
Abstract
Chemokines are a family of small cytokines, which guide a variety of immune/inflammatory cells to the site of tumor in tumorigenesis. A dysregulated expression of chemokines is implicated in different types of cancer including prostate cancer. The progression and metastasis of prostate cancer involve a complex network of chemokines that regulate the recruitment and trafficking of immune cells. The chemokine CCL2 and its main receptor CCR2 have been receiving particular interest on their roles in cancer pathogenesis. The up-regulation of CCL2/CCR2 and varied immune conditions in prostate cancer, are associated with cancer advancement, metastasis, and relapse. Here we reviewed recent findings, which link CCL2/CCR2 to the inflammation and cancer pathogenesis, and discussed the therapeutic potential of CCL2/CCR2 axis in cancer treatment based on results from our group and other investigators, with a major focus on prostate cancer.
Video Abstract
Similar content being viewed by others


Background
Chemokines and cytokines are core regulators in cancer microenvironment which has been established as one of the hallmark drivers of cancer [1]. The cellular composition of tumor microenvironment is frequently modulated by cytokine milieu secreted by cancer cells in favor of tumor progression [1, 2]. Inflammation is one of the initiating processes of carcinogenesis where inflammatory/immune cells are trafficked into the tumor microenvironment by specific cytokines termed chemokines [3]. Chemokines are a family of small cytokines with ability to induce chemotaxis, a process where directed migration of cells expressing the appropriate chemokine receptor occurs towards higher local concentrations of chemokine ligands. Chemokines guide a variety of immune cells to the site of tumor initiation and subsequently lead to an inflammatory/immune response [3]. Chemokines contribute to the development of malignancy through roles in progression, migration, angiogenesis, and metastases in multiple cancer types [4]. Elevated levels of cytokines/chemokines such as IL-8 (interleukin-8), CXCL1 (Chemokine (C-X-C motif) Ligand 1), CCL2 (Chemokine (C-C motif) ligand 2, also known as monocyte chemoattractant protein-1, MCP-1), and CXCL5 have been associated with increased growth and progression of breast, ovarian, and prostate cancer [5,6,7,8,9]. In addition to tumor cells, various cells in the host microenvironment, including infiltrating leukocytes, endothelial cells, and fibroblasts, as well as adipocytes, are able to produce cytokines/chemokines such as CCL2 for tumor growth and progression [4, 10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26].
The upregulation of CCR2 has been found to be associated with advanced cancer, metastasis, and relapse [27]. The overexpression of CCL2 and resultant promotion of tumor growth have also been observed in breast [11, 12], ovarian [13], esophageal [14], gastric [15], renal cell [16], lung [17], colon [18], and papillary thyroid cancers [19]. In breast tumors, CCL2 overexpression was associated with advanced disease, tumor progression, and angiogenesis [20], and predicts prognosis and recurrence [22]. In breast tumor bone metastases, CCL2 overexpression led to enhanced osteolysis and the release of bone matrix-bound angiogenic factors, including platelet-derived growth factor, fibroblast growth factors-1, and transforming growth factor b [21]. Several studies have also demonstrated that serum CCL2 was elevated and associated with tumor stage in patients with breast, ovarian, and lung cancers [23,24,25].
Prostate cancer (PCa) is one of the most common types of cancer and the second leading cause of cancer death in men in the United States [28]. The morbidity of PCa has still been increasing among elderly men over the last decade [29]. PCa progression and metastasis is driven by many factors including the abnormalities of many growth factors and cytokines, among others such as the mutation and/or amplification of androgen receptor and other oncogenes and the inhibition of tumor suppressor genes [30,31,32]. The overexpression of CCL2 and its main receptor CCR2 (CC chemokine receptor 2) has been observed in both primary and metastatic PCa cells [33].
In addition, Lu et al. reported that elevated serum CCL2 was associated with bone metastasis in a study of 39 prostate cancer patients at various stages, suggesting the possibility of using serum CCL2 as a prognostic biomarker [26]. Since elevated CCL2 in circulation is also one of the typical features of obesity [34,35,36,37,38], this supports the role of CCL2 in connection of obesity and cancer promotion. These results suggest the critical role of the CCL2-CCR2 axis in cancer progression and its potential use as therapeutic target.
Classification of chemokines
Chemokines are a family of small chemotactic cytokines, which are signaling proteins secreted by cells. Chemokines have been classified based on the relative position of cysteine residues near the N terminus into four major families: CC, CXC, C, and CX3C. The CC chemokine (or β-chemokine) proteins have two adjacent cysteines near their amino terminus. There have been at least 27 distinct members of this subgroup reported for mammals, called CC chemokine ligands (CCL)-1 to − 28. CXC chemokines (or α-chemokines) have two N-terminal cysteines, which are separated by one amino acid, represented in this name with an “X”. There have been 17 different CXC chemokines described in mammals. C chemokines (or γ chemokines), are unlike other chemokines in that they have only two cysteines, one N-terminal cysteine and one cysteine downstream. Two chemokines have been described for this subgroup and are called XCL1 (lymphotactin-α) and XCL2 (lymphotactin-β). CX3C chemokines (or d-chemokines) have three amino acids between the two cysteines. The only CX3C chemokine discovered to date is called fractalkine (or CX3CL1). Chemokine receptors are G-protein coupled receptors located in the cell membrane, and they transduce the extracellular signal by interacting with chemokine ligands [39]. Chemokines have substantial effects as chemotactic factors on normal development, inflammation, atherosclerosis, and angiogenesis [40]. Chemokines have been implicated in many aspects of tumorigenesis, including the regulation of cancer cell growth, angiogenesis, metastasis, and host immune response [41].
CCL2/CCR2
CCL2 (MCP-1) is a member of the CC chemokine family [42]. The CCL2 gene is located in the q11.2-q12 region of human chromosome 17, and it encodes a precursor protein of 99 amino acids that matures into 75 amino acids in size [43]. CCL2 is initially described as a “tumor-derived chemotactic factor”, and has been shown to be a potent chemoattractant for several types of immune cells, including the monocytes, natural killer cells, memory T cells, and immature dendritic cells, thereby mediating multiple proinflammatory effects and neoangiogenesis [44,45,46,47,48,49,50]. CCR2 has been found to be expressed by multiple cell types including monocytes, dendritic cells (DCs), endothelial cells, and cancer cells [51,52,53,54]. CCL2 functions through binding to CCR2, one of 19 members of the human chemokine receptor family [55]. In addition to CCL2, CCR2 has several other high-affinity ligands, including CCL7 (MCP-2), CCL8 (MCP-3), CCL13 (MCP-4), and CCL12 (MCP-5) - a murine chemokine with close homology to human CCL2 [56,57,58,59], with an order of strength in binding to CCR2 as CCL2 > > CCL13 = CCL8 > CCL7 [60]. Monocytes, immature dendritic cells, and T-cell subpopulations have high expression of CCR2 which mediates their migration towards CC chemokines such as CCL2 [61]. CCL2-CCR2 signaling axis is implicated in many inflammatory and neurodegenerative diseases such as atherosclerosis, multiple sclerosis, asthma, neuropathic pain, diabetic nephropathy, and cancer [62, 63], and therefore is explored as a potential target for the treatment of these diseases.
The role of CCL2-CCR2 axis involving immune cells in tumor progression
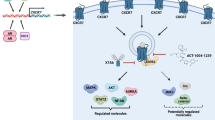
The inflammatory tumor microenvironment includes diverse host cells that are chemoattracted and induced by tumor-produced factors to generate a highly immune suppressive environment (Fig. 1), in which cross-talk between macrophages, myeloid-derived suppressor cells (MDSCs), and dendritic cells (DCs) reduces the opportunity to activate tumor-reactive T cells and thereby provides an environment for immune escape and continued tumor progression [64, 65].

Crosstalk between tumor cells and pro-inflammatory factors in the tumor microenvironment further amplifies the extravasation of tumor cells. Tumor cells produce chemokines, such as CCL2, CCL7, CCL8 and CCL13 that drive the generation of multiple types of regulatory immune cells, including T cells, B cells and myeloid-derived suppressor cells (MDSCs), and promote further TAM development by enhancing the recruitment of macrophages to the tumor site. Immune Cells in the modified tumor microenvironment subsequently produce cytokines, chemokines and other molecules that promote immune escape of tumor cells
Myeloid innate immune cells and myeloid-derived suppressor cells (MDSCs)
Monocytes, macrophages, and DCs are a heterogenous population of myeloid innate immune cells characterized by phagocytic and antigen-presenting capacities [66]. Monocytes circulate in the bloodstream for about 1 to 3 days and then typically extravasate into tissues where they differentiate into tissue resident macrophages or DCs, controlled by local environmental signals, such as colony-stimulating factor-1 (CSF-1) [67, 68].
Macrophages are a population of terminally differentiated myeloid cells and tissue-resident cells derived from monocytes circulating in peripheral blood [69]. Macrophages promote host survival by regulating adaptive immunity, eliminating infectious agents, and promoting wound healing in healthy individuals [70]. Extensive literature demonstrates that macrophages are co-opted to facilitate tumor growth during malignancy in both mice and humans [71,72,73,74]. The macrophage population is comprised of a continuous spectrum of phenotypically distinct subpopulations in their tissue microenvironment, demonstrating the complexity of this cell population [75]. The terminology ‘M1 macrophage’ and ‘M2 macrophage’ was coined to describe the different functional states of macrophages [76]. M1 or ‘classically activated’ macrophages are tumoricidal. They are activated by IFNγ or lipopolysaccharide, and characterized by high expression of IL-12 (Interleukin-12) and low expression of IL-10 (Interleukin-10). Under normal conditions, M1 macrophages are responsible for protecting the host against infection and injury and facilitating tissue remodeling [68], also suspected to be important in the formation of important organs like the heart and brain [77]. In contrast, M2 macrophages are activated by IL-4, IL-13, IL-10, and glucocorticoid hormones, produce high levels of IL-10 and low levels of IL-12, and promote tumor progression [26]. Macrophage phenotype driven by the local tumor microenvironment strongly polarizes towards an M2-like phenotype, giving rise to so-called “tumor associated macrophages” (TAMs) [78, 79]. However, studies have shown that a small number of TAM express both M1 and M2 markers [67]. The exclusive characterization of macrophage populations into M1 and M2 subtypes could be excessively simplistic, as macrophages have been found to be highly plastic cells that can demonstrate a variety of phenotypes [80]. Thus, a more comprehensive classification of TAM is needed which requires the integration of a multiparameter analysis of cell surface markers, comparison of TAM transcriptome, and consideration of the dynamic nature of macrophages [81].
TAMs enhance malignancy and promote numerous important features of tumor progression via both non-immune and immune mechanisms, including angiogenesis, motility, metastasis, and inhibition of T cell function [71]. Additionally, TAMs are also known to suppress responses to therapeutics, including chemotherapy, irradiation and angiogenic inhibitors [71, 82, 83]. In addition to malignancies, macrophages are associated with the progression of a number of other diseases such as asthma, allergic inflammation, and rheumatic inflammatory diseases [84, 85].
MDSCs are immune suppressive immature myeloid cells that are virtually elevated in all patients and experimental mice with malignancies [64]. MDSCs comprise a heterogeneous population of immature myeloid cells from the myeloid lineage characterized by co-expression of CD11b and Gr-1 and lack features of mature macrophages and dendritic cells in tumor-bearing mice. MDSCs can be divided into two distinct sub-populations: monocytic MDSCs (Mo-MDSCs) and polymorphonuclear (PMN)-MDSCs, also known as granulocytic (G)-MDSCs [75]. These two subsets differ in their gene expression profiles and immunosuppressive activities [86]. MDSCs strongly expand in pathological situations such as chronic infections and cancer, as a result of an altered haematopoiesis [86]. Marvel et al. found that activation of the immature myeloid cells via a network of regulatory mechanisms results in the accumulation of MDSCs in mice with transplantable and spontaneous tumors [87, 88]. MDSCs are discriminated from other myeloid cell types in which they possess strong immunosuppressive activities rather than immunostimulatory properties. MDSCs interact with other immune cell types including T cells, dendritic cells, macrophages and natural killer cells to regulate their functions. Accumulating evidence supports that MDSCs contribute to cancer immune evasion and tumor growth by suppressing T cell anti-tumor functions and modulating innate immune responses [86] as well as through non-immune suppressive pathway [89, 90]. MDSCs also accelerate angiogenesis, and subsequently promote tumor progression and metastasis through the expression of cytokines [91]. In many cancers, blood MDSC numbers correlate with stage and metastatic burden [92].
MDSCs and macrophages, two myeloid derived populations, are inextricably interconnected in the tumor microenvironment. They directly impact each other in a reciprocal fashion via the production of IL-10 and IL-6, respectively. Induction of one population favors the development of the other population. Clinical and experimental evidence has shown that cancer tissues with high infiltration of MDSCs and TAM are associated with poor patient prognosis and resistance to therapies [93,94,95]. Therefore, they have become a key therapeutic target.
CCL2-CCR2 axis in recruitment of monocytes and macrophages to tumor sites
Many types of cells present in the primary and metastatic tumor microenvironments, including stromal cells, leukocytes, endothelial cells, and tumor cells, produce CCL2 [11]. Prostate cancer cells LNCaP, C4-2B, PC-3, and VCaP produce higher amounts of CCL2 than primary prostate epithelial cells [96]. Tumor and stroma cells secrete CCL2 to recruit inflammatory monocytes and TAMs expressing CCR2 [2]. Monocytes recruited to tumors sites through the CCL2-CCR2 axis are polarized to TAMs, contributing to tumor cell survival [97]. Two prior studies from McClellan and Popivanova et al. suggested that CCL2 increased colon tumor numbers in mice through a CCL2-CCR2 dependent recruitment of myeloid cells [98]. Inhibition of CCL2-CCR2 signaling blocks the recruitment of inflammatory immune cells, and inhibits cancer cells metastasis in tumor-bearing mice [34,35,36,37, 63, 99,100,101,102,103,104].
Macrophage composition in different tissues or inflammatory environments depends on a dynamic equilibrium between recruited and tissue-resident macrophages [105]. Macrophages in the colonic mucosa are derived from circulating Ly6C+CCR2+ monocytes, during inflammation and under steady-state conditions [106]. In cancer, the evidence to date indicates that TAMs are dynamically replaced by circulating precursors. Both the tissue resident macrophages present in normal mammary tissues and TAMs that develop during tumor progression in the MMTV-PyMT breast cancer model are derived from blood-circulating CCR2+ monocytes, but only TAMs display self-renewal capability [107]. Elevated number of circulating blood monocytes and high macrophage infiltration into tumor tissues have been associated with poor clinical outcome in patients with various cancer types [2, 22, 71, 76, 82, 83, 108,109,110,111,112,113]. Hence, therapeutic strategies that either target TAM recruitment from inflammatory monocytes, or deplete TAMs will benefit patients with cancer or inflammatory diseases [114].
CCL2-CCR2 axis involving MDSCs in tumor progression
Overall, factors regulating MDSC accumulation and mechanisms of MDSCs’ action in cancer promotion remain underexplored. Several studies have demonstrated a role of CCL2 in recruiting MDSCs to tumor sites. Using human colorectal cancer (CRC) samples in conjunction with mouse models of colorectal carcinogenesis, Chun et al. identified a pro-neoplastic role for CCL2 in influencing MDSC accumulation and importance of MDSCs and CCL2 in tumor microenvironment during the development of CRC [115]. CCL2 and GM-CSF (Granulocyte-macrophage colony-stimulating factor) induced by oncogenic fusion protein RET/PTC3 together promote the recruitment of CD11b+GR1+ MDSCs that can promote thyroid carcinomas progression [116, 117]. Moreover, the formation of invasive squamous cell cancer and the associated production of CCL2, GM-CSF, M-CSF (Macrophage colony-stimulating factor) and TNF (Tumor Necrosis Factor) caused by conditional deletion of the gene encoding p120 catenin in mice resulted in the accumulation of immunosuppressive CD11b+GR1+CD124+ MDSCs, which activated stromal fibroblasts and promoted tumor progression [118]. Although TAMs and MDSCs are regarded as separate entities, the boundaries between them are not clearly demarcated, and they share many characteristics [119]. Intriguingly, while PMN-MDSCs increased in castrated tumors models of prostate cancer (TRAMP-C1 and MyC-CaP), the frequency of tumor infiltrating macrophages (TAMs) decreased [120], suggesting that MDSCs confers more profound suppression on the immune cells in prostatic tumor microenvironment.
The role of CCL2-CCR2 axis in prostate cancer progression
PCa is the fifth leading cause of cancer death in men worldwide. The development and progression of PCa is typically associated with an inflammatory microenvironment [121]. The involvement of CCL2-CCR2 axis in PCa progression has been consistently observed in many studies, including an enhanced CCL2-CCR2 signaling and tumor promotion under obese conditions. Therefore, PCa seems to be a good example to demonstrate the role of CCL2-CCR2 in connection of inflammation/obesity to tumor pathogenesis.
Overview of inflammation and obesity-induced inflammation in prostate cancer
Inflammation is a complex biological process and a protective response of the immune system to establish a physical barrier against the harmful stimuli, such as infection or irritation, involving molecular mediators, immune cells, and blood vessels [122]. Inflammatory cells consist of lymphocytes, neutrophils, eosinophils, plasma cells, and histiocytes, which are innate immune cells playing a major role in inflammatory process. The presence of inflammatory cells does not signify the cells themselves are inflammatory. A more exact designation is “cells entering inflammatory tissue”. Inflammation can be either acute or chronic. Acute inflammation is an immediate and innate response to harmful stimuli and efficiently minimizes impending injury by molecular, cellular events and interactions. Chronic inflammation is derived from uncontrolled acute inflammation, also known as prolonged inflammation, which is associated with various diseases, such as type 2 diabetes, atherosclerosis, rheumatoid arthritis, asthma, and cancer [123]. Currently, chronic inflammation is estimated to account for approximately 15 to 25% of human cancers [124,125,126]. Nearly all primary malignant neoplasms are associated with dense infiltrates of inflammatory cells. Macrophages, neutrophils and lymphocytes are the most abundant immune cells in the tumor microenvironment [95]. In the tumor microenvironments, the interactions among cancer cells, immune cells, endothelial cells, and fibroblasts can play important roles to contribute to tumor progression.
Obesity is characterized by an excess of body fat resulting from a chronic positive energy balance. Obesity has been associated with increased risk for metabolic diseases and cancers such as esophagus, gastric, breast, pancreas, colon, liver, endometrial, kidney, and prostate cancer [127,128,129]. In PCa, prospective studies in the United States showed that body mass index (BMI) was weakly and positively associated with PCa, and greater BMI was an independent predictor of PCa [130, 131]. Higher BMI was associated with biochemical recurrence of PCa after radical prostatectomy in an analysis of 4123 men treated by radical prostatectomy [132], and higher BMI was positivity correlated with the PCa death in a prospective study of 404,576 men [128]. A meta-analysis of advanced PCa showed a positive linear relationship with BMI for advanced PCa [133]. These epidemiological studies have shown consistent evidence of the association of obesity with advance PCa.
Adipose tissue is mainly composed of adipocytes, while stromal vascular fraction (SVF) including adipocyte derived stem cells, preadipocytes, lymphocytes, macrophages, fibroblasts and vascular endothelial cells also contribute to the growth and function of adipose tissue [134, 135]. Mature adipocytes have been considered not only as energy-storing cells, but also as highly endocrine cells which are able to secrete an heterogeneous group of molecules termed ‘adipokines’ such as chemokines, growth factors, hormones, or pro-inflammatory molecules [134, 136].
Obesity is associated with a chronic low-grade systemic inflammation that has been implicated in the development of common, medically important complications, including atherosclerosis, hepatic steatosis, and insulin resistance [137,138,139,140]. One characteristic of obesity-caused inflammation is the activation of pathways that regulate inflammation, such as JNK and NF-κB pathways [141,142,143], and immune cells infiltrating in the white adipose tissue [144,145,146,147]. Activation of these cells elevates local and systemic expression of pro-inflammatory molecules, including acute-phase reactants, procoagulant factors, chemokines, and cytokines (such as TNF, HMGB1, IL-1, and IL-6), and mediates the inflammatory response [123]. Elevated cytokine and chemokine levels are typically associated with obesity and propagate the obesity-associated inflammatory state [148,149,150,151]. Obesity also causes the accumulation of macrophages in adipose tissue [100], which have been implicated in the development and maintenance of obesity-induced adipose tissue inflammation [121, 152].
Obesity-induced inflammatory state contributes to PCa development [99]. T cells are accumulated in prostate tumor of a diet-induced obese Hi-Myc mice [153]. The cytotoxic function of NK cells to PCa cells is inhibited by humoral factors from adipocytes [154]. Myeloid differentiation is skewed towards the expansion of MDSCs under chronic inflammatory conditions or cancer [86]. These MDSCs infiltrate inflammation sites and tumors, where they stop immune responses by inhibiting T cells and NK cells. Other inflammatory cells and immune cells could be also involved in the PCa progression. These local inflammatory cells orchestrate an environment that fosters tumor proliferation and survival [155]. In a study of the relationship between inflammation and tumor progression in the prostate, Fujita et al. found that secretion of IL-6 from local macrophages was increased in prostate tissues of HFD-fed mice, and inhibition of the IL-6 pathway resulted in the suppression of tumor growth [156]. Mechanistic studies demonstrated that IL-6 might promote the proliferation of PCa cells via the STAT3 pathways and an increase of local MDSCs [157]. These results suggest that inflammation plays a central role for the progression of PCa in the studied obese state.
CCL2-CCR2 axis in mediating the interplays among microenvironment, inflammation/obesity, and prostate cancer
The cytokines and chemokines produced by prostatic tumor cells and various cells in the host microenvironment, including infiltrating leukocytes, endothelial cells, and fibroblasts [4, 10], enhance the growth, progression, migration/invasion, and metastasis of prostate cancer [8]. CCL2 has been identified as a prominent modulator in such a dynamic tumor-host interactions [8]. CCL2 was overexpressed in primary prostatic tumors as determined by immunohistochemistry [96]. In advanced prostate cancer, CCL2 expression was also notably higher in the metastatic tumor-bone microenvironment compared with that in bone marrow adjacent to the tumor as measured by cytokine arrays [158]. CCL2 acts in a paracrine and autocrine manner to stimulate PCa cell proliferation and migration. Although the molecular link between CCL2 and PCa has not been thoroughly elucidated, several studies have suggested the involvement of CCR2 in mediating the signaling of CCL2 in PCa progression [54]. High levels of CCR2 exist in prostate tumor cell surface to respond to autocrine and/or paracrine CCL2 in the microenvironment. The mRNA and protein expression of CCR2 were higher in aggressive cell lines such as DU145, PC-3, and C4-2B compared with androgen-sensitive LNCaP cells and non-neoplastic prostate epithelial cells [8, 54], and higher in prostate cancer metastatic tissues as compared with localized prostate cancer and benign prostate tissue [43]. Analysis of real-time PCR and IHC staining on tissue microarray specimens revealed that higher CCR2 expression was also associated with higher Gleason score and higher clinical pathologic stages [54], suggesting a positive association between CCR2 expression and prostate cancer progression [54]. Further, CCL2-induced prostate cancer cell chemotaxis was abolished by a CCR2 antagonist, which confirmed that CCR2 is the functional receptor of CCL2 [96].
The downstream target of CCR2 may include the PI3K/Akt signaling pathway [159]. Upon activation by CCL2-CCR2, PI3K/Akt activates mTORC1 and up-regulates survivin which is a key molecule protecting prostate cancer cells from autophagic death [160, 161]. CCL2 was also able to stimulate prostate cancer cells to extravasate into the bone through a layer of bone marrow endothelial cells partially by the activation of the small GTPase Rac through the actin-associated protein PCNT1 [162]. These observations suggest the role of CCL2 from tumor microenvironment in stimulation of prostate cancer expansion and metastasis [127,128,129, 134,135,136].
The prostate gland is surrounded by periprostatic adipose tissue (PPAT) [163]. Excess visceral adiposity around the prostate can lead to changes of the secretory pattern of adipocytes as well as to subsequent modifications in the cellular composition of periprostatic environment [134, 135]. Evidence has shown a correlation between the abundance of PPAT and tumor aggressiveness, suggesting a paracrine role of PPAT during tumorigenesis [164]. Extraprostatic spreading of PCa into PPAT is found to be a more important determinant of cancer recurrence than an invasive phenotype [165, 166]. A high infiltration of macrophages was observed in PPAT in obese animal models [163], which may be due to an increased secretion of CCL2 by adipocytes in the obese conditions. Once recruited in the tumor microenvironment by adipocytes, macrophages tend to have metabolic reprogramming and be polarized into M2 phenotype by tumor cells, favoring tumor growth and progression [167, 168]. Enhanced infiltration of activated macrophages in visceral adipose tissues was also observed in obese patients [100], and CCR2 seemed to have a direct role in the recruitment of macrophages. Ccr2-knockout animals had significantly fewer adipose tissue macrophages than wild-type mice [101]. Both Ccr2 genetic deficiency and pharmacological inhibition reduced macrophage content of adipose tissue, and improved inflammatory profile of adipose tissue including increase in adiponectin expression and amelioration in systemic glucose homeostasis [102].
Studies using high-fat diet (HFD)-induced obese mouse models have also demonstrated the tumor-promoting effect of obesity on prostate cancer. HFD promoted androgen-sensitive PCa growth and progression to androgen-independent growth in mouse models [169, 170]. Conditioned medium using serum collected from HFD-fed TRAMP mice promoted the proliferation, migration, and invasion of DU-145 cells [171]. In a prostate tumor xenograft model of mice implanted with LNCaP cells, HFD promoted tumor growth and increased blood CCL2 levels [172]. The elevated CCL2 levels in adipose tissue and blood were also observed in humans during obesity [34], and in high-fat diet-induced [35,36,37] or genetically obese rodents [103, 104]. As supported by our observations, differentiated 3 T3-L1 adipocytes cultured in vitro secret high levels of CCL2, and their co-culture with LNCaP cells significantly increased the proliferation of LNCaP cells [173]. In contrast, the restriction of caloric intake delayed PCa growth in an animal study [174].
In human studies, Platz et al. found that energy intake was positively associated with metastatic or fatal PCa in certain subsets of men in a prior prospective cohort study [175]. Huber et al. found an up-regulation of CC chemokines (CCL2, CCL3, CCL5, CCL7, CCL8, and CCL11) and their respective receptors (CCR1, CCR2, CCR3, and CCR5) in adipose tissue in obese patients [176]. The expression of CCR2, CXCR1, CXCR2 and CXCR4 are higher in human PCa tissues correlated with tumor aggressiveness [10, 54, 177, 178]. These observations further support the role of obesity-related factors in promotion of PCa progression, and underline the importance to co-target obesity in treatment of PCa and some other cancers as well.
We have conducted an in vivo study to assess the therapeutic potential of CCR2 inhibition in PCa in obese state. Male SCID mice were fed HFD starting 1 week prior to tumor inoculation. Mice were then implanted subcutaneously with androgen-sensitive LAPC-4 PCa cells. When tumors formed, mice were orally administered with a CCR2 inhibitor RS 504393 at 5 mg/kg body weight per day for 6 weeks. HFD-fed mice had elevated blood CCL2 levels compared to regular-diet fed mice (data not shown). Tumor growth in HFD-fed mice was significantly inhibited after 4 weeks of RS 504393 treatment, with a 50% inhibition at the end of the study (Fig. 2). Blood analysis revealed a significantly increased level of free fatty acids, a major source of fuel to cancer cells, in HFD-fed control mice compared to regular diet-fed control mice (LF Con), while the treatment with RS 504393 significantly reduced the adipocyte-release of free fatty acids in blood compared to HFD control (Fig. 3). This may suggest a novel role of CCL2/CCR2 in modulation of adipocytes’ metabolism and release of harmful factors.

A CCR2 inhibitor RS 504393 inhibited xenograft prostate tumor growth in SCID mice fed high fat diet. Tumors were measured weekly with caliper from the initiation of treatment. Tumor volumes were calculated and data were plotted using the geometric mean for each group vs. time. Each point represents the mean tumor volume (± SD) of measurements from the 10 mice in each group

Treatment with RS 504393 reduced blood levels of free fatty acids in high-fat diet-fed SCID mice bearing LAPC-4 xenograft prostate tumor. LF Con, regular diet-fed control mice; HF Con, high-fat diet-fed control mice; HF RS, high-fat diet-fed mice treated with RS 504393. Columns with different letters indicate significant difference between groups, P < 0.05
In addition, since CCL2-CCR2 signaling may also stimulate PCa cell migration/invasion through the layer of bone marrow endothelial cells [162], we carried out a chamber assay to test the ability of RS 504393 in inhibition of cancer cell migration in obese state (Fig. 4). Differentiated 3 T3-L1 cells were seeded on the bottom of 24-well plate. 24 h later LNCaP cells were loaded on an insert treated with or without RS 504393. Migrated cells were counted after 18 h. Co-culture with differentiated 3 T3-L1 cells significantly promotes LNCaP cell migration, while the inhibition of CCR2 signaling by RS 504393 significantly inhibited the migration of LNCaP (Fig. 4). These data provide strong support to the role of CCL2-CCR2 in prostate cancer growth and progression and indicate the therapeutic potential of this axis.

A. Schematic of LNCaP cells chemotactic assay In vitro. A. LNCaP cells migration through 8 μm pore size transwell inserts toward differentiated 3 T3-L1 cells in the bottom wells. B. Migrated LNCaP cells under a light microscope at the undersurface of the inserts were stained with crystal violet (scale bar: 20 μm). C. Cell number assessment of LNCaP cells co-cultured with differentiated 3 T3-L1 cells. **p < 0.01 versus undifferentiated 3 T3-L1 cells; ##p < 0.01 versus differentiated 3 T3-L1 cells
Conclusion
Tumor progression is regulated by various intrinsic and extrinsic (microenvironment) factors. It is now well accepted that cancer cells exist in a complex environment in which they interact with a wide variety of stromal cells, including the multiple cell types that make up the immune system of the host. Many of these interactions are mediated by chemokines. The roles of chemokines in tumorigenesis have been shown to be diverse, including both negative and positive regulation of inflammatory cells, chemoattraction of tumor cells to metastatic sites, regulation of angiogenesis, and direct regulation of proliferation of cancer cells [179]. Growth factors and cytokines are supplied to PCa cells not only in an autocrine manner but also in a paracrine manner [54]. CCL2 has been shown to have direct effects on tumor growth in an autocrine and paracrine fashion in multiple cancers, including breast, lung, cervix, ovary, sarcoma, and prostate [54]. Results from our group demonstrate the therapeutic potential of CCR2 as a novel target in treatment of PCa, and possibly other types of cancer, particularly in obese state with a host CCL2-stimulated environment. CCR2 is also the receptor of CCL2, CCL13, CCL8 and CCL7, while with the highest affinity to CCL2. Evidence indicates clearly an important role of CCL2-CCR2 axis in the development and progression of PCa, possibly through both regulating monocyte/macrophage infiltration into prostate tumors and directly stimulating PCa cells.
In summary, the cooperation between tumor-derived chemokines and host/adipose tissue-derived chemokines, particularly CCL2, through CCR2 signaling considerably contributes to tumor cell survival, proliferation, and metastasis (Fig. 5), which makes CCR2 a potential therapeutic target in cancer treatment. Further work is required to delineate the roles of host-derived CCL2 and tumor-derived CCL2 in PCa tumorigenesis and metastasis, and to elucidate the downstream signaling molecules which mediate the effect of CCR2 signaling in tumor promotion.

Interaction between obesity and tumor in promotion of tumor growth. During obesity, White Adipose Tissue WAT releases a plethora of molecules with autocrine, paracrine and endocrine functions including growth factors, adipokines, proinflammatory molecules, fatty acids (FA) and lipid metabolites and many others, which create a favorable condition for prostate cancer to develop
Availability of data and materials
The dataset(s) supporting the findings of this study are included within the article.
Abbreviations
- PCa:
-
Prostate Cancer
- CRC:
-
Human Colorectal Cancer
- CCL2:
-
chemokine (C-C motif) Ligand 2
- MCP-1:
-
Monocyte Chemoattractant Protein-1
- CCR2 :
-
CC chemokine Receptor 2
- IL-8:
-
Interleukin-8
- CXCL1:
-
Chemokine (C-X-C motif) Ligand 1
- TAMs:
-
Tumor Associated Macrophages
- MDSC:
-
Myeloid-Derived Suppressor Cells
- DCs:
-
Dendritic Cells
- NK cells:
-
Natural Killer Cells
- PPAT:
-
PeriProstatic Adipose Tissue
- SVF:
-
Stromal Vascular Fraction
- STAT3:
-
Signal Transducer and Activator of Transcription 3
- BMI:
-
Body Mass Index
- HFD:
-
High Fat Diet
- TRAMP:
-
TRansgenic Adenocarcinoma of Mouse Prostate
- SCID:
-
Severe Combined Immunodeficiency
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- M-CSF:
-
Macrophage colony-stimulating factor
- TNF:
-
Tumor Necrosis Factor
References
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Qian BZ, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475(7355):222–5.
Rani A, Dasgupta P, Murphy JJ. Prostate Cancer: the role of inflammation and chemokines. Am J Pathol. 2019;189(11):2119–37.
Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4(7):540–50.
Begley LA, et al. CXCL5 promotes prostate cancer progression. Neoplasia. 2008;10(3):244–54.
Fader AN, et al. CCL2 expression in primary ovarian carcinoma is correlated with chemotherapy response and survival outcomes. Anticancer Res. 2010;30(12):4791–8.
Singh RK, Lokeshwar BL. Depletion of intrinsic expression of Interleukin-8 in prostate cancer cells causes cell cycle arrest, spontaneous apoptosis and increases the efficacy of chemotherapeutic drugs. Mol Cancer. 2009;8:57.
Zhang J, Lu Y, Pienta KJ. Multiple roles of chemokine (C-C motif) ligand 2 in promoting prostate cancer growth. J Natl Cancer Inst. 2010;102(8):522–8.
Dutta P, et al. MCP-1 is overexpressed in triple-negative breast cancers and drives cancer invasiveness and metastasis. Breast Cancer Res Treat. 2018;170(3):477–86.
Vindrieux D, Escobar P, Lazennec G. Emerging roles of chemokines in prostate cancer. Endocr Relat Cancer. 2009;16(3):663–73.
Soria G, Ben-Baruch A. The inflammatory chemokines CCL2 and CCL5 in breast cancer. Cancer Lett. 2008;267(2):271–85.
Soria G, et al. Concomitant expression of the chemokines RANTES and MCP-1 in human breast cancer: a basis for tumor-promoting interactions. Cytokine. 2008;44(1):191–200.
Negus RP, et al. The detection and localization of monocyte chemoattractant protein-1 (MCP-1) in human ovarian cancer. J Clin Invest. 1995;95(5):2391–6.
Ohta M, et al. Monocyte chemoattractant protein-1 expression correlates with macrophage infiltration and tumor vascularity in human esophageal squamous cell carcinomas. Int J Cancer. 2002;102(3):220–4.
Ohta M, et al. Monocyte chemoattractant protein-1 expression correlates with macrophage infiltration and tumor vascularity in human gastric carcinomas. Int J Oncol. 2003;22(4):773–8.
Hemmerlein B, et al. Quantification and in situ localization of MCP-1 mRNA and its relation to the immune response of renal cell carcinoma. Cytokine. 2001;13(4):227–33.
Niiya M, et al. Induction of TNF-alpha, uPA, IL-8 and MCP-1 by doxorubicin in human lung carcinoma cells. Cancer Chemother Pharmacol. 2003;52(5):391–8.
Huang S, et al. Expression of the JE/MCP-1 gene suppresses metastatic potential in murine colon carcinoma cells. Cancer Immunol Immunother. 1994;39(4):231–8.
Tanaka K, et al. The expression of monocyte chemotactic protein-1 in papillary thyroid carcinoma is correlated with lymph node metastasis and tumor recurrence. Thyroid. 2009;19(1):21–5.
Valkovic T, et al. Macrophage level is not affected by monocyte chemotactic protein-1 in invasive ductal breast carcinoma. J Cancer Res Clin Oncol. 2005;131(7):453–8.
Cackowski FC, Roodman GD. Perspective on the osteoclast: an angiogenic cell? Ann N Y Acad Sci. 2007;1117:12–25.
Ueno T, et al. Significance of macrophage chemoattractant protein-1 in macrophage recruitment, angiogenesis, and survival in human breast cancer. Clin Cancer Res. 2000;6(8):3282–9.
Cai Z, et al. Monocyte chemotactic protein 1 promotes lung cancer-induced bone resorptive lesions in vivo. Neoplasia. 2009;11(3):228–36.
Dwyer RM, et al. Monocyte chemotactic protein-1 secreted by primary breast tumors stimulates migration of mesenchymal stem cells. Clin Cancer Res. 2007;13(17):5020–7.
Hefler L, et al. Monocyte chemoattractant protein-1 serum levels in ovarian cancer patients. Br J Cancer. 1999;81(5):855–9.
Lu Y, et al. Activation of MCP-1/CCR2 axis promotes prostate cancer growth in bone. Clin Exp Metastasis. 2009;26(2):161–9.
Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017;17(9):559–72.
Crawford ED. Epidemiology of prostate cancer. Urology. 2003;62(6 Suppl 1):3–12.
Jemal A, et al. Cancer statistics, 2007. CA Cancer J Clin. 2007;57(1):43–66.
Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004;25(2):276–308.
Pienta KJ, Bradley D. Mechanisms underlying the development of androgen-independent prostate cancer. Clin Cancer Res. 2006;12(6):1665–71.
Saraon P, Jarvi K, Diamandis EP. Molecular alterations during progression of prostate cancer to androgen independence. Clin Chem. 2011;57(10):1366–75.
Zhang J, Patel L, Pienta KJ. CC chemokine ligand 2 (CCL2) promotes prostate cancer tumorigenesis and metastasis. Cytokine Growth Factor Rev. 2010;21(1):41–8.
Christiansen T, Richelsen B, Bruun JM. Monocyte chemoattractant protein-1 is produced in isolated adipocytes, associated with adiposity and reduced after weight loss in morbid obese subjects. Int J Obes. 2005;29(1):146–50.
Takahashi K, et al. Adiposity elevates plasma MCP-1 levels leading to the increased CD11b-positive monocytes in mice. J Biol Chem. 2003;278(47):46654–60.
Chen A, et al. Diet induction of monocyte chemoattractant protein-1 and its impact on obesity. Obes Res. 2005;13(8):1311–20.
Mohanty P, et al. Evidence for a potent antiinflammatory effect of rosiglitazone. J Clin Endocrinol Metab. 2004;89(6):2728–35.
Hao Q, et al. Arctigenin inhibits prostate tumor growth in high-fat diet fed mice through dual actions on adipose tissue and tumor. Sci Rep. 2020;10(1):1403.
Zlotnik A, Burkhardt AM, Homey B. Homeostatic chemokine receptors and organ-specific metastasis. Nat Rev Immunol. 2011;11(9):597–606.
Bonecchi R, et al. Chemokines and chemokine receptors: an overview. Front Biosci (Landmark Ed). 2009;14:540–51.
Craig MJ, Loberg RD. CCL2 (monocyte Chemoattractant Protein-1) in cancer bone metastases. Cancer Metastasis Rev. 2006;25(4):611–9.
Deshmane SL, et al. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interf Cytokine Res. 2009;29(6):313–26.
Loberg RD, et al. Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Res. 2007;67(19):9417–24.
Conti I, Rollins BJ. CCL2 (monocyte chemoattractant protein-1) and cancer. Semin Cancer Biol. 2004;14(3):149–54.
Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124(2):263–6.
Peters W, Dupuis M, Charo IF. A mechanism for the impaired IFN-gamma production in C-C chemokine receptor 2 (CCR2) knockout mice: role of CCR2 in linking the innate and adaptive immune responses. J Immunol. 2000;165(12):7072–7.
Valente AJ, et al. Purification of a monocyte chemotactic factor secreted by nonhuman primate vascular cells in culture. Biochemistry. 1988;27(11):4162–8.
Carr MW, et al. Monocyte chemoattractant protein 1 acts as a T-lymphocyte chemoattractant. Proc Natl Acad Sci U S A. 1994;91(9):3652–6.
Allavena P, et al. Induction of natural killer cell migration by monocyte chemotactic protein-1, −2 and −3. Eur J Immunol. 1994;24(12):3233–6.
Sallusto F, et al. Rapid and coordinated switch in chemokine receptor expression during dendritic cell maturation. Eur J Immunol. 1998;28(9):2760–9.
Charo IF, et al. Molecular cloning and functional expression of two monocyte chemoattractant protein 1 receptors reveals alternative splicing of the carboxyl-terminal tails. Proc Natl Acad Sci U S A. 1994;91(7):2752–6.
Sozzani S, et al. Receptor expression and responsiveness of human dendritic cells to a defined set of CC and CXC chemokines. J Immunol. 1997;159(4):1993–2000.
Weber KS, et al. Expression of CCR2 by endothelial cells : implications for MCP-1 mediated wound injury repair and in vivo inflammatory activation of endothelium. Arterioscler Thromb Vasc Biol. 1999;19(9):2085–93.
Lu Y, et al. CCR2 expression correlates with prostate cancer progression. J Cell Biochem. 2007;101(3):676–85.
Zheng Y, et al. Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nature. 2016;540(7633):458–61.
Combadiere C, et al. Monocyte chemoattractant protein-3 is a functional ligand for CC chemokine receptors 1 and 2B. J Biol Chem. 1995;270(50):29671–5.
Berkhout TA, et al. Cloning, in vitro expression, and functional characterization of a novel human CC chemokine of the monocyte chemotactic protein (MCP) family (MCP-4) that binds and signals through the CC chemokine receptor 2B. J Biol Chem. 1997;272(26):16404–13.
Gong X, et al. Monocyte chemotactic protein-2 (MCP-2) uses CCR1 and CCR2B as its functional receptors. J Biol Chem. 1997;272(18):11682–5.
Sarafi MN, et al. Murine monocyte chemoattractant protein (MCP)-5: a novel CC chemokine that is a structural and functional homologue of human MCP-1. J Exp Med. 1997;185(1):99–109.
White GE, Iqbal AJ, Greaves DR. CC chemokine receptors and chronic inflammation--therapeutic opportunities and pharmacological challenges. Pharmacol Rev. 2013;65(1):47–89.
Scholten DJ, et al. Pharmacological modulation of chemokine receptor function. Br J Pharmacol. 2012;165(6):1617–43.
O'Connor T, Borsig L, Heikenwalder M. CCL2-CCR2 signaling in disease pathogenesis. Endocr Metab Immune Disord Drug Targets. 2015;15(2):105–18.
Lim SY, et al. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget. 2016;7(19):28697–710.
Ostrand-Rosenberg S, et al. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin Cancer Biol. 2012;22(4):275–81.
Wang M, et al. Role of tumor microenvironment in tumorigenesis. J Cancer. 2017;8(5):761–73.
Guilliams M, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol. 2014;14(8):571–8.
Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787–95.
Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–37.
Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: Phenotypical vs. Functional Differentiation Front Immunol. 2014;5:514.
Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–69.
Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51.
Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11(10):889–96.
Mantovani A, et al. Tumor-associated macrophages and the related myeloid-derived suppressor cells as a paradigm of the diversity of macrophage activation. Hum Immunol. 2009;70(5):325–30.
Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;22(2):231–7.
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–68.
Mantovani A, et al. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):549–55.
Nicolas-Avila JA, Hidalgo A, Ballesteros I. Specialized functions of resident macrophages in brain and heart. J Leukoc Biol. 2018;104(4):743–56.
Mantovani A, et al. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14(7):399–416.
Chanmee T, et al. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel). 2014;6(3):1670–90.
Almatroodi SA, et al. Characterization of M1/M2 tumour-associated macrophages (TAMs) and Th1/Th2 cytokine profiles in patients with NSCLC. Cancer Microenviron. 2016;9(1):1–11.
Murray PJ, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20.
Steidl C, et al. Tumor-associated macrophages and survival in classic Hodgkin's lymphoma. N Engl J Med. 2010;362(10):875–85.
Gupta, V., F. Yull, and D. Khabele, Bipolar Tumor-Associated Macrophages in Ovarian Cancer as Targets for Therapy. Cancers (Basel), 2018. 10(10).
Rana AK, et al. Monocytes in rheumatoid arthritis: Circulating precursors of macrophages and osteoclasts and, their heterogeneity and plasticity role in RA pathogenesis. Int Immunopharmacol. 2018;65:348–59.
Roberts CA, Dickinson AK, Taams LS. The interplay between monocytes/macrophages and CD4(+) T cell subsets in rheumatoid arthritis. Front Immunol. 2015;6:571.
Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–74.
Groth C, et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer. 2019;120(1):16–25.
Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest. 2015;125(9):3356–64.
Yang L, et al. Expansion of myeloid immune suppressor gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6(4):409–21.
Nagaraj S, et al. Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J Immunol. 2010;184(6):3106–16.
Singh S, et al. Initiative action of tumor-associated macrophage during tumor metastasis. Biochim Open. 2017;4:8–18.
Diaz-Montero CM, et al. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58(1):49–59.
Mantovani A. The growing diversity and spectrum of action of myeloid-derived suppressor cells. Eur J Immunol. 2010;40(12):3317–20.
Allavena P, Mantovani A. Immunology in the clinic review series; focus on cancer: tumour-associated macrophages: undisputed stars of the inflammatory tumour microenvironment. Clin Exp Immunol. 2012;167(2):195–205.
Galdiero MR, et al. Tumor associated macrophages and neutrophils in cancer. Immunobiology. 2013;218(11):1402–10.
Lu Y, et al. Monocyte chemotactic protein-1 (MCP-1) acts as a paracrine and autocrine factor for prostate cancer growth and invasion. Prostate. 2006;66(12):1311–8.
McClellan JL, et al. Linking tumor-associated macrophages, inflammation, and intestinal tumorigenesis: role of MCP-1. Am J Physiol Gastrointest Liver Physiol. 2012;303(10):G1087–95.
Popivanova BK, et al. Blockade of a chemokine, CCL2, reduces chronic colitis-associated carcinogenesis in mice. Cancer Res. 2009;69(19):7884–92.
Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415–45.
Weisberg SP, et al. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–808.
Bai Y, Sun Q. Macrophage recruitment in obese adipose tissue. Obes Rev. 2015;16(2):127–36.
Weisberg SP, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. 2006;116(1):115–24.
Kanda H, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116(6):1494–505.
Fujimoto H, et al. Stromal MCP-1 in mammary tumors induces tumor-associated macrophage infiltration and contributes to tumor progression. Int J Cancer. 2009;125(6):1276–84.
Ugel S, et al. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest. 2015;125(9):3365–76.
Bain CC, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol. 2014;15(10):929–37.
Franklin RA, et al. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344(6186):921–5.
Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4(1):71–8.
Brown JM, Recht L, Strober S. The promise of targeting macrophages in Cancer therapy. Clin Cancer Res. 2017;23(13):3241–50.
Valkovic T, et al. Expression of monocyte chemotactic protein-1 in human invasive ductal breast cancer. Pathol Res Pract. 1998;194(5):335–40.
Saji H, et al. Significant correlation of monocyte chemoattractant protein-1 expression with neovascularization and progression of breast carcinoma. Cancer. 2001;92(5):1085–91.
Rhodes DR, et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6(1):1–6.
Sun X, et al. CCL2-driven inflammation increases mammary gland stromal density and cancer susceptibility in a transgenic mouse model. Breast Cancer Res. 2017;19(1):4.
Poh AR, Ernst M. Targeting macrophages in Cancer: from bench to bedside. Front Oncol. 2018;8:49.
Chun E, et al. CCL2 promotes colorectal carcinogenesis by enhancing Polymorphonuclear myeloid-derived suppressor cell population and function. Cell Rep. 2015;12(2):244–57.
Pufnock JS, Rothstein JL. Oncoprotein signaling mediates tumor-specific inflammation and enhances tumor progression. J Immunol. 2009;182(9):5498–506.
Borrello MG, et al. Induction of a proinflammatory program in normal human thyrocytes by the RET/PTC1 oncogene. Proc Natl Acad Sci U S A. 2005;102(41):14825–30.
Stairs DB, et al. Deletion of p120-catenin results in a tumor microenvironment with inflammation and cancer that establishes it as a tumor suppressor gene. Cancer Cell. 2011;19(4):470–83.
Yu J, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol. 2013;190(7):3783–97.
Calcinotto A, et al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature. 2018;559(7714):363–9.
Fujita, K., et al., Obesity, Inflammation, and Prostate Cancer. J Clin Med, 2019. 8(2).
Gourine AV, et al. Fever in systemic inflammation: roles of purines. Front Biosci. 2004;9:1011–22.
Chen L, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. 2018;9(6):7204–18.
Mantovani A, et al. Cancer-related inflammation. Nature. 2008;454(7203):436–44.
Kawanishi, S., et al., Crosstalk between DNA Damage and Inflammation in the Multiple Steps of Carcinogenesis. Int J Mol Sci, 2017. 18(8).
Chiba T, Marusawa H, Ushijima T. Inflammation-associated cancer development in digestive organs: mechanisms and roles for genetic and epigenetic modulation. Gastroenterology. 2012;143(3):550–63.
Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer. 2004;4(8):579–91.
Calle EE, et al. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348(17):1625–38.
Ghaben AL, Scherer PE. Adipogenesis and metabolic health. Nat Rev Mol Cell Biol. 2019;20(4):242–58.
Putnam SD, et al. Lifestyle and anthropometric risk factors for prostate cancer in a cohort of Iowa men. Ann Epidemiol. 2000;10(6):361–9.
Cerhan JR, et al. Association of smoking, body mass, and physical activity with risk of prostate cancer in the Iowa 65+ rural health study (United States). Cancer Causes Control. 1997;8(2):229–38.
Freedland SJ, et al. Obesity, risk of biochemical recurrence, and prostate-specific antigen doubling time after radical prostatectomy: results from the SEARCH database. BJU Int. 2019;124(1):69–75.
Discacciati A, Orsini N, Wolk A. Body mass index and incidence of localized and advanced prostate cancer--a dose-response meta-analysis of prospective studies. Ann Oncol. 2012;23(7):1665–71.
Ouchi N, et al. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11(2):85–97.
Wang YY, et al. Adipose tissue and breast epithelial cells: a dangerous dynamic duo in breast cancer. Cancer Lett. 2012;324(2):142–51.
Choe SS, et al. Adipose tissue remodeling: its role in energy metabolism and metabolic disorders. Front Endocrinol (Lausanne). 2016;7:30.
Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res. 2005;96(9):939–49.
Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114(2):147–52.
Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115(5):1111–9.
Pickup JC. Inflammation and activated innate immunity in the pathogenesis of type 2 diabetes. Diabetes Care. 2004;27(3):813–23.
Hirosumi J, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420(6913):333–6.
Yuan M, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science. 2001;293(5535):1673–7.
Arkan MC, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11(2):191–8.
Bastard JP, et al. Evidence for a link between adipose tissue interleukin-6 content and serum C-reactive protein concentrations in obese subjects. Circulation. 1999;99(16):2221–2.
Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91.
Skurk T, Hauner H. Obesity and impaired fibrinolysis: role of adipose production of plasminogen activator inhibitor-1. Int J Obes Relat Metab Disord. 2004;28(11):1357–64.
Sha Y, et al. HMGB1 develops enhanced proinflammatory activity by binding to cytokines. J Immunol. 2008;180(4):2531–7.
Xu H, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112(12):1821–30.
Winer DA, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med. 2011;17(5):610–7.
Winer S, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med. 2009;15(8):921–9.
Feuerer M, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15(8):930–9.
Curat CA, et al. From blood monocytes to adipose tissue-resident macrophages: induction of diapedesis by human mature adipocytes. Diabetes. 2004;53(5):1285–92.
Blando J, et al. Dietary energy balance modulates prostate cancer progression in hi-Myc mice. Cancer Prev Res (Phila). 2011;4(12):2002–14.
Xu L, et al. Adipocytes affect castration-resistant prostate cancer cells to develop the resistance to cytotoxic action of NK cells with alterations of PD-L1/NKG2D ligand levels in tumor cells. Prostate. 2018;78(5):353–64.
Thompson PA, et al. Environmental immune disruptors, inflammation and cancer risk. Carcinogenesis. 2015;36(Suppl 1):S232–53.
Hayashi T, et al. High-fat diet-induced inflammation accelerates prostate Cancer growth via IL6 signaling. Clin Cancer Res. 2018;24(17):4309–18.
Jiang M, et al. Interleukin-6 trans-signaling pathway promotes immunosuppressive myeloid-derived suppressor cells via suppression of suppressor of cytokine signaling 3 in breast Cancer. Front Immunol. 2017;8:1840.
Loberg RD, et al. CCL2 is a potent regulator of prostate cancer cell migration and proliferation. Neoplasia. 2006;8(7):578–86.
Roca H, Varsos Z, Pienta KJ. CCL2 protects prostate cancer PC3 cells from autophagic death via phosphatidylinositol 3-kinase/AKT-dependent survivin up-regulation. J Biol Chem. 2008;283(36):25057–73.
Kim MS, Day CJ, Morrison NA. MCP-1 is induced by receptor activator of nuclear factor-{kappa} B ligand, promotes human osteoclast fusion, and rescues granulocyte macrophage colony-stimulating factor suppression of osteoclast formation. J Biol Chem. 2005;280(16):16163–9.
Rankine EL, et al. Brain cytokine synthesis induced by an intraparenchymal injection of LPS is reduced in MCP-1-deficient mice prior to leucocyte recruitment. Eur J Neurosci. 2006;24(1):77–86.
van Golen KL, et al. CCL2 induces prostate cancer transendothelial cell migration via activation of the small GTPase Rac. J Cell Biochem. 2008;104(5):1587–97.
Laurent V, et al. Periprostatic adipose tissue favors prostate Cancer cell invasion in an Obesity-dependent manner: role of oxidative stress. Mol Cancer Res. 2019;17(3):821–35.
van Roermund JG, et al. Periprostatic fat correlates with tumour aggressiveness in prostate cancer patients. BJU Int. 2011;107(11):1775–9.
Magi-Galluzzi C, et al. International Society of Urological Pathology (ISUP) consensus conference on handling and staging of radical prostatectomy specimens. Working group 3: extraprostatic extension, lymphovascular invasion and locally advanced disease. Mod Pathol. 2011;24(1):26–38.
Kapoor J, et al. Extraprostatic extension into periprostatic fat is a more important determinant of prostate cancer recurrence than an invasive phenotype. J Urol. 2013;190(6):2061–6.
Correa, L.H., G.S. Heyn, and K.G. Magalhaes, The Impact of the Adipose Organ Plasticity on Inflammation and Cancer Progression. Cells, 2019. 8(7).
Correa LH, et al. Adipocytes and macrophages interplay in the orchestration of tumor microenvironment: new implications in Cancer progression. Front Immunol. 2017;8:1129.
Wang Y, et al. Decreased growth of established human prostate LNCaP tumors in nude mice fed a low-fat diet. J Natl Cancer Inst. 1995;87(19):1456–62.
Ngo TH, et al. Effect of isocaloric low-fat diet on human LAPC-4 prostate cancer xenografts in severe combined immunodeficient mice and the insulin-like growth factor axis. Clin Cancer Res. 2003;9(7):2734–43.
Hu MB, et al. High-fat diet-induced adipokine and cytokine alterations promote the progression of prostate cancer in vivo and in vitro. Oncol Lett. 2018;15(2):1607–15.
Huang M, et al. A high-fat diet enhances proliferation of prostate cancer cells and activates MCP-1/CCR2 signaling. Prostate. 2012;72(16):1779–88.
Hao, Q., et al., Arctigenin inhibits prostate tumor growth in high-fat diet fed mice through dual actions on adipose tissue and tumor scientific reports, 2020. In press.
Boileau TW, et al. Prostate carcinogenesis in N-methyl-N-nitrosourea (NMU)-testosterone-treated rats fed tomato powder, lycopene, or energy-restricted diets. J Natl Cancer Inst. 2003;95(21):1578–86.
Platz EA, et al. Interrelation of energy intake, body size, and physical activity with prostate cancer in a large prospective cohort study. Cancer Res. 2003;63(23):8542–8.
Huber J, et al. CC chemokine and CC chemokine receptor profiles in visceral and subcutaneous adipose tissue are altered in human obesity. J Clin Endocrinol Metab. 2008;93(8):3215–21.
Zhang S, et al. Chemokine CXCL12 and its receptor CXCR4 expression are associated with perineural invasion of prostate cancer. J Exp Clin Cancer Res. 2008;27:62.
Murphy C, et al. Nonapical and cytoplasmic expression of interleukin-8, CXCR1, and CXCR2 correlates with cell proliferation and microvessel density in prostate cancer. Clin Cancer Res. 2005;11(11):4117–27.
Ben-Baruch A. The multifaceted roles of chemokines in malignancy. Cancer Metastasis Rev. 2006;25(3):357–71.
Acknowledgements
None.
Funding
This work was supported by NIH/NIMHD Accelerating Excellence in Translational Science Pilot Grants G0814C01 (Q. Hao); NIH/NCI 1U54CA14393, NIH/NIMHD U54MD007598 (J.V. Vadgama); NIH 1R03CA208221, 1SC1GM121202, 5S21MD000103 (P. Wang).
Author information
Authors and Affiliations
Contributions
Investigation: Q.H. and P.W.; Visualization: Q.H.; Writing-original draft preparation: Q.H; Writing-review and editing: J.V. and P.W. The author(s) read and approved the final manuscript.
Authors’ information
Qiongyu Hao: Ph.D., M.D., Postdoc, Charles R. Drew University of Medicine and Science.
Jaydutt V. Vadgama: Ph.D., Professor of Medicine, Charles R. Drew University of Medicine and Science and UCLA School of Medicine.
Piwen Wang: Ph.D., M.D., Assistant professor, Charles R. Drew University of Medicine and Science and UCLA School of Medicine.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The procedures of this study were approved by the Institutional Review Board of Charles R. Drew University of Medicine and Science.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hao, Q., Vadgama, J.V. & Wang, P. CCL2/CCR2 signaling in cancer pathogenesis. Cell Commun Signal 18, 82 (2020). https://doi.org/10.1186/s12964-020-00589-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-020-00589-8