabilizing the PD-L1 mRNA, thereby promoting PD-L1 protein synthesis by tumor cells and inhibiting the anti-tumor immune response [76]. In addition to Myc and Ras, HIF and p53 have also been shown to be involved in the regulation of glutamine metabolism in tumors, as well as in the expression of immune checkpoints by tumor cells [77, 78]. Although many researches have confirmed that some of the same regulators are involved in mediating both the regulation of glutamine metabolism and immune checkpoints, these studies were independent and did not link glutamine metabolism with immune checkpoints expression. Therefore, whether these factors regulate the expression of immune checkpoints while regulating glutamine metabolism in tumors and thus affect the anti-tumor immune response needs to be further explored.
Reprogramming of glutamine metabolism in immune cells and its impact on the immune response
Energy metabolism is an important basis for maintaining the activity and function of immune cells. In the process of immune cell activation, a large amount of energy and metabolic intermediates are required to meet the needs of macromolecule biosynthesis, so as to achieve cell proliferation, differentiation and effector functions. At the same time, the metabolic pathways of different types of immune cells during their activation, differentiation and proliferation are completely different from those in the resting state, suggesting the occurrence of “metabolic reprogramming” occurs [79]. In addition, changes in metabolic pathways further regulates the phenotype and function of immune cells, thereby affecting the body’s immune response. Glutamine is an important energy substrate for immune cells, and an important nitrogen and carbon donor for various biosynthetic precursors, and also plays a critical role in the activation and function of immune cells. Therefore, it is crucial to understand how changes in glutamine metabolism in immune cells affects their anti-tumor immune responses.
T cells
T cells are key players in the anti-tumor immune response. For example, activated CD8+T cells directly exert cytotoxic effects on tumor cells, and activated CD4+ T helper 1 (Th1) cells activate macrophages and NK cells by secreting IFN-γ, which promotes anti-tumor effects. While activated CD4+ T helper 2 (Th2) cells and regulatory T (Treg) cells promote tumor-induced immunosuppression, CD4+ T helper 17 (Th17) cells either support or inhibit tumor progression, depending on the context [80]. Usually in the resting state, the metabolic rate of the naive T cells is low; its demand for glutamine is low; and low levels of glutamine metabolism can maintain its survival [81].However, in the activated state, the Teff cells need to proliferate rapidly, thus increasing the intake of glutamine, which provides them with sufficient raw material for macromolecule synthesis, while promoting the secretion of cytokines [82].The decomposition of glutamine affects the differentiation of T cells. For example, when GlS1 is depleted, it promotes the differentiation and effector function of CD4+Th1 and CD8+ T cells by up-regulating the expression of the transcription factor T-bet, and inhibiting the mTORC1 and IL-2 signal transduction pathways, thereby inhibiting Th17 differentiation [83]. Additionally, the loss of GlS1 leads to α-kG deficiency, and impairs the differentiation of Th17 cells [84].Glutamine uptake transporters such as ASCT2, Solute carrier family 7, member 5 (SLC7A5) and Sodium-coupled Neutral Amino Acid Transporter (SNAT), when blocked, inhibit the differentiation of CD4+Th1 and Th17 cells [83], while SLC7A5-mediated glutamine uptake regulates the activation of c-MYC-dependent Teff cells. During glutamine deprivation, Teff cells show decreased c-MYC protein expression, growth restriction, and impaired immune function [85].In addition, glutamine deprivation promotes the differentiation of Treg cells through AMPK-mTORC1 signaling pathway, thereby reducing the immune function of Teff cells [86, 87].In summary, the reprogramming of glutamine metabolism in T cells regulates the differentiation and function of T cells from various aspects, thereby regulating the immune response of the body (Fig. 2).
Macrophages
Macrophages are innate immune cells. Under the stimulation of lipopolysaccharide (LPS) and IFN-γ or IL-4, naive macrophages differentiate into M1 or M2 macrophages. M1 macrophages participate in the positive immune responses and play the role in immune surveillance by secreting inflammatory cytokines and chemokines, and are involved in professional antigen presentation. M2 macrophages possess a weak antigen-presenting ability, and secrete anti-inflammatory cytokines such as IL-10 or TGF-β, and down-regulate the immune response [88,89,90].Tumor-associated macrophages (TAMs) have been shown to be functionally plastic as a special type of macrophage, which are often described as M2-like population, but there is also evidence for the existence of M1-like population [91,92,93].In fact, in the early phase of tumor establishment, TAMs display an inflammatory phenotype, but an immunosuppressive phenotype is present at the later stages of tumor progression [94]. Glutamine metabolism plays an important role in the activation of macrophages, and there are inherent differences in the dependence of different macrophage subsets on glutamine. For example, early in vivo animal experiments showed that glutamine was essential for the production of cytokines (such as IL-1, IL-6, TNFα), antigen presentation, and phagocytic functions in murine macrophages [50]. Glutaminolysis affects the polarization of M1 macrophages. The uptake and metabolism of glutamine is elevated in LPS-activated M1 macrophages, and the replenishment of α-KG by glutamine metabolism further promotes the accumulation of succinate, improving the stability of HIF-1α, which in turn drives the production of pro-inflammatory cytokines (such as IL-1) [95, 96]. M2 macrophages consume more glutamine than M1 and naive macrophages, and usually glutamine accumulates in M2 macrophages and promotes its polarization. Part of the reason for this differential effect is that the metabolite of glutamine, α-kG, alters gene expression programs that support an anti-inflammatory M2-like state. Additionally, the expression of glutamine synthase (GS) is low in M1 macrophages, but high in M2 macrophages [58, 97, 98]. Recent studies have shown that in IL-4-induced M2 macrophages, glutamine is used to support active TCA cycle, and HBP. The HBP pathway produces UDP-GlcNAc, which acts as a substrate for N-glycosylation of M2-marked proteins, such as the N-glycosylation receptor CD206, as well as KLF4, CCL22, and IRF4, thereby promoting the polarization of M2 macrophages [96]. Supporting these observations, TAMs from Lewis lung cancer (M2 phenotype) was reported to express higher levels of the glutamine metabolizing enzymes, transaminase and glutamine synthetase. However, whether and how glutamine metabolism regulates the tumor-promoting function of TAMs remains to be further demonstrated [99]. Taken together, glutamine metabolism is involved in the polarization of M1 and M2 macrophages. Since M2 macrophages consume more glutamine than M1 macrophages, in the TME, it is unclear whether inhibiting the anti-tumor immune response from M2 macrophages polarization would be greater than the effect of enhancing the anti-tumor immune response from the M1 macrophages. Or is there a homeostasis between the M1 and M2 states. Furthermore, it is not known whether glutamine metabolism affects the differentiation of naive macrophages. All the above factors would determine whether the glutamine metabolism in macrophages promotes or inhibits the anti-tumor immune response (Fig. 3).

Impact of reprogramming of glutamine metabolism in immune cells on immune response. M2 macrophages consume more glutamine, and α-KG, a metabolite of glutamine metabolism, which promote the polarization of M2 macrophages. Glutamine metabolism in M2 macrophages is essential for supporting an active TCA cycle and UDP-GlcNAc synthesis. This provides the substrate for N-glycosylation, enabling the glycosylation of M2-marked proteins, and promoting the polarization of M2 macrophages (①). Glutamine metabolism in M1 macrophages promotes the accumulation of succinate by replenishing α-KG, further improving the stability of HIF-1α, which regulates the polarization of M1 macrophages (②). Inhibition of ASCT2 and GLS in B cells reduces the production of IgG and IgM antibodies (③). Glutamine regulates neutrophil function by generating ATP and regulating the expression of components of the NADPH oxidase complex, but its pro-tumor or anti-tumor effect is unknown (④). Suppression of glutamine metabolism in NK cells inhibits its anti-tumor function (⑤)
Other immune cells
B cells modulate the function of myeloid cells to support tumor progression, by producing antibodies and immune complexes [100, 101]. Glutamine is essential for the survival of B cells in hypoxic environments [102], and also promotes the differentiation of human B cells into plasma cells and lymphocytes [57]. In addition, antibody production by B cells depends on the breakdown of glutamine. When the expression of ASCT2 and GLS are inhibited, the production of IgG and IgM antibodies is reduced [103]. Neutrophils are normally recruited by chemokines released by tumors, and tumor-associated neutrophils (TANs) promote CD8+T cells responses and anti-tumor activity in the absence of tumor-derived TGF-β, whereas in the presence of TGF-β promotes the tumor-promoting activity of CD8+T cells [104]. Neutrophils consume glutamine at the highest rates relative to other leukocytes, such as macrophages and lymphocytes [105, 106]. Glutamine enhances superoxide production in neutrophils by generating ATP and regulates the expression of components of the NADPH oxidase complex [107]. Furthermore, glutamine plays an important role in preventing adrenaline induced changes in NADPH oxidase and superoxide production in neutrophils [108]. NADPH oxidase is essential for neutrophil function, as neutrophils use extracellular traps (NETs) to perform their functions, and the action of NETs requires the activation of NADPH oxidase [109]. Therefore, glutamine metabolism is crucial for the function of neutrophils, but its pro-tumor or anti-tumor effects need to be further explored. Natural killer (NK) cells play an integral role in activating anti-tumor T cell responses and killing tumor cells by producing IFN-γ and cytotoxic molecules such as granzyme. Glutamine uptake mediated by the glutamine transporter SLC7A5, regulates the activation of c-MYC-dependent NK cells. When glutamine is deprived, NK cells exhibit reduced expression of c-MYC protein, growth restriction, and impaired immune function, while inhibition of glutamine breakdown has no effect on NK cells [85]. In addition to the immune cells discussed above, other immune cells have also been shown to play an important role in the anti-tumor immune response. However, the regulation of glutamine metabolism in these immune cells in the TME remains unknown, and further studies are needed to demonstrate its role in the body’s immune response (Fig. 3).
Glutamine metabolism inhibitors and their effect on immune response
Glutamine in the TME not just meets the metabolic needs of the rapidly proliferating tumor cells, but also does the same for the different types of immune cells. As mentioned above, the differential impact of glutamine metabolism on different types of cells in the TME would eventually determine the outcome of targeting glutamine metabolism, and its effect on tumor suppression and anti-tumor immune response. The current drugs targeting glutamine metabolism are mainly classified into three categories, namely, glutamine antimetabolites, glutaminase inhibitors and glutamine uptake inhibitors. Several recent studies have demonstrated that the above three classes of glutamine metabolism inhibitors positively impact the function of different immune cells in the TME, while inhibiting tumor cell proliferation.
Effects of glutamine antimetabolites on immune response
6-Diazo-5-oxo-L-norleucine (L-DON), as a first-generation glutamine antimetabolite, inhibits all the glutamine-utilizing enzymes. Although it promotes a strong anti-tumor effect, systemic toxicity limits its clinical application [110,111,112]. To address this problem, researchers developed JHU-083, a prodrug form of L-DON, which is selectively activated to L-DON after entering the TME, thus reducing its systemic toxicity and improving its anti-tumor immune response [113, 114]. In syngeneic mouse models treated with JHU-083, the metabolic activity of the tumor was extensively suppressed, while hypoxia was mitigated and the levels of glutamine and glucose in the TME were increased. When combined with anti-PD-1 monoclonal antibody, the complete response rates approached 100% as compared to treatment with immunotherapy alone. Therefore, it is proposed that JHU-083 may not have negative effects on immune cells, but may enhance the function of immune cells. Subsequent metabolic flux analysis showed that in vitro treatment with L-DON altered CD8+T cells towards an activated, long-lived, and memory-like state. Correspondingly, in vivo treatment of tumors with JHU-083 had an increased the number of CD8+tumor-infiltrating lymphocytes (TILs), and their transcriptional program showed an enhanced proliferative capacity and anti-cancer activity, and the expression of genes related to long-term memory CD8+T cells was up-regulated, while the expression of genes related to apoptosis was significantly down-regulated. Mechanistically, L-DON and JHU-083 suppressed tumor glutamine metabolism by inhibiting all the glutamine-utilizing enzymes, and simultaneously suppressed tumor glycolysis by activating AMP Kinase (AMPK) and inhibiting the expression of c-MYC [114, 115]. AMPK and c-MYC are recognized as key regulators of glycolytic flux [116,117,118]. Furthermore, OXPHOS in tumor cells was also suppressed due to the absence of alternative fuels as carbon sources for the TCA cycle [114]. Overall, due to the lack of plasticity in the interdependence of glycolysis, OXPHOS and glutamine metabolism in tumor cells, extensive inhibition of glutamine metabolism in tumor cells inhibits their glycolysis and OXPHOS, thereby comprehensively disintegrating the energy metabolism in tumor cells [114]. Although glutamine metabolism and glycolysis are inhibited in CD8+T cells, their OXPHOS is up-regulated, and the extracellular acetate is used as an alternative fuel to generate ATP, further activating the anti-tumor effects of CD8+T cells [114]. In addition to CD8+T cells, JHU-083 also affects the immune function of myeloid-derived suppressive cells in the TME. Generally, myeloid-derived suppressor cells (MDSCs) and TAMs in the TME inhibit the anti-tumor immune response. In tumor-bearing mouse models treated with JHU-083, tumor growth was found to be suppressed and the generation and recruitment of MDSCs was also markedly inhibited. Mechanistically, targeting tumor glutamine metabolism in the tumors promoted a decrease in CSF3secretion, and promoted the differentiation of MDSCs and TAMs into pro-inflammatory TAMs. Additionally, blocking glutamine metabolism also inhibited the expression of IDO in the tumor and myeloid derived cells, resulting in a significant reduction in the levels of kynurenine, further enhancing the anti-tumor immune response [119]. Interestingly, L-DON also enhanced the anti-tumor immune response by affecting the mechanical properties of the tumor extracellular matrix (ECM), which is responsible for the formation of the immunosuppressive TME. Hyaluronan is the main component of the ECM, and its precursor is synthesized by the HBP, and L-DON inhibits glutamine-fructose aminotransferase 1 (GFPT1), the rate-limiting enzyme in the HBP, resulting in decreased hyaluronan synthesis, affecting the mechanical properties of the ECM in the TME and enhancing the infiltration of CD8+T cells and the anti-tumor immune response [120]. In conclusion, glutamine antimetabolites effectively inhibit tumor growth while improving the anti-tumor immune response through multiple mechanisms, revealing the close interaction between glutamine metabolism and immune response in the TME (Fig. 4).

Effects of glutamine metabolism inhibitors on immune response. Glutamine antimetabolites L-DON and JHU-083 can inhibit glutamine metabolism, glycolysis, and OXPHOS in tumor cells, and comprehensively disintegrate the energy metabolism of tumors (①). Glutamine antimetabolites directly modulate the metabolism of CD8+CTLs to promote a long-lasting, activated, memory-like phenotype; enhance cytokine production; and inhibit exhaustion and apoptosis (②). Glutamine antimetabolites inhibit the generation and recruitment of MDSCs and induce the differentiation of MDSCs and TAMs into pro-inflammatory TAMs by suppressing the secretion of CSF3 (③). Glutamine antimetabolites inhibit the expression of IDO in tumor and myeloid derived cells; reduce the levels of kynurenine; and enhance anti-tumor immune response (④). Glutamine antimetabolites inhibit the HBP metabolic pathway; decrease the levels of hyaluronan; change the mechanical properties of the ECM; improve the immunosuppressive TME and enhance the anti-tumor immune response (⑤). Glutamine uptake inhibitor V-9302 has distinct effects on the differentiation of T cell subsets, favoring CD4+Th1 and CD8+CTL but reducing the levels of Treg cells (⑥, ⑧). Glutaminase inhibitor CB-839 enhances the activation of CD4 + Th1 and CD8 + CTL but suppress the differentiation of CD4+Th17 cells (⑦, ⑧)
Effects of glutaminase inhibitors on immune response
GLS is highly expressed in diverse malignancies and is essential for their survival, and tumor-targeted drugs targeting GLS have been extensively studied. BPTES and 968, are the two major classes of GLS inhibitors that have been shown to have anti-tumor activity [42, 121]. CB-839, a BPTES-based allosteric GLS inhibitor, with a better oral bioavailability and stronger inhibitory activity, is being tested in clinical trials [122]. Previous research has shown that BPTES treatment promoted pro-inflammatory M1-like activation of macrophages, thereby enhancing the body’s anti-tumor immune response [58]. However, BPTES treatment also promoted the upregulation of PD-L1 expression, which inhibited the anti-tumor function of immune cells upon PD-L1 binding to the PD-1 receptor on the surface of immune cells. Therefore, there is the possibility of immune escape after treating tumor cells with BPTES [71]. CB-839 has varied effects on the function of different types of T cells, thereby affecting the immune response. In vitro experiments showed that CB-839 treatment promoted the differentiation of CD4+Th1 cells and CD8+ cytotoxic T lymphocytes (CTLs) and induced both the cell types to secrete more cytokines. However, treatment of Th17 cells with CB-839 inhibited their differentiation, function, and cytokine production, and eventually suppressed their expansion. The reason for these opposing outcomes may be due to differences in the epigenetic response of each T cell subset to α-KG depletion after GLS blockade [65, 83]. Importantly, there is temporal heterogeneity in the response of Teff cells to CB-839 treatment, and others have shown that short-term ex vivo treatment of Teff cells with CB-839 enhanced the subsequent anti-tumor function of Th1 cells and CD8+ CTLs in vivo. However, it did not lead to a long-term effect. In addition, transient exposure to CB-839 in vitro improved the function of chimeric antigen receptor (CAR) T cells, in a mouse model receiving CAR-T cell immunotherapy for a limited time [83]. Therefore, the glutaminase inhibitor CB-839 enhances the function of Teff cells while inhibiting the function of Treg cells, thereby enhancing the anti-tumor immune response (Fig. 4).
Effects of glutamine uptake inhibitors on immune response
The glutamine transporter SLC1A5, is frequently up-regulated in the tumor cells, and its overexpression is associated with a poor prognosis in cancer patients [30, 123]. V-9302, an inhibitor targeting SLC1A5, significantly inhibits tumor cells proliferation in vitro, and suppresses tumor growth in mouse models, and also modulates the anti-tumor immune response [124]. In a spontaneous mouse model of TNBC, researchers found that V-9302 selectively blocked glutamine uptake in TNBC cells to inhibit tumor growth, but did not inhibit the T cells. Also, T cell activation was enhanced, and the levels of TILs were significantly increased, the anti-tumor function of CD8+T cells and CD4+Th1 cells were enhanced, and supported the transition of CD8+T cells to long-term memory cells, while the levels of Treg cells were reduced. Mechanistically, V-9302 sustained glutamine uptake by CD8+T cells, by promoting the compensatory upregulation of the glutamine transporter ATB0,+/Slc6a14, in CD8+T cells, to support de novo glutathione synthesis and improve redox balance in T cells. However, such compensatory upregulation of glutamine transport was not found in V-9302-treated TNBC cells [17]. Meanwhile, in human breast cancer cell lines, researchers found that V-9302 enhanced anti-tumor response by promoting ROS production, which induced the autophagic degradation of B7 homology 3 (B7H3). B7H3 is considered to act as an immune checkpoint ligand that contributes to immune escape. In mouse models of breast cancer, V-9302 treatment significantly increased autophagosome formation and decreased the expression of B7H3 in tumor cells, and enhanced CD8+ T cell infiltration and activation. The combination of V-9302 and anti-PD-1 antibody showed a greater anti-tumor effect than either of the single treatments [125, 126]. Consistent with BPTES, tumor cells treated with V-9302 up-regulated the expression of PD-L1, Thus, there is the possibility of immune escape after treating tumor cells with V-9302, and the combined targeting of glutamine metabolism and PD-L1 showed a greater anti-tumor efficacy in mouse tumor models [71]. In conclusion, V-9302, a glutamine uptake inhibitor, not just enhanced the anti-tumor function of CD8+T cells, but also induced immune escape by up-regulating the expression of PD-L1 in tumor cells, indicating that V-9302 had a complex effect on the immune response (Fig. 4).
Conclusions
The critical role of glutamine in energy generation and macromolecule synthesis underlies its importance in tumor progression and immune response. Therefore, further studies exploring the role of glutamine metabolism in the tumors and immune cells would help us to develop therapeutic strategies for targeting glutamine metabolism in the TME for cancer therapy. In fact, both tumor cells and immune cells greatly depend on the availability of glutamine to survive, proliferate, and function. Reprogramming of glutamine metabolism in the tumor cells to their biosynthetic and energy requirements to support their rapid proliferation and survival in a hypoxic TME. And reprogramming of glutamine metabolism in the immune cells maintains their survival while modulating their phenotypes and function, contributing to their pro-tumorigenic or anti-tumorigenic functions. These observations suggest that: ① there is an association in glutamine metabolism between tumor cells and immune cells in the TME, ② reprogramming of glutamine metabolism in tumor cells and immune cells is closely related to the immune response, and ③ the different rate of glutamine metabolism in the tumor and immune cells determines their differential response to glutamine metabolism inhibitors. For example, there may be a competition for glutamine between immune and tumor cells, and cell-programmed glutamine partitioning may happen between these cells in the TME. Glutamine metabolism affects immune response by regulating the differentiation and activity of Teff cells, Treg cells, and macrophages, and the expression of immune checkpoint proteins in tumor cells. Also, different types of glutamine metabolism inhibitors have been reported to have varying effects on different immune cells, while inhibiting the proliferation of tumor cells. Together, the above factors determine how glutamine metabolism in the TME affects the immune response, eventually causing tumor progression or suppression.
Combination therapy with immunotherapeutic agents and drugs targeting tumor metabolism are a major focus of current cancer research. Studies have reported a synergistic effect of targeting glutamine metabolism and anti-tumor immunity. However, the current clinical trials related to the above combination therapy have not achieved satisfactory results [127]. The reasons for this may include the inherent heterogeneity in glutamine metabolism in the tumor and immune cells, and the intricate effects of glutamine metabolism on immune responses. As discussed in our paper, glutamine uptake inhibitors not only activate the function of Teff cells in the TME, but also up-regulate the expression of PD-L1 in tumor cells, which may inhibit the function of Teff cells. This indicates the complexity of the effects of glutamine metabolism on immune responses. Although the current studies have confirmed that various types of glutamine metabolism inhibitors not just inhibit tumor proliferation effectively, but also have a positive impact on the anti-tumor function of immune cells. However, one needs to evaluate whether they also modulate the expression of immune checkpoint proteins in tumor cells and contribute to immune escape. Therefore, a deeper investigation of glutamine metabolism in tumor cells and immune cells, and the crosstalk between these cells would help us understand the mechanisms associated with immune evasion, and the glutamine requirements by immune cells, which would be crucial to fully realize the effect of combination therapy.
Availability of data and materials
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Abbreviations
- TME:
-
Tumor microenvironment
- ROS:
-
Reactive Oxygen species
- NK cells:
-
Natural killer cells
- DCs:
-
Dendritic cells
- GLS:
-
Glutaminase
- α-KG:
-
α-Ketoglutarate
- TCA:
-
Tricarboxylic acid cycle
- HIF:
-
Hypoxia-inducible factor
- LDHA:
-
Lactate dehydrogenase A
- GSH:
-
Glutathione
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NF-κB:
-
Nuclear factor-kappa B
- IFN-γ:
-
γ-Interferon
- CAFs:
-
Cancer-associated fibroblasts
- PBTLs:
-
Peripheral blood T lymphocytes
- SERCA:
-
Sarcoplasmic reticulum Ca2+-ATPase
- Th1:
-
CD4+ T helper 1
- Th2:
-
CD4+ T helper 2
- Treg:
-
Regulator T
- Th17:
-
CD4+ T helper 17
- SLC1A5:
-
Solute carrier family 1 neutral amino acid transporter member 5
- ASCT2:
-
Alanine, serine, cysteine-preferring transporter 2
- SLC7A5:
-
Solute carrier family 7, member 5
- SNAT:
-
Sodium-coupled neutral amino acid transporter
- LPS:
-
Lipopolysaccharide
- TAMs:
-
Tumor-associated macrophages
- GS:
-
Glutamine synthase
- HBP:
-
Hexosamine biosynthetic pathway
- TANs:
-
Tumor-associated neutrophils
- NETs:
-
Neutrophil extracellular traps
- L-DON:
-
6-Diazo-5-oxo-L-norleucine
- AMPK:
-
AMP kinase
- TILs:
-
CD8+tumor-infiltrating lymphocytes
- OXPHOS:
-
Oxidative phosphorylation
- MDSCs:
-
Myeloid-derived suppressor cells
- ECM:
-
Extracellular matrix
- GFPT1:
-
Glutamine-fructose aminotransferase 1
- CTLs:
-
Cytotoxic T lymphocytes
- CAR:
-
Chimeric antigen receptor
- TNBC:
-
Triple-negative breast cancer
- B7H3:
-
B7 homology 3
References
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Faubert B, Solmonson A. Metabolic reprogramming and cancer progression. Science. 2020. https://doi.org/10.1126/science.aaw5473.
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33.
McCarty MF, Whitaker J. Manipulating tumor acidification as a cancer treatment strategy. Altern Med Rev. 2010;15:264–72.
Yoo HC, Park SJ, Nam M, Kang J, Kim K, Yeo JH, Kim JK, Heo Y, Lee HS, Lee MY, et al. A variant of SLC1A5 Is a mitochondrial glutamine transporter for metabolic reprogramming in cancer cells. Cell Metab. 2020;31:267-283.e212.
Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16:619–34.
Huang W, Choi W, Chen Y, Zhang Q, Deng H, He W, Shi Y. A proposed role for glutamine in cancer cell growth through acid resistance. Cell Res. 2013;23:724–7.
Yang T, Yan X, Cao Y, Bao T, Li G, Gu S, Xiong K, Xiao T. Meta-analysis of glutamine on immune function and post-operative complications of patients with colorectal cancer. Front Nutr. 2021;8:765809.
Kaur BP, Secord E. Innate Immunity. Pediatr Clin North Am. 2019;66:905–11.
Bonilla FA, Oettgen HC. Adaptive immunity. J Allergy Clin Immunol. 2010;125:S33-40.
Terry S, Engelsen AST, Buart S, Elsayed WS, Venkatesh GH, Chouaib S. Hypoxia-driven intratumor heterogeneity and immune evasion. Cancer Lett. 2020;492:1–10.
Huang B, Song BL. Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities. Nat Metab. 2020;2:132–41.
Chen B, Gao A, Tu B, Wang Y, Yu X, Wang Y, Xiu Y, Wang B, Wan Y, Huang Y. Metabolic modulation via mTOR pathway and anti-angiogenesis remodels tumor microenvironment using PD-L1-targeting codelivery. Biomaterials. 2020;255: 120187.
Karayama M, Masuda J, Mori K, Yasui H, Hozumi H, Suzuki Y, Furuhashi K, Fujisawa T, Enomoto N, Nakamura Y, et al. Comprehensive assessment of multiple tryptophan metabolites as potential biomarkers for immune checkpoint inhibitors in patients with non-small cell lung cancer. Clin Transl Oncol. 2021;23:418–23.
Yan Y, Chang L, Tian H, Wang L, Zhang Y, Yang T, Li G, Hu W, Shah K, Chen G, Guo Y. 1-Pyrroline-5-carboxylate released by prostate Cancer cell inhibit T cell proliferation and function by targeting SHP1/cytochrome c oxidoreductase/ROS Axis. J Immunother Cancer. 2018;6:148.
Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162:1229–41.
Edwards DN, Ngwa VM, Raybuck AL, Wang S, Hwang Y, Kim LC, Cho SH, Paik Y, Wang Q, Zhang S, et al. Selective glutamine metabolism inhibition in tumor cells improves antitumor T lymphocyte activity in triple-negative breast cancer. J Clin Invest. 2021. https://doi.org/10.1172/JCI140100.
Wang JX, Choi SYC, Niu X, Kang N, Xue H, Killam J, Wang Y. Lactic acid and an acidic tumor microenvironment suppress anticancer immunity. Int J Mol Sci. 2020;21:8363.
Ma G, Li C, Zhang Z, Liang Y, Liang Z, Chen Y, Wang L, Li D, Zeng M, Shan W, Niu H. Targeted glucose or glutamine metabolic therapy combined with PD-1/PD-L1 checkpoint blockade immunotherapy for the treatment of tumors: mechanisms and strategies. Front Oncol. 2021;11:697894.
Carrascosa JM, Martínez P, Núñez de Castro I. Nitrogen movement between host and tumor in mice inoculated with Ehrlich ascitic tumor cells. Cancer Res. 1984;44:3831–5.
Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–5.
Zhang J, Pavlova NN, Thompson CB. Cancer cell metabolism: the essential role of the nonessential amino acid, glutamine. EMBO J. 2017;36:1302–15.
Daye D, Wellen KE. Metabolic reprogramming in cancer: unraveling the role of glutamine in tumorigenesis. Semin Cell Dev Biol. 2012;23:362–9.
Welbourne TC. Ammonia production and glutamine incorporation into glutathione in the functioning rat kidney. Can J Biochem. 1979;57:233–7.
DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA. 2007;104:19345–50.
Cheong H, Lindsten T, Wu J, Lu C, Thompson CB. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc Natl Acad Sci USA. 2011;108:11121–6.
Eng CH, Yu K, Lucas J, White E, Abraham RT. Ammonia derived from glutaminolysis is a diffusible regulator of autophagy. Sci Signal. 2010;3:ra31.
Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009;136:521–34.
Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol. 2007;178:93–105.
Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, Dang CV. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–5.
Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, Thompson CB. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA. 2008;105:18782–7.
Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, Lokshin M, Hosokawa H, Nakayama T, Suzuki Y, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci USA. 2010;107:7461–6.
Jackson JG, Lozano G. The mutant p53 mouse as a pre-clinical model. Oncogene. 2013;32:4325–30.
Tran TQ, Lowman XH, Reid MA, Mendez-Dorantes C, Pan M, Yang Y, Kong M. Tumor-associated mutant p53 promotes cancer cell survival upon glutamine deprivation through p21 induction. Oncogene. 2017;36:1991–2001.
Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11:761–74.
Dilshara MG, Jeong JW, Prasad Tharanga Jayasooriya RG, Neelaka Molagoda IM, Lee S, Park SR, Choi YH, Kim GY. Glutamine deprivation sensitizes human breast cancer MDA-MB-231 cells to TRIAL-mediated apoptosis. Biochem Biophys Res Commun. 2017;485:440–5.
Gaglio D, Soldati C, Vanoni M, Alberghina L, Chiaradonna F. Glutamine deprivation induces abortive s-phase rescued by deoxyribonucleotides in k-ras transformed fibroblasts. PLoS ONE. 2009;4:e4715.
Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Balestrieri C, Alberghina L, Stephanopoulos G, Chiaradonna F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol. 2011;7:523.
Talks KL, Turley H, Gatter KC, Maxwell PH, Pugh CW, Ratcliffe PJ, Harris AL. The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am J Pathol. 2000;157:411–21.
Faubert B, Vincent EE, Griss T, Samborska B, Izreig S, Svensson RU, Mamer OA, Avizonis D, Shackelford DB, Shaw RJ, Jones RG. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1α. Proc Natl Acad Sci USA. 2014;111:2554–9.
Gordan JD, Bertout JA, Hu CJ, Diehl JA, Simon MC. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell. 2007;11:335–47.
Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, Wilson KF, Ambrosio AL, Dias SM, Dang CV, Cerione RA. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 2010;18:207–19.
Biswas SK, Mantovani A. Orchestration of metabolism by macrophages. Cell Metab. 2012;15:432–7.
Ghesquière B, Wong BW, Kuchnio A, Carmeliet P. Metabolism of stromal and immune cells in health and disease. Nature. 2014;511:167–76.
Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38:633–43.
Hao S, Yan KK, Ding L, Qian C, Chi H, Yu J. Network approaches for dissecting the immune system. iScience. 2020;23:101354.
Ricciardi S, Manfrini N, Alfieri R, Calamita P, Crosti MC, Gallo S, Müller R, Pagani M, Abrignani S, Biffo S. The translational machinery of human CD4(+) T cells is poised for activation and controls the switch from quiescence to metabolic remodeling. Cell Metab. 2018;28:895-906.e895.
Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342:1242454.
Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, Pearce EJ. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115:4742–9.
Newsholme P. Why is L-glutamine metabolism important to cells of the immune system in health, postinjury, surgery or infection? J Nutr. 2001;131:2515S-2522S.
Curi R, Newsholme P, Marzuca-Nassr GN, Takahashi HK, Hirabara SM, Cruzat V, Krause M, de Bittencourt Jr PI. Regulatory principles in metabolism-then and now. Biochem J. 2016;473:1845–57.
Cruzat V, Macedo Rogero M. Glutamine: metabolism and immune function, supplementation and clinical translation. Nutrients. 2018;10:1564.
Curi R, Lagranha CJ, Doi SQ, Sellitti DF, Procopio J, Pithon-Curi TC, Corless M, Newsholme P. Molecular mechanisms of glutamine action. J Cell Physiol. 2005;204:392–401.
Curi R, Lagranha CJ, Doi SQ, Sellitti DF, Procopio J, Pithon-Curi TC. Glutamine-dependent changes in gene expression and protein activity. Cell Biochem Funct. 2005;23:77–84.
Roth E, Oehler R, Manhart N, Exner R, Wessner B, Strasser E, Spittler A. Regulative potential of glutamine–relation to glutathione metabolism. Nutrition. 2002;18:217–21.
Hiscock N, Petersen EW, Krzywkowski K, Boza J, Halkjaer-Kristensen J, Pedersen BK. Glutamine supplementation further enhances exercise-induced plasma IL-6. J Appl Physiol. 1985;2003(95):145–8.
Crawford J, Cohen HJ. The essential role of L-glutamine in lymphocyte differentiation in vitro. J Cell Physiol. 1985;124:275–82.
Liu PS, Wang H, Li X, Chao T, Teav T, Christen S, Di Conza G, Cheng WC, Chou CH, Vavakova M, et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol. 2017;18:985–94.
Kolb D, Kolishetti N. Metabolic modulation of the tumor microenvironment leads to multiple checkpoint inhibition and immune cell infiltration. ACS Nano. 2020;14:11055–66.
Palmer CS, Ostrowski M, Balderson B, Christian N, Crowe SM. Glucose metabolism regulates T cell activation, differentiation, and functions. Front Immunol. 2015;6:1.
Sukumar M, Roychoudhuri R, Restifo NP. Nutrient competition: a new axis of tumor immunosuppression. Cell. 2015;162:1206–8.
Fu Q, Xu L, Wang Y, Jiang Q, Liu Z, Zhang J, Zhou Q, Zeng H, Tong S, Wang T, et al. Tumor-associated macrophage-derived interleukin-23 interlinks kidney cancer glutamine addiction with immune evasion. Eur Urol. 2019;75:752–63.
Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, Turay AM, Frauwirth KA. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol. 2010;185:1037–44.
Yang R, Li X, Wu Y, Zhang G, Liu X, Li Y, Bao Y, Yang W. EGFR activates GDH1 transcription to promote glutamine metabolism through MEK/ERK/ELK1 pathway in glioblastoma. Oncogene. 2020;39:2975–86.
Yang WH, Qiu Y, Stamatatos O, Janowitz T, Lukey MJ. Enhancing the efficacy of glutamine metabolism inhibitors in cancer therapy. Trends Cancer. 2021;7:790–804.
Reinfeld BI, Madden MZ. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature. 2021;593:282–8.
Yang L, Achreja A, Yeung TL, Mangala LS, Jiang D, Han C, Baddour J, Marini JC, Ni J, Nakahara R, et al. Targeting Stromal Glutamine synthetase in tumors disrupts tumor microenvironment-regulated cancer cell growth. Cell Metab. 2016;24:685–700.
Fan SJ, Kroeger B. Glutamine deprivation alters the origin and function of cancer cell exosomes. EMBO J. 2020;39:e103009.
Ma G, Liang Y, Chen Y, Wang L, Li D, Liang Z, Wang X, Tian D, Yang X, Niu H. Glutamine deprivation induces PD-L1 expression via activation of EGFR/ERK/c-jun signaling in renal cancer. Mol Cancer Res. 2020;18:324–39.
Wang L, Xu T, Yang X, Liang Z, Zhang J, Li D, Chen Y, Ma G, Wang Y, Liang Y, Niu H. Immunosuppression Induced by glutamine deprivation occurs via activating PD-L1 transcription in bladder cancer. Front Mol Biosci. 2021;8:687305.
Byun JK, Park M, Lee S, Yun JW, Lee J, Kim JS, Cho SJ, Jeon HJ, Lee IK, Choi YK, Park KG. Inhibition of glutamine utilization synergizes with immune checkpoint inhibitor to promote antitumor immunity. Mol Cell. 2020;80:592-606.e598.
Qing G, Li B, Vu A, Skuli N, Walton ZE, Liu X, Mayes PA, Wise DR, Thompson CB, Maris JM, et al. ATF4 regulates MYC-mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell. 2012;22:631–44.
Csibi A, Lee G, Yoon SO, Tong H, Ilter D, Elia I, Fendt SM, Roberts TM, Blenis J. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr Biol. 2014;24:2274–80.
Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, Gouw AM, Baylot V, Gütgemann I, Eilers M, Felsher DW. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016;352:227–31.
Mukhopadhyay S, Vander Heiden MG, McCormick F. The metabolic landscape of RAS-driven cancers from biology to therapy. Nat Cancer. 2021;2:271–83.
Coelho MA, de Carné TS, Rana S, Zecchin D, Moore C, Molina-Arcas M, East P, Spencer-Dene B, Nye E, Barnouin K, et al. Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity. 2017;47:1083-1099.e1086.
Chen J, Jiang CC, Jin L, Zhang XD. Regulation of PD-L1: a novel role of pro-survival signalling in cancer. Ann Oncol. 2016;27:409–16.
Ni JM, Ni AP. Landscape of PD-1/PD-L1 regulation and targeted immunotherapy. Chin Med Sci J. 2018;33:174–82.
Kim J. Regulation of immune cell functions by metabolic reprogramming. J Immunol Res. 2018;2018:8605471.
Bailey SR, Nelson MH, Himes RA, Li Z, Mehrotra S, Paulos CM. Th17 cells in cancer: the ultimate identity crisis. Front Immunol. 2014;5:276.
Buck MD, O’Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. 2015;212:1345–60.
Swamy M, Pathak S, Grzes KM, Damerow S, Sinclair LV, van Aalten DM. Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and malignancy. Nat Immunol. 2016;17:712–20.
Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, Maseda D, Liberti MV, Paz K, Kishton RJ, et al. Distinct regulation of Th17 and Th1 cell differentiation by glutaminase-dependent metabolism. Cell. 2018;175:1780-1795.e1719.
Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y, et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26:1326–38.
Loftus RM, Assmann N, Kedia-Mehta N, O’Brien KL, Garcia A, Gillespie C, Hukelmann JL, Oefner PJ, Lamond AI, Gardiner CM, Dettmer K. Amino acid-dependent cMyc expression is essential for NK cell metabolic and functional responses in mice. Nat Commun. 2018;9:2341.
Lin SC, Hardie DG. AMPK: sensing glucose as well as cellular energy status. Cell Metab. 2018;27:299–313.
Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vázquez G, Yurchenko E, Raissi TC, van der Windt GJ, Viollet B, Pearce EL, et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity. 2015;42:41–54.
Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–96.
Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40:274–88.
Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20.
Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, Pamer EG, Li MO. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344:921–5.
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–55.
Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51.
Biswas SK, Allavena P, Mantovani A. Tumor-associated macrophages: functional diversity, clinical significance, and open questions. Semin Immunopathol. 2013;35:585–600.
Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496:238–42.
Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart KM, Ashall J, Everts B, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. 2015;42:419–30.
Ren W, Xia Y, Chen S, Wu G, Bazer FW, Zhou B, Tan B, Zhu G, Deng J, Yin Y. Glutamine metabolism in macrophages: a novel target for obesity/type 2 diabetes. Adv Nutr. 2019;10:321–30.
Palmieri EM, Menga A, Martín-Pérez R, Quinto A, Riera-Domingo C, De Tullio G, Hooper DC, Lamers WH, Ghesquière B, McVicar DW, et al. Pharmacologic or genetic targeting of glutamine synthetase skews macrophages toward an m1-like phenotype and inhibits tumor metastasis. Cell Rep. 2017;20:1654–66.
Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559–63.
Andreu P, Johansson M, Affara NI, Pucci F, Tan T, Junankar S, Korets L, Lam J, Tawfik D, DeNardo DG, et al. FcRgamma activation regulates inflammation-associated squamous carcinogenesis. Cancer Cell. 2010;17:121–34.
de Visser KE, Korets LV, Coussens LM. De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell. 2005;7:411–23.
Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012;15:110–21.
Jiang S, Yan W, Wang SE, Baltimore D. Let-7 suppresses B cell activation through restricting the availability of necessary nutrients. Cell Metab. 2018;27:393-403.e394.
Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. 2011;11:519–31.
Pithon-Curi TC, De Melo MP, Curi R. Glucose and glutamine utilization by rat lymphocytes, monocytes and neutrophils in culture: a comparative study. Cell Biochem Funct. 2004;22:321–6.
Pithon-Curi TC, Trezena AG, Tavares-Lima W, Curi R. Evidence that glutamine is involved in neutrophil function. Cell Biochem Funct. 2002;20:81–6.
Pithon-Curi TC, Levada AC, Lopes LR, Doi SQ, Curi R. Glutamine plays a role in superoxide production and the expression of p47phox, p22phox and gp91phox in rat neutrophils. Clin Sci (Lond). 2002;103:403–8.
Garcia C, Pithon-Curi TC, de Lourdes-Firmano M, Pires de Melo M, Newsholme P, Curi R. Effects of adrenaline on glucose and glutamine metabolism and superoxide production by rat neutrophils. Clin Sci (Lond). 1999;96:549–55.
Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD, Papayannopoulos V. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol. 2014;15:1017–25.
Magill GB, Myers WP, Reilly HC, Putnam RC, Magill JW, Sykes MP, Escher GC, Karnofsky DA, Burchenal JH. Pharmacological and initial therapeutic observations on 6-diazo-5-oxo-1-norleucine (DON) in human neoplastic disease. Cancer. 1957;10:1138–50.
Lynch G, Kemeny N, Casper E. Phase II evaluation of DON (6-diazo-5-oxo-L-norleucine) in patients with advanced colorectal carcinoma. Am J Clin Oncol. 1982;5:541–3.
Earhart RH, Amato DJ, Chang AY, Borden EC, Shiraki M, Dowd ME, Comis RL, Davis TE, Smith TJ. Phase II trial of 6-diazo-5-oxo-L-norleucine versus aclacinomycin-A in advanced sarcomas and mesotheliomas. Invest New Drugs. 1990;8:113–9.
Rais R, Jančařík A, Tenora L, Nedelcovych M, Alt J, Englert J, Rojas C, Le A, Elgogary A, Tan J, et al. Discovery of 6-Diazo-5-oxo-l-norleucine (DON) prodrugs with enhanced CSF delivery in monkeys: a potential treatment for glioblastoma. J Med Chem. 2016;59:8621–33.
Leone RD, Zhao L. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science. 2019;366:1013–21.
Pinkus LM. Glutamine binding sites. Methods Enzymol. 1977;46:414–27.
Dejure FR, Royla N, Herold S, Kalb J, Walz S, Ade CP, Mastrobuoni G, Vanselow JT, Schlosser A, Wolf E, Kempa S. The MYC mRNA 3’-UTR couples RNA polymerase II function to glutamine and ribonucleotide levels. EMBO J. 2017;36:1854–68.
Haikala HM, Anttila JM, Klefström J. MYC and AMPK-save energy or die! Front Cell Dev Biol. 2017;5:38.
Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006;66:8927–30.
Oh MH, Sun IH, Zhao L, Leone RD, Sun IM, Xu W, Collins SL, Tam AJ, Blosser RL, Patel CH, et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J Clin Invest. 2020;130:3865–84.
Sharma NS, Gupta VK, Garrido VT, Hadad R, Durden BC, Kesh K, Giri B, Ferrantella A, Dudeja V, Saluja A, Banerjee S. Targeting tumor-intrinsic hexosamine biosynthesis sensitizes pancreatic cancer to anti-PD1 therapy. J Clin Invest. 2020;130:451–65.
Robinson MM, McBryant SJ, Tsukamoto T, Rojas C, Ferraris DV, Hamilton SK, Hansen JC, Curthoys NP. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem J. 2007;406:407–14.
Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, Janes JR, Laidig GJ, Lewis ER, Li J, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther. 2014;13:890–901.
Hassanein M, Hoeksema MD, Shiota M, Qian J, Harris BK, Chen H, Clark JE, Alborn WE, Eisenberg R, Massion PP. SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clin Cancer Res. 2013;19:560–70.
Schulte ML, Fu A, Zhao P, Li J, Geng L, Smith ST, Kondo J, Coffey RJ. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat Med. 2018;24:194–202.
Li Q, Zhong X, Yao W, Yu J, Wang C, Li Z, Lai S, Qu F, Fu X, Huang X, et al. Inhibitor of glutamine metabolism V9302 promotes ROS-induced autophagic degradation of B7H3 to enhance antitumor immunity. J Biol Chem. 2022;298:101753.
Yang S, Wei W, Zhao Q. B7–H3, a checkpoint molecule, as a target for cancer immunotherapy. Int J Biol Sci. 2020;16:1767–73.
Meric-Bernstam Fea: a Phase 1/2 study of CB-839, a first-in-class glutaminase inhibitor, combined with nivolumab in patients with advanced melanoma (MEL), renal cell carcinoma (RCC), or non-small cell lung cancer (NSCLC). In Society for Immunotherapy of Cancer Annual Meeting 2017.
Acknowledgements
Not applicable.
Funding
This study was financially supported by the National Natural Science Foundation of China (82071750, 81772713), Taishan Scholar Program of Shandong Province (tsqn20161077), Shinan District Science and Technology Program of Qingdao (2022-2-002-YY).
Author information
Authors and Affiliations
Contributions
MG, LY and NH collected articles and designed the manuscript. MG wrote the manuscript. MG, LY, ZZL, LP and ZZ prepared tables and figures. ZM, LZ, LD, WL and CY revised and approved the manuscript. All authors read the manuscript and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ma, G., Zhang, Z., Li, P. et al. Reprogramming of glutamine metabolism and its impact on immune response in the tumor microenvironment. Cell Commun Signal 20, 114 (2022). https://doi.org/10.1186/s12964-022-00909-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-022-00909-0