
Abstract
Background
Aberrant activation of fibroblast growth factor receptor 3 (FGFR3) is frequently observed in bladder cancer, but how it involved in carcinogenesis is not well understood. The current study was aimed to investigate the underlying mechanism on the progression of bladder cancer.
Methods
The GSE41035 dataset downloaded from Gene Expression Omnibus was used to identify the differentially expressed genes (DEGs) between bladder cancer cell line RT112 with or without depletion of FGFR3, and gene ontology enrichment analysis was performed. Then, FGFR3-centered protein–protein interaction (PPI) and regulatory networks were constructed. Combined with the data retrieved from GSE31684, prognostic makers for bladder cancer were predicted.
Results
We identified a total of 2855 DEGs, and most of them were associated with blood vessel morphogenesis and cell division. In addition, KIAA1377, POLA2, FGFR3, and EPHA4 were the hub genes with high degree in the FGFR3-centered PPI network. Besides, 17 microRNAs (miRNAs) and 6 transcriptional factors (TFs) were predicted to be the regulators of the nodes in PPI network. Moreover, CSTF2, POLA1, HMOX2, and EFNB2 may be associated with the prognosis of bladder cancer patient.
Conclusions
The current study may provide some insights into the molecular mechanism of FGFR3 as a mediator in bladder cancer.
Similar content being viewed by others

Background
Bladder cancer, which refers to the cancer arising from the epithelial lining of urinary bladder, is one of the most frequent malignant cancers in the world. Its incidence has steadily increased and ranked as the sixth most common form of cancer in 2013 [1]. Though not fully known, the occurrence and development of bladder cancer is closely associated with multiple environmental factors, such as drinking, smoking, and contacting with chemical products, as well as many genetic factors [2].
Fibroblast growth factor receptor 3 (FGFR3), which belongs to the family of tyrosine kinase (RTK), is responsible for the FGF signal transduction [3]. FGFR3 signaling plays regulatory roles in cell proliferation, differentiation, and survival [4]. It is implicated in diverse physiologic and pathologic processes. Previous study revealed that FGFR3 is linked to the development of bladder cancer and FGFR3 overexpression and mutations are frequent events in patients with bladder cancer [5]. Moreover, it has been proved that bladder cancer cell proliferation in culture is inhibited when the activation of FGFR3 was blocked [6]. There is no doubt that FGFR3 takes vital roles in the process of tumorigenesis, but how FGFR3 signaling contributes to carcinogenesis is still unclear.
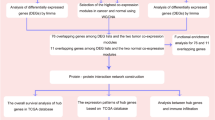
In current study, we reanalyzed the dataset GSE41035 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE41035), which provided the gene expression profiles of bladder cancer cell line RT112 with or without depletion of FGFR3, to identify the differentially expressed genes (DEGs) upon loss of FGFR3 in bladder cancer. Based on this dataset, Du et al. (another contributor of GSE41035) have documented that FGFR3 can stimulate stearoyl CoA desaturase 1 activity to promote bladder tumor growth [7]. Protein–protein interaction (PPI) and regulatory networks related to FGFR3 were constructed to investigate the possible mechanisms underlying the oncogenic role of FGFR3 in bladder cancer. Besides, we predicted the potential prognostic makers from the DEGs in the FGFR3-centered PPI network for bladder cancer combined with the data retrieved from the dataset GSE31684 [8].
Methods
Affymetrix microarray data
In current study, gene expression data of GSE41035 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE41035) and GSE31684 [8] were downloaded from Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/), respectively. Both of these two datasets were sequenced on [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array. In GSE41035 uploaded by Modrusan et al., total of 24 cell samples, ranging from GSM1007032 to GSM1007055, were included. In this dataset, RT112 cells, a bladder cancer cell line, were transduced with a doxycycline-inducible control EGFP short hairpin (shRNA) or three independent FGFR3 shRNAs and then treated with or without doxycycline for depletion of FGFR3 protein. Consequently, gene expression alterations induced by FGFR3 loss would be identified by microarray analysis. The dataset GSE31684 uploaded by Riester et al. contained the gene expression patterns associated with clinical and prognostic variables of 93 bladder cancer patients managed by radical cystectomy. For these 93 patients, the median ages were ranged from 32.1 to 91.1 years and the male/female ratio was 68/25. Meanwhile, 95% of them were transitional cell carcinoma (TCC), and the remains were TCC/squamous. In addition, the radical cystectomy stages of them were 5 cases of pTa, 10 cases of pT1, 17 cases of pT2, 42 cases of pT3, and 19 cases of pT4, and the follow-up times of them were ranged from 1 to 175 months. This study was approved by Memorial Sloan-Kettering Cancer Center Institutional Review Board.
Data preprocessing and DEG identification
RankProd is a simple and rapid method for DEG identification, which is based on the rank of gene expression and reliably utilized to explore difference of dataset with small sample size. After normalized by robust multi-array average (RMA) [9], the probe-level data in CEL files were converted into expression measures. Then, RankProd [10] was applied to identify the differentially expressed genes between the FGFR3-depleted RT112 cells and the control cells. False discovery ratio (FDR) <0.05 was set as the threshold.
GO functional enrichment analysis
Gene Ontology (GO) is a tool for functional annotation of large-scale genomic data [11]. In the current study, we used GO function [12] to identify the over-represented GO biology processes among the DEGs. p value <0.05 was chosen as cutoff criterion.
Construction of FGFR3-centered PPI network
The PPI data was collected from the Human Protein Reference Database (HPRD) [13], a centralized resource for information about human proteins, their interactions with other human proteins, and protein–disease relationships. The PPI network was generated by mapping the previously identified DEGs to the PPI data. FGFR3 was employed as a query node to construct the FGFR3-centered PPI network. First, the direct interacting DEGs with FGFR3 were selected, and then, the neighbors of these DEGs were also selected. The subnetwork was generated based on these proteins and visualized by Cytoscape [14].
Construction of regulatory network
The experimentally validated dataset of human miRNAs/targets and transcriptional factors (TFs)/targets were respectively retrieved from miRTarBase [15], a database listing the miRNA–mRNA interactions collected from the literatures in PubMed, and TRANSFAC (TRANScription FACtor database) [16], a manually curated database of eukaryotic transcription factors, their genomic binding sites, and DNA binding profiles. Based on these two databases, miRNAs, which have regulatory targets among DEGs, and TFs included in DEGs were screened. The regulatory network associated with the genes in the above sub-PPI network was constructed using these data and visualized by Cytoscape [14].
Exploring the potential prognostic makers for bladder cancer
The gene expression profiles and prognostic variables were retrieved from GSE31684 [8]. According to the median expression values of the genes in FGFR3-centered PPI network, patients with bladder cancer were divided into high- and low-expression groups. Prognosis-related genes were predicted by Cox regression model based on univariate analysis and multivariate analyses. Survival analysis was conducted by using the bioconductor splines package and survival package in R. Log-rank test was applied for comparison, and p value <0.05 was chosen as the threshold.
Results
Identification of DEGs
Genes that were differentially expressed after doxycycline induction (FDR <0.05) in the control cell line were considered as the DEG1 group, and those in 3 FGFR3-depleted cell lines were considered as the DEG2 group. Finally, we identified a total of 1428 upregulated and 1427 downregulated genes in the DEG2 group while there were only 28 upregulated and 58 downregulated genes in the DEG1 group.
GO enrichment analysis
To determine the potential processes mediated by the DEG2 group, we separately mapped the upregulated and downregulated to the GO BP database. With p value <0.05 as the threshold, 5 GO terms were over-represented by the upregulated genes (Table 1 (a)) and the most significant one was regulation of transcription, DNA dependent (GO:0006355) with p value = 1.50E−06. The others were negative regulation of biological process (GO:0048519, p value = 1.69E−06), response to type I interferon (GO:0034340, p value = 1.42E−05), blood vessel morphogenesis (GO:0048514, p value = 2.42E−05), and epithelium development (GO:0060429, p value = 0.000125). However, 85 GO terms were enriched among the downregulated genes. Table 1 (b) lists the top 5 terms, from which we found that these terms mainly associated with cell division and carboxylic acid metabolic process.
FGFR3-cented PPI network
The genes in DEG2 group were mapped to the PPI data retrieved from the HPRD, and a network with 920 nodes and 1514 edges were created. Then, 7 DEGs, which were directly interacted with FGFR3, and other 31 interactional DEGs neighboring of these 7 nodes were selected to construct the FGFR3-centered PPI network (Fig. 1). The top 5 genes with the greatest node degree ≥4 are listed in Table 2, which were KIAA1377, POLA2 (polymerase (DNA directed), alpha 2), FGFR3, EPHA4 (ephrin type A receptor 4), and KRT8 (keratin 8).

The FGFR3-centered protein–protein interaction network. The pink nodes represent upregulated genes, and yellow nodes represent downregulated genes
The regulatory network related to FGFR3
The potential regulatory miRNAs and TFs targeting to the genes included in the FGFR3-centered PPI network were screened out, and a regulatory network was constructed. There were 17 miRNAs/targets, and 8 TF/target regulatory relationships were predicted (Fig. 2).

FGFR3-related regulatory network. The pink and yellow nodes represent upregulated and downregulated genes, respectively. The square and triangle nodes represent miRNAs and transcription factors, respectively. The arrows represent regulatory relationships between the differentially expressed genes and miRNAs while the dotted line represents regulatory relationships between differentially expressed genes and transcription factors
Prediction of potential prognostic makers for bladder cancer
Cox regression model was applied to identify the potential genes in the FGFR3-centered PPI network that were associated with bladder cancer prognosis. HMOX2 (heme oxygenase-2), CSTF2 (cleavage stimulation factor, 3′ pre-RNA, subunit 2, 64 kDa), and POLA1 (polymerase (DNA directed), alpha 1) were the three obviously significant related genes, and a marginal significant correlation was found between EFNB2 (ephrin-B2) and prognosis of bladder cancer (Table 3). However, a multivariate analysis about these 4 genes showed that only CSTF2 and EFNB2 were significant related to the recurrence of bladder cancer (p value = 0.005 and 0.030) (Table 4). Moreover, the survival curves displayed in Fig. 3 also obviously presented that patients with high expression level of CSTF2 and POLA1 showed relative favorable prognosis. However, HMOX2 and EFNB2 may be the risk factors of bladder cancer. All of these results might indicate that CSTF2 and EFNB2 were two important protective factors in the prognosis of bladder cancer.

Kaplan–Meier survival curves of patients with bladder cancer according to the expression of HMOX2, CSTF2, POLA1, and EFNB2
Discussion
Bladder cancer is a complex process, implicated with multiple genetic abnormalities. A body of studies evidenced that FGFR3 acts as an important oncogenic driver in bladder cancer. Here, we explored the possible mechanism how FGFR3 signaling mediates bladder cancer development and progression by bioinformatics methods. We found that the expression levels of 2855 genes were altered upon the depletion of FGFR3 in bladder cancer cell line. These downregulated genes were mainly involved in cell division and carboxylic acid metabolic process while the upregulated were related to blood vessel morphogenesis and response to type I interferon. Increased cell division is a well-known cause and essential for development and progression of human cancer [17]. Adequate blood supply, such as angiogenesis, is also required for the growth and metastasis of tumor [18]. Fibroblast growth factors (FGFs) and FGF receptors can act as angiogenesis inducers and have contribution to tumor vascularization [19]. Moreover, it has shown that IFN-a, as one type of I interferon inhibits the expression of basic fibroblast growth factor (bFGF), reduces angiogenesis and thus inhibits tumor growth in the bladder wall [20]. Therefore, FGFR3 may promote the development of bladder cancer by regulating cell division, response to type I interferon signaling, and blood vessel morphogenesis.
To gain insight into the oncogenic mechanism of FGFR3 in bladder cancer, we constructed FGFR3-related molecular networks. First, a FGFR3-centered PPI network was generated and KIAA1377, POLA2, FGFR3, EPHA4, and KRT8 were the hub genes. KIAA1377, also known as Cep126, regulates microtubule organization at the centrosome by modulating the transport of pericentriolar satellites [21]. Centrosome takes vital roles in several fundamental cellular functions, including cell division. POLA2, encoding the B-subunit of DNA polymerase α, has been reported to be involved in cell proliferation by mediating DNA replication, recombination, and repair [22]. EPHA4, a member of the EPH receptors, is closely related to tumor progression [23]. But based on the previous studies, the investigations of relationships among these hub genes were still absent. The researches of these genes were only concentrated on bioinformatics, except that EPHA4 has been confirmed to interact with FGFR3 in vitro [24]. Thus, it was very essential to explore the correlations among FGFR3, KIAA1377, POLA2, and EPHA4 in bladder cancer, as well as other cancers or diseases. Next, we predicted several regulators and constructed a regulatory network based on the PPI network. Total 17 miRNAs and 6 TFs were involved. Among these TFs, TP53 is a well-known tumor suppressor gene while MITF (microphthalmia-associated transcription factor) is an oncogene [25]. ATF2 (activating transcription factor 2) and CREB1 (CAMP responsive element binding protein 1) are both CREB-related proteins and share the same cAMP responsive element sequence. They are also involved in cancer progression [26]. From the regulatory, we found that 2 miRNAs, hsa-miR-100 and hsa-miR-99a, directly interacted with FGFR3. Song et al. have found the aberrant expression of these 2 miRNAs in bladder cancer by microarray analysis of 25 cases of bladder urothelial carcinomas and adjacent normal bladder tissue [27]. Moreover, in fact, the negative regulation of FGFR3 by mir-100 has been demonstrated in clear cell ovarian cancer cells [28]. Thus, the above molecules may be responsible for the carcinogenesis of FGFR3 in bladder cancer.
In addition to the molecule mechanism how FGFR3 contributes to the bladder cancer progression, we tried to explore the clinical value of FGFR3. Thus, we screened the bladder cancer recurrence-related genes from the FGFR3-centered PPI network. The results indicated that patients with high expression level of CSTF2 and POLA1 showed relative favorable prognosis. However, HMOX2 and EFNB2 may be the risk factors of bladder cancer. CSTF2 encodes a nuclear protein which contains a ribonucleoprotein-type binding domain in the N-terminal region. There is a significant correlation between CSTF2 and poor prognosis for lung cancer patients [29]. POLA1, encoding the catalytic subunit of DNA polymerase, is responsible for the initiation of DNA replication. Inducible HOMX1 and constitutive HOMX2 are 2 structurally related isozymes that compose heme oxygenase. The expression level of HOMX1 is associated with cervical lymph node metastasis of tongue squamous cell carcinomas [30]. EFNB2 has been evidenced to be the prognostic maker for esophageal squamous cell carcinoma [31]. In current study, we revealed that there were correlations between these 4 genes and the prognosis for patients with bladder cancer. The results further confirmed that the genes in the PPI network may be responsible for the carcinogenesis of FGFR3 in bladder cancer.
Conclusions
In conclusion, we analyzed gene expression profiles of the bladder cancer cell line RT112 with or without depletion of FGFR3 and conducted further analyses using a computational bioinformatics approach based on the publicly available data. This study provided some insights on the mechanism underlying the carcinogenesis of FGFR3 in bladder cancer. However, further experimental studies are needed.
Abbreviations
- bFGF:
-
Basic fibroblast growth factor
- DEGs:
-
Differentially expressed genes
- FGFR3:
-
Fibroblast growth factor receptor 3
- FGFs:
-
Fibroblast growth factors
- GO:
-
Gene Ontology
- HPRD:
-
Human Protein Reference Database
- PPI:
-
Protein–protein interaction
- RMA:
-
Robust multi-array average
- RTK:
-
Family of tyrosine kinase
References
HOURS AC. Care of the patient with bladder cancer. Nurs Made Incred Easy. 2014;12:40-48.
Volanis D, Kadiyska T, Galanis A, Delakas D, Logotheti S, Zoumpourlis V. Environmental factors and genetic susceptibility promote urinary bladder cancer. Toxicol Lett. 2010;193:131–7.
Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10:116–29.
Dailey L, Ambrosetti D, Mansukhani A, Basilico C. Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor Rev. 2005;16:233–47.
Tomlinson D, Baldo O, Harnden P, Knowles M. FGFR3 protein expression and its relationship to mutation status and prognostic variables in bladder cancer. J Pathol. 2007;213:91–8.
Tomlinson DC, Hurst CD, Knowles MA. Knockdown by shRNA identifies S249C mutant FGFR3 as a potential therapeutic target in bladder cancer. Oncogene. 2007;26:5889–99.
Du X, Wang QR, Chan E, Merchant M, Liu J, French D, et al. FGFR3 stimulates stearoyl CoA desaturase 1 activity to promote bladder tumor growth. Cancer Res. 2012;72:5843.
Riester M, Taylor JM, Feifer A, Koppie T, Rosenberg JE, Downey RJ, et al. Combination of a novel gene expression signature with a clinical nomogram improves the prediction of survival in high-risk bladder cancer. Clin Cancer Res. 2012;18:1323–33.
Irizarry RA, Hobbs B, Collin F, Beazer‐Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–64.
Hong F, Breitling R, McEntee CW, Wittner BS, Nemhauser JL, Chory J. RankProd: a bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics. 2006;22:2825–7.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–9.
Wang J, Zhou X, Zhu J, Gu Y, Zhao W, Zou J, et al. GO-function: deriving biologically relevant functions from statistically significant functions. Brief Bioinform. 2012;13:216–27.
Prasad TK, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, et al. Human protein reference database—2009 update. Nucleic Acids Res. 2009;37:D767–72.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504.
Hsu S-D, Lin F-M, Wu W-Y, Liang C, Huang W-C, Chan W-L, et al. miRTarBase: a database curates experimentally validated microRNA–target interactions. Nucleic Acids Res. 2010;39:163-169.
Wingender E, Chen X, Hehl R, Karas H, Liebich I, Matys V, et al. TRANSFAC: an integrated system for gene expression regulation. Nucleic Acids Res. 2000;28:316–9.
Preston-Martin S, Pike MC, Ross RK, Jones PA, Henderson BE. Increased cell division as a cause of human cancer. Cancer Res. 1990;50:7415–21.
Folkman J. Angiogenesis: initiation and modulation. In: Symp. Fundam. Cancer Res; 1983. p. 201–8.
Presta M, Dell’Era P, Mitola S, Moroni E, Ronca R, Rusnati M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005;16:159–78.
Dinney CP, Bielenberg DR, Perrotte P, Reich R, Eve BY, Bucana CD, et al. Inhibition of basic fibroblast growth factor expression, angiogenesis, and growth of human bladder carcinoma in mice by systemic interferon-α administration. Cancer Res. 1998;58:808–14.
Bonavita R, Walas D, Townley AK, Luini A, Stephens DJ, Colanzi A. Cep126 is required for pericentriolar satellite localisation to the centrosome and for primary cilium formation. Biol Cell. 2014;106:254-267.
Flotho C, Coustan-Smith E, Pei D, Cheng C, Song G, Pui C-H, et al. A set of genes that regulate cell proliferation predicts treatment outcome in childhood acute lymphoblastic leukemia. Blood. 2007;110:1271–7.
Liu C, Huang H, Wang C, Kong Y, Zhang H. Involvement of ephrin receptor A4 in pancreatic cancer cell motility and invasion. Oncol Lett. 2014;7:2165–9.
Fujita K, Schlessinger J, Sakaguchi K. Trans-activation of EphA4 and FGF receptors mediated by direct interactions between their cytoplasmic domains. Proc Natl Acad Sci U S A. 2005;102:18866–71.
Davis IJ, Kim JJ, Ozsolak F, Widlund HR, Rozenblatt-Rosen O, Granter SR, et al. Oncogenic MITF dysregulation in clear cell sarcoma: defining the MiT family of human cancers. Cancer Cell. 2006;9:473–84.
Deng X, Liu H, Huang J, Cheng L, Keller ET, Parsons SJ, et al. Ionizing radiation induces prostate cancer neuroendocrine differentiation through interplay of CREB and ATF2: implications for disease progression. Cancer Res. 2008;68:9663–70.
Song T, Xia W, Shao N, Zhang X, Wang C, Wu Y, et al. Differential miRNA expression profiles in bladder urothelial carcinomas. Asian Pac J Cancer Prev. 2010;11:905–11.
Nagaraja AK, Creighton CJ, Yu Z, Zhu H, Gunaratne PH, Reid JG, et al. A link between mir-100 and FRAP1/mTOR in clear cell ovarian cancer. Mol Endocrinol. 2010;24:447–63.
Aragaki M, Takahashi K, Akiyama H, Tsuchiya E, Kondo S, Nakamura Y, et al. Characterization of a cleavage stimulation factor, 3′ pre-RNA, subunit 2, 64 kDa (CSTF2) as a therapeutic target for lung cancer. Clin Cancer Res. 2011;17:5889–900.
Yanagawa T, Omura K, Harada H, Nakaso K, Iwasa S, Koyama Y, et al. Heme oxygenase-1 expression predicts cervical lymph node metastasis of tongue squamous cell carcinomas. Oral Oncol. 2004;40:21–7.
Tachibana M, Tonomoto Y, Hyakudomi R, Hyakudomi M, Hattori S, Ueda S, et al. Expression and prognostic significance of EFNB2 and EphB4 genes in patients with oesophageal squamous cell carcinoma. Dig Liver Dis. 2007;39:725–32.
Acknowledgements
Not applicable.
Funding
None.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors’ contributions
WC and EGM analyzed and interpreted the data. LZ and TY performed the acquisition of data. CYZ was a major contributor in writing and revising the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional information
Wei Cao and Enguang Ma are co-first author.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Cao, W., Ma, E., Zhou, L. et al. Exploring the FGFR3-related oncogenic mechanism in bladder cancer using bioinformatics strategy. World J Surg Onc 15, 66 (2017). https://doi.org/10.1186/s12957-017-1125-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12957-017-1125-4