
Abstract
Introducing oxygen-vacancy into the surface of the non-enzymatic sensor is supposed to be an effective way to improve inherently low catalytic activity and specificity of non-enzymatic sensors. In this work, CuO/C was synthesized at different temperatures using metal-organic frameworks as sacrificial templates to receive additional content of oxygen-vacancy. The product with the highest oxygen vacancy was found at 400 °C (named CuO/C-400 °C), which increased catalytically active sites and enhanced the charge-transfer efficiency. The sensing performance was afterward explored by amperometry under an optimal applied potential at 0.5 V (vs. SCE), presenting a broad detection range from 5.0 µM to 25.325 mM (R2 = 0.9998) with a sensitivity of 244.71 µA mM− 1 cm− 2, and a detection limit of 1 µM. Furthermore, the reliability and selectivity of CuO/C-400 °C sensors were extensively explored in the presence of artificial serum/saliva samples with gradient glucose concentrations. The human blood samples were also detected with high recoveries compared with the clinical Hexokinase method. Hence, the prepared CuO/C-400 °C sensor with a broad detection range and high selectivity can be applied for the diabetes diagnosis ex vivo without further dilution for real-time analysis in practical applications.
Similar content being viewed by others


Introduction
Diabetes is a chronic disease that occurs when insulin is neither well-produced nor effectively utilized. Over 4.2 million people died from diabetes in 2019 [1], and this number is projected to constantly increase to about 700 million by 2040 s, according to the estimation of the International Diabetes Federation (IDF) [2]. Precise control of blood glucose levels in daily life can not only reduce the symptom and increase the survival rates of diabetes, but also prevent or delay long-term, serious health problems, such as heart disease, vision loss, and kidney disease, so the development of glucose monitoring devices in real-time is necessary [3]. Although commercial glucose oxidase-based glucose meters have been widely used, the sensing performance is easily interfered with by the external storage and transport conditions, which hinders their applications in glucose determination [4]. Non-enzymatic glucose sensors (such as metal oxides), in that case, have attracted much attention for their high chemical stability and easy operation. Nevertheless, compared with glucose oxidase enzymes, the catalytic activity and specificity of non-enzymatic materials are usually lower, resulting in a poorer sensing performance [5,6,7].
CuO, which is expected to be an attractive non-enzymatic sensor for its p-type semiconductor with a narrow bandgap of 1.2 eV, has been widely explored [8, 9], especially for non-enzymatic glucose sensors [10,11,12]. Still, the detection range of most as-prepared sensors is 0–5 mM which is narrower than the glucose in human blood (4–7 mM for healthy people and ≥ 9 mM for diabetic patients) due to the weak conductivity of CuO [13, 14]. While introducing oxygen vacancies by adjusting defect structures and electronic states, such as heating the material in a reductive atmosphere, and doping nonmetals (halogen, etc.) to replace lattice oxygen [15, 16], has been suggested to improve the conductivity of CuO [17, 18]. However, these methods often require secondary heating, and the reductive atmosphere is prone to make crystal agglomerate and collapse. Thus it is urgent to develop a technique to increase the oxygen-vacancy content in CuO without secondary heating.
Metal-organic frameworks (MOFs) are composed of metal ions and organic links through coordination bonds with regular pores. These have been widely applied in biomedicine, such as sensors, bioimaging materials, and drug delivery agents [19,20,21,22]. Recently, metal oxide/C-derived MOFs have attracted much attention in the electrochemical field for designing appropriate structures with plenty of active species [23]. For example, thermal treatment can reduce the reaction time with no post-treatment, whilst the product keeps the morphology of the initial MOF with a large surface area [24, 25]. More importantly, such treatment allows more atomically active sites to be exposed, resulting in better MOF performance. Recently, many groups found that oxygen-vacancy in MOF-derived materials can enhance electrochemical performance [26]. Hence, exploring MOF-derived CuO with more atoms active-sites and oxygen-vacancy will be a promising way to achieve a more comprehensive linear range for glucose sensing. However, the change of oxygen-vacancy under different processing temperatures is seldom reported for the MOF-derived sensors.
Given the above-mentioned advantages and consideration, in this study, the Cu-MOF was synthesized through the coordination of copper ions and homophenolic acid, followed by the calcination in the furnace at different temperatures (350 oC, 400 oC, and 450 oC) to receive the CuO/C core-shell nanoparticles with oxygen vacancy. The thermal treatment of Cu-MOF under different temperature were further found to create active sites and increase the oxygen-vacancy for the electrocatalytic oxidation of glucose. Compared with CuO/C-350oC and CuO/C-450oC, the developed CuO/C-400 °C has the most oxygen-vacancy and the highest response toward glucose oxidation in primary media. The wide detection range of glucose was explored using CuO/C-400 °C modified glassy carbon electrode (GCE) by amperometry under an optimal applied potential at 0.5 V due to the oxygen-vacancy and adsorbed hydroxyl ions. The sensing performance was then verified in artificial serum/saliva and human blood sample in real time analysis with remarkable reproducibility.
Experimental
The synthesis of CuO/C at different temperatures
Cu-MOF was first synthesized at room temperature (RT) by liquid phase method. In detail, 3 mmol Cu(NO3)2·6H2O was dissolved in 25 mL deionized water, while 2 mmol 1,3,5-benzenetricarboxylic acid and 6 mmol triethylamine were mixed in 25 mL ethanol. Then two solution was mixed under stirring and kept for 24 h at RT. The final blue powder was received after the purification with ethanol and dry at 60 oC for 24 h.
For the preparation of CuO/C nanoparticles, the synthesized Cu-MOF (100 mg) was afterward placed in a porcelain boat and calcined in a muffle furnace with the increased temperature at a rate of 1 oC/min. After reaching the specified temperature, the temperature was kept constant for 2 h and then cooled down to RT. The final samples were black and named CuO/C-XoC (X = 350, 400, 450), depending on the calcination temperature.
The preparation of the working electrode
0.3 μm and 0.05 μm alumina slurries were used sequentially to polish the glassy carbon electrodes (GCE; diameter, 3 mm), followed by the immersion in ethanol and ultrapure water in sequence for ultrasonic cleaning. For the preparation of the CuO/C-X oC (X = 350, 400, 450) based working electrode, 2 mg of prepared CuO/C-X oC powder was dispersed in 1 ml of ultrapure water under ultrasound for 30 min. Then 5 µL of the CuO/C-X oC dispersion was directly dropped onto the surface of the pre-treated GCE and dried under 30 oC. The GCE /CuO/C-X oC electrode was obtained by repeating the drip - drying process for four times.
Real samples detection
To further study the practical application of the sensor, 5 µL of the CuO/C-400 °C dispersant was dropwise added to the working electrode area of the electrode (diameter: 3 mm) through the drip-drying process four times. The simulated serum sample was prepared using PBS containing 10% FBS, while simulated saliva was received from Chuangfeng Technology Company (Dongguan, China). The human serum samples (2 diabetics and 2 healthy) were kindly donated by Anzhen Hospital (Beijing, China) and tested using by Hexokinase method (Roche COBAS INTEGRA 800 automatic biochemical analyzer). The performance of the synthesized biosensor in complex biological samples containing different concentrations of glucose was investigated by amperometry under an applied voltage of 0.5 V.
Results and discussion
Synthesis and characterization of Cu-MOF
After the successful preparation of the Cu-MOF, the morphology and properties were examined by commonly used material characterization methods, including transmission electron microscope (TEM), EDS mapping, X-ray diffractometer (XRD), X-ray photoelectron spectroscopy (XPS), etc. TEM results showed that the prepared Cu-MOF was nanoparticles with the size of 50–80 nm, and energy dispersive spectrometer (EDS)-mapping represented that the synthesized nanoparticles were mainly composed of Cu, C, and O with uniform dispersion (Additional file 1: Fig. S1). The powder XRD patterns of the as-prepared Cu-MOF (Additional file 1: Fig. S2) further identified a good crystalline state that was in line with the previous literature [27], indicating the successful synthesis of Cu-MOF. Meanwhile, the high purity and good crystal quality of the Cu-MOF can be further confirmed by the observation of sharp and intense diffraction peaks. To further confirm the element and the chemical state, the prepared Cu-MOF was tested by XPS (Additional file 1: Figs. S3 and S4), revealing the presence of Cu, C, and O without other elemental contaminants. Furthermore, the high-resolution spectra of XPS can also confirmed the successful preparation of Cu-MOF containing Copper ion (II) and 1,3,5-benzenetricarboxylic acid.
In addition, the heat treatment mechanism of Cu-MOF under air was simulated by means of thermogravimetric-differential thermal analysis (TG-DTA) between 25 oC and 600 oC (Additional file 1: Fig. S5) with two obvious weight loss steps. The first mass loss, with 30.47 wt% from 25 to 296 oC, relating to the removal of water and other molecules for negative of DTA. While the weight loss of 42.36 wt% from 296 oC to 350 oC indicated framework of Cu-MOF began to collapse at this stage. After 350 oC, there was no noticeable weight loss could be observed, representing that the Cu-MOF was decomposed entirely and totally converted to CuO directly. Hence the 350 oC, 400 oC, and 450 oC were selected to calcine Cu-MOF in this study to explore the changes of oxygen-vacancy content caused by thermal treatment temperature.
Synthesis and characterization of CuO/C at different temperature
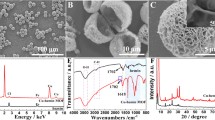
The samples obtained after thermal treatment under air at different temperatures (350 oC, 400 oC, and 450 oC) were named as CuO/C-350 oC, CuO/C-400 oC and CuO/C-450 oC, respectively (Fig. 1a). In addition, a commercial CuO with a size of 40 nm was bought for comparison. The prepared samples at different temperatures and commercial CuO were first observed by scanning electron microscope (SEM). As shown in the SEM images (Fig. 1b and S6), the average size of prepared samples were calculated to be 50.78, 59.28, 40.88, and 44.95 nm for CuO/C-350 oC, CuO/C-400 oC, CuO/C-450 oC, and commercial CuO, respectively. TEM, HR-TEM together with the mapping were used to further confirm the detail morphology of the prepared samples (Fig. 1c–e). Similar with the results from SEM, the size of CuO/C-400 oC in TEM image was about 60 nm. Moreover, an amorphous carbon layer was observed to wrap on the surface of copper oxide, forming a core-shell structure of CuO/C by HR-TEM in Fig. 1d. Further well-defined lattice fringes with the d-spacing of the lattice fringes were measured to be 0.25 nm attributing to the (111) reflections of monoclinic CuO. Meanwhile, the EDS (Additional file 1: Fig. S7) and EDS-mapping analysis (Fig. 1e) were confirmed that the architecture of CuO/C-400oC contained Cu and O elements distributing homogenously in the entire architecture of CuO/C-400 oC.

a Scheme illustration of the synthesis of CuO/C; b SEM images of CuO/C-350oC, CuO/C-400 °C and CuO/C-450 °C, the inset is the particle size statistics; c TEM, d HR-TEM and e Cu, O, C element mapping of CuO/C-400 °C; f XRD patterns of CuO/C-350 °C, CuO/C-400 °C, CuO/C-450 °C and the standard PDF of CuO; g XRD patterns for the 2Ɵ from 30˚ to 40˚; h EPR spectra of CuO/C-350 °C, CuO/C-400 °C and CuO/C-450 °C
The crystal structures of the prepared samples and commercial CuO were also characterized by XRD (Fig. 1f and Additional file 1: Fig. S8). All the diffraction peaks can be indexed to the monoclinic-phase of CuO (JCPDS 48-1548). Specifically, the peaks with 2Ɵ of 32.406˚, 35.489˚, 38.694˚, 48.841˚, 53.403˚, 58.194˚, 61.498˚, 66.101˚, 67.909˚, 72.301˚, and 74.998˚ matched the crystal surface of monoclinic CuO without any signal from copper acetate or other precursor compounds, indicating the production of high purity single-phase CuO. Furthermore, the results of different samples with 2Ɵ from 30˚ to 40˚ were identified in a narrow range in Fig. 1g. As the under bound orbital of atom located on the nonbonding orbital of the transition metal due to the extra electrons generated with the oxygen-vacancy, causing the peaks of the crystal planes shift to lower angles. The tested angle of (111) was 35.48˚, 35.44˚, 35.493˚, and 35.579˚ for CuO/C-350 oC, CuO/C-400 °C, CuO/C-450 oC, and commercial CuO, respectively, demonstrating more oxygen-vacancies of CuO/C-400 oC. In addition, the electron paramagnetic resonance (EPR) spectrum (Fig. 1h), which was proved to be an effective tool for manifesting the presence of atomic vacancy [28], was used to examine the formation of oxygen vacancies in the samples. All the samples displayed a symmetrical EPR signal at g = 2.005. Compared with the calcination at other temperatures, the signal strength of CuO/C-400 oC was the strongest, indicating the highest content of oxygen vacancies. Herein, the CuO/C-400 °C might have the most oxygen-vacancy among all samples, indicating a potentially good electrochemical activity.
To further investigate the surface element chemical states and oxygen-vacancy, the prepared CuO samples and commercial CuO were subjected to XPS testing. The peaks of C, O, and Cu could be observed in all prepared samples (Fig. 2a–c), whilst only Cu and O existed in commercial CuO (Fig. 2d). Moreover, the binding energy of Cu for all samples were similar, which indicated the Cu oxidation state with no noticeable difference. The spectra of C1 s for the prepared samples showed there were a carbon layer with sp3 bonding on the surface which was consistent with the result of TEM images. For the high-resolution spectra of O1 s, there were two O1 s surface peaks that could be fitted by two bands. The band with lower binding energy was ascribed to the lattice oxygen (Cu-O) of the CuO crystal lattice, corresponding to 529.7 eV, 529.6 eV, 529.9 eV, and 529.9 eV of CuO/C-350 °C, CuO/C-400 °C, CuO/C-450 °C, and commercial CuO, respectively. A shoulder band with higher binding energy was ascribed to the adsorbed oxygen or oxygen in hydroxyl-like groups on the surface of CuO (denoted as oxygen-vacancy). The band of oxygen-vacancy was related to the bands at 531.4 eV, 531.7 eV, 531.6 eV, and 531.8 eV of CuO/C-350oC, CuO/C-400oC, CuO/C-450 °C, and commercial CuO, respectively. Peak areas were used to calculate the relative content of different elemental states of O on the surface. The ratio of oxygen-vacancy to Cu-O was 1.15, 1.62, 1.31, and 1.05 for CuO/C-350 °C, CuO/C-400 °C, CuO/C-450 °C, and commercial CuO, respectively, indicating that CuO/C-400 °C had the highest oxygen-vacancy. The possible mechanism was that the increase of temperature might cause a carbon-mediated local reduction reaction at the surface of CuO/C, bringing an improvement in oxygen vacancies without disrupting the lattice. However, excessive temperature would lead to the structure collapse to reduce the oxygen-vacancy [29]. The experiment showed that with the increase of temperature (from 350 to 400 °C), oxygen dissociation is caused, leading to the generation of more oxygen vacancy. As the temperature further increased (400 to 450 °C), the structure collapsed, finally deducing the oxygen vacancy of CuO/C. The TG-DTA results indicated that the temperature increase did not lead to further loss of the mass or the collapse of the structure. However, the XPS results demonstrated that the change of temperature would lead to the oxygen-vacancy content change, together with the higher density of the surface defects, the surface adsorption sites, and the catalytic activity. Hence, the CuO/C-400 °C was expected to show good electrocatalytic ability toward glucose oxidation.

XPS spectra of a CuO/C-350oC, b CuO/C-400 °C, and c CuO/C-450 °C: survey scan, C1 s, O1 s and Cu2p; d Survey scan, O1 s and Cu2p for commercial CuO (40 nm); e The ratio of oxygen-vacancy and Cu-O for CuO/C-350 °C, CuO/C-400 °C, CuO/C-450 °C and commercial CuO. (MEAN ± SD, n = 3, *p < 0.05)
Electrocatalytic performance of the obtained electrodes for glucose detection
Electrochemical Impedance Spectroscopy (EIS) was used to analyze the mass transfer characteristics and the charge of components in sensors. Figure 3a showed the Nyquist plot obtained for the GCE, GCE/CuO, GCE/CuO/C-350 °C, GCE/CuO/C-400 °C, GCE/CuO/C-450 °C electrodes in 0.1 M KCl containing 5 mM Fe(CN)63− /4− with frequency from 0.1 to 105 Hz at 0.2 V. The CuO electrode showed the smallest resistance value (Rct) was about 1544 Ω, while the Rct of GCE/CuO/C-350oC, GCE/CuO/C-400 °C, and GCE/CuO/C-450oC were 2003 Ω, 7244 Ω, and 2515 Ω, respectively, owning to the increase of surface oxygen vacancies increase can destroy the crystal structure in the nanoparticles, resulting in an increased conductivity of the sensors.

a Nyquist plots of the CuO/C-350oC, CuO/C-400 °C, CuO/C-450 °C and commercial CuO in 0.1 M KCl electrolyte containing 5 mM Fe(CN)63− /4− and an applied AC frequency range of 0.1–105 Hz at 0.2 V (vs. Ag/AgCl) with an amplitude of 5 mV; b Cyclic voltammograms of the CuO/C-400 °C electrodes in 0.1 M NaOH with/without 0.2 mM glucose at a scan rate of 100 mV·s− 1; c Amperometric i-t response of the CuO/C-350 °C, CuO/C-400 °C and CuO/C-450 °C electrodes in 0.1 M NaOH at 0.5 V (vs. SCE) with stirring, insert is the response current density of 1.0 mM glucose on the CuO/C-350 °C, CuO/C-400 °C and CuO/C-450 °C electrodes derived from Fig. 3c (MEAN ± SD, n = 3, *p < 0.05); d CV curves of CuO/C-400 °C in 0.5 mM K3Fe(CN)6/0.1 M KCl electrolyte at different scan rate and (e), f the corresponding fitting curves
The CV curves were used to evaluate the performance of the prepared GCE/CuO/C-XoC (X = 350, 400, 450) sensors for catalyzing glucose. All electrodes were tested in 0.1 M NaOH solution with or without 0.2 mM glucose. As shown in Fig. 3b, the CV curves of the GCE/CuO/C-400 °C electrode showed a distinct oxidation peak in glucose, while no oxidation peak was observed without glucose. The catalytic oxidation ability of the other three materials was also tested under the same conditions (Additional file 1: Fig. S9). The largest catalytic oxidation current of the GCE/CuO/C-400 °C electrode indicated that the CuO/C-400 °C had the strongest catalytic oxidation capacity for glucose in comparison, which was related to the highest oxygen vacancy of the CuO/C-400 °C and can not only increase the charge transfer efficiency but also enhance the interaction between oxygen-containing species and the metal oxide surface effectively. The 0.5 V was selected as the applied potential for chronoamperometry detection as it had the highest response with the gradual addition of glucose under different potentials and enough driving force for the glucose oxidation reaction. The amperometric curves of different electrodes were carried out under 0.5 V with an addition of 1.0 mM glucose. The current responses of GCE/CuO/C-400 °C, GCE/CuO/C-350 °C, and GCE/CuO/C-450 °C to glucose were 17, 10, and 7 µA, respectively (Fig. 3c). It proved that the catalytic ability of the CuO/C-400oC material was significantly higher than that of the other two materials. Figure 3d showed the CVs at different scan rates for GCE/CuO/C-400 °C in electrolytes contained 0.2 mM glucose. In the range of 30 mV/s to 200 mV/s, the current increased with the aggrandizement of the scan rate. As shown in Fig. 3e, f, and Additional file 1: Fig. S10, the oxidation current had a linear relationship with the scan rate and the square root of the scan rate, indicating the co-existence of surface confinement and diffusion control in the CuO/C-X °C [30,31,32]. Moreover, as the slopes of the regression equation of GCE/CuO/C-350oC and GCE/CuO/C-450 oC were significantly lower than that of GCE/CuO/C-450 oC, which was related to the catalytic performance of the material.
Figure 4a, b showed the i-t curve of GCE/CuO/C-400 °C along with the corresponding linear plots of the calibration curve. According to the results, the electrode exhibited a rapid response with the addition of glucose, indicating CuO/C-400 °C has high catalytic activity. The sensitivity and linear range of these modified electrodes could be obtained from calibration curves. Based on the catalytic ability of CuO/C, the electrode showed a prominent characteristic that the current response value gradually increases with the increase of glucose concentration. In addition, the concentration of oxygen vacancies on the surface of the CuO/C was controlled by changing the calcination temperature during the calcination process, and the catalytic ability of CuO/C-X °C (X = 350, 400, 450) to glucose was different, which in turn leaded to different sensitivity among GCE/CuO/C-X oC (X = 350, 400, 450) electrodes. Among them, the GCE/CuO/C-400 °C electrode exhibited the highest sensitivity at about 244.71 µA mM− 1 cm− 2, while the sensitivities of GCE/CuO/C-350 oC and GCE/CuO/C-450 °C electrodes were 140.69 and 79.06 µA mM− 1 cm− 2, respectively. Furthermore, the linear correlation between the response current (µA) and glucose concentration (mM) of the GCE/CuO/C-400 °C electrode was y = 17.13x + 0.7648 (R2 = 0.9998) with the linear range from 5.0 µM to 25.325 mM, and the limit of detection (LOD) of 1.0 µM (S/N = 3). Compared with other reported CuO-based non-enzymatic sensor in Table 1, our prepared CuO/C-400 oC with abundant oxygen-vacancy through a simple preparation process without secondary heating. The MOF derived material offers larger surface area for more activated species and oxygen-vacancy leading to the enhanced charge-transfer efficiency. Furthermore, the thin carbon layer on the surface of the copper oxide during the formation process may facilitate the electron transfer, reduce the physical changes on the surface of CuO during the catalytic process, so as to better maintain the detection activity and repeatability of biosensor. Herein, the GCE/CuO/C-400 °C electrode showed remarkable electrocatalytic activity toward glucose oxidation with a wider detection range than other reported CuO-based nonenzymatic glucose probes.

a Amperometric responses of CuO/C-400 °C in 0.1 M NaOH upon consecutive addition of glucose at 0.5 V (vs. Ag/AgCl); b the corresponding calibration curves of the CuO/C-400 °C for the glucose detection; c, d influence of interfering substances (0.1 mM NaCl, 0.1 mM KCl, 0.1 mM AA, 0.1 mM maltose and 0.1 mM GSH) on the amperometric response to glucose by CuO/C-400 °C sensor (MEAN ± SD, n = 3, **p < 0.01)
Selectivity and stability of the resulting electrodes
It is considered that multiple possible interferents (NaCl, KCl, AA, fructose, and GSH) may coexist with glucose in a real serum environment. Figure 4c showed i-t curves of the sensor with successfully adding 1.0 mM glucose and 0.1 mM interferences in the electrolyte. The response current changed significantly after the addition of glucose. In contrast, the changed response currents were almost negligible when interfering substances were added to the electrolyte. Figure 4d showed the comparison of perturbation responsiveness and glucose responsiveness, and it can be clearly seen that the effect of perturbation was not obvious. Thus, the resulting electrodes demonstrated acceptable resistance to interference measurements. For the stability test, the sensors were stored in RT and tested every 2 days. As shown in Fig. 5f, the sensors still maintained good detection performance within 28 days, which was about 95% of the initial value, indicating that the obtained sensor can be harnessed to sensitively detect the glucose concentration after being placed for a long time.

Amperometric responses of CuO/C-400 °C upon the consecutive addition of glucose in a artificial serum and c artificial saliva at 0.5 V (vs. Ag/AgCl); the corresponding calibration curves of the CuO/C-400 °C for the glucose detection in b artificial serum and d artificial saliva; e Amperometric i-t response of the CuO/C-400 °C electrodes at 0.5 V (vs. SCE) for different samples; f The relative current strength of the electrode within 28 days
Real sample detection
As the CuO/C-400 °C nanoparticles containing oxygen vacancies with a remarkable ability to catalyze glucose oxidation and a good anti-interference ability. The prepared biosensor has also been explored with artificial serum/saliva (Fig. 5a–d) and real blood to establish its applicability in real-time detection. Specifically, the detection performance of the sensor in 0.1 M NaOH solution containing different biological samples was investigated by continuously adding an artificial serum and saliva containing 1 M and 0.1 M glucose, respectively, under an applied voltage of 0.5 V. The current response was found to increase with the increase of added artificial samples with perfect regression equations of I(µA) = 16.3 C(mM) + 3.670 µA (R2 = 0.9996) and I(µA) = 16.23 C(mM) + 0.6128 µA (R2 = 1.0000) from 1 to 16 mM and 0.1–1.5 mM, respectively.
In addition, the detection performance of the sensor was also verified by dropping different samples, including artificial and actual samples, directly on the surface without dilution (Fig. 5e). As shown in Table 2, the concentrations of the pieces were calculated referring to different equations mentioned above with high recovery from 97% to 103% in Table 2. In addition, the comparison of the glucose levels estimated with the fabricated biosensor and concentration recorded from the clinically available Hexokinase method was shown in Table 3. Herein, the designed CuO/C-400 °C sensor can be applied in different conditions with remarkable sensing performance. Notably, the screen-printed-based sensors can also be converted into portable detection platforms with miniature electrochemical workstations.
Conclusion
In this work, the CuO/C with the most abundant content of oxygen-vacancy was synthesized through pyrolyzing MOF as a sacrificial template at 400 °C with fast electron transfer, low overpotential, and high response current, which contributed to its excellent electrocatalytic activity toward glucose oxidation. The electrooxidation of glucose on the CuO/C-400 °C sensor was correlated with the pairing of vacancies (h+) and adsorbed OH−, resulting in high accumulated energy and oxidation of the glucose molecules. The sensing performance of CuO/C-400 °C was then applied with a wide linear range from 5.0 µM to 25.325 mM (R2 = 0.9998) and a good sensitivity of 244.71 µA mM− 1 cm− 2. In order to verify the accurate sample analysis, the CuO/C-400 °C sensor was also performed in simulated saliva/serum and human blood samples, and the results indicated that our prepared biosensor could effectively detect the glucose with high selectivity and recovery without further dilution, which opens up possibilities for ex vivo diagnostic applications for clinical and domestic use.
References
Saeedi P, Salpea P, Karuranga S, Petersohn I, Malanda B, Gregg EW, Unwin N, Wild SH, Williams R. Mortality attributable to diabetes in 20–79 years old adults, 2019 estimates: results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res Clin Pract. 2020;162:108086.
Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, Colagiuri S, Guariguata L, Motala AA, Ogurtsova K, et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res Clin Pract. 2019;157:107843.
Shrivastava SR, Shrivastava PS, Ramasamy J. Role of self-care in management of diabetes mellitus. J Diabetes Metab Disord. 2013;12:14.
Ferri S, Kojima K, Sode K. Review of glucose oxidases and glucose dehydrogenases: a bird’s eye view of glucose sensing enzymes. J Diabet Sci Technol. 2011;5:1068–76.
Zhuang Z, Su X, Yuan H, Sun Q, Xiao D, Choi MMF. An improved sensitivity non-enzymatic glucose sensor based on a CuO nanowire modified Cu electrode. Analyst. 2008;133:126–32.
Zhang Y, Nie J, Wei H, Xu H, Wang Q, Cong Y, Tao J, Zhang Y, Chu L, Zhou Y, Wu X. Electrochemical detection of nitrite ions using Ag/Cu/MWNT nanoclusters electrodeposited on a glassy carbon electrode. Sens Actuators B. 2018;258:1107–16.
Sehit E, Altintas Z. Significance of nanomaterials in electrochemical glucose sensors: an updated review (2016–2020). Biosens Bioelectron. 2020;159:112165.
Amaniampong PN, Trinh QT, Wang B, Borgna A, Yang Y, Mushrif SH. Biomass oxidation: formyl C-H bond activation by the surface lattice oxygen of regenerative CuO nanoleaves. Angew Chem Int Ed. 2015;54:8928–33.
Gao D, McCrum IT, Deo S, Choi Y-W, Scholten F, Wan W, Chen JG, Janik MJ, Roldan Cuenya B. Activity and selectivity control in CO2 electroreduction to multicarbon products over CuOx catalysts via electrolyte design. ACS Catal. 2018;8:10012–20.
Yuan M, Guo X, Pang H. Derivatives (Cu/CuO, Cu/Cu2O, and CuS) of Cu superstructures reduced by biomass reductants. Mater Today Chem. 2021;21:100519.
Li J, Wu Y, Qin Y, Liu M, Chen G, Hu L, Gu W, Zhu C. AgCu@CuO aerogels with peroxidase-like activities and photoelectric responses for sensitive biosensing. Chem Comm. 2021;57:13788–91.
Ashok A, Kumar A, Tarlochan F. Highly efficient nonenzymatic glucose sensors based on CuO nanoparticles. Appl Surf Sci. 2019;481:712–22.
Naikoo GA, Awan T, Salim H, Arshad F, Hassan IU, Pedram MZ, Ahmed W, Faruck HL, Aljabali AAA, Mishra V, et al. Fourth-generation glucose sensors composed of copper nanostructures for diabetes management: a critical review. Bioeng Transl Med. 2022;7:e10248.
Oliver NS, Toumazou C, Cass AEG, Johnston DG. Glucose sensors: a review of current and emerging technology. Diabet Med. 2009;26:197–210.
Zhang L, Zhou J, Li J, Liu G, Lin X, Mao B, Liu R, Zhang S, Wang J-Q. Surface structural reconstruction for optical response in iodine-modified TiO2 photocatalyst system. J Phys Chem C. 2014;118:13726–32.
Huang J, Zhu Y, Yang X, Chen W, Zhou Y, Li C. Flexible 3D porous CuO nanowire arrays for enzymeless glucose sensing: in situ engineered versus ex situ piled. Nanoscale. 2015;7:559–69.
Lin W-J, Lin Y-S, Chang H-T, Unnikrishnan B, Huang C-C. Electrocatalytic CuBr@CuO nanoparticles based salivary glucose probes. Biosens Bioelectron. 2021;194:113610.
Zhao Y, Chang C, Teng F, Zhao Y, Chen G, Shi R, Waterhouse GIN, Huang W, Zhang T. Defect-engineered ultrathin δ-MnO2 nanosheet arrays as bifunctional electrodes for efficient overall water splitting. Adv Energy Mater. 2017;7:1700005.
Jiang Z, Wang Y, Sun L, Yuan B, Tian Y, Xiang L, Li Y, Li Y, Li J, Wu A. Dual ATP and pH responsive ZIF-90 nanosystem with favorable biocompatibility and facile post-modification improves therapeutic outcomes of triple negative breast cancer in vivo. Biomaterials. 2019;197:41–50.
Zhang T, Jiang Z, Chen L, Pan C, Sun S, Liu C, Li Z, Ren W, Wu A, Huang P. PCN-Fe(III)-PTX nanoparticles for MRI guided high efficiency chemo-photodynamic therapy in pancreatic cancer through alleviating tumor hypoxia. Nano Res. 2020;13(1):273–81.
Jiang Z, Yuan B, Qiu N, Wang Y, Sun L, Wei Z, Li Y, Zheng J, Jin Y, Li Y, et al. Manganese-zeolitic imidazolate frameworks-90 with high blood circulation stability for MRI-guided tumor therapy. Nano-Micro Lett. 2019;11:61.
Qin J, Cho M, Lee Y. Ferrocene-encapsulated Zn zeolitic imidazole framework (ZIF-8) for optical and electrochemical sensing of Amyloid-β oligomers and for the early diagnosis of alzheimer’s disease. ACS Appl Mater Interfaces. 2019;11:11743–8.
Wang Q, Luo Y, Hou R, Zaman S, Qi K, Liu H, Park HS, Xia BY. Redox tuning in crystalline and electronic structure of bimetal–organic frameworks derived cobalt/nickel boride/sulfide for boosted faradaic capacitance. Adv Mater. 2019;31:1905744.
Guo Y, Tian X, Wang X, Sun J. Fe2O3 nanomaterials derived from prussian blue with excellent H2S sensing properties. Sens Actuators B. 2019;293:136–43.
Huang Z, Fan L, Zhao F, Chen B, Xu K, Zhou S-F, Zhang J, Li Q, Hua D, Zhan G. Rational engineering of multilayered Co3O4/ZnO nanocatalysts through chemical transformations from matryoshka-type ZIFs. Adv Funct Mater. 2019;29:1903774.
Wen L, Li X, Zhang R, Liang H, Zhang Q, Su C, Zeng Y-J. Oxygen vacancy engineering of MOF-derived Zn-doped Co3O4 nanopolyhedrons for enhanced electrochemical nitrogen fixation. ACS Appl Mater Interfaces. 2021;13:14181–8.
Zhao Q, Zhu L, Lin G, Chen G, Liu B, Zhang L, Duan T, Lei J. Controllable synthesis of porous Cu-BTC@polymer composite beads for iodine capture. ACS Appl Mater Interfaces. 2019;11:42635–45.
Ma L, Gao X, Zhang W, Yuan H, Hu Y, Zhu G, Chen R, Chen T, Tie Z, Liu J, et al. Ultrahigh rate capability and ultralong cycling stability of sodium-ion batteries enabled by wrinkled black titania nanosheets with abundant oxygen vacancies. Nano Energy. 2018;53:91–6.
Fu L, Chen H, Wang K, Wang X. Oxygen-vacancy generation in MgFe2O4 by high temperature calcination and its improved photocatalytic activity for CO2 reduction. J Alloys Compd. 2022Jan;25(891).
Barsan MM, Ghica ME, Brett CMA. Electrochemical sensors and biosensors based on redox polymer/carbon nanotube modified electrodes: a review. Anal Chim Acta. 2015;881:1–23.
Ali SR, Iqbal MZ, Faisal MM, Alzaid M. Diffusion control and surface control mechanism in hierarchical nanostructured porous zinc-based MOF material for supercapattery. Int J Energy Res. 2022;46(10):14424–35.
Chen GZ. Linear and non-linear pseudocapacitances with or without diffusion control. Prog Nat Sci Mater Int. 2021;31:792–800.
He Y, Zheng J. One-pot ultrasonic-electrodeposition of copper–graphene nanoflowers in Ethaline for glucose sensing. Anal Methods. 2013;5:767–72.
Balasubramanian P, Velmurugan M, Chen S-M, Hwa K-Y. Optimized electrochemical synthesis of copper nanoparticles decorated reduced graphene oxide: application for enzymeless determination of glucose in human blood. J Electroanal Chem. 2017;807:128–36.
Bao J, Qi Y, Huo D, Hou J, Geng X, Samalo M, Liu Z, Luo H, Yang M, Hou C. A sensitive and selective non-enzymatic glucose sensor based on AuNPs/CuO NWs-MoS2 modified electrode. J Electroanal Chem. 2019;166:B1179–85.
Ding R, Jiang J, Wu F, Gong M, Zhu J, Huang X. Cu@C composite nanotube array and its application as an enzyme-free glucose sensor. Nanotechnology. 2011;22:375303.
Gao Y, Yang F, Yu Q, Fan R, Yang M, Rao S, Lan Q, Yang Z, Yang Z. Three-dimensional porous Cu@Cu2O aerogels for direct voltammetric sensing of glucose. Microchim Acta. 2019;186:192.
Liu X, Sui Y, Yang X, Jiang L, Wang F, Wei Y, Zou B. A feasible approach to synthesize Cu2O microcrystals and their enhanced non-enzymatic sensor performance. RSC Adv. 2015;5:59099–105.
Sun Y, Li Y, Wang N, Xu QQ, Xu L, Lin M. Copper-based metal-organic framework for non-enzymatic electrochemical detection of glucose. Electroanalysis. 2018;30:474–8.
Luo Y, Wang Q, Li J, Xu F, Sun L, Bu Y, Zou Y, Kraatz H-B, Rosei F. Tunable hierarchical surfaces of CuO derived from metal–organic frameworks for non-enzymatic glucose sensing. Inorg Chem Front. 2020;7:1512–25.
Wu L, Lu Z, Ye J. Enzyme-free glucose sensor based on layer-by-layer electrodeposition of multilayer films of multi-walled carbon nanotubes and Cu-based metal framework modified glassy carbon electrode. Biosens Bioelectron. 2019;135:45–9.
Yang Z, Yan X, Li Z, Zheng X, Zheng J. Synthesis of Cu2O on AlOOH/reduced graphene oxide for non-enzymatic amperometric glucose sensing. Anal Methods. 2016;8:1527–31.
Tian K, Baskaran K, Tiwari A. Nonenzymatic glucose sensing using metal oxides – comparison of CuO, Co3O4, and NiO. Vacuum. 2018;155:696–701.
Acknowledgements
The authors also want to thank the Analysis & Testing Center at the Beijing Institute of Technology.
Funding
The authors acknowledge funding from the National Natural Science Foundation of China (32101153, 82003150) and the China Postdoctoral Science Foundation (2020M680395), the Shanghai Sailing Program (20YF1453400), Shanghai Medical Innovation Project (21Y11905800) and the “Chenguang Program” supported by Shanghai Education Development Foundation & Shanghai Municipal Education Commission (20CG25).
Author information
Authors and Affiliations
Contributions
CZ was in charge of the data curation and wrote original draft preparation, XT and JC were visualized and investigated the original draft and involved in planning and supervised the work, JZ was carried out the partial experiment, ZJ and JQ were conceived of the presented idea, investigating, reviewing and editing the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
For experiments related to electrochemical monitoring based on human blood samples, we have the corresponding ethical approval and informed consent form approved by the Ethics Committee of Shanghai Tenth People’s Hospital (2020-KN115-01), and the ethical approval document and consent form approve that project-related experimental studies can involve biomedical research on human subjects.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Experimental. Figure S1. a TEM and b element mapping of as-prepared Cu-MOF. Figure S2. XRD pattern of as-prepared Cu-MOF from 5o to 80o. Figure S3. Survey scan of as-prepared Cu-MOF. Figure S4. High resolution XPS spectra of C1s,O1s and Cu2pof the as-prepared Cu-MOF. Figure S5. TG curves and DTA of as-prepared Cu-MOFin the air. Figure S6. The SEM image of commercial CuO (inset: size distribution histogram). Figure S7. EDS result of CuO/C-400 oC. Figure S8. XRD pattern of commercial CuO. Figure S9. Cyclic voltammograms of the a CuO/C-350 oC, b CuO/C-450 oC and c commercial CuO electrodes in 0.1 M NaOH with/without 0.2 mM glucose at a scan rate of 100 mV s−1. Figure S10. CV curves of a CuO/C-350 oC and b CuO/C-450 oC in 0.5 mM K3Fe(CN)6/0.1 M KCl electrolyte at different scan rate and c, d the corresponding fitting curves. Figure S11. Amperometric i-t response of the CuO/C-400 oCelectrodes in 0.1 M NaOH at different voltage (vs. SCE) with stirring. Figure S12. a Amperometric responses of CuO/C-350 oC in 0.1 M NaOH upon consecutive addition of glucose at 0.5 V (vs. Ag/AgCl) and b correspondingcalibration curves of the CuO/C-350 oC for glucose detection. Figure S13. a Amperometric responses of CuO/C-450 oC in 0.1 M NaOH upon consecutive addition of glucose at 0.5 V (vs. Ag/AgCl) and b the corresponding calibration curves of CuO/C-450 oC for glucose detection..
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhao, C., Tang, X., Zhao, J. et al. MOF derived core-shell CuO/C with temperature-controlled oxygen-vacancy for real time analysis of glucose. J Nanobiotechnol 20, 507 (2022). https://doi.org/10.1186/s12951-022-01715-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12951-022-01715-z