
Abstract
Programmed cell death 1 ligand 1 (PD-L1) is the ligand for programmed death protein-1 (PD-1), is associated with immunosuppression. Signaling via PD-1/PD-L1 will transmits negative regulatory signals to T cells, inducing T-cell inhibition, reducing CD8+ T-cell proliferation, or promoting T-cell apoptosis, which effectively reduces the immune response and leads to large-scale tumor growth. Accordingly, many antibody preparations targeting PD-1 or PD-L1 have been designed to block the binding of these two proteins and restore T-cell proliferation and cytotoxicity of T cells. However, these drugs are ineffective in clinical practice. Recently, numerous of studies have shown that, in addition to the surface of tumor cells, PD-L1 is also found on the surface of extracellular vesicles secreted by these cells. Extracellular vesicle PD-L1 can also interact with PD-1 on the surface of T cells, leading to immunosuppression, and has been proposed as a potential mechanism underlying PD-1/PD-L1-targeted drug resistance. Therefore, it is important to explore the production, regulation and tumor immunosuppression of PD-L1 on the surface of tumor cells and extracellular vesicles, as well as the potential clinical application of extracellular vesicle PD-L1 as tumor biomarkers and therapeutic targets.
Video Abstract
Similar content being viewed by others

Background
PD-L1 (also known as B7H1 and CD274) is a 40-kDa type 1 transmembrane protein, expressed in a variety of cells and has the greatest immunosuppressive effect when it is expressed on tumor cells [1]. Under normal conditions, the immune system reacts to foreign antigens collected in lymph nodes or the spleen and promote the proliferation and differentiation of cytotoxic T cells. When PD-L1 on tumor cells is highly expressed under the regulation of various factors, it can combine with PD-1 on the surface of T cells and transmit inhibitory signals, leading to T cell dysfunction or depletion, and then promote tumor immune escape [2, 3].
Intercellular communication is crucial under both physiological and pathological conditions. Cells mainly communicate through direct contact and the release of soluble factors, including growth factors, cytokines, and hormones. Recently, a novel method of intercellular communication involving the release of extracellular vesicles (EVs) was identified and has attracted increasing research interest [4, 5]. Many drugs targeting PD-1/PD-L1 have been developed aiming to attenuate their immunosuppressive effects; to date, however, their therapeutic benefits have been limited [6, 7], likely due to a special form of PD-L1 that is present on the surface of EVs [8, 9]. EVs comprise any type of membrane-bound vesicle that is released by cells and can be generally divided into two subgroups—exosome and microvesicle—depending on the diameter and method of formation [10].
The physiology of EVs
Exosome biogenesis and secretion
The diameter of exosomes ranges from 40 to 160 nm [10]. Invaginated plasma membrane buds off to form early-sorting endosomes (ESEs), which fuse with trans-Golgi network-derived vesicles that can contain cytoplasmic molecules. ESEs mature into late-sorting endosomes (LSEs) through exchanging materials, and then into multivesicular bodies (MVBs) which contain intraluminal vesicles (ILVs) formed by the inward invagination of the endosomal limiting membrane. MVB maturation occurs through at least two mechanisms, one involves endosomal sorting complex required for transport (ESCRT)-0, -I, -II, and -III and proteins associated with ESCRT-III (e.g., VPS4 and ALIX); in the other, which is independent of ESCRT, ILVs and MVBs are generated through lipids, ceramides, four transmembrane protein families, heat shock proteins, and others [11,12,13]. Once MVBs have been produced, they can either fuse with lysosomes or autophagic lysosomes, which leads to the degradation of MVBs; or they can fuse with the plasma membrane and be released to out of the cells as exosomes [14, 15]. Exosome secretion mainly depends on the auxiliary activity of the Rab and soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) protein families [16]. The Rab protein family comprises small GTPases, several of which regulate vesicle transport and fusion through GTP/GDP cycling [17, 18]. For instance, the regulators of endosomal recycling, Rab11 was shown to regulate the secretion of exosomes in K562 cells [19, 20] and Rab35 regulates exosomes release in oligodendroglial cells by controlling the docking/tethering of vesicles to the plasma membrane [21]. Additionally, our group demonstrated that CA-IKKβ reduces the expression of Rab7 and induces the phosphorylation of SNAP23 at Ser95, which further promotes small EV (sEV) secretion [22]. SNARE proteins form complexes that can mediate the fusion of adjacent plasma membranes as well as that of MVBs with the cell membrane [23]. In K562 cells, VAMP7, a constituent of the SNARE complex, is required for the secretion of exosomes into the extracellular space [24]. In several other cell types, Ca2+ can regulate the secretion of exosomes, which may be achieved by activating the SNARE complex [25]. Other SNARE complexes may also be involved in exosome secretion; however, these are not listed here [26, 27].
Microvesicle biogenesis and secretion
Microvesicles have diameters that range from 50 nm to 1 mm [28]. The plasma membrane undergoes several molecular rearrangements at the sites of microvesicle biogenesis, including changes in Ca2+ levels and lipid and protein composition, leading to membrane budding [29, 30]. Changes in Ca2+ concentrations lead to the recruitment and activation of calcium-dependent enzymes, which can result in the asymmetric rearrangement of membrane phospholipids and changes in the lipid composition of the plasma membrane [31, 32]. A recent study has also shown that in the resting state, Ca2+ mobilization and calpain activation can lead to higher vesiculation levels in malignant (MCF-7) cells than in non-malignant (hCMEC-D3) cells, further confirming that calcium levels play a role in microvesicle formation [33]. Moreover, microvesicles may also arise from cholesterol-rich lipid rafts [34]. Among the proteins involved in microvesicle biogenesis, members of the small GTPase family, such as RhoA, participate in a Rho GTPase-dependent signaling pathway, which triggers the activation of Rho kinase and Lim kinase, finally leading to cofilin phosphorylation and, subsequently, enhanced microvesicle production [35]. Surprisingly, RhoC, similar to RhoA, is also a member of the small GTPase family, cannot induce microvesicle formation in cells, indicating that the signaling mechanism leading to microvesicle biogenesis is highly specific [36]. The release of microvesicles requires them to split from the plasma membrane, which is promoted by the reorganization of the actin–myosin cytoskeleton in a process that may also involve small GTP-binding proteins [37]. Muralidharan-Chari et al. found that GTP/GDP cycling on ARF6 regulates an actomyosin-based membrane abscission mechanism in tumor cells to promote microvesicle release [38]. Similarly, ARF1 affects myosin light-chain (MLC) phosphorylation through modulating RhoA and RhoC activity, which, in turn, promotes the release of microvesicles [39]. Additionally, a recent study reported that lipotoxicity-induced EV release is mediated by the DR5 proapoptotic signaling cascade (CHOP → DR5 → caspase-8 → caspase-3), leading to ROCK1 activation [40].
The role of PD-L1 in tumor immune escape
PD-L1 promotes tumor cell immune escape
PD-L1 and PD-L2 (B7DC and CD273) are both ligands for PD-1, an immune checkpoint receptor expressed on the surface of T cells [41, 42]. PD-L1 is mainly expressed on human tumor-associated antigen-presenting cells including tumor environmental dendritic cells (DCs) [43], monocyte-derived myeloid DCs [44], macrophages [43], neutrophils [45], fibroblasts [46], mast cells [47], and other non-tumor cells such as vascular endotheliocytes, keratinocytes, pancreatic islet cells, astrocytes, and corneal epithelial cells [48]. PD-L2 is found on macrophages [49] and DCs [50], among other cells. Importantly, PD-L1 and PD-L2 are co-expressed in a variety of tumor cells. To date, however, evidence to show that blocking PD-L2 or PD-L1 and PD-L2 simultaneously has greater therapeutic efficacy than blocking PD-L1 alone is lacking. As PD-L1 is the main immune checkpoint ligand for PD-1 on T cells in the tumor immune microenvironment [48], we will concentrate more on PD-1/PD-L1-related research, and how this axis mediates tumor immune escape. It is known that PD-L1 on tumor cells interact with PD-1 on T cells, resulting in T-cell dysfunction. Under normal conditions, antigen-activated T lymphocytes can specifically recognize tumor cells and directly kill them, while the combination of PD-L1 and PD-1 may induce T-cell apoptosis, anergy, exhaustion [51,52,53], and the expression of interleukin 10 (IL-10), a negative regulator of cellular immune responses [54]. However, little is known about the mechanism underlying how PD-1 mediates T-cell dysfunction. T-cell activation requires two types of signals, namely, a T-cell receptor (TCR) signal and a signal from a costimulatory factor, such as CD28. Yokosuka and colleagues showed that PD-1 and TCR can form microclusters, which can reduce the phosphorylation of signaling molecules downstream of TCR by recruiting Src homology 2 domain-containing tyrosine phosphatase 2 (SHP2), resulting in the weakening of T-cell activation [55]. However, Hui et al. found that the combination can lead to the phosphorylation of two tyrosine residues (Y224 and Y248) in the PD-1 cytosolic domain by the lymphocyte-specific protein tyrosine kinase Lck and the subsequent recruitment of SHP2 to dephosphorylate PD-1 and CD28, thereby inactivating CD28 and suppressing T-cell function [3].
Regulation of PD-L1 expression on tumor cells
Numerous factors influence the expression of the immunosuppressive ligand PD-L1 in the tumor microenvironment, including genomic alterations and epigenetic, transcriptional, post-transcriptional, and post-translational regulatory mechanisms (Table 1 and Fig. 1).

Regulation of PD-L1 expression on the surface of tumor cells. Many factors affect the expression of PD-L1 on tumor cell surface, including genomic alterations and epigenetic, transcriptional, post-transcriptional, and post-translational regulatory mechanisms
Genomic alternations and epigenetic regulation
Three main genomic alternations have been associated with increased PD-L1 expression, namely, amplification, translocation, and disruption of its 3'UTR region. PD-L1 is located on Chromosome 9p24.1, and when this genomic region is amplified, the expression of PD-L1 is significantly increased, as evidenced by poor prognosis and short progression-free survival for patients with Hodgkin's lymphoma, small cell lung cancer (SCLC), non-small cell lung cancer (NSCLC), and other malignant tumors [56,57,58]. In primary mediastinal large B-cell lymphoma, the PD-L1 locus was specifically rearranged, resulting in the increased expression of PD-L1 [59], and disruption of its 3'UTR region such as delete 3′UTR of PD-L1 through genome editing can increase its protein expression in many tumors [60]. The expression of PD-L1 on the surface of tumor cells can also be upregulated through epigenetic mechanisms such as histone acetylation and H3K4 trimethylation [61, 62].
Transcriptional regulation
Inflammatory cytokines
Many inflammatory cytokines are involved in coordinating anti-tumor immunity. These inflammatory cytokines and related inflammatory pathways can also increase the expression of PD-L1 on tumor cells, thereby inhibiting tumor immunity [99]. The interferon family includes two major cytokine-related classes, i.e., type I interferons (IFN-α, IFN-β, and IFN-ω) and type II interferons (IFN-γ), among IFN-γ is a proinflammatory cytokine produced by activated T cells and natural killer (NK) cells, is known to exert effective antiviral and growth-inhibitory effects [100]. Several studies have shown that IFN-γ can induce PD-L1 expression through the IFN-γ/JAK/STAT1 signaling pathway, thereby promoting the immune escape of cancer cells [101, 102]. In addition to IFN-γ, several other inflammatory cytokines also enhance PD-L1 expression on cancer cells or tumor-associated stromal cells, such as IFN-α/β [66], Toll-like receptors3/4 [63, 73], TNF-α [103], TGF-β [104], and IL-4/6/10/17/27 [64, 65, 71, 72, 105]. Interestingly, the detection of the expression of inflammatory cytokines (e.g., IFN-γ, TNF-α, and several ILs) is a predictor of immune checkpoint therapy outcome for advanced NSCLC, while high expression of inflammatory cytokines is positively correlated with anti-PD-1 therapeutic effectiveness [106]. Nevertheless, more data is required to confirm (1) that there is indeed a correlation between inflammatory cytokines and PD-L1 expression in the tumor microenvironment; (2) the specific mechanism underlying a potential correlation; and (3), the effect of inflammatory cytokines on PD-L1 expression in vivo.
Oncogenic signaling pathways
In addition to promoting tumor progression in the inherent way of tumor cells, oncogenic signaling pathways can also promote tumor growth by up regulating the expression of PD-L1, thus potentially promoting immune escape. Elucidating how oncogenic signals drive PD-L1 expression can help determine the associated mechanism and provide a therapeutic basis for combining the inhibition of these oncogenic signaling pathways with immune checkpoint therapies for cancer treatment [48]. MYC is one of the most common contributors to tumorigenesis, and its expression is estimated to be elevated or dysregulated in up to 70% of human cancers [107]. MYC has been reported to positively regulate PD-L1 expression in a variety of cancers, including esophageal squamous cell carcinoma [80], NSCLC [79], and lymphoma [75], with evidence indicating that MYC directly regulates the expression of PD-L1 at the transcriptional level [108]. One study reported that RAS also significantly boosted the expression of PD-L1 through a redox-mediated mechanism that RAS activation promoted reactive oxygen species (ROS) production and induced FGFR1 expression, leading to a significant up regulation of PD-L1 expression [76]. Interestingly, Coelho and colleagues demonstrated that RAS can also upregulate PD-L1 expression by increasing PD-L1 mRNA stability via the modulation of the AU-rich element-binding protein tristetraprolin (TTP) [109]. In addition to MYC and RAS, PD-L1 expression can also be upregulated by HIF-1α/2α [81, 85, 110], anaplastic lymphoma kinase (ALK) [82], epidermal growth factor receptor (EGFR) [84], phosphatidylinositol 3-kinase (PI3K) [78], and mitogen-activated protein kinase (MAPK) [83] when they are mutated or overexpressed. Remarkably, many inhibitors that target these oncogenic signaling pathways have been approved by the United States Food and Drug Administration (FDA). These findings highlight the feasibility of combining the inhibition of these oncogenes with immune checkpoint therapy to obtain better treatment effects.
Post-transcriptional regulation
MiRNA is a non-coding single-stranded RNA molecule, with about 22–24 nucleotides encoded by endogenous genes, which can regulate post-transcriptional gene expression in animals and plants and play a significant role in intracellular homeostasis and disease [111]. Studies have shown that miRNAs can regulate the expression of PD-L1 either by directly interacting with PD-L1 mRNA or affecting the expression of PD-L1 regulators [48]. Cortez et al. showed that p53 can downregulate the expression of PD-L1 in NSCLC cell lines, an effect that is mediated by the direct binding of miR-34 with the 3′UTR of PD-L1 [88]. Similarly, a recent report demonstrated that miR-34a negatively modulates PD-L1 expression, thereby suppressing the proliferation, metastasis, and invasion of gastric tumor cells [93].Moreover, miR-200 in NSCLC [87]and breast cancer [90], miR-152 in gastric cancer [92], miR-217 in laryngeal cancer [89], miR-124-3p in colorectal cancer [91], and miR-383-5p in breast cancer [86] are thought to play a role in the inhibition of PD-L1 expression.
Post-translational modifications
Protein post-translational modification is to increase the functional diversity of proteome through the covalent addition of functional groups or proteins, the proteolytic cleavage of regulatory subunits or the degradation of the whole protein, including phosphorylation, glycosylation, ubiquitination, nitrosylation and methylation, which plays a key role in regulating protein stability, translocation and protein–protein interaction. The post-translational modification of PD-L1 is considered to be an important mechanism of its tumor immunosuppression [2]. Studies have shown that the interaction of glycogen synthase kinase 3 β (GSK3 β) with PD-L1 can induce phosphorylation-dependent proteasome degradation of PD-L1 [94]. Furthermore, EGF can upregulate the expression of β-1,3-galactosyl-O-glycosyl-glycoprotein (B3GNT3) in triple-negative breast cancer cells, thereby promoting the N-glycosylation of PD-L1 and contributing to its interacting with PD-1, finally leading to T cells dysfunction [95]. Chan et al. reported that JAK1 can bind with PD-L1 in the endoplasmic reticulum and indicated PD-L1 Tyr phosphorylation through the IL-6/JAK1 pathway is necessary for the combination of PD-L1 and the N-glycosyltransferase STT3A to upregulate PD-L1 expression [96]. Moreover, COP9 signalosome 5 (CSN5), induced by NF-κB p65, as well as the deubiquitinase USP22, inhibit PD-L1 ubiquitination and degradation, whereas their depletion inhibits tumorigenesis and promotes T-cell cytotoxicity [97, 98].
The role of EV PD-L1 in tumor immune microenvironment
PD-L1 loading on EVs
Nucleic acids (including DNA, RNA [mRNA, miRNA, lncRNA]), proteins (including MHC-I, MHC-II, PMEL, TCR, and FasL), and lipids (phosphatidylserine, cholesterol, ceramide) can be delivered to receptor cells as EV contents [4, 28]. The composition of EVs is largely dependent on the cell type and can also be affected by different cellular conditions, including cytoplasmic content [10, 112]. Here, we mainly focus on how PD-L1 is attached to EVs. A recent study identified differences in PD-L1 levels among different cancer cell lines, which could not be explained by the speed of protein translation or protein degradation levels, and the authors speculated that PD-L1 could be secreted from cells in EVs, either in the form of more EVs or as single vesicles carrying more PD-L1 [9]. This suggests that the process involved in loading PD-L1 on EVs may be related to the EV biogenesis process. When the ESCRT-related protein ALG-2 interacting protein X (ALIX) is deleted, the level of PD-L1 on exosomes decreases, and that on cell surface increases, possibly because ALIX is required for the transfer of PD-L1 from the endosomal limiting membrane into MVBs [113]. Similarly, the ESCRT subunit HRS can mediate the identification and sorting of exosome contents, and its knockdown can lead to the decrease of exosomal PD-L1(Exo-PD-L1) level and an increase in that of cellular PD-L1 [8]. In the prostate cancer cell line PC3, when the Rab27a gene, which is related to exosome secretion, and the neutral sphingomyelinase 2 (nSMase2) gene, which promotes the budding of intravesicular vesicles, are knocked out, the levels of PD-L1 and the exosomal marker CD63 are significantly decreased. These data show that Rab27a and nSMase2 play a significant role in the production and secretion of PD-L1-containing exosomes [9]. Many other mechanisms involved in how PD-L1 is loaded onto EVs are currently under investigation, and targeting these mechanisms in combination with anti-PD-L1/ PD-1 therapy has potential as an effective treatment for PD-L1-related cancers.
The regulation of PD-L1 expression on EVs
The expression of PD-L1 can be influenced by IFN-γ, which is involved in tumor immune escape. Chen et al. found that Exo-PD-L1 has the same membrane topology as PD-L1 on the surface of tumor cells, and the amount of Exo-PD-L1 secreted by tumor cells increased significantly following IFN-γ treatment [8]. Similarly, Ricklefs and colleagues reported that under IFN-γ stimulation, EVs with low PD-L1 expression can inhibit T-cell activation, implying that IFN-γ can also promote PD-L1 expression on EVs [114]. Recently, Chatterjee et al. showed that TGF-β increase the expression of PD-L1 on the exosomes secreted by breast cancer cells in a dose-dependent manner, while blocking exosome release and inhibiting the expression of TGF-β reduced the tumor burden and enhanced T cell toxicity [115]. Also, under TGF-β stimulation, the numbers of PD-L1-containing EVs produced by fibroblasts are increased [116]. Mitochondrial Lon, which functions as a chaperone and DNA-binding protein, plays a role in protein quality control and stress responses. Lon can regulate the metabolism of mitochondrial DNA (mtDNA) and the production of mitochondrial ROS [117]. When Lon is overexpressed, oxidized mtDNA is released into the cytoplasm, IFN production is induced through the cGAS-STING-TBK1 pathway, and the expression of PD-L1 and indoleamine2,3-dioxygenase1(IDO-1) is upregulated, finally leading to the inhibition of T cells activation. Surprisingly, Lon upregulation also induces the secretion of EVs carrying mtDNA and PD-L1 [118]. Radium-223 (Ra-223) was the first bone-homing radiopharmaceutical developed that improved median overall survival (OS) in metastatic prostate cancer patients [119]. The latest findings show that several immune-related factors are enriched in EVs derived from mice treated with Ra-223, including PD-L1, and that ICB/Ra-223 combination therapy can improve the curative effect of anti-tumor therapy [120]. 5-Fluorouracil (5-FU), another key drug for advanced gastric cancer chemotherapy, has also been shown to dose- and time-dependently augment Exo-PD-L1 expression [121]. Recent studies also found that microvesicles from breast cancer cells exposed to radiation carry cargos containing different immunomodulatory proteins, including PD-L1, that inhibit T-cell function and promote tumor growth [122]. Many other cytokines, proteins, and drugs can also affect the expression of PD-L1 on EVs. These merit further investigation because the inhibitors of these factors or the combination of some of these drugs have the potential to improve the curative effect of tumor therapy.
EV PD-L1 is involved in inducing immune escape in different types of tumors
Because immune escape is a major driver of tumor progression, PD-1 and PD-L1, both immune checkpoint-associated proteins, have become the subject of intense investigation. Indeed, immune checkpoint suppressors, mainly those targeting PD-1 and PD-L1, have shown unprecedented prospects and impressive efficacy in the treatment of various human cancers. Nevertheless, the response of a considerable number of cancer patients to this treatment is still poor. Chen et al. revealed that PD-L1 present on EVs displays the same extracellular domain topology as its cell-surface counterpart [8]. As EV PD-L1 may exert functions similar to those of tumor cell surface protein PD-L1 upon PD-1 binding (Fig. 2), EV PD-L1 and their role in tumor immunity have been widely studied over recent years. Several studies have recently reported that PD‐L1 is also detected on EVs in many cancer types, such as prostate cancer [9], melanoma [8], breast cancer [123], head and neck cancer [124], pancreatic cancer [125], glioblastoma [114], gastric cancer [126], and NSCLC [127] (Table 2).

Abbreviated drawing of the formation process of EV-PD-L1 and its direct and indirect inhibitory effects against T cells. ① The process of Exo-PD-L1 production. ②Microvesicles produced by budding can also carry PD-L1. ③ PD-L1 present on the surface of exosomes secreted by tumor cells directly binds to PD-1 on T cells, inducing an immune checkpoint response that inhibits the activation of T cells and disrupts their function, thus inhibiting antitumor immunity. ④ Exosomes released by tumor cells can mediate the increase of PD-L1 expression on the surface of macrophages, neutrophils or monocytes, and then combine with PD-1 on the surface of T cells to inhibit T cells. ⑤ IFN-γ secreted by T cells can promote the expression of PD-L1 on the surface of tumor cells and exosomes
Prostate cancer
Most immune checkpoint inhibitors targeting PD-L1 have no effect on prostate cancer patients. Until recently, it was believed that the low PD-L1 expression in prostate cancer cell lines and tissue samples tissues could explain this phenomenon [128]. However, it has since been suggested that prostate cancer cells may secrete PD-L1-carrying EVs, leading to the association of anti-PD-L1 drugs to PD-L1 on the EVs, thereby allowing immune escape to occur. As PD-L1 undergoes endocytosis from the cell surface, the authors premised that PD-L1 was discretely released from in exosomes. Relative to other vesicles, exosomes were enriched by sucrose density gradient centrifugation, and PD-L1 and HRS were found to colocalize with the exosomal marker CD63 in the same sucrose fraction. As previously mentioned, when Rab27a and nSMase2, genes that are related to exosome biogenesis, are knocked out, the PD-L1 and CD63 levels are significantly decreased [9]. These evidences suggested that PD-L1 could be transported in exosomes. To then elucidate the role of this special form of PD-L1 in the tumor microenvironment, the authors injected wild-type (WT), Rab27a null, and PD-L1 null TRAMP-C2 cells into the flanks of C57BL6/J syngeneic mice, and, after excluding the key factors of exosomes production, found that Exo-PD-L1 promoted tumor progression. Meanwhile, PD-L1 deletion significantly increased the number, effector function (GzmB), and proliferation (Ki67) of CD8+ T cells, and also reduced CD8+ T cell exhaustion (Tim-3) in the TRAMP-C2 cancer cells-administered murine draining lymph nodes. Interestingly, when PD-L1-knockout TRAMP-C2 cells were introduced into one side of a mouse, and WT TRAMP-C2 cells into the bilateral side after a few days, WT TRAMP-C2 cells did not develop into tumors in the mouse, indicating that to the presence of PD-L1-deficient tumor cells could generate a strong memory, even to tumor cells that release Exo-PD-L1 [9].
Melanoma
PD-L1 was also evident in EVs produced by murine metastatic melanoma B16-F10 cells. The correlation between exosomes and PD-L1 was further confirmed by iodixanol density gradient centrifugation. Because of the low PD-L1 expression in microvesicles, this study focused on Exo-PD-L1. PD-L1 levels in exosomes of metastatic melanoma cells was remarkably elevated, compared to the primary melanoma cells, indicating that the Exo-PD-L1 was strongly correlated with the malignant degree of the tumor [8]. Subsequently, to evaluate the function of Exo-PD-L1 in vivo, the authors generated a murine model of melanoma in C57BL/6 mice utilizing PD-L1-depleted B16-F10 cells. They found that the exosome administration from parental B16-F10 cells could significantly promote tumor growth and reduce quantity of tumor-infiltrating CD8+ T lymphocytes (TIL) [8]. Collectively, these data suggest that PD-L1 suppresses antitumor immunity. Recently, Liu et al. demonstrated that the disruption of the glutamic acid–cystine metabolic balance can lead to elevated PD-L1 levels in melanoma via the transcription factors IRF4 and EGR1, promote PD-L1-containing exosomes release, induce M2 macrophage polarization, and reduce PD-1/PD-L1 inhibitor efficacy [129]. These data indicate that glutamic acid–cystine metabolic balance is crucial for immunotherapy, and that the targeted suppression of Exo-PD-L1 release may represent an important means of improving the therapeutic activity of PD-1/PD-L1 suppression.
Breast cancer
Yang et al. demonstrated that PD-L1 and CD63 colocalized in MVBs originating from the human breast cancer cell line MDA-MB-231 and human breast cancer tissues via immunofluorescence staining and immunohistochemistry, respectively, thereby confirming that breast cancer cells can also secrete PD-L1-carrying exosomes [123]. Additionally, Exo-PD-L1 dose-dependently inhibited the expression of markers of T-cell activation, such as CD3/CD28-driven ERK phosphorylation and NF-κB activation, as well as PHA-induced IL-2 secretion. The authors further showed that Exo-PD-L1 could interact with PD-1 and suppress T-cell cytotoxicity, thereby promoting tumor growth in vivo [123]. Interestingly, in addition to the role played by breast cancer cell-originating Exo-PD-L1 in tumorigenesis and tumor development, Sun et al. suggested that the Exo-PD-L1 derived from bone marrow significantly increased the lung metastasis of cells from the murine breast cancer line 4T1 by damaging the antitumor CD8+ T cell responses at metastatic sites [130].In addition, as the most malignant type of breast cancer, the triple-negative breast cancer cell derived microparticles can also load PD-L1, especially in patients receiving chemoradiotherapy. Microparticles PD-L1 can negatively regulate CD8+T cells and polarizing macrophages to M2, resulting in an immunosuppressive microenvironment that promotes tumor progression [131].
Glioblastoma
Ricklefs et al. reported finding PD-L1 in glioblastoma-derived EVs, which significantly downregulated levels of early and late activation markers CD69 and CD25 on T cells, respectively, and also reduced the proliferative ability of T cells. Interestingly, further studies revealed that the low PD-L1 levels in plasmoblastoma was upregulated following IFN-γ stimulation, which elevates PD-L1 levels on the EVs, thus inhibiting T-cell activation [114]. Exosomes have also been known to indirectly regulate the immune system via induction of PD-L1 production in a secondary cell type [132]. For example, glioblastoma stem cell-derived exosomes upregulate the expression of PD-L1 in human monocytes, which may correlate with STAT3 phosphorylation [133].
Gastric cancer
A multivariate analysis demonstrated that Exo-PD-L1 present in peripheral blood was related to high immunosuppressive activity and poor prognosis in gastric cancer (GC) patients. Furthermore, Exo-PD-L1 was found to exert a more enhanced immunosuppressive response, compared to soluble PD-L1, possibly because exosomal MHC-I promotes Exo-PD-L1-driven T-cell dysfunction [126]. Similar to that seen in glioblastoma, GC-derived EVs transport high-mobility group box-1 (HMGB1) activates signal transducer and activator of transcription 3 (STAT3) and elevates the expression of PD-L1 in neutrophils, thereby inhibiting T-cell immunity [134]. Strategies that interfere with the EV-related HMGB1/STAT3/PD-L1 network have potential as treatments for GC. 5-FU is the main chemotherapeutic agent currently used for the treatment of advanced GC [135], however, recent studies have found that 5-FU promotes a rise in Exo-PD-L1 via the miR-940/Cbl-b/STAT5A network, leading to immunosuppression in patients with late-stage disease [121].
Head and neck cancer
PD-L1 was detected in exosomes from patients with head and neck squamous cell carcinoma (HNSCC) by confocal microscopy and flow cytometry, and this form of PD-L1 was associated with disease activity, UICC staging, and lymph node status. In contrast, PD-L1 expressions were not associated with any clinicopathological parameter. Unlike PD-L1low exosomes, exposure to PD-L1high exosomes inhibited CD69 levels, which interferes with the activation of effector T cells [124].
Pancreatic cancer
Lux et al. found that Exo-PD-L1 levels were inversely proportional to postsurgical survival duration in patients with pancreatic ductal adenocarcinoma; however, whether pancreatic cancer cells evade the immune response via Exo-PD-L1 requires further investigation [125].
NSCLC
PD-L1 was found to be present in extracted plasma exosomes of NSCLC patients in vitro, in vivo, and preclinical models. Moreover, Exo-PD-L1 has been reported to inhibit the synthesis of IL-2 and IFN-γ by CD8+ T cells, and dose-dependently reduce the overall quantity of CD8+ T cells, indicating that Exo-PD-L1 promotes the apoptosis of CD8+ T cells and tumor progression via PD-1/PD-L1 interaction [127, 136].In addition, a recent study has shown that exosomes derived from NSCLC can also promote the expression of PD-L1 on macrophages through a NF-κB dependent and glycolysis dominated metabolic reprogramming mechanism, so as to promote tumor metastasis [137].
Apart from to the endogenous Exo-PD-L1 derived directly from solid tumors, chronic lymphocytic leukemia (CLL)-derived exosomes also regulate PD-L1 levels in monocytes. Seiffert et al. identified that, compared with healthy controls, the serum of CLL patients was richer in exosomes from B cells. Sequencing and analysis of the RNA and proteins in the exosomes, respectively, indicated that exosomes from patients with CLL were rich in non-coding Y RNA hY4 (Y RNA is a highly conserved, short, non-coding RNA related to DNA replication and RNA quality control). They also found that hY4 in exosomes derived from CLL patients can bind to TLR7 on the surface of monocytes, thereby promoting the transcription of various inflammatory factors and PD-L1 in monocytes. These events led to the inhibition of tumor immunity and provided a good microenvironment for cancer cell survival [138]. These findings present a potential new direction for CLL immunotherapy. Similarly, endoplasmic reticulum stress promotes the secretion of exosomal miR-23a-3p and the upregulation of PD-L1 levels in macrophages via the phosphatase and tensin homolog (PTEN)/phosphoinositide-4,5-bisphosphate 3-kinase (PI3K)/protein kinase B (AKT) axis [139].
Potential application of EV PD-L1 in tumors
EV PD-L1 as biomarker
Tumor immunotherapy has achieved remarkable results over recent years and many clinical studies have shown that immunotherapy can improve the prognosis and significantly prolong the survival time of tumor patients. At the same time, in the context of the trend for precise treatment associated with modern oncology, predictive biomarker detection before treatment can help match individual patients with the most beneficial treatment scheme and reduce the cost of immunotherapy. Consequently, the identification of biomarkers for tumor immunotherapy has gained increasing research interest. PD-L1 has been evaluated as a biomarker of tumor response to immunotherapy [140,141,142]. However, analysis of the results of preliminary study related to 45 FDA drug approvals encompassing 15 tumor types and carried out from 2011 to April 2019 indicated that PD-L1 is predictive in only 28.9% of these cases, while the number of cases in which PD-L1 is not predictive is as high as 53.3% [143]. Several reasons were proposed to explain the heterogeneity of PD-L1 predictions. First, the type of tumor tissue detected (fresh vs. archived), the type of PD-L1 detection method, and PD-L1 expression cutoffs displayed substantial heterogeneity. Secondly, the expression of PD-L1 is regulated by a variety of molecular mechanisms in the tumor microenvironment, and the ability of PD-L1 to drive immunogenicity varies with tumor type [144]. Thirdly, the expression of PD-L1 has temporal and spatial heterogeneity [145] and may also be influenced by prior treatment [144]. In summary, PD-L1 does not appear to be a clinically useful biomarker, meanwhile, people are also actively exploring the role of EVs as tumor immune markers. Therefore, a better biomarker–-EV PD-L1 is needed to guide the choice of immunotherapy (Table 3).
EV PD-L1 can be used as a diagnostic marker for various tumors, such as the Exo-PD-L1 levels in metastatic melanoma and NSCLC patients (especially those in advanced stages) were reported to be higher than those in healthy controls [8, 136]. Based on the role of EV PD-L1 as a diagnostic biomarker, some efficient methods for detecting Exo-PD-L1 have gradually emerged. For instance, Fe3O4@TiO2 nanoparticles were designed to enrich and separate exosomes from solution, and displayed a capture rate of 96.5% within 5 min. Subsequently, anti-PD-L1 antibody-modified Au@Ag@MBA SERS tags were utilized to quantify Exo-PD-L1 levels [148]. Additionally, a new PD-L1 aptamer, which not only has good selectivity but also interacts with natural PD-L1 more efficiently than antibodies, was combined with thermophoresis to yield a uniform, low volume, efficient, and sensitive method for the quantitation of Exo-PD-L1 (HOLMES-ExoPD-L1) [149]. Wang et al. used gold-silver (Au@Ag) core–shell nanobipyramids and gold nanorods to produce a unique plasma signal pattern, allowing rapid and highly sensitive exosome detection and accurate identification of Exo-PD-L1 [150].
In patients with HNSCC, the relative fluorescence values (RFVs) for Exo-PD-L1 were higher in patients with active disease than those without evidences of disease, and were also higher in patients with high UICC staging (III and IV) compared with those with low UICC staging (I and II) [124]. Similarly, in NSCLC patients, higher levels of Exo-PD-L1, rather than soluble PD-L1, were reported to be interrelated to larger tumor size, positive lymph node status, distant metastasis, and late TNM staging [136]. These observations indicate that Exo-PD-L1, rather than soluble PD-L1, may be a clinically relevant variable with potential predictive value in HNSCC and NSCLC. In addition, compared with those who have low levels of Exo-PD-L1, the OS of pancreatic cancer and GC patients with high Exo-PD-L1 levels was markedly lower; thus, PD-L1 expression on exosomes can be considered to be a negative prognostic factor for these cancers [125, 126]. Moreover, the amount of PD-L1 DNA contained in serum and plasma EVs obtained from glioblastoma patients was positively correlated with the size of the tumor [114]. Osteosarcoma is a major malignant bone tumor, with approximately 15%–20% of patients exhibiting lung metastasis [151]. Osteosarcoma cells can stimulate lung metastasis by releasing exosomes carrying PD-L1 and N-cadherin, indicating that the test of exosomes carrying PD-L1 and N-cadherin in serum can be used as a predictor of lung metastasis in osteosarcoma patients [146].
In addition to tumor diagnosis and progression, PD-L1 is also the marker of the efficacy of tumor immune checkpoint inhibitors. The levels of Exo-PD-L1 were remarkably higher in melanoma patients who did not respond to anti-PD-1 drugs than in those who did [8]. Cordonnier and colleagues suggested that PD-L1 may also be a predictor of curative effect in melanoma patients, and has the advantages of noninvasive collection and real-time monitoring [152]. Furthermore, Exo-PD-L1 expression levels were significantly lower in melanoma and NSCLC patients responding to anti-PD-1 treatment compared with those in patients with disease progression [147]. In addition to lower levels of Exo-PD-L1, patients who responded to anti-PD-1 therapy also exhibited a higher level of CD28 expression, so the combination of Exo-PD-L1 with serum CD28 may be an effective marker for predicting the response to anti-PD-1 therapy [153].
The significance of EV PD-L1 in therapy
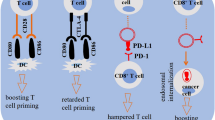
In recent years, immune checkpoint therapy has been increasingly used as an important treatment for various cancer. Although immune checkpoint therapy improves the prognosis of patients with various types of cancer, only fews of these patients have achieved long-term benefits. Why most patients do not respond to or cannot maintain their response to immune checkpoint therapy is the subject of intense investigation. The mechanisms underlying drug resistance are usually divided into two types—primary and acquired. With primary drug resistance, patients have no initial response to immune checkpoint blockade, while with acquired resistance, patients initially respond to immune checkpoint therapy but later become refractory to treatment [154, 155]. A substantial amount of convincing evidence exists for the occurrence of primary resistance to drugs targeting EV PD-L1, which may underlie the relatively low response rate to anti-PD-L1/PD-1 therapy [8, 126]. Although the specific mechanisms involved in the EV PD-L1-mediated resistance to anti-PD-L1/PD-1 therapy remain largely unclear, we can make a guess about it, as shown in Fig. 3.

The mechanism of action of anti-PD-1/PD-L1 drugs and the potential mechanism underlying the exosomal PD-L1-mediated resistance to anti-PD-L1 drugs. ① Anti-PD-1/PD-L1 drugs can interact with PD-1/PD-L1, free T cells from the checkpoint block, and restore immune responses. ② and ③ Exo-PD-L1 is thought to contribute to resistance during immunotherapy through two mechanisms. In one, exo-PD-L1 binds to anti-PD-L1 monoclonal antibody (mAb), leading to PD-L1 exposure on the tumor surface (②); in the other, Although anti-PD-L1 mAb can interact with PD-L1 on the surface of tumor, exo-PD-L1 can directly interact with PD-1 on the surface of T cells to inhibit immunity (③)
EVs, as an essential material information exchange carrier in the tumor microenvironment, can also play a double-edged sword role. It is undeniable that EVs released by DCs and genetically engineered T cells expressing chimeric antigen receptor (CAT) play an emerging role in tumor immunotherapy [156]. However, EV PD-L1 or EV carrying another immune checkpoint such as LGALS9 can play a negative role in tumor immunity [157]. Similarly, EV also carries some monoclonal antibody targets or RNA, leading to failure of treatment, such as EV released from breast cancer cells contains HER2 protein or AFAP1-AS1 (actin filament associated protein 1 antisense RNA 1), which will affect the efficacy of trastuzumab [158, 159]. In addition, exosomes can also promote the formation of tumor cell pre-metastatic niche [160]. Therefore, exosomes may become the target of tumor therapy. One method is to block the release of exosomes, in which GW4869 is the most commonly used exosome inhibitor [161]. Dai et al. developed the assembled nanounits of GW4869 and iron death inducer (Fe3+) to reduce the secretion of tumor derived exosomes and weaken the immunosuppressive effect of exo-PD-L1, which induce anti-tumor immune response of melanoma cells and stimulate cytotoxic T lymphocytes and immune memory [162]. Similarly, amiloride, a calcium exchanger inhibitor, is considered to be an inhibitor of exosome release [163, 164]. At the experimental level, Rab27A null TRAMP-C2 cells were generated through CRISPR/cas9-mediated deletion and injected into C57BL6/J mice. No tumor growth was detected in these animals, even after more than 4 months [9]. Similarly, in a murine model of breast cancer, Rab27A knockdown in a 4T1 cell line markedly inhibited tumor progression and enhanced anti-PD-L1 curative effect [123]. Notably, in MC38 cell lines, the combination of exosome removal and anti-PD-L1 therapy prolonged the survival time of mice to a degree similar to that of PD-L1 null cell lines [9]. Another method is to target the released exosomes, such as, a recent study designed and synthesized a new compound called Exoblock, which has the characteristics of high affinity binding to phosphatidylserine, was found to significantly block the immunosuppressive activity of human ovarian cancer and melanoma related exosomes [165].In conclusion, these findings suggest that the elimination of EVs has potential for use as an effective adjunctive therapy for improving the efficiency of anti-tumor therapy. Interestingly, a recent study constructed an engineered MDA-MB-231 cell line, which overexpressed high-affinity mutant human PD-1 protein (havPD-1) and knocked out endogenous PD-L1 and β-2 microglobulin microglobulin. HavPD-1 EVs produced by this cell line reduced the proliferation of PD-L1 overexpressed cancer cells and induced apoptosis [166].
In eukaryotes, cells can respond to external stimuli through autophagy and exosome secretion to maintain cell homeostasis. Autophagy is a lysosome-dependent mechanism for the degradation of cellular components and can be induced by oxidative stress, starvation, or protein aggregation. Amphisomes are intermediate organelles formed through the fusion of autophagosomes and exosomes [167]. It was recently reported that PD-L1 on exosomes can influence tumor autophagy. For example, glioblastoma stem cell-derived, PD-L1-containing exosomes can activate AMPK1/ULK1 signaling cascade-mediated autophagy, thus increasing the resistance of glioblastoma to temozolomide [168]. Importantly, this study may lead to an alternative strategy for the treatment of glioblastoma.
The immunosuppressive effect of EV PD-L1 is well known; however, this type of PD-L1 has also been associated with positive effects. A recent study suggested the inhibitory effect of PD-L1 may promote tissue repair [169] as excessive and persistent proinflammatory activity after trauma can aggravate tissue damage [170, 171]. Exo-PD-L1 can promote the migration of epidermal cells and dermal fibroblasts, markedly accelerating wound contraction and reepithelization in a mouse model of skin excision injury. Additionally, Exo-PD-L1 also inhibited the production of cytokines as well as the number of CD8+ T-cells in the spleen and peripheral lymph nodes [169]. In summary, Exo-PD-L1 plays an immunosuppressive role and promotes tissue repair.
Conclusions
Tumor immune escape plays a significant role in tumor occurrence and development, and can also partly explain the failure of immunotherapy. PD-L1 derived from tumor cells can interact with PD-1 on immune cells, thereby inhibiting the activity of T cells. Meanwhile, EVs can carry nucleic acids, proteins, lipids, and other molecules into the systemic circulation and transport them to all parts of the body, thereby participating in intercellular communication. Many studies involving tumor models have shown that EV PD-L1 plays a significant role in the immune escape of several cancer types. Tumor cells release EV PD-L1, which can interact with PD-1 on the surface of T cells, thereby inhibiting their effector function and reducing the release of the pro-inflammatory cytokines IFN-γ, IL-2, and granzyme-b. There are two main forms of EVs—exosomes and microvesicles—both of which can carry PD-L1. However, due to the complexity of methods associated with the separation and purification of microvesicles, most research to date has focused on Exo-PD-L1. However, with the progress of research methods, this is likely to change in the future. EV PD-L1 can also exert its immunosuppressive activity through indirect mechanisms (Table 2 and Fig. 2). These observations indicate that the mechanisms underlying immune escape are very complex and that PD-L1 released in EVs is merely the tip of the iceberg. There is still a long way to go to achieve effective cancer treatment.
In conclusion, we summarized the immunosuppressive effect of EV PD-L1 in many tumor models as well as its potential role as the marker of early cancer diagnosis, tumor progression, and tumor-targeting immunotherapy. However, many questions remain unanswered, including whether all tumors produce and secrete PD-L1-carrying EVs, whether all EV PD-L1 will lead to immunosuppression, and whether other mechanisms or signaling pathways are also involved in this inhibition. Furthermore, although EV PD-L1 can sequester anti-PD-L1 drugs, thus contributing to drug resistance, the specific mechanism underlying its involvement in the resistance to anti-PD-1/PD-L1 immunotherapy is still unclear. Moreover, to date, EV PD-L1 has not been widely used as the biomarker in clinical practice.
Availability of data and materials
No data involved.
Abbreviations
- ALIX:
-
ALG-2 interacting protein X
- ALK:
-
Anaplastic lymphoma kinase
- AFAP1-AS1:
-
Actin filament associated protein 1 antisense RNA 1
- ARF6:
-
ADP-ribosylation factor 6
- ARF1:
-
ADP- ribosylation factor 1
- B3GNT3:
-
β-1,3-Galactosyl-O-glycosyl -glycoprotein
- CLL:
-
Chronic lymphocytic leukemia
- CSN5:
-
COP9 signalosome 5
- DCs:
-
Dendritic cells
- EVs:
-
Extracellular vesicles
- Exo-PD-L1:
-
Exosomal PD-L1
- ESEs:
-
Early-sorting endosomes
- EGFR:
-
Epidermal growth factor receptor;
- ESCRT:
-
Endosomal sorting complex required for transport
- FDA:
-
Food and Drug Administration
- GC:
-
Gastric cancer
- GTP:
-
Guanosinetriphosphate
- GDP:
-
Guanosine diphosphate
- GSK3B:
-
Glycogen synthase kinase 3B
- HNSCC:
-
Head and neck squamous cell carcinoma
- havPD-1:
-
High affinity mutant human PD-1 protein
- HMGB1:
-
High-mobility group box-1
- HRS:
-
Hepatocyte growth factor-regulated tyrosine kinase substrate
- IDO-1:
-
Indoleamine2,3-dioxygenase1
- IFN-γ:
-
Interferon-gamma
- IL-10:
-
Interleukin 10
- ILVs:
-
Intraluminal vesicles
- LSEs:
-
Late-sorting endosomes
- MAPK:
-
Mitogen-activated protein kinase
- MLC:
-
Myosin light-chain
- MVBs:
-
Multivesicular bodies
- mtDNA:
-
Mitochondrial DNA
- NK:
-
Natural killer
- nSMase2:
-
Neutral sphingomyelinase 2
- NSCLC:
-
Non-small cell lung cancer
- OS:
-
Overall survival
- PD-L1:
-
Programmed cell death 1 ligand 1
- PD-1:
-
Programmed death protein-1
- PI3K:
-
Phosphatidylinositol 3-kinase
- PTEN:
-
Phosphatase and tensin homolog
- RFVs:
-
Relative fluorescence values
- Ra-223:
-
Radium-223
- ROS:
-
Reactive oxygen species
- STAT3:
-
Signal transducer and activator of transcription 3
- SHP2:
-
Src homology 2 domain-containing tyrosine phosphatase 2
- SCLC:
-
Small cell lung cancer
- SNARE:
-
Soluble N-ethylmaleimide-sensitive factor attachment protein receptor
- sEV:
-
Small EV
- TCR:
-
T-cell receptor
- TNF-α:
-
Tumor necrosis factor α
- TGF-β:
-
Transforming growth factor β
- TTP:
-
Tristetraprolin
- VAMP7:
-
Synaptic vesicle-associated membrane protein 7
- VPS4:
-
Vacuolar protein sorting 4
- WT:
-
Wild-type
- 5-FU:
-
5- Fluorouracil
References
Han Y, Liu D, Li L. PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res. 2020;10:727–42.
Cha JH, Chan LC, Li CW, Hsu JL, Hung MC. Mechanisms controlling PD-L1 expression in cancer. Mol Cell. 2019;76:359–70.
Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, Sasmal DK, Huang J, Kim JM, Mellman I, Vale RD. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017;355:1428–33.
Xu R, Rai A, Chen MS, Suwakulsiri W, Greening DW, Simpson RJ. Extracellular vesicles in cancer—implications for future improvements in cancer care. Nat Rev Clin Oncol. 2018;15:617–38.
Pitt JM, Kroemer G, Zitvogel L. Extracellular vesicles: masters of intercellular communication and potential clinical interventions. J Clin Investig. 2016;126:1139–43.
Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science. 2013;342:1432–3.
Xu-Monette ZY, Zhang M, Li J, Young KH. PD-1/PD-L1 blockade: have we found the key to unleash the antitumor immune response? Front Immunol. 2017;8:1597.
Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, Yu Z, Yang J, Wang B, Sun H, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018;560:382–6.
Poggio M, Hu T, Pai CC, Chu B, Belair CD, Chang A, Montabana E, Lang UE, Fu Q, Fong L, Blelloch R. Suppression of exosomal PD-L1 Induces systemic anti-tumor immunity and memory. Cell. 2019;177:414–27.
Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367:eaau6977.
Hessvik NP, Llorente A. Current knowledge on exosome biogenesis and release. Cell Mol Life Sci. 2018;75:193–208.
Peng X, Yang L, Ma Y, Li Y, Li H. Focus on the morphogenesis, fate and the role in tumor progression of multivesicular bodies. Cell Commun Signal. 2020;18:122.
Stuffers S, Sem Wegner C, Stenmark H, Brech A. Multivesicular endosome biogenesis in the absence of ESCRTs. Traffic. 2009;10:925–37.
McAndrews KM, Kalluri R. Mechanisms associated with biogenesis of exosomes in cancer. Mol Cancer. 2019;18:52.
Kalluri R. The biology and function of exosomes in cancer. J Clin Investig. 2016;126:1208–15.
Cai H, Reinisch K, Ferro-Novick S. Coats, tethers, Rabs, and SNAREs work together to mediate the intracellular destination of a transport vesicle. Dev Cell. 2007;12:671–82.
Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10:513–25.
Jin H, Tang Y, Yang L, Peng X, Li B, Fan Q, Wei S, Yang S, Li X, Wu B, et al. Rab GTPases: central coordinators of membrane trafficking in cancer. Front Cell Dev Biol. 2021;9:648384.
Savina A, Fader CM, Damiani MT, Colombo MI. Rab11 promotes docking and fusion of multivesicular bodies in a calcium-dependent manner. Traffic. 2005;6:131–43.
Savina A, Vidal M, Colombo MI. The exosome pathway in K562 cells is regulated by Rab11. J Cell Sci. 2002;115:2505–15.
Hsu C, Morohashi Y, Yoshimura S, Manrique-Hoyos N, Jung S, Lauterbach MA, Bakhti M, Gronborg M, Mobius W, Rhee J, et al. Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A-C. J Cell Biol. 2010;189:223–32.
Peng X, Yang L, Ma Y, Li X, Yang S, Li Y, Wu B, Tang S, Zhang F, Zhang B, et al. IKKbeta activation promotes amphisome formation and extracellular vesicle secretion in tumor cells. Biochim Biophys Acta Mol Cell Res. 2021;1868:118857.
Jahn R, Scheller RH. SNAREs–engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–43.
Fader CM, Sanchez DG, Mestre MB, Colombo MI. TI-VAMP/VAMP7 and VAMP3/cellubrevin: two v-SNARE proteins involved in specific steps of the autophagy/multivesicular body pathways. Biochim Biophys Acta. 2009;1793:1901–16.
Sharma P, Mesci P, Carromeu C, McClatchy DR, Schiapparelli L, Yates JR 3rd, Muotri AR, Cline HT. Exosomes regulate neurogenesis and circuit assembly. Proc Natl Acad Sci U S A. 2019;116:16086–94.
Gross JC, Chaudhary V, Bartscherer K, Boutros M. Active Wnt proteins are secreted on exosomes. Nat Cell Biol. 2012;14:1036–45.
Hyenne V, Apaydin A, Rodriguez D, Spiegelhalter C, Hoff-Yoessle S, Diem M, Tak S, Lefebvre O, Schwab Y, Goetz JG, Labouesse M. RAL-1 controls multivesicular body biogenesis and exosome secretion. J Cell Biol. 2015;211:27–37.
van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19:213–28.
Minciacchi VR, Freeman MR, Di Vizio D. Extracellular vesicles in cancer: exosomes, microvesicles and the emerging role of large oncosomes. Semin Cell Dev Biol. 2015;40:41–51.
Tricarico C, Clancy J, D’Souza-Schorey C. Biology and biogenesis of shed microvesicles. Small GTPases. 2017;8:220–32.
Muralidharan-Chari V, Clancy JW, Sedgwick A, D’Souza-Schorey C. Microvesicles: mediators of extracellular communication during cancer progression. J Cell Sci. 2010;123:1603–11.
Piccin A, Murphy WG, Smith OP. Circulating microparticles: pathophysiology and clinical implications. Blood Rev. 2007;21:157–71.
Taylor J, Azimi I, Monteith G, Bebawy M. Ca(2+) mediates extracellular vesicle biogenesis through alternate pathways in malignancy. J Extracell Vesicles. 2020;9:1734326.
Del Conde I, Shrimpton CN, Thiagarajan P, Lopez JA. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood. 2005;106:1604–11.
Li B, Antonyak MA, Zhang J, Cerione RA. RhoA triggers a specific signaling pathway that generates transforming microvesicles in cancer cells. Oncogene. 2012;31:4740–9.
Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000;406:532–5.
D’Souza-Schorey C, Clancy JW. Tumor-derived microvesicles: shedding light on novel microenvironment modulators and prospective cancer biomarkers. Genes Dev. 2012;26:1287–99.
Muralidharan-Chari V, Clancy J, Plou C, Romao M, Chavrier P, Raposo G, D’Souza-Schorey C. ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr Biol. 2009;19:1875–85.
Schlienger S, Campbell S, Claing A. ARF1 regulates the Rho/MLC pathway to control EGF-dependent breast cancer cell invasion. Mol Biol Cell. 2014;25:17–29.
Hirsova P, Ibrahim SH, Krishnan A, Verma VK, Bronk SF, Werneburg NW, Charlton MR, Shah VH, Malhi H, Gores GJ. Lipid-induced signaling causes release of inflammatory extracellular vesicles from hepatocytes. Gastroenterology. 2016;150:956–67.
Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11:3887–95.
Tseng SY, Otsuji M, Gorski K, Huang X, Slansky JE, Pai SI, Shalabi A, Shin T, Pardoll DM, Tsuchiya H. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med. 2001;193:839–46.
Peng Q, Qiu X, Zhang Z, Zhang S, Zhang Y, Liang Y, Guo J, Peng H, Chen M, Fu YX, Tang H. PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat Commun. 2020;11:4835.
Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, Krzysiek R, Knutson KL, Daniel B, Zimmermann MC, et al. Blockade of B7–H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003;9:562–7.
Cheng Y, Li H, Deng Y, Tai Y, Zeng K, Zhang Y, Liu W, Zhang Q, Yang Y. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis. 2018;9:422.
Teramoto K, Igarashi T, Kataoka Y, Ishida M, Hanaoka J, Sumimoto H, Daigo Y. Clinical significance of PD-L1-positive cancer-associated fibroblasts in pN0M0 non-small cell lung cancer. Lung Cancer. 2019;137:56–63.
Hirano T, Honda T, Kanameishi S, Honda Y, Egawa G, Kitoh A, Nakajima S, Otsuka A, Nomura T, Dainichi T, et al. PD-L1 on mast cells suppresses effector CD8(+) T-cell activation in the skin in murine contact hypersensitivity. J Allergy Clin Immunol. 2021;148:563–73.
Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity. 2018;48:434–52.
Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci U S A. 2003;100:5336–41.
Garo LP, Ajay AK, Fujiwara M, Beynon V, Kuhn C, Gabriely G, Sadhukan S, Raheja R, Rubino S, Weiner HL, Murugaiyan G. Smad7 controls immunoregulatory PDL2/1-PD1 signaling in intestinal inflammation and autoimmunity. Cell Rep. 2019;28:3353–66.
Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Investig. 2015;125:3384–91.
Tsushima F, Yao S, Shin T, Flies A, Flies S, Xu H, Tamada K, Pardoll DM, Chen L. Interaction between B7–H1 and PD-1 determines initiation and reversal of T-cell anergy. Blood. 2007;110:180–5.
Zou W, Wolchok JD, Chen L. PD-L1 (B7–H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv324.
Dong H, Zhu G, Tamada K, Chen L. B7–H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–9.
Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. 2012;209:1201–17.
George J, Saito M, Tsuta K, Iwakawa R, Shiraishi K, Scheel AH, Uchida S, Watanabe SI, Nishikawa R, Noguchi M, et al. Genomic amplification of CD274 (PD-L1) in small-cell lung cancer. Clin Cancer Res. 2017;23:1220–6.
Ikeda S, Okamoto T, Okano S, Umemoto Y, Tagawa T, Morodomi Y, Kohno M, Shimamatsu S, Kitahara H, Suzuki Y, et al. PD-L1 is upregulated by simultaneous amplification of the PD-L1 and JAK2 genes in non-small cell lung cancer. J Thorac Oncol. 2016;11:62–71.
Roemer MG, Advani RH, Ligon AH, Natkunam Y, Redd RA, Homer H, Connelly CF, Sun HH, Daadi SE, Freeman GJ, et al. PD-L1 and PD-L2 genetic alterations define classical hodgkin lymphoma and predict outcome. J Clin Oncol. 2016;34:2690–7.
Twa DD, Chan FC, Ben-Neriah S, Woolcock BW, Mottok A, Tan KL, Slack GW, Gunawardana J, Lim RS, McPherson AW, et al. Genomic rearrangements involving programmed death ligands are recurrent in primary mediastinal large B-cell lymphoma. Blood. 2014;123:2062–5.
Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, Maeda T, Nagata Y, Kitanaka A, Mizuno S, et al. Aberrant PD-L1 expression through 3’-UTR disruption in multiple cancers. Nature. 2016;534:402–6.
Lu C, Paschall AV, Shi H, Savage N, Waller JL, Sabbatini ME, Oberlies NH, Pearce C, Liu K. The MLL1-H3K4me3 axis-mediated PD-L1 expression and pancreatic cancer immune evasion. J Natl Cancer Inst. 2017;109:djw283.
Zhu H, Bengsch F, Svoronos N, Rutkowski MR, Bitler BG, Allegrezza MJ, Yokoyama Y, Kossenkov AV, Bradner JE, Conejo-Garcia JR, Zhang R. BET bromodomain inhibition promotes anti-tumor immunity by suppressing PD-L1 expression. Cell Rep. 2016;16:2829–37.
Boes M, Meyer-Wentrup F. TLR3 triggering regulates PD-L1 (CD274) expression in human neuroblastoma cells. Cancer Lett. 2015;361:49–56.
Carbotti G, Barisione G, Airoldi I, Mezzanzanica D, Bagnoli M, Ferrero S, Petretto A, Fabbi M, Ferrini S. IL-27 induces the expression of IDO and PD-L1 in human cancer cells. Oncotarget. 2015;6:43267–80.
Deng XX, Jiao YN, Hao HF, Xue D, Bai CC, Han SY. Taraxacum mongolicum extract inhibited malignant phenotype of triple-negative breast cancer cells in tumor-associated macrophages microenvironment through suppressing IL-10/STAT3/PD-L1 signaling pathways. J Ethnopharmacol. 2021;274:113978.
Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017;19:1189–201.
Ju X, Zhang H, Zhou Z, Chen M, Wang Q. Tumor-associated macrophages induce PD-L1 expression in gastric cancer cells through IL-6 and TNF-ɑ signaling. Exp Cell Res. 2020;396:112315.
Liu C, Gao AC. IFNγ, a double-edged sword in cancer immunity and metastasis. Cancer Res. 2019;79:1032–3.
Mandai M, Hamanishi J, Abiko K, Matsumura N, Baba T, Konishi I. Dual faces of IFNγ in cancer progression: a role of PD-L1 induction in the determination of pro- and antitumor immunity. Clin Cancer Res. 2016;22:2329–34.
Ni XY, Sui HX, Liu Y, Ke SZ, Wang YN, Gao FG. TGF-β of lung cancer microenvironment upregulates B7H1 and GITRL expression in dendritic cells and is associated with regulatory T cell generation. Oncol Rep. 2012;28:615–21.
Quandt D, Jasinski-Bergner S, Müller U, Schulze B, Seliger B. Synergistic effects of IL-4 and TNFα on the induction of B7–H1 in renal cell carcinoma cells inhibiting allogeneic T cell proliferation. J Transl Med. 2014;12:151.
Stein S, Henze L, Poch T, Carambia A, Krech T, Preti M, Schuran FA, Reich M, Keitel V, Fiorotto R, et al. IL-17A/F enable cholangiocytes to restrict T cell-driven experimental cholangitis by upregulating PD-L1 expression. J Hepatol. 2021;74:919–30.
Wang K, Wang J, Liu T, Yu W, Dong N, Zhang C, Xia W, Wei F, Yang L, Ren X. Morphine-3-glucuronide upregulates PD-L1 expression via TLR4 and promotes the immune escape of non-small cell lung cancer. Cancer Biol Med. 2021;18:155–71.
XXXX
Atsaves V, Tsesmetzis N, Chioureas D, Kis L, Leventaki V, Drakos E, Panaretakis T, Grander D, Medeiros LJ, Young KH, Rassidakis GZ. PD-L1 is commonly expressed and transcriptionally regulated by STAT3 and MYC in ALK-negative anaplastic large-cell lymphoma. Leukemia. 2017;31:1633–7.
Glorieux C, Xia X, He YQ, Hu Y, Cremer K, Robert A, Liu J, Wang F, Ling J, Chiao PJ, Huang P. Regulation of PD-L1 expression in K-ras-driven cancers through ROS-mediated FGFR1 signaling. Redox Biol. 2021;38:101780.
Guo R, Li Y, Wang Z, Bai H, Duan J, Wang S, Wang L, Wang J. Hypoxia-inducible factor-1α and nuclear factor-κB play important roles in regulating programmed cell death ligand 1 expression by epidermal growth factor receptor mutants in non-small-cell lung cancer cells. Cancer Sci. 2019;110:1665–75.
Huang TY, Chang TC, Chin YT, Pan YS, Chang WJ, Liu FC, Hastuti ED, Chiu SJ, Wang SH, Changou CA, et al. NDAT targets PI3K-mediated PD-L1 upregulation to reduce proliferation in Gefitinib-resistant colorectal cancer. Cells. 1830;2020:9.
Kim EY, Kim A, Kim SK, Chang YS. MYC expression correlates with PD-L1 expression in non-small cell lung cancer. Lung Cancer. 2017;110:63–7.
Liang MQ, Yu FQ, Chen C. C-Myc regulates PD-L1 expression in esophageal squamous cell carcinoma. Am J Transl Res. 2020;12:379–88.
Messai Y, Gad S, Noman MZ, Le Teuff G, Couve S, Janji B, Kammerer SF, Rioux-Leclerc N, Hasmim M, Ferlicot S, et al. Renal cell carcinoma programmed death-ligand 1, a new direct target of hypoxia-inducible factor-2 alpha, is regulated by von Hippel-Lindau gene mutation status. Eur Urol. 2016;70:623–32.
Ota K, Azuma K, Kawahara A, Hattori S, Iwama E, Tanizaki J, Harada T, Matsumoto K, Takayama K, Takamori S, et al. Induction of PD-L1 expression by the EML4-ALK oncoprotein and downstream signaling pathways in non-small cell lung cancer. Clin Cancer Res. 2015;21:4014–21.
Stutvoet TS, Kol A, de Vries EG, de Bruyn M, Fehrmann RS, van Scheltinga AGT, de Jong S. MAPK pathway activity plays a key role in PD-L1 expression of lung adenocarcinoma cells. J Pathol. 2019;249:52–64.
Yan J, Zhou B, Guo L, Chen Z, Zhang B, Liu S, Zhang W, Yu M, Xu Y, Xiao Y, et al. GOLM1 upregulates expression of PD-L1 through EGFR/STAT3 pathway in hepatocellular carcinoma. Am J Cancer Res. 2020;10:3705–20.
You L, Wu W, Wang X, Fang L, Adam V, Nepovimova E, Wu Q, Kuca K. The role of hypoxia-inducible factor 1 in tumor immune evasion. Med Res Rev. 2021;41:1622–43.
Azarbarzin S, Hosseinpour-Feizi MA, Banan Khojasteh SM, Baradaran B, Safaralizadeh R. MicroRNA -383-5p restrains the proliferation and migration of breast cancer cells and promotes apoptosis via inhibition of PD-L1. Life Sci. 2021;267:118939.
Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA, Zhang X, Yi X, Dwyer D, Lin W, et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun. 2014;5:5241.
Cortez MA, Ivan C, Valdecanas D, Wang X, Peltier HJ, Ye Y, Araujo L, Carbone DP, Shilo K, Giri DK, et al. PDL1 Regulation by p53 via miR-34. J Natl Cancer Inst. 2016;108:djv303.
Miao S, Mao X, Zhao S, Song K, Xiang C, Lv Y, Jiang H, Wang L, Li B, Yang X, et al. miR-217 inhibits laryngeal cancer metastasis by repressing AEG-1 and PD-L1 expression. Oncotarget. 2017;8:62143–53.
Noman MZ, Janji B, Abdou A, Hasmim M, Terry S, Tan TZ, Mami-Chouaib F, Thiery JP, Chouaib S. The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. Oncoimmunology. 2017;6:e1263412.
Roshani Asl E, Rasmi Y, Baradaran B. MicroRNA-124–3p suppresses PD-L1 expression and inhibits tumorigenesis of colorectal cancer cells via modulating STAT3 signaling. J Cell Physiol. 2021;236:7071–87.
Xie G, Li W, Li R, Wu K, Zhao E, Zhang Y, Zhang P, Shi L, Wang D, Yin Y, et al. Helicobacter pylori promote B7–H1 expression by suppressing miR-152 and miR-200b in gastric cancer cells. PLoS ONE. 2017;12:e0168822.
Yong H, Fu J, Gao G, Shi H, Zheng D, Zhou X. MiR-34a suppresses the proliferation and invasion of gastric cancer by modulating PDL1 in the immune microenvironment. Mol Cell Probes. 2020;53:101601.
Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, Khoo KH, Chang SS, Cha JH, Kim T, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7:12632.
Li CW, Lim SO, Chung EM, Kim YS, Park AH, Yao J, Cha JH, Xia W, Chan LC, Kim T, et al. Eradication of triple-negative breast cancer cells by targeting glycosylated PD-L1. Cancer Cell. 2018;33:187–201.
Chan LC, Li CW, Xia W, Hsu JM, Lee HH, Cha JH, Wang HL, Yang WH, Yen EY, Chang WC, et al. IL-6/JAK1 pathway drives PD-L1 Y112 phosphorylation to promote cancer immune evasion. J Clin Investig. 2019;129:3324–38.
Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, Chang SS, Lin WC, Hsu JM, Hsu YH, et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell. 2016;30:925–39.
Wang Y, Sun Q, Mu N, Sun X, Wang Y, Fan S, Su L, Liu X. The deubiquitinase USP22 regulates PD-L1 degradation in human cancer cells. Cell Commun Signal. 2020;18:112.
Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–61.
Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–86.
Liu C, Gao AC. IFNgamma, a double-edged sword in cancer immunity and metastasis. Cancer Res. 2019;79:1032–3.
Mandai M, Hamanishi J, Abiko K, Matsumura N, Baba T, Konishi I. Dual faces of IFNgamma in cancer progression: a role of PD-L1 induction in the determination of pro- and antitumor immunity. Clin Cancer Res. 2016;22:2329–34.
Ju X, Zhang H, Zhou Z, Chen M, Wang Q. Tumor-associated macrophages induce PD-L1 expression in gastric cancer cells through IL-6 and TNF-a signaling. Exp Cell Res. 2020;396:112315.
Ni XY, Sui HX, Liu Y, Ke SZ, Wang YN, Gao FG. TGF-beta of lung cancer microenvironment upregulates B7H1 and GITRL expression in dendritic cells and is associated with regulatory T cell generation. Oncol Rep. 2012;28:615–21.
Zhang GQ, Jiao Q, Shen CT, Song HJ, Zhang HZ, Qiu ZL, Luo QY. Interleukin 6 regulates the expression of programmed cell death ligand 1 in thyroid cancer. Cancer Sci. 2021;112:997–1010.
Boutsikou E, Domvri K, Hardavella G, Tsiouda D, Zarogoulidis K, Kontakiotis T. Tumour necrosis factor, interferon-gamma and interleukins as predictive markers of antiprogrammed cell-death protein-1 treatment in advanced non-small cell lung cancer: a pragmatic approach in clinical practice. Ther Adv Med Oncol. 2018;10:1758835918768238.
Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35.
Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, Gouw AM, Baylot V, Gutgemann I, Eilers M, Felsher DW. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016;352:227–31.
Coelho MA, de Carne Trecesson S, Rana S, Zecchin D, Moore C, Molina-Arcas M, East P, Spencer-Dene B, Nye E, Barnouin K, et al. Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity. 2017;47:1083–99.
Guo R, Li Y, Wang Z, Bai H, Duan J, Wang S, Wang L, Wang J. Hypoxia-inducible factor-1alpha and nuclear factor-kappaB play important roles in regulating programmed cell death ligand 1 expression by epidermal growth factor receptor mutants in non-small-cell lung cancer cells. Cancer Sci. 2019;110:1665–75.
Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15:509–24.
Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal-Bengtson B, Dingli F, Loew D, Tkach M, Thery C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A. 2016;113:E968-977.
Monypenny J, Milewicz H, Flores-Borja F, Weitsman G, Cheung A, Chowdhury R, Burgoyne T, Arulappu A, Lawler K, Barber PR, et al. ALIX regulates tumor-mediated immunosuppression by controlling EGFR activity and PD-L1 presentation. Cell Rep. 2018;24:630–41.
Ricklefs FL, Alayo Q, Krenzlin H, Mahmoud AB, Speranza MC, Nakashima H, Hayes JL, Lee K, Balaj L, Passaro C, et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci Adv. 2018;4:eaar2766.
Chatterjee S, Chatterjee A, Jana S, Dey S, Roy H, Das MK, Alam J, Adhikary A, Chowdhury A, Biswas A, et al. Transforming growth factor beta orchestrates PD-L1 enrichment in tumor-derived exosomes and mediates CD8 T-cell dysfunction regulating early phosphorylation of TCR signalome in breast cancer. Carcinogenesis. 2021;42:38–47.
Kang JH, Jung MY, Choudhury M, Leof EB. Transforming growth factor beta induces fibroblasts to express and release the immunomodulatory protein PD-L1 into extracellular vesicles. FASEB J. 2020;34:2213–26.
Pinti M, Gibellini L, Nasi M, De Biasi S, Bortolotti CA, Iannone A, Cossarizza A. Emerging role of Lon protease as a master regulator of mitochondrial functions. Biochim Biophys Acta. 2016;1857:1300–6.
Cheng AN, Cheng LC, Kuo CL, Lo YK, Chou HY, Chen CH, Wang YH, Chuang TH, Cheng SJ, Lee AY. Mitochondrial Lon-induced mtDNA leakage contributes to PD-L1-mediated immunoescape via STING-IFN signaling and extracellular vesicles. J Immunother Cancer. 2020;8:e001372.
Parker C, Nilsson S, Heinrich D, Helle SI, O’Sullivan JM, Fossa SD, Chodacki A, Wiechno P, Logue J, Seke M, et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med. 2013;369:213–23.
Vardaki I, Corn P, Gentile E, Song JH, Madan N, Hoang A, Parikh N, Guerra L, Lee YC, Lin SC, et al. Radium-223 treatment increases immune checkpoint expression in extracellular vesicles from the metastatic prostate cancer bone microenvironment. Clin Cancer Res. 2021;27:3253–64.
Zhang M, Fan Y, Che X, Hou K, Zhang C, Li C, Wen T, Wang S, Cheng Y, Liu Y, Qu X. 5-FU-induced upregulation of exosomal PD-L1 causes immunosuppression in advanced gastric cancer patients. Front Oncol. 2020;10:492.
Timaner M, Kotsofruk R, Raviv Z, Magidey K, Shechter D, Kan T, Nevelsky A, Daniel S, de Vries EGE, Zhang T, et al. Microparticles from tumors exposed to radiation promote immune evasion in part by PD-L1. Oncogene. 2020;39:187–203.
Yang Y, Li CW, Chan LC, Wei Y, Hsu JM, Xia W, Cha JH, Hou J, Hsu JL, Sun L, Hung MC. Exosomal PD-L1 harbors active defense function to suppress T cell killing of breast cancer cells and promote tumor growth. Cell Res. 2018;28:862–4.
Theodoraki MN, Yerneni SS, Hoffmann TK, Gooding WE, Whiteside TL. Clinical significance of PD-L1(+) exosomes in plasma of head and neck cancer patients. Clin Cancer Res. 2018;24:896–905.
Lux A, Kahlert C, Grutzmann R, Pilarsky C. c-Met and PD-L1 on circulating exosomes as diagnostic and prognostic markers for pancreatic cancer. Int J Mol Sci. 2019;20:3305.
Fan Y, Che X, Qu J, Hou K, Wen T, Li Z, Li C, Wang S, Xu L, Liu Y, Qu X. Exosomal PD-L1 retains immunosuppressive activity and is associated with gastric cancer prognosis. Ann Surg Oncol. 2019;26:3745–55.
Kim DH, Kim H, Choi YJ, Kim SY, Lee JE, Sung KJ, Sung YH, Pack CG, Jung MK, Han B, et al. Exosomal PD-L1 promotes tumor growth through immune escape in non-small cell lung cancer. Exp Mol Med. 2019;51:1–13.
Martin AM, Nirschl TR, Nirschl CJ, Francica BJ, Kochel CM, van Bokhoven A, Meeker AK, Lucia MS, Anders RA, DeMarzo AM, Drake CG. Paucity of PD-L1 expression in prostate cancer: innate and adaptive immune resistance. Prostate Cancer Prostatic Dis. 2015;18:325–32.
Liu N, Zhang J, Yin M, Liu H, Zhang X, Li J, Yan B, Guo Y, Zhou J, Tao J, et al. Inhibition of xCT suppresses the efficacy of anti-PD-1/L1 melanoma treatment through exosomal PD-L1-induced macrophage M2 polarization. Mol Ther. 2021;29:2321–34.
Sun Y, Guo J, Yu L, Guo T, Wang J, Wang X, Chen Y. PD-L1(+) exosomes from bone marrow-derived cells of tumor-bearing mice inhibit antitumor immunity. Cell Mol Immunol. 2020. https://doi.org/10.1038/s41423-020-0487-7.
Li C, Qiu S, Jin K, Zheng X, Zhou X, Jin D, Xu B, Jin X. Tumor-derived microparticles promote the progression of triple-negative breast cancer via PD-L1-associated immune suppression. Cancer Lett. 2021;523:43–56.
Morrissey SM, Yan J. Exosomal PD-L1: roles in tumor progression and immunotherapy. Trends Cancer. 2020;6:550–8.
Gabrusiewicz K, Li X, Wei J, Hashimoto Y, Marisetty AL, Ott M, Wang F, Hawke D, Yu J, Healy LM, et al. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. Oncoimmunology. 2018;7:e1412909.
Shi Y, Zhang J, Mao Z, Jiang H, Liu W, Shi H, Ji R, Xu W, Qian H, Zhang X. Extracellular vesicles from gastric cancer cells induce PD-L1 expression on neutrophils to suppress T-Cell immunity. Front Oncol. 2020;10:629.
Ilson DH. Advances in the treatment of gastric cancer. Curr Opin Gastroenterol. 2018;34:465–8.
Li C, Li C, Zhi C, Liang W, Wang X, Chen X, Lv T, Shen Q, Song Y, Lin D, Liu H. Clinical significance of PD-L1 expression in serum-derived exosomes in NSCLC patients. J Transl Med. 2019;17:355.
Morrissey SM, Zhang F, Ding C, Montoya-Durango DE, Hu X, Yang C, Wang Z, Yuan F, Fox M, Zhang HG, et al. Tumor-derived exosomes drive immunosuppressive macrophages in a pre-metastatic niche through glycolytic dominant metabolic reprogramming. Cell Metab. 2021;33:2040–58.
Haderk F, Schulz R, Iskar M, Cid LL, Worst T, Willmund KV, Schulz A, Warnken U, Seiler J, Benner A, et al. Tumor-derived exosomes modulate PD-L1 expression in monocytes. Sci Immunol. 2017;2:1–12.
Liu J, Fan L, Yu H, Zhang J, He Y, Feng D, Wang F, Li X, Liu Q, Li Y, et al. Endoplasmic reticulum stress causes liver cancer cells to release exosomal miR-23a-3p and up-regulate programmed death ligand 1 expression in macrophages. Hepatology. 2019;70:241–58.
Liu D, Wang S, Bindeman W. Clinical applications of PD-L1 bioassays for cancer immunotherapy. J Hematol Oncol. 2017;10:110.
Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. 2015;14:847–56.
Doroshow DB, Bhalla S, Beasley MB, Sholl LM, Kerr KM, Gnjatic S, Wistuba II, Rimm DL, Tsao MS, Hirsch FR. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat Rev Clin Oncol. 2021;18:345–62.
Davis AA, Patel VG. The role of PD-L1 expression as a predictive biomarker: an analysis of all US Food and Drug Administration (FDA) approvals of immune checkpoint inhibitors. J Immunother Cancer. 2019;7:278.
Zhang J, Dang F, Ren J, Wei W. Biochemical aspects of PD-L1 regulation in cancer immunotherapy. Trends Biochem Sci. 2018;43:1014–32.
Mansfield AS, Aubry MC, Moser JC, Harrington SM, Dronca RS, Park SS, Dong H. Temporal and spatial discordance of programmed cell death-ligand 1 expression and lymphocyte tumor infiltration between paired primary lesions and brain metastases in lung cancer. Ann Oncol. 2016;27:1953–8.
Wang J, Zhang H, Sun X, Wang X, Ren T, Huang Y, Zhang R, Zheng B, Guo W. Exosomal PD-L1 and N-cadherin predict pulmonary metastasis progression for osteosarcoma patients. J Nanobiotechnol. 2020;18:151.
Del Re M, Marconcini R, Pasquini G, Rofi E, Vivaldi C, Bloise F, Restante G, Arrigoni E, Caparello C, Bianco MG, et al. PD-L1 mRNA expression in plasma-derived exosomes is associated with response to anti-PD-1 antibodies in melanoma and NSCLC. Br J Cancer. 2018;118:820–4.
Pang Y, Shi J, Yang X, Wang C, Sun Z, Xiao R. Personalized detection of circling exosomal PD-L1 based on Fe3O4@TiO2 isolation and SERS immunoassay. Biosens Bioelectron. 2020;148:111800.
Huang M, Yang J, Wang T, Song J, Xia J, Wu L, Wang W, Wu Q, Zhu Z, Song Y, Yang C. Homogeneous, low-volume, efficient, and sensitive quantitation of circulating exosomal PD-L1 for cancer diagnosis and immunotherapy response prediction. Angew Chem Int Ed Engl. 2020;59:4800–5.
Wang C, Huang CH, Gao Z, Shen J, He J, MacLachlan A, Ma C, Chang Y, Yang W, Cai Y, et al. Nanoplasmonic sandwich immunoassay for tumor-derived exosome detection and exosomal PD-L1 profiling. ACS Sens. 2021;6:3308–19.
Bacci G, Longhi A, Fagioli F, Briccoli A, Versari M, Picci P. Adjuvant and neoadjuvant chemotherapy for osteosarcoma of the extremities: 27 year experience at Rizzoli Institute, Italy. Eur J Cancer. 2005;41:2836–45.
Cordonnier M, Nardin C, Chanteloup G, Derangere V, Algros MP, Arnould L, Garrido C, Aubin F, Gobbo J. Tracking the evolution of circulating exosomal-PD-L1 to monitor melanoma patients. J Extracell Vesicles. 2020;9:1710899.
Zhang C, Fan Y, Che X, Zhang M, Li Z, Li C, Wang S, Wen T, Hou K, Shao X, et al. Anti-PD-1 therapy response predicted by the combination of exosomal PD-L1 and CD28. Front Oncol. 2020;10:760.
Bagchi S, Yuan R, Engleman EG. Immune checkpoint inhibitors for the treatment of cancer: clinical impact and mechanisms of response and resistance. Annu Rev Pathol. 2021;16:223–49.
Schoenfeld AJ, Hellmann MD. Acquired resistance to immune checkpoint inhibitors. Cancer Cell. 2020;37:443–55.
Massaro C, Min W, Pegtel DM, Baglio SR. Harnessing EV communication to restore antitumor immunity. Adv Drug Deliv Rev. 2021;176:113838.
Wang M, Cai Y, Peng Y, Xu B, Hui W, Jiang Y. Exosomal LGALS9 in the cerebrospinal fluid of glioblastoma patients suppressed dendritic cell antigen presentation and cytotoxic T-cell immunity. Cell Death Dis. 2020;11:896.
Ciravolo V, Huber V, Ghedini GC, Venturelli E, Bianchi F, Campiglio M, Morelli D, Villa A, Della Mina P, Menard S, et al. Potential role of HER2-overexpressing exosomes in countering trastuzumab-based therapy. J Cell Physiol. 2012;227:658–67.
Han M, Gu Y, Lu P, Li J, Cao H, Li X, Qian X, Yu C, Yang Y, Yang X, et al. Exosome-mediated lncRNA AFAP1-AS1 promotes trastuzumab resistance through binding with AUF1 and activating ERBB2 translation. Mol Cancer. 2020;19:26.
Feng W, Dean DC, Hornicek FJ, Shi H, Duan Z. Exosomes promote pre-metastatic niche formation in ovarian cancer. Mol Cancer. 2019;18:124.
Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, Schwille P, Brugger B, Simons M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science. 2008;319:1244–7.
Wang G, Xie L, Li B, Sang W, Yan J, Li J, Tian H, Li W, Zhang Z, Tian Y, Dai Y. A nanounit strategy reverses immune suppression of exosomal PD-L1 and is associated with enhanced ferroptosis. Nat Commun. 2021;12:5733.
Savina A, Furlan M, Vidal M, Colombo MI. Exosome release is regulated by a calcium-dependent mechanism in K562 cells. J Biol Chem. 2003;278:20083–90.
Zhou L, Zhang T, Shao W, Lu R, Wang L, Liu H, Jiang B, Li S, Zhuo H, Wang S, et al. Amiloride ameliorates muscle wasting in cancer cachexia through inhibiting tumor-derived exosome release. Skelet Muscle. 2021;11:17.
Bhatta M, Shenoy GN, Loyall JL, Gray BD, Bapardekar M, Conway A, Minderman H, Kelleher RJ Jr, Carreno BM, Linette G, et al. Novel phosphatidylserine-binding molecule enhances antitumor T-cell responses by targeting immunosuppressive exosomes in human tumor microenvironments. J Immunother Cancer. 2021;9:e003148.
Chen Y, Wang L, Zheng M, Zhu C, Wang G, Xia Y, Blumenthal EJ, Mao W, Wan Y. Engineered extracellular vesicles for concurrent Anti-PDL1 immunotherapy and chemotherapy. Bioact Mater. 2021;9:251–65.
Xu J, Camfield R, Gorski SM. The interplay between exosomes and autophagy—partners in crime. J Cell Sci. 2018;131:jcs215210.
Zheng Y, Liu L, Wang Y, Xiao S, Mai R, Zhu Z, Cao Y. Glioblastoma stem cell (GSC)-derived PD-L1-containing exosomes activates AMPK/ULK1 pathway mediated autophagy to increase temozolomide-resistance in glioblastoma. Cell Biosci. 2021;11:63.
Su D, Tsai HI, Xu Z, Yan F, Wu Y, Xiao Y, Liu X, Wu Y, Parvanian S, Zhu W, et al. Exosomal PD-L1 functions as an immunosuppressant to promote wound healing. J Extracell Vesicles. 2019;9:1709262.
Eming SA, Martin P, Tomic-Canic M. Wound repair and regeneration: mechanisms, signaling, and translation. Sci Transl Med. 2014;6:265sr266.
Martin P, Leibovich SJ. Inflammatory cells during wound repair: the good, the bad and the ugly. Trends Cell Biol. 2005;15:599–607.
Acknowledgements
Not applicable.
Funding
This study was supported by grants from National Natural Science Foundation of China (No. 81472302/No. 82003040), Natural Science Foundation of Liaoning Province (2020-BS-103), China Postdoctoral Science Foundation (2020M681016), Xingliaoyingcaijihua Project of Liaoning Province (XLYC1902050) and Qingnianyingcaijihua Project of China Medical University.
Author information
Authors and Affiliations
Contributions
JL and XP drafted this review and designed the tables and figures; LY gave some valuable suggestions; SY, XL, SW, MH provided guidance and revised this manuscript; Critical revision of the manuscript for important intellectual content: SZ, GH, HZ, QF. LY and HL conceived the final approval of the version to be submitted and obtaining of the funding. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The authors declare that their participation in writing this review as well as its publication is voluntary.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, J., Peng, X., Yang, S. et al. Extracellular vesicle PD-L1 in reshaping tumor immune microenvironment: biological function and potential therapy strategies. Cell Commun Signal 20, 14 (2022). https://doi.org/10.1186/s12964-021-00816-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-021-00816-w