
Abstract
Background
Protein disulfide isomerase A3 (PDIA3, also named GRP58, ER-60, ERp57) is conserved across species and mediates protein folding in the endoplasmic reticulum. PDIA3 is, reportedly, a chaperone for STAT3. However, the role of PDIA3 in regulating mitochondrial bioenergetics and STAT3 phosphorylation at serine 727 (S727) has not been described.
Methods
Mitochondrial respiration was compared in immortalized human cerebral microvascular cells (CMEC) wild type or null for PDIA3 and in whole organism C. Elegans WT or null for pdi-3 (worm homologue). Mitochondrial morphology and cell signaling pathways in PDIA3-/- and WT cells were assessed. PDIA3-/- cells were subjected to oxygen–glucose deprivation (OGD) to determine the effects of PDIA3 on cell survival after injury.
Results
We show that PDIA3 gene deletion using CRISPR-Cas9 in cultured CMECs leads to an increase in mitochondrial bioenergetic function. In C. elegans, gene deletion or RNAi knockdown of pdi-3 also increased respiratory rates, confirming a conserved role for this gene in regulating mitochondrial bioenergetics. The PDIA3-/- bioenergetic phenotype was reversed by overexpression of WT PDIA3 in cultured PDIA3-/- CMECs. PDIA3-/- and siRNA knockdown caused an increase in phosphorylation of the S727 residue of STAT3, which is known to promote mitochondrial bioenergetic function. Increased respiration in PDIA3-/- CMECs was reversed by a STAT3 inhibitor. In PDIA3-/- CMECs, mitochondrial membrane potential and reactive oxygen species production, but not mitochondrial mass, was increased, suggesting an increased mitochondrial bioenergetic capacity. Finally, PDIA3-/- CMECs were more resistant to oxygen–glucose deprivation, while STAT3 inhibition reduced the protective effect.
Conclusions
We have discovered a novel role for PDIA3 in suppressing mitochondrial bioenergetic function by inhibiting STAT3 S727 phosphorylation.
Similar content being viewed by others


Background
PDIA3 is a protein disulfide isomerase that promotes protein folding in the ER [1,2,3,4]. PDIA3 is also present in the cytoplasm, nucleus, mitochondria and plasma membrane caveolae [5,6,7], where it mediates glucose stress responses [8] and modulates transcriptional regulation [8]. Human PDIA3 has homology with proteins in distant eukaryotes, with approximately 50% primary sequence homology with PDI-3 of C. elegans, suggesting vital conserved roles between species. PDIA3 deletion in mice is lethal at E13.5 [9] and deletion of pdi-3 in C. elegans causes morphological defects (“dumpy”) and dysregulated collagen deposition [10]. In humans, PDIA3 is ubiquitously expressed and has been implicated in pathology such as cancer progression [11], Huntington’s disease [12], amyotrophic lateral sclerosis [13], and traumatic brain injury [14]. Mitochondrial dysfunction is a hallmark of these conditions [15,16,17,18], but little is known about PDIA3’s role in mitochondrial homeostasis and function.
Within mitochondria, PDIA3 promotes the intrinsic apoptotic pathway and mitochondrial permeabilization [6, 19, 20]. Furthermore, PDIA3 can increase mitochondrial calcium uptake by transcriptionally upregulating the mitochondrial calcium uniporter [21]. Finally, PDIA3 is present at ER-mitochondrial contact sites, suggesting a possible role for non-transcriptional regulation between these organelles [22]. A general protein disulfide isomerase inhibitor can protect against reductions in ATP levels in an in vitro Huntington disease model [12], supporting the notion that PDIA3 might inhibit mitochondrial bioenergetic function.
A potential mechanism through which PDIA3 might modulate mitochondrial bioenergetics is through its interaction with STAT3. PDIA3 is a chaperone for STAT3 and can modulate STAT3 transcriptional activity by inhibiting or promoting phosphorylation of the Y705 residue [8, 9, 23,24,25] and inhibiting STAT3 S727 phosphorylation [23, 26]. Phosphorylation of only the S727 residue targets STAT3 for mitochondrial import [27], where it promotes bioenergetic function and reduces reactive oxygen species (ROS) [28, 29]. In cardiomyocytes, STAT3 prevents mitochondrial permeabilization through inhibiting mitochondrial permeability transition pore opening and reduces toxic ROS levels during ischemia/reperfusion injury [30, 31]. Phosphorylated S727 (pS727) STAT3 also preserves mitochondrial function during post-ischemic conditioning after injury [32]. We have shown that reduced mitochondrial pS727 STAT3 in endothelial cells is associated with ER stress-induced cell death [33], while increased mitochondrial pS727 STAT3 improves bioenergetic function and endothelial cell survival [34]. Brain endothelial cells have very high concentrations of mitochondria [35, 36], which are important for maintaining the blood–brain-barrier [37, 38]. Here, we investigated whether PDIA3 suppresses mitochondrial bioenergetic function through STAT3 by using CMECs, a well-differentiated human brain endothelial cell line. We used C. elegans to test the role of the homologue, PDI3, in an intact animal because the PDIA3 knockout in mice is embyronically lethal [9]. Additionally, we tested whether PDIA3 deletion would be protective against OGD injury in cultured CMECs.
Methods
CMECs and C. elegans
CMECs are an immortalized clone of well-differentiated microvascular endothelial cells originally obtained from brain tissue of a female epilepsy patient [39, 40] and were expanded and maintained at 37 °C and 5% CO2 as described [41]. We chose to use CMEC’s as these cells are highly transfectable which allows for genetic manipulation and brain endothelial cells have robust expression of PDIA3 [42]. N2 and TP66 C. elegans strains were obtained from the Caenorhabditis Genetics Center (CGC). C. elegans were chosen as a model because a knockout is available (TP66) while PDIA3 KO in mice leads to a lethal phenotype [9]. TP66 worms have a pdi-3 gene deletion, originally identified by genetic screens of mutants generated by random UV/trimethylpsoralen and were backcrossed four times [10]. TP66 worms were found to be homozygous for the pdi-3 deletion with genotyping performed by PCR of isolated genomic DNA (primers: 5′-CGTGTCTTGAAAGTTGCTC-3′, 3′-CCCTCTAACTTCGAACATTGG-5′). No difference was observed between the strains in the developmental time from L1 larvae to adulthood. Worms were grown on Nematode Growth Media agar plates with a surface coating of live OP50 E. coli feeder bacteria [43]. The worms were synchronized by bleaching followed by overnight suspension in M9 buffer to obtain L1 stage larva and experiments were performed at the L3/L4 stage.
Bioenergetic measurements
Oxygen consumption rate (OCR) assays were performed as described [44] with minor modifications. Briefly, we used an XF HS mini-Seahorse analyzer (Agilent) with Flux Packs (103,022–100, Agilent). For quantification of bioenergetic functions, basal respiration was calculated as baseline OCR minus OCR level after antimycin A, ATP-linked respiration as the level after baseline minus oligomycin, reserve (spare) capacity as the level after FCCP minus baseline, and maximal respiration as the OCR level reached after FCCP minus the antimycin A level. CMECs were plated onto collagen type 1 (3440–005-01, Cultrex, 150 µg/ml in H2O) at ~ 20,000 cells/well. L3/4 stage worms were fed heat-killed OP50 E. coli bacteria for 48 h to avoid background respiration from E. coli that may have stuck to C. elegans after washing. Bacteria were heat killed by two cycles of flash freezing in liquid nitrogen followed by heating to 70 °C for 2 h. Immediately prior to analysis, worms were washed 5× with 0.1 M NaCl to remove excess bacteria before counting and plating ~ 75 worms/well in M9 medium. For worms, 40 mM sodium azide was used instead of antimycin A and oligomycin was excluded because it cannot readily diffuse through the worm cuticle. C. elegans were counted by an investigator who was blinded to the genotype and treatment of the worms from images taken after the assays were performed.
Fluorometric and morphological measurements
Fluorometric measurements were carried out as described [34]. CMECs were plated at 43 cells/mm2 (~ 20,000 cells/well) for 48 h on type 1 rat collagen, then incubated with 1 µg/ml Hoechst 33,342 (H1399, Fisher) to stain the nuclei, 0.5 µM MitoTracker Red CMXRos (M7512, Invitrogen) to assess mitochondrial membrane potential, 5 µM MitoSox Red (M36008, Thermo) for superoxide measurements or 0.2 µM LysoTracker Red (L7528, Thermo) to assess lysosomal mass. Mitochondrial surface area and volume were measured using IMARIS software [45] using CMECs after incubation with MitoTracker Green (M7514, Invitrogen) and imaging by confocal microscopy (20–30 image planes in the Z-axis, Leica TCS SP8).
Western protein detection
Western blots were performed with primary antibodies (Table 1) as described [46]. Quanititative capillary western (WES) for small amounts of mitochondrial protein were performed as described [41]. Lysates were loaded at 1 μg per capillary into plates (SM-W002, ProteinSimple) [41]. Detection was performed by chemiluminescence (DM-001, ProteinSimple) and data analysis was performed using Compass software (Version 4.0.0).
CRISPR-Cas9-mediated PDIA3 deletion
To create permanent PDIA3-/- CMECs, we used CRISPR-Cas9 as described [34]. We chose to perform genetic ablation to enable specific knockdown of targeted genes. Pharmacological inhibition of PDIA3 is difficult because available PDI inhibitors lack specificity [47, 48]. Cas9 guide RNAs were designed using CHOPCHOP [49] targeting exon 4 of the human PDIA3 gene. Oligonucleotides (5′-GATCGTGTCAGCCACTTGAAGAAGC-3′, 3′-AAAACGCTTCTTCAAGTGGCTGACA-5′) were annealed and directionally cloned with BamHI and BsmBI into a pCas-Guide plasmid (Ge100002, Origene) encoding Cas9 downstream of a CMV promoter. Genotyping was performed by standard PCR using primers flanking the predicted cut site (5′-GATCGGGTAACGAGTTGGTCCCGC-3′, 3′-AAAACGTAGGCCCAACAGTTCCCC-5′).
siRNA and PDIA3 transfections
CMECs were transfected with an siRNA targeting PDIA3 (L-003674, Horizon) or a non-targeting control siRNA (D-001810–10, Horizon), using 0.25% [v/v] Lipofectamine 3000 and 25 nM siRNA, as described [41]. Cells were maintained for 6 days before protein isolation. PDIA3 overexpression was performed by transfection of mammalian expression cassettes (pcDNA 3.1 + , V79020, Thermo) 72 h before OCR measurements. First, PDIA3 was cloned by PCR-amplification using cDNA as template material, reverse transcribed (Cat# 18,091,050, Invitrogen) from purified CMEC RNA. Amplified product (5′-CGCGGATCCCACCTCGCCGCCATGCGC-3′, 3′-CGGGAATTCTTAGAGATCCTCCTGTGCCTTCTTC-5′) was directionally cloned into the pcDNA 3.1 ( +) backbone downstream of a CMV promotor, using BamHI and EcoRI sites at the 5′ and 3′ ends, respectively and confirmed by Sanger Sequencing. Transfections of plasmid DNA isolated from endotoxin free midi-prep kits (D6915, Omega Biotek) were performed as described [34], with 0.5 µg DNA/well in Seahorse plates. Control cells were transfected with green fluorescent protein (GFP) constructs. For siRNA knockdown of PDI-3, N2 WT strain worms were fed HT115(DE3) bacteria expressing an siRNA construct targeting PDI-3 (RCE1182-202297109, Clone: H06O01.1, Horizon Discovery) up to L3/4 or a control empty vector, in the presence of ampicillin (100 μg/ml), tetracycline (10 μg/ml) and IPTG (Isopropyl β-D-1-thiogalactopyranoside, 1 mM) to induce RNAi expression.
RT-qPCR
RT-qPCR was performed using Taqman probes as described [46]. Analyses were performed using the delta-delta-Ct method with relative gene expression first normalized to GAPDH mRNA and then expressed relative to the experimental controls. Primers for human PDIA3 (Hs04194196_g1) and GAPDH (Hs02786624_g1) or mouse PDIA3 (Mm00433130_m1) and GAPDH (Mm99999915_g1) were from ThermoFisher and designed to cross exon borders to specifically target cDNA.
OGD
OGD was performed as described [50]. CMECs were plated at 200,000 cells/well and maintained for 48 h before a 24 h incubation in glucose-free deoxygenated DMEM in a hypoxia chamber (5% CO2 in 95% nitrogen). Afterwards, Hoechst was added to the medium and CMECs were imaged with a 20 × objective for quantification with ImageJ software, using a grid set to 100,000-pixel size, with nine squares per image counted blindly. Live cells were characterized by large diffuse nuclei and were counted blindly. Lactate dehydrogenase release was used as an indicator of cell death and measured according to manufacturer’s protocol (786-210, G-Biosciences).
Statistics
Sample sizes were based upon power analysis (G*power) and indicate independent experiments in the results. Statistical analyses used GraphPad Prizm with Student’s t-test for two groups, one-way ANOVA for more than two groups, and a two-way ANOVA for multiple variables in more than two groups with normally (gaussian) distributed data.
Results
PDIA3 inhibits mitochondrial bioenergetic function in CMECs
PDIA3 is known to regulate STAT3 signaling but its role in bioenergetic function is unknown. Genetic deletion of PDIA3 in CMECs with CRISPR-Cas9 was confirmed by qPCR (Fig. 1a, n = 3), western blotting (Fig. 1b) and genomic PCR with primers flanking the targeted Cas9 cut site (Fig. 1c). WT control CMECs were transfected with Cas9 and a scrambled gRNA. PDIA3-/- CMECs had increased oxygen consumption rates (OCRs) (Fig. 1d) with an increase in reserve capacity (Fig. 1e, n = 3, p < 0.01), indicating a greater spare ATP-generating capacity and an increased ability to respond to stress or higher energy demand [51]. The maximum respiratory capacity was also increased in PDIA3-/- relative to control-transfected WT cells (Fig. 1e, n = 3, p < 0.01), indicating these cells have increased maximum respiratory rates. As a confirmation, PDIA3 overexpressed in PDIA3-/- CMECs suppressed bioenergetic function (Fig. 1f, n = 3, p < 0.001), particularly maximal respiratory capacity (Fig. 1g, n = 3, p < 0.01) relative to GFP-transfected controls.

PDIA3 inhibits mitochondrial respiratory function in cultured human brain endothelial cells. a PDIA3 gene deletion or PDIA3-/- (KO) was achieved in immortalized hCMEC/D3 cells (CMECs) by CRISPR-Cas9 and confirmed by RT-qPCR (delta–delta-Ct normalized first to GAPDH) and western blotting (b). WT CMECs were transfected with non-targeting gRNA. c Genotype was further confirmed by PCR from genomic CMEC DNA surrounding the target cut site. d Respiratory rates, as measured by oxygen consumption rate (OCR) by Seahorse analysis, were increased in PDIA3-/- CMECs relative to WT cells (repeated measures ANOVA. n = 3 per condition). e Reserve (spare) capacity (Res. Cap.) and maximal respiratory capacity (Max. Resp.) were increased but not basal respiratory rate and ATP-linked respiration (n = 3 each, two-way ANOVA). f The PDIA3-/- phenotype of increased bioenergetic function was largely reversed by overexpressing WT human PDIA3 compared to GFP control transfection (g)
PDI-3 inhibits bioenergetic function in C. elegans
We determined whether PDIA3 inhibited mitochondrial function in an evolutionarily distant species and in an intact multicellular organism. The pdi-3 gene is the C. elegans worm ortholog of mammalian PDIA3 and has ~ 50% amino acid homology and conserved catalytic thioredoxin domains (Fig. 2a). TP66 worms have a pdi-3 deletion [10] as confirmed by PCR showing a shorter amplicon due to a lesion around exon 3 [10] (Fig. 2b). OCR was increased in TP66 relative to WT N2 strain worms (Fig. 2c), showing increased reserve capacity and maximal respiration (Fig. 2d, n = 6 independent plates, p < 0.05). Moreover, RNAi knockdown of pdi-3 in WT N2 worms increased OCR, particularly maximal respiration, relative to control-transfected N2 worms (Fig. 2e).

PDI-3, a PDIA3 ortholog, inhibits respiratory function in whole C. elegans worms. a PDIA3 is highly conserved in the eukaryotic genome, having approximately 50% amino acid sequence homology between humans and worms (thioredoxin domains highlighted in yellow). b The PDI-3 gene deleted TP66 strain C. elegans genotype was confirmed by PCR targeting genomic DNA surrounding the excision site. c, d PDI-3 is deficient in the TP66 strain of C. elegans and leads to an increase in reserve capacity and maximal respiration compared to N2 WT worms after treatment with the mitochondrial oxidative phosphorylation uncoupler FCCP (with ~ 75 worms from n = 6 plates, two-way ANOVA, p < 0.05). e–f Acute knockdown of PDI-3 by siRNA in N2 strain worms increased maximal respiration (n = 3, two-way ANOVA, p < 0.05) relative to control (empty vector) fed worms
PDIA3 reduces mitochondrial activity
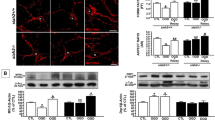
MitoTracker Red CMXRos dye accumulation depends on mitochondrial membrane potential [52]. PDIA3-/- CMECs treated with this dye had greater red fluorescence than WT CMECs (Fig. 3a, n = 6, p < 0.001) when normalized to nuclear Hoechst stain to account for variability in cell numbers [33]. In contrast, the signal from LysoTracker Red dye, which fluoresces when localized to acidic organelles such as lysosomes and autophagosomes, was not different between genotypes (Fig. 3b, n = 6), suggesting that PDIA3-/- CMECs did not have alterations in lysosomal or autophagosomal mass or mitophagy [53]. PDIA3-/- cells had increased MitoSox Red fluorescence, indicating increased mitochondrial superoxide production (Fig. 3c, n = 4, p < 0.01), likely due to the increased mitochondrial electron transport chain activity as indicated by the by the increased OCRs.

Mitochondrial membrane potential and ROS, but not mitochondrial mass, is increased in CMECs. a MitoTracker Red CMXRos dye is a potentiometric dye that accumulates in active mitochondria. Fluorometry showed that MitoTracker Red CMXRos intensity was greater in PDIA3-/- CMECs relative to WT (~ 20%, p < 0.001, n = 4, Student’s t-test). Values were normalized to nuclear Hoechst dye to account for potential variations in cell number and expressed as a fold of WT. b The fluorescence of LysoTracker Red, a dye that accumulates in lysosomes, was not different between genotypes. c Superoxide was measured using MitoSox Red. PDIA3-/- CMECs had higher levels of superoxide (n = 4, p < 0.01) relative to WT CMECs. d Synthetic bands derived from peak chemiluminescent intensity of e capillary western blotting of proteins from isolated mitochondria show that the amount of pyruvate dehydrogenase (PDHA1) was not different between PDIA3-/- and WT CMECs. The chemiluminescence peak amount is indicated by the number 55 K indicating 55,000. f, g GAPDH, a cytoplasmic protein, was largely absent, indicating the purity of the mitochondrial sample. h, i PDIA3 was present in mitochondrial fractions of WT, but not PDIA3-/-, CMECs. j, k Regular western blots of whole cell lysates show that the PDIA3-/- and WT CMECs have similar amounts of the mitochondrial protein PDHA1, indicating a similar mitochondrial mass. The cytoplasmic household protein α-tubulin was used as loading control
PDIA3-/- CMECs have normal mitochondrial mass
To exclude the possibility that bioenergetic increases were caused by changes in mitochondrial mass, protein from purified mitochondria were quantified using capillary western analysis. Pyruvate dehydrogenase subunit A1 (PDHA1) is essential for metabolizing the end product of glycolysis into the mitochondrial citric acid cycle and altered PDH activity affects OCR [54]. PDHA1 expression was not different between PDIA3-/- and WT CMECs (Fig. 3d, e). Cytosolic GAPDH was not detected, confirming the purity of the mitochondrial fractions (Fig. 3f, g). PDIA3 was present in WT mitochondrial extracts, but not those from PDIA3-/- CMECs (Fig. 3h, i), confirming PDIA3 is present in the mitochondria. Finally, PDHA1 was equally abundant in whole cell lysates of both genotypes, further suggesting that mitochondrial mass is not altered by PDIA3 knockout (Fig. 3j, k). Moreover, the mitochondrial tubular networks of MitoTracker Green-treated cells appeared similar in PDIA3-/- and WT CMECs, and their estimated mitochondrial surface area (Fig. 4b) and volume visualized by by confocal microscopy were not different (Fig. 4c).

Mitochondrial surface area or volume is not altered in PDIA3-/- CMECs. a WT or PDIA3-/- (KO) CMECs were stained with MitoTracker Green dye and imaged by confocal microscopy. A total of 32 cells were imaged across 3 independent experiments and mitochondrial surface area and volume were analyzed using IMARIS software. Neither total mitochondrial surface area (b) or volume (c) were different between WT and PDIA3-/- CMECs
PDIA3 reduces bioenergetic function through increased STAT3 S727 phosphorylation
STAT3 S727 phosphorylation increases mitochondrial respiration [55]. PDIA3 binds to STAT3 [9] to modulate Y705 phosphorylation [8, 23, 56] and can also inhibit S727 phosphorylation [23, 26]. Here, PDIA3-/- CMECs showed increased levels of STAT3 S727 phosphorylation (Fig. 5a, b; n = 3 for WT and n = 4 PDIA3-/-, p < 0.05). Levels of STAT3 Y705 did not appear to be altered in PDIA3-/- CMECs relative to WT (Fig. 5a). We have shown that integrin-FAK signaling promotes mitochondrial function through STAT3 S727 phosphorylation [34]. However, phosphorylation of FAK Y397 was not decreased in PDIA3-/- CMECs (Fig. 5a, c). The increase in STAT3 pS727 in PDIA3-/- CMECs was reversed by overexpression of PDIA3 compared to control GFP (Fig. 5d, e). Conversely, PDIA3 siRNA reduced PDIA3 protein expression and increased STAT3 pS727 (Fig. 5f, g). We next determined whether PDIA3-/- increased bioenergetic function through STAT3 signaling by using the STAT3 inhibitor, stattic [57], which suppresses STAT3 pS727 and bioenergetic function in bEnd5 endothelial cells [34]. Stattic reduced OCR in PDIA3-/- CMECs (Fig. 5h), with the cells showing lower basal and ATP-linked respiration rates (Fig. 5i, p < 0.05, n = 3).

PDIA3 inhibits mitochondrial function by suppressing pS727 STAT3. a STAT3 S727 phosphorylation was increased in PDIA3-/- CMECs, as confirmed by densitometry (b), * p < 0.05, Student’s t-test). Phosphorylation of STAT3 Y705 was not obviously different in PDIA3-/- CMECs, considering the total amounts of STAT3 and the β-actin loading control. We previously found that FAK drives mitochondrial bioenergetics via STAT3 S727 phosphorylation [34] but pFAK was not different in PDIA3-/- (KO) CMECs, as confirmed by densitometry (c). d Overexpression of WT human PDIA3 in PDIA3-/- CMECs reduced pSTAT3 S727, as seen by western blotting and densitometry (e). f) The inhibitory effect of PDIA3 on STAT3 S727 phosphorylation was confirmed by siRNA mediated knockdown of PDIA3 (siPDIA3) in WT CMECs relative to a non-targeting control siRNA (siCtrl). Knockdown increased STAT3 S727 phosphorylation (g). h Finally, we tested whether increased STAT3 S727 phosphorylation was necessary for PDIA3-/- increased respiratory function. Inhibition of STAT3 with stattic in PDIA3-/- CMECs, reduced respiratory function compared within 1 h, as measured by Seahorse assay (n = 6, p < 0.01, one-way ANOVA). i Quantification showed decreased basal and ATP-linked respiration (n = 6, p < 0.01, two-way ANOVA), which indicates PDIA3 suppresses mitochondrial function through inhibition of STAT3
PDIA3 promotes cell growth
Mitochondrial function is often linked to proliferation and survival [58], which we controlled for as a potential confounding factor in our analyses. PDIA3-/- CMECs grew more slowly between 48 and 72 h after plating (Additional file 1: Fig. S1A, B, p < 0.05, n = 3). Release of lactate dehydrogenase, a measure for cell death, was not different between genotypes between 48 and 72 h (Additional file 1: Fig. S1C). Thus, PDIA3 might promote proliferation, but not cell death, under normal conditions. The late differences in proliferation between PDIA3-/- and WT CMECs should not confound the OCR measurements made 24 h after plating or compared within the same genotype. Moreover, the OCR data were normalized to protein content.
PDIA3 reduces resistance to OGD by inhibiting pS727 STAT3
PDIA3 can promote cell death by stabilizing mitochondrial calpains and activating apoptosis inducing factor [6]. To test whether PDIA3 alters cell survival under conditions which mimic key aspects of ischemia, we performed OGD, which causes cell death in part by inhibiting mitochondrial bioenergetic function [59]. The role of PDIA3 in OGD is unclear because its expression increased in response to glucose depletion [1], but decreased upon oxygen depletion [60]. PDIA3-/- CMECs had less cell death than WT CMECs after OGD (Fig. 6a, n = 3, p < 0.001), with approximately 40% survival in PDIA3-/- and 10% survival in WT CMECs after 24 h of OGD (Fig. 6b, p < 0.001, n = 3). Inhibition of STAT3 abolished the protective effect of PDIA3 gene deletion (Fig. 6c). PDIA3-/- CMECs had better preserved pS727 and total STAT3 than WT CMECs after a 24 h OGD (Fig. 6d, e). STAT3 pY705 was completely abolished by OGD treatment in both genotypes (Fig. 6d). Upstream FAK was strongly phosphorylated in PDIA3-/-, but not WT, CMECs following OGD (Fig. 6d). Finally, PDIA3 expression was not altered in WT control CMECs relative to OGD (Fig. 6f–g). These data suggest that PDIA3 decreases survival of CMECs in vitro under OGD conditions through STAT3 inhibition.

PDIA3 contributes to cell death from oxygen–glucose deprivation. a A 24 h period of OGD caused a loss of live (yellow arrows) and appearance of apoptotic (white arrowheads) CMECs compared to normoxic control conditions, as revealed by blue nuclear Hoechst staining. b Cell counts from 3 fields/well show that the number of live PDIA3-/- cells was four times higher than WT cells after OGD (p < 0.001, n = 3, Student’s t-test). c Pharmacological inhibition of STAT3 with stattic reduced the protective effect of PDIA3-/- against OGD in CMECs (Student’s t-test, n = 3, p < 0.05). d Western blotting shows that PDIA3-/- CMECs had more phosphorylated STAT3 S727, as measured by densitometry (e) as well as increased pFAK Y397 which was completely absent in control conditions or WT CMECs undergoing OGD. STAT3 Y705 phosphorylation was lost after OGD in both PDIA3-/- and WT CMECs, indicating Y705 plays no part in PDIA3-/- cell survival. Beta actin was used as a loading control to account for differences in cell survival and STAT3 and actin blots were run on the same gel. f PDIA3 expression was not different in WT control (Normoxia) relative to WT OGD CMECs as measured by densitometry (g)
Discussion
Our most important and novel findings are that PDIA3 inhibits mitochondrial respiratory function, S727 STAT3 phosphorylation and cell survival under OGD conditions. The novel role of PDIA3 in downregulating mitochondrial bioenergetics is consistent with proteomics data indicating its potential involvement in modulating aerobic capacity in skeletal muscle [26]. PDIA3 had been shown to inhibit STAT3 S727 phosphorylation and our genetic and pharmacological results demonstrate that this downregulates mitochondrial respiratory function and cell survival under OGD conditions [23]. STAT3 regulation of mitochondrial respiration is dependent upon S727 phosphorylation and subsequent interaction with retinoid-interferon mortality-19 (GRIM19), which mediates mitochondrial import and integration with complex I of the electron transport chain [27, 55]. Thus, loss of PDIA3 and consequently increased pS727 may lead to increased availability of STAT3 for mitochondrial import. Although PDIA3 binds to STAT3 by direct protein–protein interaction [23] and modulates its signaling [61, 62], the mechanism by which it suppresses pS727 is unclear, as PDIA3 is not a phosphatase. PDIA3 is known to chaperone proteins [7] and it might act as a chaperone for serine phosphatases that dephosphorylate S727 STAT3 and bring them in close proximity to STAT3 S727. PDIA3-STAT3 association typically occurs in either the cytosol [63] or the nucleus [8]. However, like others [20], we found PDIA3 within mitochondria as well, suggesting that PDIA3 could promote dephosphorylation of pS727 within mitochondria. We have shown that STAT3 S727 is dephosphorylated in isolated mitochondria, indicating the presence of mitochondrial-resident serine phosphatases that can perform this function [34]. It remains to be determined whether PDIA3 exerts its observed influence on mitochondrial respiration mostly from its binding to complex I on the inner membrane of the organelle or indirectly from elsewhere.
PDIA3 might also contribute to cell survival and death through other pathways that affect mitochondria. Depletion of the related PDIA1 in endothelial cells causes increased mitochondrial fission and ROS production, leading to cellular senescence [64], although bioenergetic function was not tested. Mitochondrial fragmentation and increases in ROS have been observed in hyperglycemic conditions [65]. A potential role of PDIA3 in decreasing mitophagy is suggested by its role in autophagy in pancreatic beta cells leading to apoptosis [66]. Here, PDIA3 deletion did not affect mitochondrial mass or morphology, but, like depletion of PDIA1, it increased ROS production. Thus, increased bioenergetics upon PDIA3 deletion in CMECs and C. elegans is likely the result of increases in the activity of key electron transport chain complexes. Vitamin D restores autolysosomal function in gut epithelial cells through activation of PDIA3 [24]. Although changes in mitophagy and mitochondrial turnover could result in altered bioenergetic capacity, we did not detect differences in lysosomal content between WT and PDIA3-/- CMECs. On the other hand, PDIA1, like PDIA3, contains the prototypical -CGHC- thioredoxin amino acid sequence at its amino and carboxyl termini, which can reduce ROS accumulation. Loss of either one may reduce ROS scavenging capacity within a cell. In our study, PDIA3-/- CMECs showed an increased mitochondrial membrane potential, as well as greater ROS production. This is consistent with a role for PDIA3 as an inhibitor of bioenergetic capacity.
Our study also suggests that PDIA3 inhibition of mitochondrial function is not restricted to mammalian cells. In C. elegans, a homolog of mammalian STAT proteins STA-1 has only 30% homology to STAT3 and no data exists on whether PDI-3 interacts with STA-1 [67]. The shortened (dumpy) morphological phenotype of pdi-3 deleted worms [10] is likely caused by decreased collagen crosslinking and extracellular matrix stabilization due to the lack of PDI-3 transglutaminase activity, as well as the loss of chaperone function in the ER. However, the dumpy phenotype may be enhanced by the increased respiration which produces increased amounts of ROS that are known to modulate cuticle remodeling during molting [68, 69]. PDIA3 may also play a role in suppressing muscle cell functions in which PDIA3 has a potential involvement in aerobic capacity [26]. PDIA3 expression is increased in the gastrocnemius muscle of 30 month old relative to younger 7 month old rats [70] and in chronological muscle ageing in post-menopausal women [71], potentially explaining in part why aged skeletal muscle has reduced oxidative capacity relative to young muscle [72].
Our data also suggest that PDIA3 plays a detrimental role under pathological conditions such as OGD, which mimics key aspects of ischemia. Importantly, both OGD and ischemia are characterized by mitochondrial bioenergetic failure [33]. Long-term bioenergetic dysfunction leads to apoptosis [73]. PDIA3 protein expression is upregulated after OGD in SHSY-5Y neuronal cells [74], but was not altered in the CMECs. Mitochondria play a key role in apoptosis [73], releasing cytochrome C through the mitochondrial permeability transition pore, which STAT3 can suppress [75]. PDIA3 is known to stabilize expression of mitochondrial calpains and cleavage of apoptosis inducing factor, thereby contributing to increased apoptosis [6], so abrogation of PDIA3 functions could result in decreases in pro-apoptotic pathway activation. In addition to this, PDIA3 interacts directly with STAT3 to suppress pY705 [23]. It is likely that during this interaction, pS727 is inhibited [76]. By reducing the amount of pSTAT3 Y705 in the nucleus, PDIA3 may increase the available pool of cytoplasmic STAT3 capable of being phosphorylated on S727 and, after translocation to the mitochondria, promoting mitochondrial function and cell survival. Alternatively, reduced PDIA3 can function as a ROS scavenger [77], and increased ROS promotes STAT3 phosphorylation on S727 and mitochondrial recruitment [78]. Thus, the ROS increases observed in PDIA3-/- cells could be the mechanistic link between PDIA3 and pSTAT3 S727. Following OGD, we found that pY705 was completely abolished in both WT and PDIA3-/- CMECs, suggesting that Y705 is not important for cell survival in this model. Pharmacological inhibition of STAT3 with stattic resulted in a loss of PDIA3-/- protective effects against OGD. Other studies found that inhibition of STAT3 can be protective against hypoxic injury by blocking translocation to the nucleus [79]. STAT3 has also been found in the ER, where it has a role in regulating calcium homeostasis as key to its anti-apoptotic activities [76]. We observed increased mitochondrial superoxide in PDIA3-/- cells and this may lead to decreased rates of cell growth (Additional file 1: Fig. S1) through downregulation of cyclins involved in cell cycle checkpoints. P21 and P53 are upregulated in response to ROS production, delaying progression from G1 to S phases of the cell cycle [80]. At the same time, it is possible that loss of PDIA3 leads to decreased cell death through increased STAT3 S727 phosphorylation and reduced calcium overload [76]. Further, reduced FAK-S727 STAT3 activation following ER stress in mouse bEnd5 endothelial cells leads to decreased cell survival [33], suggesting an important role for this pathway in preventing apoptosis under injurious conditions. Here, both FAK pY397 and STAT3 pS727 were reduced following OGD, suggesting that partial preservation of this signaling pathway protects these cells. However, the mechanism for how PDIA3-/- increases FAK pY397 under OGD, but not under normoxic conditions requires further investigation.
Conclusions
In conclusion, our study defines a novel role for PDIA3 in inhibiting mitochondrial bioenergetics (Fig. 7), whereby decreases in PDIA3 increases respiratory function and reduces cell loss during OGD. This presents an important step forward in understanding PDIA3 functions related to STAT3 signaling and new avenues for therapeutic intervention for diseases such as stroke.

PDIA3 reduces mitochondrial function: Overview. In endothelial cells, PDIA3 inhibits phosphorylation of the S727 residue of STAT3, which promotes mitochondrial bioenergetics. PDIA3 gene knockout with CRISPR or knockdown with siRNA increases STAT3 activation, mitochondrial bioenergetics, cellular function and cell survival under OGD conditions. In C. elegans, the PDI-3 homolog plays a similar role in mitochondrial bioenergetics as shown in the TP66 mutant strain
Availability of data
All data and analyses of this study have been included in the manuscript. Detailed methods are available upon reasonable request.
Abbreviations
- ER:
-
Endoplasmic reticulum
- ERp57:
-
Endoplasmic reticulum protein 57
- GRP57:
-
Glucose regulated protein 58
- CMEC:
-
Cerebral microvascular endothelial cell
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- FAK:
-
Focal adhesion kinase
- FCCP:
-
Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- GFP:
-
Green fluorescent protein
- OCR:
-
Oxygen consumption rate
- OGD:
-
Oxygen–glucose deprivation
- N2:
-
Wild-type C. elegans strain
- PCR:
-
Polymerase chain reaction
- PDHA1:
-
Pyruvate dehydrogenase-A1
- PDI-3:
-
Protein disulfide isomerase 3
- PDIA3:
-
Protein disulfide isomerase A member 3
- pS727:
-
Phosphorylated serine 727
- ROS:
-
Reactive oxygen species
- S727:
-
Serine 727
- siRNA:
-
Short-interfering RNA
- STA-1:
-
Signal transducer and activator of transcription 1
- STAT3:
-
Signal transducer and activator of transcription 3
- TP66:
-
C. elegans Strain with a pdi-3 deletion
- WT:
-
Wild-type
- Y705:
-
Tyrosine 705
References
Lee AS. The accumulation of three specific proteins related to glucose-regulated proteins in a temperature-sensitive hamster mutant cell line K12. J Cell Physiol. 1981;106(1):119–25.
Lindquist JA, Jensen ON, Mann M, Hämmerling GJ. ER-60, a chaperone with thiol-dependent reductase activity involved in MHC class I assembly. EMBO J. 1998;17(8):2186–95.
Hughes EA, Cresswell P. The thiol oxidoreductase ERp57 is a component of the MHC class I peptide-loading complex. Curr Biol. 1998;8(12):709–12.
Morrice NA, Powis SJ. A role for the thiol-dependent reductase ERp57 in the assembly of MHC class I molecules. Curr Biol. 1998;8(12):713–6.
Chen J, Olivares-Navarrete R, Wang Y, Herman TR, Boyan BD, Schwartz Z. Protein-disulfide isomerase-associated 3 (Pdia3) mediates the membrane response to 1,25-dihydroxyvitamin D3 in osteoblasts. J Biol Chem. 2010;285(47):37041–50.
Ozaki T, Yamashita T, Ishiguro S-I. ERp57-associated mitochondrial μ-calpain truncates apoptosis-inducing factor. Biochim Biophys Acta. 2008;1783(10):1955–63.
Hettinghouse A, Liu R, Liu C-J. Multifunctional molecule ERp57: From cancer to neurodegenerative diseases. Pharmacol Ther. 2018;181:34–48.
Kondo R, Ishino K, Wada R, Takata H, Peng W-X, Kudo M, et al. Downregulation of protein disulfide-isomerase A3 expression inhibits cell proliferation and induces apoptosis through STAT3 signaling in hepatocellular carcinoma. Int J Oncol. 2019;54(4):1409–21.
Coe H, Jung J, Groenendyk J, Prins D, Michalak M. ERp57 modulates STAT3 signaling from the lumen of the endoplasmic reticulum. J Biol Chem. 2010;285(9):6725–38.
Winter AD, McCormack G, Page AP. Protein disulfide isomerase activity is essential for viability and extracellular matrix formation in the nematode Caenorhabditis elegans. Dev Biol. 2007;308(2):449–61.
Shimoda T, Wada R, Kure S, Ishino K, Kudo M, Ohashi R, et al. Expression of protein disulfide isomerase A3 and its clinicopathological association in gastric cancer. Oncol Rep. 2019;41(4):2265–72.
Zhou X, Li G, Kaplan A, Gaschler MM, Zhang X, Hou Z, et al. Small molecule modulator of protein disulfide isomerase attenuates mutant huntingtin toxicity and inhibits endoplasmic reticulum stress in a mouse model of Huntington’s disease. Hum Mol Genet. 2018;27(9):1545–55.
Woehlbier U, Colombo A, Saaranen MJ, Pérez V, Ojeda J, Bustos FJ, et al. ALS-linked protein disulfide isomerase variants cause motor dysfunction. EMBO J. 2016;35(8):845–65.
Wang W-T, Sun L, Sun C-H. PDIA3-regulted inflammation and oxidative stress contribute to the traumatic brain injury (TBI) in mice. Biochem Biophys Res Commun. 2019;518(4):657–63.
Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12(10):685–98.
Carmo C, Naia L, Lopes C, Rego AC. Mitochondrial dysfunction in huntington’s disease. Adv Exp Med Biol. 2018;1049:59–83.
Joshi AU, Mochly-Rosen D. Mortal engines: mitochondrial bioenergetics and dysfunction in neurodegenerative diseases. Pharmacol Res. 2018;138:2–15.
Kulbe JR, Hall ED. Chronic traumatic encephalopathy-integration of canonical traumatic brain injury secondary injury mechanisms with tau pathology. Prog Neurobiol. 2017;158:15–44.
Ozaki T, Yamashita T, Ishiguro S-I. Mitochondrial m-calpain plays a role in the release of truncated apoptosis-inducing factor from the mitochondria. Biochim Biophys Acta. 2009;1793(12):1848–59.
Zhao G, Lu H, Li C. Proapoptotic activities of protein disulfide isomerase (PDI) and PDIA3 protein, a role of the Bcl-2 protein Bak. J Biol Chem. 2015;290(14):8949–63.
He J, Shi W, Guo Y, Chai Z. ERp57 modulates mitochondrial calcium uptake through the MCU. FEBS Lett. 2014;588(12):2087–94.
Paillusson S, Stoica R, Gomez-Suaga P, Lau DHW, Mueller S, Miller T, et al. There’s something wrong with my MAM; the ER-mitochondria axis and neurodegenerative diseases. Trends Neurosci. 2016;39(3):146–57.
Choe MH, Min JW, Jeon HB, Cho D-H, Oh JS, Lee HG, et al. ERp57 modulates STAT3 activity in radioresistant laryngeal cancer cells and serves as a prognostic marker for laryngeal cancer. Oncotarget. 2015;6(5):2654–66.
Hu W, Zhang L, Li MX, Shen J, Liu XD, Xiao ZG, et al. Vitamin D3 activates the autolysosomal degradation function against Helicobacter pylori through the PDIA3 receptor in gastric epithelial cells. Autophagy. 2019;15(4):707–25.
Chichiarelli S, Gaucci E, Ferraro A, Grillo C, Altieri F, Cocchiola R, et al. Role of ERp57 in the signaling and transcriptional activity of STAT3 in a melanoma cell line. Arch Biochem Biophys. 2010;494(2):178–83.
Burniston JG, Kenyani J, Gray D, Guadagnin E, Jarman IH, Cobley JN, et al. Conditional independence mapping of DIGE data reveals PDIA3 protein species as key nodes associated with muscle aerobic capacity. J Proteomics. 2014;106:230–45.
Tammineni P, Anugula C, Mohammed F, Anjaneyulu M, Larner AC, Sepuri NBV. The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J Biol Chem. 2013;288(7):4723–32.
Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324(5935):1713–6.
Meier JA, Hyun M, Cantwell M, Raza A, Mertens C, Raje V, et al. Stress-induced dynamic regulation of mitochondrial STAT3 and its association with cyclophilin D reduce mitochondrial ROS production. Sci Signal. 2017;10(472).
Boengler K, Hilfiker-Kleiner D, Heusch G, Schulz R. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res Cardiol. 2010;105(6):771–85.
Wu L, Tan J-L, Chen Z-Y, Huang G. Cardioprotection of post-ischemic moderate ROS against ischemia/reperfusion via STAT3-induced the inhibition of MCU opening. Basic Res Cardiol. 2019;114(5):39.
Heusch G, Musiolik J, Gedik N, Skyschally A. Mitochondrial STAT3 activation and cardioprotection by ischemic postconditioning in pigs with regional myocardial ischemia/reperfusion. Circ Res. 2011;109(11):1302–8.
Banerjee K, Keasey MP, Razskazovskiy V, Visavadiya NP, Jia C, Hagg T. Reduced FAK-STAT3 signaling contributes to ER stress-induced mitochondrial dysfunction and death in endothelial cells. Cell Signal. 2017;36:154–62.
Visavadiya NP, Keasey MP, Razskazovskiy V, Banerjee K, Jia C, Lovins C, et al. Integrin-FAK signaling rapidly and potently promotes mitochondrial function through STAT3. Cell Commun Signal. 2016;14(1):32.
Oldendorf WH, Cornford ME, Brown WJ. The large apparent work capability of the blood-brain barrier: a study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann Neurol. 1977;1(5):409–17.
Doll DN, Hu H, Sun J, Lewis SE, Simpkins JW, Ren X. Mitochondrial crisis in cerebrovascular endothelial cells opens the blood-brain barrier. Stroke. 2015;46(6):1681–9.
Prakash R, Carmichael ST. Blood-brain barrier breakdown and neovascularization processes after stroke and traumatic brain injury. Curr Opin Neurol. 2015;28(6):556–64.
Grammas P, Martinez J, Miller B. Cerebral microvascular endothelium and the pathogenesis of neurodegenerative diseases. Expert Rev Mol Med. 2011;13:e19.
Weksler BB, Subileau EA, Perrière N, Charneau P, Holloway K, Leveque M, et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J Off Publ Fed Am Soc Exp Biol. 2005;19(13):1872–4.
Weksler B, Romero IA, Couraud P-O. The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS. 2013;10(1):16.
Keasey MP, Jia C, Pimentel LF, Sante RR, Lovins C, Hagg T. Blood vitronectin is a major activator of LIF and IL-6 in the brain through integrin-FAK and uPAR signaling. J Cell Sci. 2018;131(3).
Landel V, Stephan D, Cui X, Eyles D, Feron F. Differential expression of vitamin D-associated enzymes and receptors in brain cell subtypes. J Steroid Biochem Mol Biol. 2018;177:129–34.
Edwards CB, Copes N, Brito AG, Canfield J, Bradshaw PC. Malate and fumarate extend lifespan in Caenorhabditis elegans. PLoS ONE. 2013;8(3):e58345.
Visavadiya NP, Keasey MP, Razskazovskiy V, Banerjee K, Jia C, Lovins C, et al. Correction to: Integrin-FAK signaling rapidly and potently promotes mitochondrial function through STAT3. Cell Commun Signal. 2020;18(1):64.
Chevrollier A, Cassereau J, Ferré M, Alban J, Desquiret-Dumas V, Gueguen N, et al. Standardized mitochondrial analysis gives new insights into mitochondrial dynamics and OPA1 function. Int J Biochem Cell Biol. 2012;44(6):980–8.
Keasey MP, Kang SS, Lovins C, Hagg T. Inhibition of a novel specific neuroglial integrin signaling pathway increases STAT3-mediated CNTF expression. Cell Commun Signal. 2013;11:35.
Kaplan A, Gaschler MM, Dunn DE, Colligan R, Brown LM, Palmer AG, et al. Small molecule-induced oxidation of protein disulfide isomerase is neuroprotective. Proc Natl Acad Sci USA. 2015;112(17):E2245–52.
Hoffstrom BG, Kaplan A, Letso R, Schmid RS, Turmel GJ, Lo DC, et al. Inhibitors of protein disulfide isomerase suppress apoptosis induced by misfolded proteins. Nat Chem Biol. 2010;6(12):900–6.
Labun K, Montague TG, Krause M, Torres Cleuren YN, Tjeldnes H, Valen E. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 2019;47(W1):W171–4.
Keasey MP, Scott HL, Bantounas I, Uney JB, Kelly S. MiR-132 is upregulated by ischemic preconditioning of cultured hippocampal neurons and protects them from subsequent OGD toxicity. J Mol Neurosci. 2016;59(3):404–10.
Pfleger J, He M, Abdellatif M. Mitochondrial complex II is a source of the reserve respiratory capacity that is regulated by metabolic sensors and promotes cell survival. Cell Death Dis. 2015;6(7):e1835.
Kholmukhamedov A, Schwartz JM, Lemasters JJ. Isolated mitochondria infusion mitigates ischemia-reperfusion injury of the liver in rats: mitotracker probes and mitochondrial membrane potential. Vol. 39, Shock (Augusta, Ga.). 2013. p. 543.
Ebrahimi-Fakhari D, Saffari A, Wahlster L, Di Nardo A, Turner D, Lewis TLJ, et al. Impaired mitochondrial dynamics and mitophagy in neuronal models of tuberous sclerosis complex. Cell Rep. 2016;17(4):1053–70.
Zhong Y, Li X, Ji Y, Li X, Li Y, Yu D, et al. Pyruvate dehydrogenase expression is negatively associated with cell stemness and worse clinical outcome in prostate cancers. Oncotarget. 2017;8(8):13344–56.
Wegrzyn J, Potla R, Chwae Y-J, Sepuri NBV, Zhang Q, Koeck T, et al. Function of mitochondrial Stat3 in cellular respiration. Science. 2009;323(5915):793–7.
Li S, Zhao X, Chang S, Li Y, Guo M, Guan Y. ERp57-small interfering RNA silencing can enhance the sensitivity of drug-resistant human ovarian cancer cells to paclitaxel. Int J Oncol. 2019;54(1):249–60.
Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13(11):1235–42.
Antico Arciuch VG, Elguero ME, Poderoso JJ, Carreras MC. Mitochondrial regulation of cell cycle and proliferation. Antioxid Redox Signal. 2012;16(10):1150–80.
Park H-H, Han M-H, Choi H, Lee YJ, Kim JM, Cheong JH, et al. Mitochondria damaged by oxygen glucose deprivation can be restored through activation of the PI3K/Akt pathway and inhibition of calcium influx by amlodipine camsylate. Sci Rep. 2019;9(1):15717.
Capitanio D, Fania C, Torretta E, Viganò A, Moriggi M, Bravatà V, et al. TCA cycle rewiring fosters metabolic adaptation to oxygen restriction in skeletal muscle from rodents and humans. Sci Rep. 2017;7(1):9723.
Eufemi M, Coppari S, Altieri F, Grillo C, Ferraro A, Turano C. ERp57 is present in STAT3-DNA complexes. Biochem Biophys Res Commun. 2004;323(4):1306–12.
Guo GG, Patel K, Kumar V, Shah M, Fried VA, Etlinger JD, et al. Association of the chaperone glucose-regulated protein 58 (GRP58/ER-60/ERp57) with Stat3 in cytosol and plasma membrane complexes. J Interf cytokine Res Off J Int Soc Interf Cytokine Res. 2002;22(5):555–63.
Gaucci E, Raimondo D, Grillo C, Cervoni L, Altieri F, Nittari G, et al. Analysis of the interaction of calcitriol with the disulfide isomerase ERp57. Sci Rep. 2016;6:37957.
Kim Y-M, Youn S-W, Sudhahar V, Das A, Chandhri R, Cuervo Grajal H, et al. Redox regulation of mitochondrial fission protein Drp1 by protein disulfide isomerase limits endothelial senescence. Cell Rep. 2018;23(12):3565–78.
Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci U S A. 2006;103(8):2653–8.
Yamamoto E, Uchida T, Abe H, Taka H, Fujimura T, Komiya K, et al. Increased expression of ERp57/GRP58 is protective against pancreatic beta cell death caused by autophagic failure. Biochem Biophys Res Commun. 2014;453(1):19–24.
Wang Y, Levy DEC. C. elegans STAT: evolution of a regulatory switch. FASEB J Off Publ Fed Am Soc Exp Biol. 2006;20(10):1641–52.
Stenvall J, Fierro-González JC, Swoboda P, Saamarthy K, Cheng Q, Cacho-Valadez B, et al. Selenoprotein TRXR-1 and GSR-1 are essential for removal of old cuticle during molting in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2011;108(3):1064–9.
Thein MC, Winter AD, Stepek G, McCormack G, Stapleton G, Johnstone IL, et al. Combined extracellular matrix cross-linking activity of the peroxidase MLT-7 and the dual oxidase BLI-3 is critical for post-embryonic viability in Caenorhabditis elegans. J Biol Chem. 2009;284(26):17549–63.
Piec I, Listrat A, Alliot J, Chambon C, Taylor RG, Bechet D. Differential proteome analysis of aging in rat skeletal muscle. FASEB J Off Publ Fed Am Soc Exp Biol. 2005;19(9):1143–5.
Gueugneau M, Coudy-Gandilhon C, Gourbeyre O, Chambon C, Combaret L, Polge C, et al. Proteomics of muscle chronological ageing in post-menopausal women. BMC Genom. 2014;15:1165.
Conley KE, Jubrias SA, Esselman PC. Oxidative capacity and ageing in human muscle. J Physiol. 2000 Jul;526 Pt 1(Pt 1):203–10.
Mattson MP, Kroemer G. Mitochondria in cell death: novel targets for neuroprotection and cardioprotection. Trends Mol Med. 2003;9(5):196–205.
Herrmann AG, Deighton RF, Le Bihan T, McCulloch MC, Searcy JL, Kerr LE, et al. Adaptive changes in the neuronal proteome: mitochondrial energy production, endoplasmic reticulum stress, and ribosomal dysfunction in the cellular response to metabolic stress. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2013;33(5):673–83.
Szczepanek K, Chen Q, Derecka M, Salloum FN, Zhang Q, Szelag M, et al. Mitochondrial-targeted Signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. J Biol Chem. 2011;286(34):29610–20.
Avalle L, Camporeale A, Morciano G, Caroccia N, Ghetti E, Orecchia V, et al. STAT3 localizes to the ER, acting as a gatekeeper for ER-mitochondrion Ca(2+) fluxes and apoptotic responses. Cell Death Differ. 2019;26(5):932–42.
Yoo DY, Cho SB, Jung HY, Kim W, Choi G-M, Won M-H, et al. Tat-protein disulfide-isomerase: a possible candidate for preventing ischemic damage in the spinal cord. Cell Death Dis. 2017;8(10):e3075.
Mohammed F, Gorla M, Bisoyi V, Tammineni P, Sepuri NBV. Rotenone-induced reactive oxygen species signal the recruitment of STAT3 to mitochondria. FEBS Lett. 2020;594(9):1403–12.
D’Angelo B, Ek CJ, Sun Y, Zhu C, Sandberg M, Mallard C. GSK3β inhibition protects the immature brain from hypoxic-ischaemic insult via reduced STAT3 signalling. Neuropharmacology. 2016;101:13–23.
Shackelford RE, Kaufmann WK, Paules RS. Oxidative stress and cell cycle checkpoint function. Free Radic Biol Med. 2000;28(9):1387–404.
Acknowledgements
We express our gratitude to Henry Gong for assistance with C. elegans protocols and to Jaeden Pyburn for cell counts. Additionally, we are grateful to Dr. Valentin Yakubenko and Dr. Jonathon Moorman for providing the use of the Fluorometer and Seahorse instrument, respectively.
Funding
Funding was provided by National Institutes of Health (NS116310 to MPK, NS102745 to TH), and in part by C06RR0306551 and the Quillen College of Medicine at East Tennessee State University.
Author information
Authors and Affiliations
Contributions
Conceptualization: M.P.K., TH; Methodology: M.P.K. V.R.; Validation: M.P.K; Formal analysis: M.P.K.; Investigation: M.P.K., VR., J.C., P.C.B., E.D.P., T.H.; Resources: M.P.K., T.H.; Data curation: M.P.K.; Writing—original draft: M.P.K. V.R.; Writing—review and editing: M.P.K., V.R., J.C., P.C.B., E.D.P., T.H.; Visualization: M.P.K.; Supervision: M.P.K; Project administration: M.P.K.; Funding acquisition: M.P.K., T.H. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to publication
All experiments were performed according to East Tennessee State University guidelines.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplemental Figure S1.
PDIA3−/− reduces cellular growth rate in immortalized hCMEC/d3 human endothelial brain cells. A) We tested whether PDIA3−/− changed cell replication in CMECs. Counts were performed from brightfield images taken at 24 h intervals (100x magnification). Fields were overlaid with a 225 x 225 μm grid and a total area of 0.4 mm2 was counted per field over a total of 3 random fields per well, each field covered 1.47 mm2. Cell counts were normalized to cell numbers at 24 h after plating to account for slight differences in plating and cells that did not adhere. B) PDIA3−/− and WT cells grew at the same rate over the initial 24–48 h for both genotypes. For WT cells, cell growth continued to increase at 24 h until the cells reached confluence. On the other hand slowed PDIA3−/− growth rate slowed. C) We ruled out that cell death was increased in PDIA3−/− which might account for differences in cell number. Lactate dehydrogenase (LDH), released upon cell membrane damage, was not different between WT and PDIA3−/− cells between 48–72 h, a period which showed differing growth rates between the two genotypes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Keasey, M.P., Razskazovskiy, V., Jia, C. et al. PDIA3 inhibits mitochondrial respiratory function in brain endothelial cells and C. elegans through STAT3 signaling and decreases survival after OGD. Cell Commun Signal 19, 119 (2021). https://doi.org/10.1186/s12964-021-00794-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-021-00794-z