
Abstract
Objective
Lung adenocarcinoma (LUAD) is one of the major subtypes of lung cancer that is associated with poor prognosis. The aim of this study was to identify useful biomarkers to enhance the treatment and diagnosis of LUAD.
Methods
GEO2R was used to identify common up-regulated differentially expressed genes (DEGs) in the GSE32863, GSE40791, and GSE75037 datasets. The DEGs were submitted to Metascape for gene ontology and pathway enrichment analysis as well as construction of the protein-protein interaction (PPI) network, while the molecular complex detection (MCODE) plug-in was employed to filter important subnetworks. The expression levels of the hub genes and their prognostic values were evaluated using the UALCAN, GEPIA2, and Kaplan-Meier plotter databases. The timer algorithm was utilized to determine the correlation between immune cell infiltration and the expression levels of hub genes in LUAD tissues. In addition, the hub gene mutation landscape and the correlation analysis with tumor mutational burden (TMB) score were evaluated using maftools package and ggstatsplot package in R software, respectively.
Results
We identified 156 common up-regulated DEGs, with gene ontology and pathway enrichment analysis indicating that they were mostly enriched in mitotic cell cycle process and cell cycle pathway. DEGs in the subnetwork with the largest number of genes were AURKB, CCNB2, CDC20, CDCA5, CDCA8, CENPF, and KNTC1. The seven hub genes were highly expressed in LUAD tissues and were associated with poor prognosis. These hub genes were negatively correlated with most immune cells. The somatic mutation landscape showed that AURKB, CDC20, CENPF, and KNTC1 had mutations and were positively correlated with TMB scores.
Conclusions
Our findings demonstrate that increased expression of seven hub genes is associated with poor prognosis for LUAD patients. Additionally, the TMB score indicates that the high expression of hub gene increases immune cell infiltration in patients with lung adenocarcinoma which may significantly improve response to immunotherapy.
Similar content being viewed by others
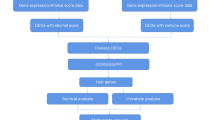


Introduction
Lung cancer is the leading cause of cancer-related morbidity and mortality worldwide and is mainly divided into non-small cell lung cancer and small cell lung cancer [1]. The proportion of lung adenocarcinoma (LUAD) in non-small cell lung cancer is approximately 55% [2]. Recently, there has been a significant increase in the incidence of LUAD, but the specific pathogenesis of LUAD still remains unclear [3]. Despite the availability of several therapeutic options for patients with LUAD, including surgery, targeted therapy, and immunotherapy, the overall survival rate of patients is still poor [4, 5]. As a result, it is critical to identify effective biomarkers associated with the progression of LUAD that could serve as therapeutic targets.
The recent advancement in chip sequencing technology has generated a lot of microarray data from various tumor samples [6]. Bioinformatics techniques have been extensively utilized in cancer research to identify useful biomarkers using large amounts of microarray datasets [7, 8]. The purpose of this research was to analyze three LUAD microarray datasets from the GEO database, namely GSE32863, GSE40791, and GSE75037. GEO2R algorithms were used to identify differentially expressed genes (DEGs) between tumor and normal tissues. The common up-regulated genes were then analyzed in Metascape for gene ontology and pathway enrichment [9, 10]. Metascape was also used for PPI network construction, and the MCODE online plug-in was utilized to identify the significant subnetworks of the PPI network [9]. The subnetwork with the greatest number of hub genes was chosen for further analysis. We compared the expression levels of key genes between tumor and normal samples using the GEPIA2 and UALCAN datasets [11, 12] and determined their prognostic value for patients with LUAD using the Kaplan-Meier Plotter database [13]. Finally, the Timer database was used to explore the correlation between hub genes and the infiltration of six different types of immune cells in LUAD tissues, including B cells, CD4+ T cells, CD8+ T cells, neutrophils, macrophages, and dendritic cells [14]. Our aim was to identify useful prognostic markers and immune-related therapeutic targets.
Methods
Acquisition of data
All GSE datasets analyzed were downloaded from the GEO database—an open access database for storing microarray and high-throughput sequencing data [15]. The GSE32863 dataset contained data for 58 LUAD tissues and 58 adjacent normal lung tissues analyzed using the Illumina HumanWG-6 v3.0 expression beadchip; the GSE40791 dataset contained data for 94 LUAD samples and 100 normal samples analyzed using the Affymetrix Human Genome U133 Plus 2.0 Array, while the GSE75037 dataset contained data for 83 LUAD tissues and 83 adjacent normal tissues analyzed using the Illumina HumanWG-6 v3.0 expression beadchip. In GSE32863 and GSE75037, each pair of tissues came from the same patient, but in GSE40791, each sample was from each patient. Therefore, a total of 335 patients were included in this study.
Analysis of DEGs
GEO2R was used to identify DEGs in the three GSE datasets with the parameters set as P > 0.05 and |logFC| > 2. A volcano map was used to visualize the DEGs, while a Venn diagram was used to identify up-regulated and down-regulated DEGs in the three GSE datasets. In this study, we were interested in the up-regulated DEGs that were common among the three datasets.
Analysis of gene ontology and pathway enrichment
Gene ontology (GO) has been widely used to analyze specific functions of genes classified into molecular function (MF), biological process (BP), and cellular component (CC) after annotating a given gene list. The purpose of pathway enrichment analysis is to use statistical methods to find significantly enriched pathway analysis in the target gene list. Metascape is an online tool for GO and biological pathway enrichment analysis, which presents results in form of high-quality charts and sententious explanation [9]. We used Metascape for GO analysis of the common up-regulated DEGs with the cutoff of P value, min overlap, and min enrichment set as less than 0.01, 3, and 1.5, respectively.
Construction of the PPI network
We also used Metascape to construct the PPI network—the interaction network of all proteins based on the relevance and similarity of the submitted gene list. We then selected the significant subnetworks from the overall PPI for subsequent analysis using the MCODE plug-in. We investigated the subnetwork with the largest number of genes and explored the characteristics of its constituent hub genes.
Verification of the expression level of hub genes
GEPIA2 is a database that can analyze RNA sequencing expression data from 9736 tumor samples and 8587 normal samples from the TCGA and GTEx projects [11], while UALCAN is a comprehensive and interactive online database that validates the expression level of candidate genes in different tumors in the TCGA database [12]. The GEPIA2 database was used to verify the differential expression of hub genes between the tumor tissues and adjacent tissues of LUAD patients, while the UALCAN database was used to analyze the expression level of hub genes in different stages in patients with LUAD. In addition, we further analyzed the co-expression correlation between hub genes and the expression level of hub genes in different cancers using GEPIA2.
Analysis of prognosis
The Kaplan-Meier Plotter is an online survival analysis database for 54,000 genes in 21 malignant tumors [16]. We used the Kaplan-Meier Plotter to determine the ability of the hub genes to predict OS of LUAD patients. In addition, we also analyzed the prognostic significance of the hub genes in different types of cancer using the GEPIA2 database.
Analysis of immune infiltration, somatic mutation, and TMB score
We collected mRNA-seq data from 513 lung adenocarcinoma patients in the TCGA database. We then used the Timer algorithm to explore the infiltration of six immune cells, including B cell, macrophage, myeloid dendritic cell, neutrophil, CD4+ T cell CD4+, and T cell CD8+ in specific tumor tissues [14]. The multi-gene correlation map was displayed using the pheatmap and ggstatsplot packages in R software. To investigate the mutation frequency of hub genes in lung adenocarcinoma, we downloaded the hub gene mutation data from the TCGA database and visualized the somatic mutation landscape using the maftools package in R software. The ggstatsplot package in R software was used to explore the correlation between the TMB score and the hub genes.
Results
Identification of DEGs in LUAD
We identified 307 up-regulated and 591 down-regulated DEGs in the GSE32863 dataset (Fig. 1A), 2298 DEGs including 721 up-regulated and 1250 down-regulated genes in the GSE40791 dataset (Fig. 1B), and 2550 DEGs including 1122 up-regulated and 1428 down-regulated genes in the GSE75037 dataset (Fig. 1C). Venn diagram analysis identified 156 common up-regulated genes and 434 down-regulated genes among the three GSE datasets (Fig. 1D, E).

Screening of differentially expressed genes (DEGs). A–C Volcano maps showing the distribution of DEGs in three datasets. D, E Venn diagrams showing the intersection of common up-regulated and common down-regulated genes, respectively
Gene ontology functional and pathway enrichment analysis of DEGs
In this study, Metascape was used to perform gene ontology functional annotation and pathway enrichment analysis of the common up-regulated genes from the three datasets. Gene ontology was explored according to the following three categories, namely biological processes (BP), cellular components (CC), and molecular functions (MF). BP terms were most significantly enriched in the mitotic cell cycle process, spindle assembly, meiotic spindle assembly, collagen fibril organization, and double-strand break repair via break-induced replication (Fig. 2A, Table 1). CC terms were enriched in midbody, apical plasma membrane, cell-cell junction, lateral plasma membrane, and fibrillar collagen trimer (Fig. 2B, Table 2). MF terms were most significantly enriched in cell adhesion molecule binding, kinase binding, extracellular matrix structural constituents, protein homodimerization activity, and calcium ion binding (Fig. 2C, Table 3). In addition, the results of pathway enrichment analysis revealed that all up-regulated DEGs were mainly enriched in cell cycle, cell cycle checkpoints, integrin cell surface interactions, APC/C:Cdh1-mediated degradation of Cdc20, and other APC/C:Cdh1-targeted proteins in late mitosis/early G1 and SUMOylation of DNA replication proteins (Fig. 2D, Table 4).

GO and pathway enrichment analysis of common up-regulated DEGs. A–C The enrichment analysis results of biological processes (BP), cellular components (CC), and molecular functions (MF). D The results of pathway enrichment analysis
PPI network construction and hub gene screening
The PPI network was constructed using Metascape and each code represented the specific common up-regulated DEG in the network (Fig. 3A). The molecular complex detection (MCODE) algorithm was applied to identify densely connected network components. In the whole screening process, seven clusters of MCODE with closely related functions were found and displayed in different colors, namely MCODE1, MCODE2, MCODE3, MCODE4, MCODE5, MCODE6, and MCODE7 (Fig. 3B). MCODE1 consisted of AURKB, CCNB2, KNTC1, CENPF, CDCA8, CDCA5, and CDC20; MCODE2 consisted of COL3A1, COL1A1, COL5A2, and COL10A1; MCODE3 consisted of TUBB2B, MCM4, PRC1, and KIF20A; MCODE4 consisted of EEF1A2, MCM2, and CDC45; MCODE5 consisted of LCN2, MMP9, and TCN1; MCODE6 consisted of AURKA, PTTG1, and UBE2C; MCODE7 consisted of MUC20, GALNT6, and MUC16, respectively. MCODE1 had the largest number of genes and further analysis was carried out to determine the clinical significance of each gene in LUAD.

The protein-protein interaction (PPI) network constructed using common up-regulated DEGs. A The landscape of PPI network. B The significant subnetworks originating from the overall PPI network
Expression analysis
Using the expression analysis function in the GEPIA2 database, we found that all the genes in MCODE1 apart from KNTC1 were significantly up-regulated in tumor tissues compared to normal samples (Fig. 4). Furthermore, we found that the expression levels of the seven genes were significantly higher in the tumor tissue than in normal tissues regardless of tumor stage using UALCAN (Fig. 5). The seven genes were also highly expressed in most cancers (Supplementary Fig. 1).

Comparison of the expression levels of hub genes between tumor tissues and normal tissues in LUAD patients using the GEPIA2 database. A–G The expression levels of AURKB, CCNB2, CDC20, CDCA5, CDCA8, CENPF, and KNTC1 in LUAD tissues are significantly higher in tumor tissues than in normal lung tissues

The analysis of expression levels of hub genes in different stages of LUAD patients using the UACALN database. A–G The expression levels of AURKB, CCNB2, CDC20, CDCA5, CDCA8, CENPF, and KNTC1 were significantly higher in tumor tissues than in normal tissues during the different stages of LUAD
Survival analysis
Kaplan-Meier analysis showed that high expression levels of the hub genes were associated with shorter overall survival of patients with LUAD (Fig. 6). The specific data is as follows: The HR of AURKA is 2.78, 95% CI is 2.2–3.51, and Logrank P is less than 1e−16; the HR of CCNB2 is 2.68, 95% CI is 2.05–3.49, and Logrank P = 4.1e−14; the HR of CDC20 is 2.46, 95% CI is 1.92–3.15, and Logrank P = 1.2e−13; the HR of CDCA5 is 2.32, 95% CI is 1.8–2.99, and Logrank P = 2.5e−11; the HR of CDCA8 is 1.99, 95% CI is 1.58–2.51, and Logrank P = 3.5e−09; the HR of CENPF is 1.66, 95% CI is 1.31–2.10, and Logrank P = 2.1e−05; the HR of KNTC1 is 1.47, 95% CI was 1.16–1.86, and Logrank P = 0.0015. High expression of the hub genes was also associated with poor prognosis in the majority of cancers, although the results in LUAD and lung squamous cell carcinoma were inconclusive (Supplementary Fig. 2).

The survival analysis of hub genes based on the Kaplan-Meier database. A–G High expression levels of AURKB, CCNB2, CDC20, CDCA5, CDCA8, CENPF, and KNTC1 were associated with short OS in patients with LUAD
Analysis of immune infiltration, somatic mutation, and TMB score
To investigate the potential function of the hub genes in LUAD patients, we used the TIMER algorithm to determine if the expression levels of the hub genes were associated with the infiltration levels of six types of immune cells in the tumor microenvironment of LUAD. AURKB was negatively correlated with B cell, macrophage, myeloid dendritic cell, CD4+ T cell, and CD8+ T cell infiltration; CCNB2 was negatively correlated with B cell, macrophage, myeloid dendritic cell, and CD4+T cell infiltration; CDC20 was negatively correlated with B cell, macrophage, myeloid dendritic cell, and CD4+ T cell infiltration; CDCA5 was negatively correlated with neutrophil, but positively correlated with B cell and CD4+T cell infiltration; CDCA8 was negatively correlated with B cell and CD4+ T cell, but positively correlated with neutrophil infiltration; CENPF were negatively correlated with B cell and myeloid dendritic cell infiltration; KNTC1 was negatively correlated with B cell but positively correlated with neutrophil infiltration (Fig. 7A, Table 5).

The correlation between the expression levels of hub genes and the infiltration of six immune cells and the TMB score in LUAD patients. A CDCA5 was positively correlated with B cell and CD4+ T cell; CDCA8 and KNTC1 were positively correlated with neutrophil; the rest of hub genes were negatively correlated with most immune cells. B Oncoplot displaying the somatic landscape of the LUAD cohort. The mutation frequencies of AURKB, CDC20, CENPF, and KNTC1 in LUAD patients were 1%, 1%, 6%, and 3%, respectively. In addition, missense mutation is the main mutation type. C–F Correlation analysis between hub gene expression and TMB score. AURKB, CDC20, CENPF, and KNTC1 were all positively correlated with the TMB score, and the Spearman correlation coefficients were 0.46, 0.47, 0.40, and 0.39, respectively
Somatic mutation analysis showed that AURKB, CDC20, CENPF, and KNTC1 had different types of mutations in patients with lung adenocarcinoma, and the main type was missense mutation (Fig. 7B). In addition, an increase in the expression of the four genes was associated with an increase in the TMB score (Fig. 7C–F). High TMB scores are associated with good response to immune therapy [17].
Discussion
LUAD is one of the most important subtypes of non-small cell lung cancer, with high incidence and mortality [18]. Currently, there are no effective biomarkers for the accurate diagnosis of LUAD patients [19]. Therefore, we used bioinformatics tools to screen candidate genes for the diagnosis and prognosis of LUAD from the GEO database. In addition, we explored the correlation among the immune cell infiltration and TMB score and the expression levels of these genes in the LUAD tissues.
First, we identified 156 common up-regulated genes and 434 common down-regulated genes among the three GSE datasets obtained from the GEO database. The 156 common up-regulated genes were then submitted to Metascape for GO and pathway enrichment analysis. Biological processes (BP), molecular functions (MF), and cellular components (CC) were included in the GO analysis. Subsequently, the PPI network was constructed based on common up-regulated genes using Metascape and the MCODE plug-in was used to screen out seven important subnetworks in the PPI network, namely MCODE 1, MCODE 2, MCODE 3, MCODE 4, MCODE 5, MCODE 6, and MCODE 7 [20]. MCODE 1 contained the largest number of key genes, including AURKB, CDC20, CDCA5, CDCA8, CENPF, KNTC1, and CCNB2. Expression analysis using GEPIA2 and UALCAN databases showed that all the hub genes in MCODE 1 except KNTC1 were highly expressed in LUAD tissues compared to normal tissues. In addition, survival analysis using the Kaplan-Meier Plotter database showed that high expression of these key genes was associated with poor prognosis in patients with LUAD. Furthermore, we explored the correlation of these candidate genes with the infiltration of six immune cells in patients with LUAD to determine the potential response of the tumors to immunotherapy. It is worth noting that through somatic mutation analysis, we found that AURKB, CDC20, CENPF, and KNTC1 had different frequencies of mutations in patients with lung adenocarcinoma, and mainly missense mutation type. Furthermore, we analyzed the correlation between the expression of four genes and the TMB score and found that with the increase of the expression of these genes in patients with lung adenocarcinoma, the TMB score also increased. It is well known that a high TMB score indicates that cancer patients have better immunotherapy effects. Therefore, if we can improve the proportion of immune cell infiltration in lung adenocarcinoma tissues in future clinical studies, it may be able to effectively improve the efficacy of immunotherapy and prolong the survival period of patients with lung adenocarcinoma.
AURKB (aurora kinase B) is a protein-coding gene that acts as a key regulator of mitosis [21]. Many studies have confirmed that AURKB is a crucial carcinogenic factor in different kinds of carcinoma. For example, AURKB was found to be expressed at higher levels in renal cell carcinoma tissues than in normal kidney tissues, suggesting that it may regulate renal cell carcinoma progression by modulating the intestinal immune network for IgA production and signaling pathways involving cytokine-cytokine receptor interactions [22]. Furthermore, AURKB was overexpressed in gastric cancer and was strongly linked to clinicopathological features of the disease. Silencing of AURKB may decrease the invasive and migratory capacities of gastric cancer cells by disrupting the VEGFA/Akt/mTOR and Wnt/-catenin/Myc pathways [23]. Additionally, AURKB activation was associated with acquired resistance to EGFR TKIs, suggesting that AURKB should be targeted in NSCLC patients scheduled for anti-EGFR treatment but who lack resistance mutations [24].
CDC20 (cell division cycle 20) appears to act as a regulatory protein interacting with several other proteins at multiple points in the cell cycle [25]. Min Shi et al. found that CDC20 played a crucial role in the development of hepatocellular carcinoma by regulating the PHD3 protein [26]. Besides, Yang Gao et al. found that targeting CDC20 sensitized colorectal cancer cells to radiotherapy through mitochondrial-dependent apoptotic signaling [27]. Qin Zhang et al. found that CDC20 combined with CD44 or β-catenin could serve as an important indicator for the prognosis of patients with prostate cancer [28]. Furthermore, Huan Deng et al. found that CDC20 was up-regulated in LUSC at the mRNA and protein levels [29]. However, the role and mechanisms of action of CDC20 in LUAD remain unclear.
CDCA5 (cell division cycle associated 5) is another protein-coding gene involved in DNA repair [30]. CDCA5 promotes the progression of bladder cancer by dysregulating mitochondria-mediated apoptosis, cell cycle regulation, and activation of the PI3k/AKT/mTOR pathway [31]. Additionally, CDCA5 aids in the development of esophageal squamous cell carcinoma and may be an important target for esophageal squamous cell carcinoma immunotherapy [32]. Moreover, CDCA5 phosphorylation and activation by mitogen-activated protein kinase are critical for human lung cancer [33].
CDCA8 (cell division cycle associated 8) is a component of the chromosomal passenger complex, which is required for mitosis and cell division [34]. Increased CDCA8 expression in ovarian tissues probably plays a critical role in the development of ovarian cancer through the PLK1 pathway [35]. Besides, CDCA8 is involved in the construction of meiotic spindles and chromosomal segregation during human oocyte meiosis [36]. CDCA8 overexpression has been shown to accelerate the development of cutaneous melanoma and is associated with poor prognosis [37]. Additionally, aurora kinase B-mediated phosphorylation and activation of CDCA8 plays a major role in human lung cancer [38]. Moreover, miR-133b suppressed cell proliferation, motility, and invasion in lung adenocarcinoma by targeting CDCA8 [39].
CENPF is a gene that encodes a protein that is involved in the centromere-kinetochore complex association [40]. Overexpression of CENPF in breast cancer was associated with poor prognosis and tumor bone metastases by controlling parathyroid hormone-related peptide (PTHrP) production via activating PI3K-AKT-mTORC1 [41]. Besides, the HnRNPR-CCNB1/CENPF axis was involved in the proliferation and metastasis of gastric cancer [42]. Additionally, silencing CENPF substantially reduced LUAD cell tumor development in an experimental xenograft lung cancer model using naked mice armpits of the right forelimb. However, there are no sufficient studies on the mechanism of CENPF in LUAD [43].
KNTC1 encodes a protein participating in the processes that guarantee correct chromosomal segregation during cell division [44]. A recent study indicated that silencing KNTC1 with shRNA inhibited cell viability and caused apoptosis in esophageal squamous cell carcinoma [45]. Moreover, relevant bioinformatics publications demonstrated that KNTC1 was associated with a poor outcome in patients with hepatocellular carcinoma and cervical cancer [46, 47]. However, there has been no research on the mechanism of action of KNTC1 in patients with LUAD.
CCNB2 (cyclin B2) is a member of the B-type cyclins family that can interact with p34cdc2, and is a critical component of the cell cycle regulation [48]. CCNB2 has been shown to promote the proliferation of triple-negative breast cancer cells in vitro and in vivo [49]. As validated by a comprehensive bioinformatics study, CCNB2 has been identified as a promising therapeutic target for ovarian cancer [50]. Additionally, miR-335-5p disrupts the cell cycle and increases lung adenocarcinoma metastasis by targeting CCNB2 [51]. Moreover, CCNB2 had been identified as a marker of responsiveness to immune checkpoint inhibitors (ICI) in NSCLC and overexpression of CCNB2 is a poor prognostic indicator in Chinese patients with NSCLC [52, 53].
In recent years, an increasing number of studies have revealed that diverse immune components in the tumor microenvironment play a significant role in the molecular process of various tumors and development. A study on the advanced lung cancer inflammation index, for example, discovered that it can predict shorter overall survival not only in patients with lung adenocarcinoma, but also in patients with other tumors at a low level of expression [54]. TMED2, MOESIN, DPYSL2, and LncRNA MEG3 have been discovered to enhance the occurrence and development of patients with lung adenocarcinoma via several immune-related regulatory mechanisms [55,56,57,58]. Furthermore, the hunt for efficient immune checkpoint inhibitors and indicators to predict the efficacy of immunotherapy is critical. It is well known that pembrolizumab and chemotherapy are currently the first-line treatments for small cell lung cancer, and their therapeutic effectiveness has been widely accepted [59]. In the studies related to lung adenocarcinoma patients, it was found that individuals with smoking history could benefit more from the treatment of immune checkpoint inhibitors and that the combination of immune checkpoint inhibitors nivolumab and ipilimumab was much more effective than monotherapy [60, 61]. However, there is currently a scarcity of effective indicators of tumor immunotherapy. We expect that further indicators similar to PD-L1 will emerge in the future to predict the efficacy of NSCLC immunotherapy [62]. Therefore, we expect that the results of this study will provide reference value to the immunotherapy of lung adenocarcinoma patients from different molecular subtypes. The findings of our research were acquired through data mining of an online database using bioinformatics techniques. The limitations of this study include the absence of specific in vitro or in vivo experiments to validate the significance of the selected hub genes in LUAD patients. Additionally, the results of our study may include partial bias owing to the issue of data quantity and quality. As a result, there is a need for further studies to validate our findings and identify the role of these genes in lung adenocarcinoma.
Conclusion
In conclusion, the high expression of the candidate genes screened in this study is associated with poor prognosis in LUAD patients. High expression of the candidate genes combined with the TMB score indicates a better response to immunotherapy in patients with lung adenocarcinoma. However, there is a need for more experiments to validate the significance and mechanism of action of these genes in LUAD patients.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- LUAD:
-
Lung adenocarcinoma
- DEGs:
-
Differentially expressed genes
- GO:
-
Gene ontology
- PPI:
-
Protein-protein interaction
- MCODE:
-
Molecular complex detection
- MF:
-
Molecular function
- BP:
-
Biological process
- CC:
-
Cellular component
- GEO:
-
Gene Expression Omnibus
- OS:
-
Overall survival
- AURKB:
-
Aurora kinase B
- CDC20:
-
Cell division cycle 20
- CDCA5:
-
Cell division cycle associated 5
- CDCA8:
-
Cell division cycle associated 8
- CCNB2:
-
Cyclin B2
- NSCLC:
-
Non-small cell lung cancer
- ICI:
-
Immune checkpoint inhibitor
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Hespanhol V, Queiroga H, Magalhães A, Santos AR, Coelho M, Marques A. Survival predictors in advanced non-small cell lung cancer. Lung Cancer. 1995;13(3):253–67.
Tang Q, Li W, Zheng X, Ren L, Liu J, Li S, et al. MELK is an oncogenic kinase essential for metastasis, mitotic progression, and programmed death in lung carcinoma. Signal Transduct Target Ther. 2020;5(1):279.
Liu Z, Sun D, Zhu Q, Liu X. The screening of immune-related biomarkers for prognosis of lung adenocarcinoma. Bioengineered. 2021;12(1):1273–85.
Jones GD, Brandt WS, Shen R, Sanchez-Vega F, Tan KS, Martin A, et al. A genomic-pathologic annotated risk model to predict recurrence in early-stage lung adenocarcinoma. JAMA Surg. 2021;156(2):e205601.
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res. 2013;41(Database issue):D991–5.
Wang J, Shi W, Miao Y, Gan J, Guan Q, Ran J. Evaluation of tumor microenvironmental immune regulation and prognostic in lung adenocarcinoma from the perspective of purinergic receptor P2Y13. Bioengineered. 2021;12(1):6286–304.
Qu Y, Cheng B, Shao N, Jia Y, Song Q, Tan B, et al. Prognostic value of immune-related genes in the tumor microenvironment of lung adenocarcinoma and lung squamous cell carcinoma. Aging (Albany NY). 2020;12(6):4757–77.
Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523.
Zhou Q, Zhang F, He Z, Zuo M-Z. E2F2/5/8 serve as potential prognostic biomarkers and targets for human ovarian cancer. Front Oncol. 2019;9:161.
Tang Z, Kang B, Li C, Chen T, Zhang Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019;47(W1):W556–60.
Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVSK, et al. UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19(8):649–58.
Hou G-X, Liu P, Yang J, Wen S. Mining expression and prognosis of topoisomerase isoforms in non-small-cell lung cancer by using Oncomine and Kaplan-Meier plotter. PLoS One. 2017;12(3):e0174515.
Li T, Fan J, Wang B, Traugh N, Chen Q, Liu JS, et al. TIMER: a web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 2017;77(21):e108–10.
Hadley D, Pan J, El-Sayed O, Aljabban J, Aljabban I, Azad TD, et al. Precision annotation of digital samples in NCBI’s gene expression omnibus. Sci Data. 2017;4:170125.
Sun C-C, Li S-J, Hu W, Zhang J, Zhou Q, Liu C, et al. Comprehensive analysis of the expression and prognosis for E2Fs in human breast cancer. Mol Ther. 2019;27(6):1153–65.
Gandara DR, Paul SM, Kowanetz M, Schleifman E, Zou W, Li Y, et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med. 2018;24(9):1441–8.
Shao J, Zhang B, Kuai L, Li Q. Integrated analysis of hypoxia-associated lncRNA signature to predict prognosis and immune microenvironment of lung adenocarcinoma patients. Bioengineered. 2021;12(1):6186–200.
Seijo LM, Peled N, Ajona D, Boeri M, Field JK, Sozzi G, et al. Biomarkers in lung cancer screening: achievements, promises, and challenges. J Thorac Oncol. 2019;14(3):343–57.
Li L, Lv J, He Y, Wang Z. Gene network in pulmonary tuberculosis based on bioinformatic analysis. BMC Infect Dis. 2020;20(1):612.
Marima R, Hull R, Penny C, Dlamini Z. Mitotic syndicates aurora kinase B (AURKB) and mitotic arrest deficient 2 like 2 (MAD2L2) in cohorts of DNA damage response (DDR) and tumorigenesis. Mutat Res Rev Mutat Res. 2021;787:108376.
Wan B, Huang Y, Liu B, Lu L, Lv C. AURKB: a promising biomarker in clear cell renal cell carcinoma. PeerJ. 2019;7:e7718.
Wang Z, Yu Z, Wang G-H, Zhou Y-M, Deng J-P, Feng Y, et al. AURKB promotes the metastasis of gastric cancer, possibly by inducing EMT. Cancer Manag Res. 2020;12:6947–58.
Bertran-Alamillo J, Cattan V, Schoumacher M, Codony-Servat J, Giménez-Capitán A, Cantero F, et al. AURKB as a target in non-small cell lung cancer with acquired resistance to anti-EGFR therapy. Nat Commun. 2019;10(1):1812.
Wang L, Zhang J, Wan L, Zhou X, Wang Z, Wei W. Targeting Cdc20 as a novel cancer therapeutic strategy. Pharmacol Ther. 2015;151:141–51.
Shi M, Dai W-Q, Jia R-R, Zhang Q-H, Wei J, Wang Y-G, et al. APC-mediated degradation of PHD3 stabilizes HIF-1a and promotes tumorigenesis in hepatocellular carcinoma. Cancer Lett. 2021;496:144–55.
Gao Y, Wen P, Chen B, Hu G, Wu L, Xu A, et al. Downregulation of CDC20 increases radiosensitivity through Mcl-1/p-Chk1-mediated DNA damage and apoptosis in tumor cells. Int J Mol Sci. 2020;21(18):6692.
Zhang Q, Huang H, Liu A, Li J, Liu C, Sun B, et al. Cell division cycle 20 (CDC20) drives prostate cancer progression via stabilization of β-catenin in cancer stem-like cells. EBioMedicine. 2019;42:397–407.
Deng H, Hang Q, Shen D, Ying H, Zhang Y, Qian X, et al. High expression levels of CDK1 and CDC20 in patients with lung squamous cell carcinoma are associated with worse prognosis. Front Mol Biosci. 2021;8:653805.
Xu T, Ma M, Dai J, Yu S, Wu X, Tang H, et al. Gene expression screening identifies CDCA5 as a potential therapeutic target in acral melanoma. Hum Pathol. 2018;75:137–45.
Fu G, Xu Z, Chen X, Pan H, Wang Y, Jin B. CDCA5 functions as a tumor promoter in bladder cancer by dysregulating mitochondria-mediated apoptosis, cell cycle regulation and PI3k/AKT/mTOR pathway activation. J Cancer. 2020;11(9):2408–20.
Xu J, Zhu C, Yu Y, Wu W, Cao J, Li Z, et al. Systematic cancer-testis gene expression analysis identified CDCA5 as a potential therapeutic target in esophageal squamous cell carcinoma. EBioMedicine. 2019;46:54–65.
Nguyen M-H, Koinuma J, Ueda K, Ito T, Tsuchiya E, Nakamura Y, et al. Phosphorylation and activation of cell division cycle associated 5 by mitogen-activated protein kinase play a crucial role in human lung carcinogenesis. Cancer Res. 2010;70(13):5337–47.
Shuai Y, Fan E, Zhong Q, Chen Q, Feng G, Gou X, et al. CDCA8 as an independent predictor for a poor prognosis in liver cancer. Cancer Cell Int. 2021;21(1):159.
Chen C, Chen S, Luo M, Yan H, Pang L, Zhu C, et al. The role of the gene family in ovarian cancer. Ann Transl Med. 2020;8(5):190.
Zhang C, Zhao L, Leng L, Zhou Q, Zhang S, Gong F, et al. CDCA8 regulates meiotic spindle assembly and chromosome segregation during human oocyte meiosis. Gene. 2020;741:144495.
Ci C, Tang B, Lyu D, Liu W, Qiang D, Ji X, et al. Overexpression of CDCA8 promotes the malignant progression of cutaneous melanoma and leads to poor prognosis. Int J Mol Med. 2019;43(1):404–12.
Hayama S, Daigo Y, Yamabuki T, Hirata D, Kato T, Miyamoto M, et al. Phosphorylation and activation of cell division cycle associated 8 by aurora kinase B plays a significant role in human lung carcinogenesis. Cancer Res. 2007;67(9):4113–22.
Hu C, Wu J, Wang L, Liu X, Da B, Liu Y, et al. miR-133b inhibits cell proliferation, migration, and invasion of lung adenocarcinoma by targeting CDCA8. Pathol Res Pract. 2021;223:153459.
Mahmoud AD, Ballantyne MD, Miscianinov V, Pinel K, Hung J, Scanlon JP, et al. The human-specific and smooth muscle cell-enriched LncRNA SMILR promotes proliferation by regulating mitotic CENPF mRNA and drives cell-cycle progression which can be targeted to limit vascular remodeling. Circ Res. 2019;125(5):535–51.
Sun J, Huang J, Lan J, Zhou K, Gao Y, Song Z, et al. Overexpression of CENPF correlates with poor prognosis and tumor bone metastasis in breast cancer. Cancer Cell Int. 2019;19:264.
Chen E-B, Qin X, Peng K, Li Q, Tang C, Wei Y-C, et al. HnRNPR-CCNB1/CENPF axis contributes to gastric cancer proliferation and metastasis. Aging (Albany NY). 2019;11(18):7473–91.
Li M-X, Zhang M-Y, Dong H-H, Li A-J, Teng H-F, Liu A-L, et al. Overexpression of CENPF is associated with progression and poor prognosis of lung adenocarcinoma. Int J Med Sci. 2021;18(2):494–504.
Han H-Y, Mou J-T, Jiang W-P, Zhai X-M, Deng K. Five candidate biomarkers associated with the diagnosis and prognosis of cervical cancer. Biosci Rep. 2021;41(3):BSR20204394.
Liu C-T, Min L, Wang Y-J, Li P, Wu Y-D, Zhang S-T. shRNA-mediated knockdown of KNTC1 suppresses cell viability and induces apoptosis in esophageal squamous cell carcinoma. Int J Oncol. 2019;54(3):1053–60.
Shen S, Kong J, Qiu Y, Yang X, Wang W, Yan L. Identification of core genes and outcomes in hepatocellular carcinoma by bioinformatics analysis. J Cell Biochem. 2019;120(6):10069–81.
Chen H, Wang X, Jia H, Tao Y, Zhou H, Wang M, et al. Bioinformatics analysis of key genes and pathways of cervical cancer. Onco Targets Ther. 2020;13:13275–83.
Shubbar E, Kovács A, Hajizadeh S, Parris TZ, Nemes S, Gunnarsdóttir K, et al. Elevated cyclin B2 expression in invasive breast carcinoma is associated with unfavorable clinical outcome. BMC Cancer. 2013;13:1.
Wu S, Su R, Jia H. Cyclin B2 (CCNB2) stimulates the proliferation of triple-negative breast cancer (TNBC) cells in vitro and in vivo. Dis Markers. 2021;2021:5511041.
Yang D, He Y, Wu B, Deng Y, Wang N, Li M, et al. Integrated bioinformatics analysis for the screening of hub genes and therapeutic drugs in ovarian cancer. J Ovarian Res. 2020;13(1):10.
Wang X, Xiao H, Wu D, Zhang D, Zhang Z. miR-335-5p regulates cell cycle and metastasis in lung adenocarcinoma by targeting CCNB2. Onco Targets Ther. 2020;13:6255–63.
Pabla S, Conroy JM, Nesline MK, Glenn ST, Papanicolau-Sengos A, Burgher B, et al. Proliferative potential and resistance to immune checkpoint blockade in lung cancer patients. J Immunother Cancer. 2019;7(1):27.
Qian X, Song X, He Y, Yang Z, Sun T, Wang J, et al. CCNB2 overexpression is a poor prognostic biomarker in Chinese NSCLC patients. Biomed Pharmacother. 2015;74:222–7.
Hua X, Chen J, Wu Y, Sha J, Han S, Zhu X. Prognostic role of the advanced lung cancer inflammation index in cancer patients: a meta-analysis. World J Surg Oncol. 2019;17(1):177.
Feng L, Cheng P, Feng Z, Zhang X. Transmembrane p24 trafficking protein 2 regulates inflammation through the TLR4/NF-κB signaling pathway in lung adenocarcinoma. World J Surg Oncol. 2022;20(1):32.
Li Y-Q, Zheng Z, Liu Q-X, Lu X, Zhou D, Zhang J, et al. Moesin as a prognostic indicator of lung adenocarcinoma improves prognosis by enhancing immune lymphocyte infiltration. World J Surg Oncol. 2021;19(1):109.
Wu Y-J, Nai A-T, He G-C, Xiao F, Li Z-M, Tang S-Y, et al. DPYSL2 as potential diagnostic and prognostic biomarker linked to immune infiltration in lung adenocarcinoma. World J Surg Oncol. 2021;19(1):274.
Wang C, Tao X, Wei J. Effects of LncRNA MEG3 on immunity and autophagy of non-small cell lung carcinoma through IDO signaling pathway. World J Surg Oncol. 2021;19(1):244.
Liu Q, Zhang Y, Liu M, Xu R, Yi F, Wei Y, et al. The benefits and risks of pembrolizumab in combination with chemotherapy as first-line therapy in small-cell lung cancer: a single-arm meta-analysis of noncomparative clinical studies and randomized control trials. World J Surg Oncol. 2021;19(1):298.
Mo J, Hu X, Gu L, Chen B, Khadaroo PA, Shen Z, et al. Smokers or non-smokers: who benefits more from immune checkpoint inhibitors in treatment of malignancies? An up-to-date meta-analysis. World J Surg Oncol. 2020;18(1):15.
Chen J, Li S, Yao Q, Du N, Fu X, Lou Y, et al. The efficacy and safety of combined immune checkpoint inhibitors (nivolumab plus ipilimumab): a systematic review and meta-analysis. World J Surg Oncol. 2020;18(1):150.
Peng Z, Lin H, Zhou K, Deng S, Mei J. Predictive value of pretreatment PD-L1 expression in EGFR-mutant non-small cell lung cancer: a meta-analysis. World J Surg Oncol. 2021;19(1):145.
Acknowledgements
We would like to express our gratitude to the team members for their contributions to this article, and then we will work diligently to do relevant research in the future.
Funding
This study was supported by the grants from the National Natural Science Foundation of China (81903992), Youth Foundation of Jiangsu Commission of Health (No. Q2017004), Jiangsu Provincial Medical Youth Talent (No. QNRC2016656), and Six talent peaks project in Jiangsu Province (No.WSN-042).
Author information
Authors and Affiliations
Contributions
BR, SZ, and ZX participated in the whole study design. The manuscript was drafted by ZX, SW, ZR, and LX and revised by ZX, XG, and LX. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Fig. 1.
The expression landscape of hub genes in different cancers using GEPIA2 database. The graph demonstrated that the hub genes were highly expressed in the majority of cancers.
Additional file 2: Supplementary Fig. 2.
The prognostic values of the hub genes in different cancers. The higher the red intensity of the square color, the higher the gene expression level, indicating a worse prognosis for patients.
Additional file 3: Supplementary Table 1.
Up and down regulation of differentially expressed genes (DEGs) in three GSE datasets.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xu, Z., Wang, S., Ren, Z. et al. An integrated analysis of prognostic and immune infiltrates for hub genes as potential survival indicators in patients with lung adenocarcinoma. World J Surg Onc 20, 99 (2022). https://doi.org/10.1186/s12957-022-02543-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12957-022-02543-z