
Abstract
Radiopharmaceuticals have proven to be effective agents, since they can be successfully applied for both diagnostics and therapy. Effective application of relevant radionuclides in pre-clinical and clinical studies depends on the choice of a sufficient delivery platform. Herein, we provide a comprehensive review on the most relevant aspects in radionuclide delivery using the most employed carrier systems, including, (i) monoclonal antibodies and their fragments, (ii) organic and (iii) inorganic nanoparticles, and (iv) microspheres. This review offers an extensive analysis of radionuclide delivery systems, the approaches of their modification and radiolabeling strategies with the further prospects of their implementation in multimodal imaging and disease curing. Finally, the comparative outlook on the carriers and radionuclide choice, as well as on the targeting efficiency of the developed systems is discussed.
Similar content being viewed by others


Introduction
Since the discovery of radioactivity by Henri Becquerel and Marie Curie, and first medical application of radium by Henri Alexandre Danlos and Eugene Bloch for the treatment of tuberculous skin lesion, the radionuclide therapy has made significant progress. Nowadays, it is hard to overestimate the importance of radionuclides application in medicine, as they are extensively used in both diagnostics and therapy [1]. The reason for such demand is explained by two main factors. First of all, the radionuclides are highly effective and universal cytotoxic agents that are capable of destroying cells with ionizing radiation, which cannot be avoided or negated by any cellular means [2]. Secondly, the use of γ and β+ emitters is crucial for diagnostics, because it allows one to take advantage of superior imaging methods utilizing the emission for precise detection of the radionuclide distribution in the whole organism. Although radionuclides possess unique diagnostic and therapeutic features, they are not able to selectively target tumor sites (with only few exceptions, e.g. 131I can passively accumulate at the tumor tissues, expressing sodium/iodide symporter (NIS) [3,4,5]), thus, issues related to the radionuclide delivery become an area of a significant interest and growth.
As a matter of fact, drug delivery systems promote the concentration of the drug reaching the target site and the enhanced pharmacokinetic profiles [6]. The addressable delivery of the drug can usually occur via active or passive targeting. The passive targeting is performed via accumulation of drug carriers at a particular site due to the inherent pathophysiological, physicochemical or pharmacological factors [7,8,9]. While the active targeting is occurred when the drug carriers are modified with active targeting ligands, possessing a high affinity for binding to a specific cell type or tissue in the organism [10,11,12]. In contrast to the off-target drug delivery, the active targeting enables enhanced therapeutic efficiency and reduced side effects associated with the systemic toxicity [13]. The same principles are true when it comes to the delivery of radionuclides. The use of delivery systems, which can ensure the addressable delivery of the loaded cargo, is crucial for both therapeutic and diagnostic radionuclides. The more effective delivery of radionuclides will ensure a lowering in demand of these agents, which allow decreasing isotope dose administrated per patient to reduce a risk of exposure and costs [14,15,16]. Also, selective targeting of diagnostic radionuclides at the site of interest will provide increasing image quality when positron emission tomography (PET) or single-photon emission computed tomography (SPECT) is performed. The targeting delivery of radionuclides becomes the main priority since the high amounts of irradiation during the treatment in unfavorable delivery areas can result in possible side effects. Additionally, delivery systems can be modified to provide further possibilities of detection and visualization. Such augmentation can further enhance the diagnostic value of radiopharmaceuticals and is important for the delivered dose estimation when it comes to the therapeutic radionuclides. The concept of active targeting delivery of radionuclides is presented in Fig. 1.

Administration of radionuclide carriers in the tumor site and their further accumulation via active targeting approach
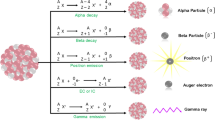
The application of radionuclide is determined by its nature. Radioisotopes commonly used for the therapeutic radiopharmaceuticals emit α-, β-particles, or Auger electrons, which cause cytotoxic deoxyribonucleic acids (DNA) damage through numerous mechanisms such as reactive oxygen species, single and double stranded breaks, and inhibition of DNA repair mechanisms [17, 18]. The emissions of β-, α-, and Auger particles vary in penetrating range (Fig. 2) and linear energy transfer (LET), thus the choice of radionuclide depends on multiple factors, such as the type and size of the targeted cancer, density of the target and its heterogeneity.

Types of radioactive decay with the demonstration of the soft tissue penetration range
The β− emitters have a low LET (0.2 keV/mm) and a relatively long penetrating range in tissue (one to several mm), as a result, they can penetrate deep in the tumor and can also partially damage the surrounding normal tissues. Common β− emitters include 177Lu, 90Y and 131I, which has already been used for thyroid cancer treatment. Both iodine and lutetium radioisotopes co-emit a γ photon that can be detected by SPECT [19].
The radionuclides with β+ decay emit positron and are used exclusively for diagnostical purposes and can be visualized via PET. Commonly used β+ emitters are usually short-lived (e.g.15O, 13N, 11C and 18F) and used to estimate the location of disease and quantitatively examine various biochemical and physiological processes. However, the interest in the application of the long lived β+ emitters (89Zr, 64Cu and 52Mn) has arisen, since they can be used to monitor the course of treatment for long period of time (2–3 weeks) [20].
The emitted α-particles can penetrate only a few cell diameters (50–100 µm in soft tissue) and have a high LET (80–100 keV/µm); therefore, they can be more effective for treatment of smaller lesions and metastases. Commonly used α-emitters include 213Bi, 223Ra, radiohalogen 211At, radiometal 225Ac [21]. One of the most frequently used in nuclear medicine radioisotopes is 225Ac, which consecutively undergoes four α-decays, during two of which γ ray is emitted. This isotope has been successfully applied for tumor therapy in preclinical and clinical studies [22,23,24]. Auger emitters (125I and 99mTc) have high LET (4–26 keV/µm) with a very short penetration depth of 2 to 500 nm [25]. Therefore, Auger emitters like α-emitters are more suitable for minimizing damage of the normal tissues compared to β-emitters [26]. However, due to the small penetration depth of Auger electrons, the radionuclides should be delivered at the closest proximity to the cell. For this reason, the development of the radionuclide delivery systems is required.
This review is focused on known radionuclide delivery systems traditionally applied in nuclear medicine, which are categorized into four main groups: (i) antibodies and antibody fragments, (ii) organic and (iii) inorganic nanoparticles (NPs), and finally (iv) microspheres (Fig. 1). We describe the design considerations of recently developed radionuclide delivery systems, their modifications and radiolabeling approaches, as well as a current state in clinical and preclinical studies, or their potential to be implemented into the clinical practice.
Antibodies and antibody fragments
Immunoglobulin type G (IgG) monoclonal antibodies (mAbs) are the most commonly used type of targeting molecules for pharmaceutical application, including radiomedicine. Their size is approximately 150 kDa, and mAbs are composed of two identical polypeptide “heavy chains” paired with two “light chains”. MAbs consist of an antigen-binding fragment (Fab), a fragment crystallizable (Fc), two disulfide bonds in the hinge region and a conserved glycosylation site at amino acid N297 of each heavy chain (Fig. 3) [27]. MAbs can specifically bind antigens without further modifications, and thus they already possess targeting properties.

(this figure was reproduced from Scott et al. [29] with the required copyright permission)
a Correlation between blood clearance, tumor penetration, plasma half-life and renal uptake and the previously discussed antibodies and antibody fragments of different size: bsAbs, intact MAbs, scFv, sdAbs and alternative protein scaffolds. Uptake is expressed as percentage of injected dose per gram (ID/g). b PET/computed tomography (CT) images of mice bearing prostate stem-cell antigen (PSCA)–expressing LAPC-9 prostate cancer xenografts. PSCA expression was visualized with intact mAbs, single-chain Fv–Fc (scFV-Fc) wild type (WT) mAbs, scFV-Fc double mutant (DM) mAbs, Minibodies and Diabody labeled with N-succinimidyl-4-[18F]-fluorobenzoate (18F-SFB). All microPET images were scaled individually to best show tumor targeting
The mAbs labeled with a γ- or positron-emitting radionuclide are actively used for quantitative biodistribution imaging using SPECT or PET [28]. The use of mAbs-based PET and SPECT has been implemented in cancer treatment to guide therapy selection, to estimate dosimetry for radioimmunotherapy (RIT), and to track response to therapy [29, 30]. Apart from imaging of oncological targets, immunoPET has been employed for detection and tracking of immune cells [31]. The immunoPET can also be used to monitor inflammation and immune responses [32].
Given the nature of attached radionuclide, the same antibody can be used for both diagnostic imaging and RIT, thus, providing a theranostic platform [33]. A number of works showed efficiency of RIT [34,35,36] in hematopoietic cancers and to a lesser extent, solid tumors [37].
Additionally, there is an approach in radiomedicine in which theranostic radionuclide pairs such as 86Y and 90Y are used [38, 39]. For instance, mAbs labeled with diagnostic radionuclides (β+ emitter 86Y) were administered first in order to localize the tumor site and determine the specificity of the applied mAbs. Since the 86Y and 90Y are the same element, the distribution and pharmacokinetics of mAbs labeled with these radionuclides is the same. This allows a very precise estimation of the required dosage of therapeutic radionuclide for RIT and, thus, improvement of the treatment [38, 39].
Nonetheless, mAbs possess certain disadvantages, which are common to any therapeutic radionuclide carriers. For example, a decay is a continuous process, which occurs without any regard to whether the mAbs are bonded to their antigens or not. Therefore, it is important to shorten the free circulation period of the radioconjugates as much as possible. MAbs themselves typically exhibit long circulation times in blood (which contributes to bone marrow toxicity) and a reduced diffusion into the tumoral mass, which can lead to the accumulation of radionuclides in critical organs, especially, in liver. There are some possibilities to address it such as the optimization of antibody pharmacokinetics, biodistribution, and clearance in vivo. At the present moment, antibody-based delivery optimization can be achieved using modern protein engineering, which allows to reformate intact mAbs into smaller derivatives of antibodies. These derivatives include smaller monovalent fragments such as single domain antibodies (sdAbs), diabodies, minibodies, protein scaffolds, and more complex bispecific antibodies (bsAbs). Their structures are represented in Fig. 3. The smaller antibody fragments are able to clear more rapidly from the circulation compared to the intact mAbs, resulting in better contrast images after shorter periods of time. For imaging, antibody fragments that lack the Fc region are desired due to the removal of the biological function and neonatal Fc receptor (FcRn) recycling, which allows to obtain the optimal contrast at shorter time periods. Wu et al. have shown that the reduction of the molecular weight of antibodies below ~ 60 kDa can dramatically accelerate clearance. For example, high contrast images can be obtained for small single‐domain antibodies or diabodies within the same day (4–8 h), compared to the larger minibodies, which are usually formed by the next day (24–48 h) [29]. Thus, smaller antibodies offer certain advantages regarding better pharmacokinetics. For therapeutic applications, the circulation time of the radiolabeled antibody fragments, on one hand, needs to be sufficient for the dose deposition in tumors, while limiting toxicity to normal tissues. The plasma half‐life of small fragments and scaffolds (targeting molecules of protein nature [40]) can be extended by chemical or recombinant approaches such as poly(ethylene glycol) (PEG) modification (PEGylation) or fusion to Fc domain, albumin, or albumin‐binding proteins. The influence of antibody variations created by joining antibody variable light (VL) and variable heavy (VH) domains on their plasma half-lives detected by PET is presented in Fig. 3. The tumor penetration and renal uptake is increased with the size reduction of antibodies. However, the smaller antibody fragments for RIT have a lower size than renal filtration cutoff and, therefore, they are cleared through the kidneys, which is less radioresistant compared to liver.
Minibodies
The minibody is a bivalent dimer comprising of a single-chain variable fragments (scFv) with a human IgG constant heavy chain 3 domain (Fig. 3) with a serum half-life of 5–12 h, and due to its size (~ 80 kDa) is excreted via hepatic clearance [41]. Radiolabeled minibodies were used as diagnostic PET imaging tracers targeting carcinoembryonic antigen (CEA) [42, 43], CD8 T cells [44], CD20 B Cells, PSCA and prostate-specific membrane antigen (PSMA) [45].
Minibody, composed of the recombinant scFv of L19 antibody and the constant heavy chain 4 domain of human IgE (L19-SIP), was engineered to target extra domain B of fibronectin. 131I-L19-SIP demonstrated superior therapeutic efficacy compared with dimeric scFv or mAbs in teratocarcinoma tumor-bearing mice [32].
For imaging, 124I-L19-SIP (radretumab) was developed as an immunoPET tracer to perform patient selection in clinical trials, which later received 131I-L19-SIP RIT. Thus, 124I-L19-SIP was used to predict dosage delivered to tumors in patients with brain metastases, while 131I-L19-SIP was tested as RIT in combination with external beam radiation in patients with multiple brain metastases. The results showed reduction in 18F-fluorodeoxyglucose (FDG) uptake in common patients. Radretumab was also tested in patients with relapsed hematological cancers, where its diagnostic dose was preliminary used to determine eligibility for further treatment. In cases when tumor uptake was sufficient, three of ten patients who received radretumab showed complete response (clinical trial NCT01125085). Thus, patients with advanced lymphomas could benefit from concurrent chemotherapy.
Diabodies
The diabody is a noncovalent scFv dimer (~ 55 kDa) in which a shortened linker prevents the VH and VL from self-pairing, forcing cross-pairs in trans with a second chain to form a dimer, reconstituting two binding sites (Fig. 3). Radiolabeled diabodies are widely used for high-contrast PET imaging of a variety of targets due to the improved tumor penetration compared to intact mAbs. The fast clearance of the diabodies with a plasma half-life of 2–5 h is advantageous for the same-day PET imaging of immune cell subsets, such as CD20, CD4 and CD8 T cells [46, 47], as well as solid tumors, including prostate cancer [48].
Single-domain antibodies
Single-domain antibodies (sdAbs, 12–15 kDa) (Fig. 3) include small monomeric fragments derived from human VH or VL domains, linked with short peptide sequence, and has been given the commercial name Nanobody™ [49, 50]. These sdAbs have high stability, low immunogenicity and more compact structure for better targeting compared to IgG [51].
The camelid and shark domains have long complementarity-determining region 3 (CDR3), which allow for improved extension into cavities of target antigens [52]. More recent radiolabeled sdAbs studies have focused on the camelid VHH (Nanobody, registered trademark of “Ablynx NV”), and common targets include PSMA [53], CEA [54], and epidermal growth factor receptor (EGFR) [55], human epidermal growth factor receptor 2 (HER2) [56], and immune cell markers [57].
There are some nanobodies labeled with therapeutic radionuclides. For instance, the sdAbs commercially available as Nanobody 2Rs15d labeled with 177Lu via diethylenetriaminepentaacetic acid (DTPA) and targeting tumor biomarker HER2 was evaluated in theranostic studies. 177Lu-DTPA-2Rs15d was injected in mice bearing luciferase expressing ovarian cancer cell line SKOV-3 xenografts. By the day 125 tumor growth was halted, 5 of 8 mice were completely tumor-free and 3 other mice had small, but unpalpable tumors [58].
Nanobodies are also employed for diagnostics. For instance, Xavier et al. reported synthesis and preclinical validation of a novel anti-HER2 tracer, 68Ga-NOTA-2Rs15d, for immunoPET. The described agents were capable of highly specific accumulation in the tumor areas and reduced kidney retention compared to the previously reported Nanobodies [59]. This was achieved by the removal of histidine tag. Overall, the reported tracer proved to be safe in vivo. Further, Keyaert et al. performed clinical trials employing reported immunotracer [60]. In this work, 20 female patients with HER2-positive primary or metastatic breast carcinoma were involved. The studies revealed no adverse effects of tracer administration and the radiolabeled Nanobody was able to accumulate at HER2 overexpressing tumor sites at much higher concentrations compared to the healthy surrounding tissues. Therefore, authors continued their work in a Phase II clinical trials.
Alternative non-antibody protein scaffolds
Alternative protein scaffolds (targeting molecule of protein nature) are easy to produce in bacteria or yeast, and are typically stable in harsh labeling conditions such as high temperature. They range from 2 to 20 kDa, which enables tissue penetration to access binding sites easier than mAbs (Fig. 3). However, the small size also results in a fast renal filtration, and certain applications may require additional engineering or other modifications to increase the plasma half-life. This can be achieved by PEGylation, Fc fusion, albumin fusion, or use of albumin binding domains [61]. A widely explored scaffold for radionuclide delivery is the Affibody (Affibody AB™), small (6.5 kDa) scaffolds composed of alpha helices originally based on the Z domain of staphylococcal protein A [62]. Affibodies rapidly penetrate tumors for early high-contrast images due to rapid extravasation and tumor penetration [63], and they are compatible with short-lived radionuclides such as 68Ga and 18F for PET, or 111In for SPECT. Affibody-based imaging and therapy have been developed to target EGFR [64] and HER2 [65].
RIT studies in HER2-expressing tumor models support the development of the HER2-binding Affibody ZHER2:342 to treat trastuzumab-resistant HER2 positive tumors. Due to the small size, the ZHER2:342 clears rapidly and biodistribution studies with residualizing radionuclides show high renal reabsorption. Therefore, fusion to an albumin-binding domain was used to reduce renal toxicity and increase circulation for therapeutic radionuclides [66]. For example, 177Lu labeled ZHER2:342 successfully targeted HER2 positive microxenografts as confirmed by gamma-camera imaging, and the treatment extended survival in mice with high and low HER2 expressing tumors, however, overall mortality was caused by bone marrow toxicity [67]. Another study describes biodistribution of anti-HER2 Affibody ZHER2:395-TCO and residualizing radiometals 111In and 177Lu [65].
Bispecific antibodies
Antibody engineering enables novel functionality such as bispecific binding. Antibodies which contain two different antigen-binding sites in one molecule are called bispecific [68]. BsAbs can target two or more antigens resulting in improved delivery to a tumor for better imaging or therapeutic outcome. Brinkmann et al. have recently reviewed the development and clinical implementation of bsAbs [69, 70]. BsAbs can be particularly useful for radionuclide delivery as part of a pretargeting strategy. According to pretargeting approach, one arm of the bispecific targets the tumor, and the other arm recognizes a radiolabeled hapten (typically a small molecule or peptide for imaging or RIT). The bsAbs are administered first and only after sufficient time, they accumulate at the tumor and clear from the circulation. Further, the radiolabeled hapten is injected, which rapidly binds to the already pre-localized bsAbs [71]. Another effective pretargeting strategy uses a trivalent bsAb with two target-specific Fabs and an anti-histamine-succinyl-glycine Fab, which are linked to each other via disulfide bonds. Example targets for bsAbs pretargeting include CD105 and EGFR [72], CEA [73], trophoblast antigen 2 (TROP2) [74].
Example of bispecific antibodies usage for pretargeting strategy was reported in the recent work of Heskamp et al. [75]. The authors demonstrated the usage of Tri-Fab antibody TF2 consisting of anti-CEA with variable fragments against histaminesuccinyl-glycine (HSG), which allow pretargeting of radiolabeled hapten–peptide IMP-288. TF2 was used for pretargeted RIT in mice bearing CEA-positive colorectal cancer xenografts, where α-emitting hapten 213Bi-IMP288 was shown to be at least as effective as β-emitting hapten 177Lu-IMP288.
Antibody labeling approaches
Generally, the radiolabeling of antibodies is occurred through the use of chelating agents [76,77,78,79,80,81]. The attaching chelators to antibodies is performed covalently via complementary reactive functional groups for the conjugation to each other. The N-hydroxysuccinimide esters (NHS), isothiocyanates (SCN) and anhydrides can be considered as the most conventional reactive electrophilic groups that can react with the ε-amino group of lysines on the antibody at alkaline conditions (pH 7.2–9). In such conditions, NHS- or SCN-containing chelators can be easily conjugated with antibodies to form strong covalent bond. After the chelator attachment is achieved, the radiolabeling is occurred via the complexation process. However, in case of conjugation reaction using NHS- or SCN chemistry, the presence of multiple amino acids in antibody structure may lead to a lack of both stoichiometric control and site-specificity [82,83,84]. The spontaneous chelator-antibody conjugation can decrease affinity for target receptors and provide non-optimal pharmacokinetics. Therefore, the development of more desirable chemoselective approach to bind chelators with antibodies is required and urgent.
The “click chemistry” is found to be powerful method for rapid antibody chelation that provides an appropriate site-selective conjugation of chelators with antibodies. However, this method requires the antibody pre-modification. There are many examples where the reactive “click” groups have been site-selectively introduced into antibodies [85, 86]. The site-selective conjugation methods for preparation of radiolabeled antibodies include the modification of cysteines/disulfide bonds and the glycan region of the antibody and enzyme-mediated conjugation [28].
The cysteine is widely used a single amino acid or thiol-containing small molecule for antibody modification. The most popular method of antibodies conjugation via cysteines involves so-called a Michael reaction of a thiol groups with a maleimide, resulting in formation of a succinimidyl thioether product. The maleimide groups have been widely used to incorporate the chelators via cysteine thiol groups [28]. However, maleimide-thiol modification can be unstable in vivo and, therefore, other new cysteine-reactive reagents that provide enhanced conjugate stability have been explored including employing phenyloxadiazole sulfones, dibromomaleimides, and dithiophenolmaleimides [87]. Also, monobromo maleimide has been used to generate stable antibody conjugates [88].
Considering the glycan region of the antibody, they can be chemically modified to provide site-selective attachment of chelators. One widely used example of such modification includes the generation of an aldehyde group by oxidizing the cis-glycol groups of terminal hexoses using sodium periodate [89,90,91]. Further, the generated aldehydes can react with chelators, containing amine groups to form stable imine conjugates.
The application of enzyme-mediated conjugation also provides site-specific binding of chelators to target antibodies. This approach involves the use of enzymes that recognize two complementary motifs on the antibody and the chelator functional groups [85, 92]. The enzymes catalases covalent binding of the chelator to antibody using these two complementary motifs. The β-1,4-galactosyltransferase (Y289L), transglutaminase and, sortase A are typical enzymes used in site-directed enzyme-mediated conjugation of chelators to antibodies. Such technique was applied to incorporate 89Zr-labeled desferrioxamine (DFO) chelator and 64Cu-labeled 4-(1,4,8,11-tetraazacyclotetradec-1-yl)-methyl benzole acid tetrahydrochloride (CPTA) chelator [85, 93]. Another work described the 18F-labeling reaction using sortase [94]. It should be noted that enzyme-mediated conjugation is occurred under mild conditions that do not denature the antibody.
Besides chelation techniques, the direct radioiodination of antibodies is a powerful method for the preparation of antibody-based radiopharmaceuticals [95]. The radioiodination can be achieved by directed radiolabeling the iodine radioisotope to tyrosines on the antibody or antibody fragment using well-established procedures that reported early starting from 1980s [96]. The good example of such radiopharmaceutical is tositumomab (131I-labeled anti-CD20 antibody), which received Food and Drug Administration (FDA) approval for the treatment of Non-Hodgkin’s lymphoma in 2003 [97].
In case of radioactive 18F, the antibody labeling is performed by directed binding to a tyrosine residue via electrophilic reaction [98]. Moreover, the incorporation of 18F can be achieved via conjugation of 18F-labeled reactive precursors, containing prosthetic groups [99].
Another interesting approach of antibody radiolabeling involves photoradiochemical methods that use light-induced antibody modification with chelators derivatized with aryl azide (ArN3) groups [100, 101]. Recently, Holland et al. demonstrated the photochemical conjugation of mAbs with 68Ga via an ArN3 functionalized 2-[4,7-bis(carboxymethyl)-1,4,7-triazonan-1-yl]pentanedioic acid chelate (NODAGA-PEG3-ArN3). The method works via a conventional two-step, preconjugation and radiolabeling pathway, and also by a one-pot, pre-radiolabeling route. As the authors reported, the speed and simplicity of this photoradiochemical route renders the method suitable for automation [101].
Outlooks
mAbs were historically first trial instrument to provide targeting delivery of bioactive compounds into the site of interest. Many radionuclide labeled mAbs are actively used for diagnostic and therapy of malignant neoplasms while the protein engineering opportunities allows to control plasma half-life time and biodistribution parameters of radionuclides. As a result, the change of interest from the traditional radionuclide-full antibody conjugates to the antibody-based fragments is now observed. The use of antibody derivates allow to increase the specifity, targeting ability and distribution parameters of radionuclides. Besides, employing bispecific antibodies can reduce radioisotope dosage in pretargeting strategy. Also, the antibody fragments have great potential as targeting ligands for nano- and microparticle based therapeutics with the resulting constructs demonstrating enhanced selectivity, specificity and pharmacokinetics. Combination of high specifity of antibodies based targeting ligands with high loading capacity of nano- and microparticle allows to obtain modern form of radiopharmaceuticals. Moreover, the use of NPs as a radionuclide carrier guarantee the stability of radionuclide during its delivery and save the biological properties of targeting ligands, which results in minimizing antibody-radionuclide interactions. The recent studies on radionuclide delivery using antibodies and antibody fragments were presented in Table 1.
Organic NPs
The organic NPs are actively used to transport various kinds of biologically active substances, including radionuclides [102]. At present, many types and variations of organic NPs were designed and fabricated. There are basically five main categories of organic NPs that have been utilized in nuclear medicine for therapy and diagnostics: (i) liposomes, (ii) albumin-based NPs, (iii) dendrimers, (iv) polymeric micelles and (v) polymeric NPs. A large number of techniques have been developed for radionuclide labeling and surface functionalization of organic NPs by targeting ligands in order to provide addressable delivery of radionuclides at the desired site, i.e., tumor area. In this section, we consider the organic nanocarriers used for radiolabeling and discuss the principles and approaches in targeting ligand modification and radionuclides incorporation with appropriate examples (Fig. 4).

The main categories of organic NPs used for radiopharmaceutical formulations with schematic representation of their radiolabeling strategies. Below the commonly used macrocyclic and acylic chelators for radiolabeling are depicted
Liposomes
Liposomes are phospholipid vesicles with one or more bilipid layers that contain an aqueous phase within them. They were firstly discovered by Banghman and colleagues [103]. The sizes of these carriers can vary from the smallest (up to 100 nm) to gigantic (more than 1 micron) depending on the methods of synthesis. In the case of nanoscale liposomes, their important feature is the tendency to accumulate in the tumor tissues, providing passive drug delivery mechanism [104]. The current strategy of radionuclide delivery to the tumor sites is based on the active targeting of liposomes via their surface functionalization with specific ligands, i.e. peptides, antibodies, small molecules, proteins and so forth [105]. As an example, Anti-HER2 antibodies were successfully conjugated onto the liposome surface with conventional thiol-maleimide chemistry technique to deliver bioactive compounds into HER2-overexpressing tumor [103]. Another clinically used target is the EGFR that is constitutively activated in many solid tumors. Recently, Shargh and colleagues published the comprehensive review article describing the different conjugation techniques for preparation of antibody-targeted liposomes [103].
Cell-targeting peptides are also used to conjugate with liposomes through covalent bond by a variety of linkages (maleimide linkage bond, peptide bond, sulfanyl bond, disulfide bond and phosphatidylethanolamine-linker bond) [106]. For example, Zhang et al. reported on liposomes containing cantharidin and a tumor specific cell-penetrating peptide BR2. BR2-directed liposomes demonstrated enhanced cellular-uptake in hepatocellular carcinoma as compared to non-targeted liposomes [107]. Recently, Backer et al. developed a new method for covalent coupling of targeting proteins to liposomes via so-called “dock and lock” strategy, which exploits the natural interaction between the dimerization and docking domain that provides “safe” protein conjugation. Many small molecules (e.g. folate, affibody, carbohydrate and so forth) were used as targeting ligands for functionalization of liposome surface. However, a recent trend in functionalization of liposomes is the combination of peptides and antibodies in a single formulation for surface modification. Such dual modification provides enhanced selectivity compared to the single type of modification.
In 1993 Goins with colleagues firstly investigated the labeling of liposomes with radionuclides [108]. The addition of radionuclides was performed after the liposome formation and not during the liposome synthesis. Then, the new methods of radiolabeling with liposomes have been developed. Nowadays, four general approaches can be highlighted for incorporating radionuclides into liposomes: (i) passive encapsulation, (ii) membrane labeling, (iii) surface chelation, and (iv) remote loading via lipophilic chelator or ionophore (Fig. 5) [108]. In case of passive encapsulation, the radionuclide pre-associated with chelator is included in the buffer solution, where liposomes are formed. In this method, the incorporation efficiency of radionuclides does not usually exceed 10% in case of nanoscale liposomes (< 100 nm), therefore, this method is rarely used for incorporation of highly cost therapeutic radionuclides such as 225Ac and 177Lu. According to the membrane labeling, the radionuclides can be conjugated to a lipid layer that is formulated into the liposomes via simple incubation of liposome with radionuclides. The limitation of this approach is the instability of liposome radiolabeling because of non-covalent bonding of radionuclides with liposome membrane. The use of chelating agents for radionuclide labeling can improve the stability of radionuclide biding, but not in all cases. For example, the incorporating lipid chelator conjugated onto liposome membrane does not provide an appropriate in vivo stability and the radionuclides, which are attached onto the liposome surface, interact with the components of the blood stream. This may result in the detachment of the radionuclides from the liposomes even at the labeling efficiency 90%. In general, DOTA and 1,4,7-Triazacyclononane-1,4,7-triacetic acid (NOTA) are widely used chelating agents for surface modification of liposomes. The use of lipophilic chelating agents such as N,N-bis(2-mercaptoethyl)-Nʹ,Nʹ-diethylethylenediamine (BMEDA), and hexamethylpropyleneamine oxime (HMPAO), as well as ionophores can promote penetration of radionuclides through the lipid bilayer, providing a high stability of radiolabeling [109]. Moreover, by employing lipophilic chelator or ionophore, the radionuclides can be carried through the lipid bilayer inside the aqueous phase of liposome, where the radionuclides are bound with pre-loaded metal chelator. This concept of radiolabeling has been successfully realized by Edmonds et al. Authors developed a simple and efficient radiolabeling method providing excellent radiolabeling yields, purities, and stabilities with 89Zr, 52Mn, and 64Cu, without the modification of the delivery system components [110].

Schematic representation of various radiolabeling approaches of liposomes, targeting ligand modifications with further application in different field of nuclear medicine
The high flexibility of chemical structure and wide range of radiolabeling approaches allow to design the liposomes for various biomedical applications, including multimodal imaging, radiochemotherapy, RIT, α therapy, infection and inflammation imaging, neurological imaging and so forth [110]. Recently, Kleynhans et al. reported on a detailed description about application of liposomes in different fields of nuclear medicine [111].
Apart from conjugation with radionuclides, liposomes can additionally modified to provide imaging properties. As an example, Kim et al. designed trimodal liposomes for optical, nuclear, and magnetic resonance imaging (MRI) [112]. These liposomes were capable of incorporating 124I emitting both nuclear and optical imaging signals, and simultaneously lipophilic gadolinium complex as MRI contrast agent. Considering the theranostic approach, such liposomes could be additionally loaded with antitumor drugs (e.g. doxorubicin, vincristine and so forth). The combination of therapeutic radionuclides (e.g. 186/188Re) with chemotherapeutic agents (e.g. doxorubicin) in one single liposome provided radiochemotherapeutical effect [113, 114]. Interesting results were obtained for liposomes labeled with 225Ac, which is promising therapeutic radionuclide for targeted α-therapy [115,116,117,118]. An example for employment of radiolabeled liposomes in visualization of infection and inflammation was demonstrated by Ferreira with co-workers, who developed a long-circulating and pH sensitive liposomes containing a 99mTc-labeled antibiotic (ceftizoxime) for identification of osteomyelitis foci [119]. Also the 99mTc-labeled liposomes were used as blood-pool imaging agents [120]. A novel application of liposomal 18F is the imaging of synaptic density in the brain by targeting voltage-dependent calcium channels (N-type Ca2+). This tracer can image neurodegeneration and evaluate cognitive function during therapy [121].
The liposome technologies are the most studied delivery systems in humans [122]. We refer to recently published review articles by Petersen et al. [108] and Lamichhane et al. [123], which highlighted the examples of the clinical studies focused on the application of liposomal-based radiopharmaceuticals in nuclear imaging using SPECT or PET. For example, Lopez-Berstein et al. administrated 99mTc-labeled liposomes to seven cancer patients as imaging agents for tumor detection and staging [124]. In addition, there are clinical studies with 111In-labeled liposomes that demonstrated their safety and utility for tumor detection [125]. Later, Koukourakis et al. performed the first clinical studies of 99mTc-labeled liposomes containing a therapeutic drug (doxorubicin) on seven patients with Kaposi’s sarcoma and head and neck cancer [126]. Recently, Lee et al. reported a clinical trial of MM-302, a PEGylated liposomal doxorubicin, labeled with 64Cu radioisotope targeted against HER2 ([64Cu]MM-302) [127]. The 19 patients with metastatic breast cancer for imaging study were selected for testing MM-302 radiolabeled with 64Cu. The results from the PET imaging (Fig. 6) demonstrated that [64Cu]MM-302 remained in the circulation for over 24 h, and thereafter accumulated mostly in the liver and spleen.

(this figure was reproduced from Lee et al. [127] with the required copyright permission)
PET and PET/CT images of [64Cu]MM-302 in lesions at different anatomic locations. The regions of interest used to measure tumor deposition of [64Cu]-MM-302 are shown in blue or turquoise outlines
Another recent example of clinical use of liposomal radiopharmaceuticals was performed by the Institute of Nuclear Energy Research (Taiwan) that has already advanced a lipid theranostic agent into Phase I clinical testing (clinical trial NCT02271516). The tested liposome system was radiolabeled with 188Re as therapeutic and diagnostic isotope linked to BMEDA, which acts as a chelating agent. These 188Re-BMEDA liposomes exhibited a higher therapeutic efficacy in rodent xenografts [111].
Albumin-based NPs
The albumin is a major protein in blood plasma and actively used in formation of albumin-based drug delivery systems because of selective accumulation into the solid tumor via so-called the enhanced permeation and retention (EPR) effect [128]. At the moment, there is already existed the first albumin based antineoplastic agent approved by the FDA, which is called Abraxane. Especially, human serum albumin (HSA) and bovine serum albumin (BSA) have driven extensive research in nuclear medicine [129]. Since the HSA molecule is more preferable than BSA for further preclinical and clinical application due to the low immunogenicity, we further review only HSA-based formulations. The HSA molecule has active chemical centers for radionuclide labeling (carboxyl, amine and thiol groups) [130]. HSA was covalently labeled with various radionuclides (68Ga, 64Cu, 89Zr and so forth) by employing different chelators. 99mTc-labeled macroaggregated albumins are already commercially available as SPECT imaging agents such as 99mTc-Pulmolite® and 99mTc-HSA microspheres B20® for lung perfusion imaging; 99mTc-Albures® for liver and spleen imaging and 99mTc-Nanocoll® as well as 99mTc-Nanotop® for bone marrow and sentinel lymph node (SLN) imaging. Several techniques can be employed for effective radiolabeling of albumins. Usually, introduction of macrocyclic or acyclic chelators (DOTA, DTPA) into albumin structure is used to achieve the desired stability and suitable pharmacokinetic properties. Another promising approach to label radionuclides with HSA is the employment of low molecular weight albumin-binding molecules (HSA binders), which were further identified as reversible albumin binders. These HSA binders possess a high affinity towards HSA and can be coated with targeting ligands and radionuclides, forming HSA-binding conjugates. The concept of using reversible HSA-binders to modulate the blood circulation time and hence the pharmacokinetic profile of radiolabeled molecules was adapted and applied by other groups [128]. Besides HSA in an individual form, the radionuclide (usually 99mTc) labeled HSA aggregates are widely used in clinics. For example, 99mTc-labeled HSA-based NPs were prepared by radiolabeling of 99mTcO4− to protein using SnCl2 as the reducing agent at a wide pH range of 3.8–8.0 [131]. The size of formed HSA-based aggregates depended on the amount of initial reagents and reaction time, and varied from nanometers to micrometers. The HSA-based NPs, which are generally formed via cross-linking reaction with glutarealdehyde, can be decorated with active targeting ligands and simultaneously labeled with radionuclides. The radiolabeling HSA-based NPs were also prepared using 99mTc(CO) +3 precursor, possessing the high affinity binding to the histidine residues on the protein surface [132]. The more recent method for obtaining radiolabeled HSA NPs was reported by Charkavarty et al. [133]. Authors developed one-pot synthesis protocol fabrication of small sized (4–5 nm diameter), water-soluble, intrinsically radiolabeled 64Cu metal NPs capped within HSA. The synthesis was based on the formation of strong complex between Cu and HSA at pH ~ 12 with the following heating at ~ 55 °C that led to the formation of 64Cu-HSA nanocomposites. Also Tian with co-workers developed 131I-labeled HSA coated manganese dioxide NPs through the mixing HSA solution with manganese chloride forming HSA-MnO2 NPs with further labeling radionuclide 131I using a standard iodogen oxidation method [134].
Currently, there is a significant interest in fabrication of multifunctional HSA delivery systems, which combine several properties in one drug carrier. For example, HSA-based NPs can simultaneously contain contrast agents for MRI imaging, radionuclides for PET, antitumor drugs for combined thermotherapy and targeted ligands for active targeted delivery.
Dendrimers
Dendrimers can be considered as well-defined monodisperse and globular nanovectors, possessing explicit architecture and composition with highly controllable size and surface properties [135]. They consist of a core and several layers with active terminal groups. The Vögtle, Newkome, and Tomalia scientific groups firstly reported on dendrimers in the late of 1970s and early 1980s independently. Nowadays, the application of dendimers in biomedicine as drug delivery platform is rapidly growing. The design principle of dendrimers, including surface/interior chemistry, size generation, shape, flexibility and composition, has been comprehensively reviewed and described in the work [136]. Various types of dendrimers including polyamidoamine (PAMAM), polypropyleneimine (PPI), poly(glycerol-co-succinic acid), poly-l-lysine (PLL), melamine, triazine, poly(glycerol), poly(2,2-bis(hydroxymethyl)-propionic acid) and PEG, as well as carbohydrate-based and citric-acid-based ones, have been developed as drug delivery platforms [137,138,139,140]. Among them, PAMAM- and PPI-based dendrimers have been some of the most widely investigated vectors that have gained tremendous attention [141]. The surface of dendrimers is rich of reactive (terminal) groups, which can be functionalized with targeting ligands. A wide selection of ligands (biotin, folic acid, amino acids, peptides, aptamers and mAbs) was successfully conjugated with dendrimers surfaces. The biodistribution of dendrimers has been investigated by administration of radiolabeled dendrimers in animals. The charge of dendrimers has a significant impact on the biodistribution and toxicity of dendrimers. As demonstrated, the cationic dendrimers possess low circulation times compared to anionic dendrimers [142]. Moreover, the cationic dendrimers were more toxic than anionic dendrimers, which can be related with membrane disruption due to interactions with negatively charged cell membranes [143].
Considering the physical half-life of radionuclides and radiolabeling strategies, dendrimers are mainly modified with chelators attached to the surface with further labeling with radionuclides (post-labeling approach) [144]. Medically relevant radionuclides were successfully applied to form stable dendrimers-radionuclide conjugates. Most of the data are associated with the preclinical studies of radiolabeled dendrimers. For example, PAMAM dendrimers functionalized with 10-[(4-carboxy-1-oxidopyridin-2-yl)methyl]-1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid (H4do3a-pyNO‑C) can be mentioned. This formulation was used for radiolabeling with 177Lu with high radiochemical purity [145]. The pioneering work in dendrimer radiolabeling was performed by Mukhtar et al., who synthesized two water soluble dendritic porphyrins for radiolabeling with 99mTc [146]. Agashe et al. also showed the potential of 99mTc-labeled dendrimers using PPI dendrimers for the biodistribution investigations [147]. For solely nuclear imaging purposes, dendrimers were radiolabeled with 76Br to monitor angiogenesis using PET imaging [148]. It should be noted that radiolabeling with dendrimers could occur at room temperature [149]. Recently, Garrigue et al. reported on self-assembling supramolecular dendrimers bearing NOTA for complexing with 68Ga [150]. The 68Ga-labeled dendrimers showed an effective tumor targeting in case of prostate, glioblastoma, colorectal and pancreatic cancers. The radionuclide-based theranostic dendrimers labeled with radionuclides emitting both β and γ rays simultaneously (177Lu, 131I, 188Re) were investigated in References [151,152,153,154]. For instance, the theranostic 131I-labeled PAMAM dendrimers modified with PEG and chlorotoxin (CTX) as the targeting agent were employed for targeted SPECT imaging and radiotherapy of a matrix metalloproteinase-2 (MMP-2) overexpressing xenografted glioma model in vivo [152]. It should be noted that iodinated dendrimers are excellent computer tomography (CT) contrast agents, what suit for multimodal imaging (combination of SPECT with CT). The 188Re-labeled PLL dendrimers (ImDendrim) is into Phase I clinical testing (clinical trial NCT03255343) for treatment of colorectal cancer.
For improved diagnosis accuracy, two or more modalities of contrast imaging are combined to overcome the disadvantages of each individual technique. For this reason, radionuclide-labeled dendrimers conjugated with other imaging agents (e.g. fluorescent moieties). The development of dual SPECT/fluorescence imaging dendrimers platform is quiet useful, because radionuclide-based SPECT imaging allows increased depth penetration whereas near infrared (NIR) optical imaging provides excellent real-time spatial resolution. Urano et al. employed a PAMAM dendrimer platform that can be linked to both radionuclides and optical probes, enabling dual-modality scintigraphic and five-color NIR optical lymphatic imaging [155]. In case of PET/NIR optical imaging, Wang et al. developed PAMAM dendrimer platform simultaneously conjugated with 64Cu and NIR fluorescence-emitting dye Cyanine5.5 (Cy5.5) for dual-modality imaging of ovarian cancer [156].
Polymeric micelles
The polymeric micelles are defined as organized self-assembly composed of amphiphilic macromolecules (amphiphilic, di- or tri-block copolymers) in a block-selective solvent [157]. They consist of a hydrophobic core and hydrophilic corona shell, exhibiting a wide range of sizes (usually 5–100 nm). As described in many works, the hydrophobic core serves as anchors for poorly water soluble drugs. The polymeric micelles possess the ultra-stability and can accumulate in tumor tissue through the EPR effect. A number of polymeric micelles with various structural organization as carriers for anticancer therapeutic and diagnostic agents was reported in literature [157].
Similar to other delivery systems, polymeric micelles can be functionalized with targeting ligands. Commonly used ligands are mAbs, carbohydrate moieties, aptamers, proteins, and peptides [158]. Widely used targets include the EGFR, Her2, folate receptor, and PSMA.
To incorporate radionuclide in micelle structure, the chelation techniques can be employed. In 1995, Trubetskoy and Torchilin firstly incorporated amphichilic chelating agents such as DTPA-phosphatidylethanolamine (DTPA-PE) and DTPA-stearylamine (DTPA-SA) into PEG-modified micelles and used these micelles for γ-scintigraphy of the lymphatic system after subcutaneous administration in rabbits. Nowadays, with the development of micelle fabrication, the different chelators for the radionuclide labeling can be easily conjugated with the end-groups of micelle polymeric shell. Mainly, the radiolabeling of polymeric micelles is occurred on the hydrophilic shell, while hydrophobic drugs are encapsulated in the core of the micelles. In this case, post-labeling approach is used, when chelators (DTPA or DOTA) are conjugated to the hydrophilic corona shell and further bind the radionuclides. Also, radionuclides can be encapsulated in the hydrophobic core of the micelles using a lipophilic radionuclide-ligand complex during the micelle formation. The micelles are usually functionalized with diagnostic radionuclides such as 18F [159, 160], 111In [161,162,163,164] and 64Cu [165,166,167] for further evaluation of in vivo biodistribution and targeting efficiency. There are only a few works, where micelles were labeled with therapeutic radionuclides such as 90Y [168], 131I [169]. The micelles loaded with γ emitters (99mTc and 111In) have extensively been investigated for in vivo biodistribution studies using SPECT imaging [162, 170]. Another more relevant example was reported by Miura, who developed DOTA-functionalized polymeric micelles for labeling with 111In [171]. Later, Jensen with co-workers developed triblock PEG‐pHEMA‐PCMA micelle modified with macrocyclic chelator 2,2′‐(1,4,8,11‐tetraazabicyclo[6.6.2]hexadecane‐4,11‐diyl)diacetic acid (CB‐TE2A) using pre-labeling approach for complexation with radioisotope 64Cu. The conjugation of CB‐TE2A chelator was performed to the primary alcohols of the pHEMA block through 4‐dimethylaminopyridine–catalyzed 1‐ethyl‐3‐(3‐dimethylaminopropyl)carbodiimide coupling in dimethylformamide (DMF) [172].
In clinical trials polymeric micelles received authorization for evaluation mainly as anticancer drug carriers [173]. Almost all of them can be prepared by self-assembly of amphiphilic block copolymers containing hydrophilic PEG blocks and hydrophobic polyester, poly(aspartic acid), or poly(glutamic acid) blocks. The drug molecules used in these systems are mostly classical cancerostatics (paclitaxel, camptothecin and its analogue doxorubicin, or platinum complexes). Paclitaxel loaded polymeric micelles (Genexol PM) have reached the most advanced testing stages and received approval for the use in Europe and Korea for the treatment of breast and small cell lung cancer. Ulbrich et al. gave a clear description on clinical studies of polymeric micelles as anticancer drug carriers used in cancer therapy [173]. As mentioned above, the hydrophobic core of micelles is preferable for incorporating of hydrophobic drugs while hydrophilic corona shell suits for radiolabeling. From this point of view, these so-called theranostic micelles are generally considered as multifunctional platform that combines drugs and imaging agents for PET or SPECT.
Polymeric-based NPs
Polymeric-based NPs can be defined as solid polymer particles or aggregates (micro- and nanosized ranges), where the bioactive compounds are encapsulated into the polymer matrix or coated with the surface of polymer NPs. There is no real classification of polymeric NPs and each literature review uses its own grouping. In this section, we did not include polymeric micelles, which have already been described above. The polymeric NPs are currently applying for different biomedical purposes, including drug and gene delivery, tissue engineering, bioimaging and so forth [174]. They are able to form highly stable complexes with radionuclides as well as permit a rapid labeling of targeting ligands. The techniques of incorporating radionuclides into polymer structure were adapted from the strategies for radiolabeling of proteins. In general, pre-labeling and post-labeling approaches are used for incorporating radionuclides with chelating agents. As for the pre-labeling method, the radionuclides are conjugated with chelating agent before their attachment to the polymer structure. In case of post-labeling approach, the polymeric NPs can be decorated with chelators via conjugation reaction or polymerization of monomers before their radiolabeling [173]. The DOTA and DTPA are most frequently used chelators that demonstrated high stability complexation with 68Ga, 64Cu and 111In. The attachment of chelating agents to the polymer structure is usually achieved via functional groups in chelators that can react with free amines (active esters, isothiocyanates), sulfhydryl- (maleimides, iodoacetamides), carboxylate- (amines, alcohols) or alkynyl- (azide) groups of the polymer to form stable polymer-chelator conjugates [80]. The classical organic iodination procedure is applied for polymer radiolabeling. The direct electrophilic iodination of tyrosine residues using so-called the Bolton–Hunter reagent (i.e., radiolabeled N-succinimidyl-3-(4-hydroxyphenyl) propionate) can be considered as the mostly varied radiolabeling method in case of polymer-based structures. Another smart and straightforward method for radiolabeling of polymeric NPs involves the formation of polyvinyl phenol particles surrounded by PEG [175]. The polyvinyl phenol is able to form a stable core, where 125I radionuclides can be introduced via electrophilic aromatic substitution.
Polylactic acid (PLA), polyglycolic acid (PGA) or their copolymer poly(lactic-co-glycolic acid) (PLGA) belong to the most advanced biodegradable polymers used in the preparation of polymer-based nanocarriers. Moreover, PLGA and PGA are already approved by FDA for clinical use in macroformulations. The surface of polymeric NPs is frequently coated with hydrophilic polymers (e.g. PEO) to prevent the adsorption of plasma proteins. In case of nuclear imaging, radionuclides such as 11C, 18F, 64Cu, 76Br, 99mTc, 111In, and 90Y have been used with a wide range of synthetic polymers to formulate nanosized carriers [159, 176, 177]. For example, the PLGA NPs, labeled with 99mTc, are successfully used in diagnostics for imaging of lungs [178] and sentinel lymph nodes [177]. 111In-labeled galactosylated PLGA NPs have been developed as trackable carriers for the liver specific delivery of drugs [179]. Sirianni et al. developed radiolabeled PLGA NPs with biotinylated 18F prosthetic groups for their tracking imaging using PET [180]. As for PLA, Banerjee et al. fabricated PSMA-targeted PLA-based NPs radiolabeled with 111In for SPECT imaging of PSMA-expressing tissues [181].
Natural polymers such as chitosan and dextran are also actively used for delivery of diagnostic radionuclides. Various formulations based on radiolabeled chitosan were reported. The 99mTc/131I-labeled water-soluble chitosan derivatives were used as SPECT/PET imaging agents [182]. Akhlaghi et al. performed the radiolabeling of chitosan derivatives with 66Ga using chelator DTPA. The use of DTPA for complexation with 66Ga allowed to achieve the highest efficiency to prevent radionuclide leakage [183]. Also, Lee et al. reported a facile method to label PEOylated-chitosan NPs with 64Cu, using azide-functionalized chitosan and DOTA derivatives with appending dibenzyl cyclooctyne moieties. This formulation allowed fast labeling (~ 30 min) with high radiolabeling yield (98%) and showed a tumor uptake in adenocarcinomic human alveolar basal epithelial cells (A549) tumor-bearing mice. The nanosized glycol chitosan NPs were labeled with 64Cu via copper-free click reaction for study of in vivo biodistibution. The dextran is also effectively used for targeted delivery of radionuclides. Moreover, dextran-based carriers was proved to be effective in clinical studies (Phase I and II), and a clinical Phase III study with oral cavity squamous cell carcinoma patients confirmed the detection and accurate prediction of the pathologic nodal status with 0% false negative results [184].
Outlooks
At present moment, most of the described organic based NPs are used to optimize radionuclide delivery: increase circulation time, avoid non-specific activity and provide tumor accumulation. The advances in surface chemistry enable different functionalization of organic NPs with targeting ligands. Several issues should be taken into account before implementation of organic NPs into clinics. First, the appropriate and convenient approach of radiolabeling should be provided to guarantee sufficient radiochemical stability. Second, radiolabeling should not influence the particle size, shape, surface charge and composition in order to provide optimal pharmacokinetics and biodistribution. Besides, the individual characteristics of radionuclide carriers should be additionally considered: biocompatibility, biodegradability, utilization of NPs components and their cytotoxicity. The recent studies on radionuclide delivery using organic NPs were presented in Table 2.
Inorganic NPs
Another type of radionuclide delivery system is NPs of inorganic nature. Such NPs can consist of different inorganic materials with the sizes ranging from the nanometers to micrometers [185]. The composition and structure of the NPs core determine their unique properties. For example, the use of gold NPs (Au NPs) as a core makes it possible to perform X-ray and photoacoustic imaging using such delivery systems. Whereas the utilization of an iron core allows NPs to be used for MRI imaging (Fig. 7) [186]. Despite the unique physicochemical properties of NPs and their diagnostic potential, the use of such carriers for the delivery of radionuclides in clinical practice is rather limited. It is associated with undesired toxicity, low targeting efficiency, short circulation time in the bloodstream and so forth [187]. In this regard, only a few inorganic NPs-based delivery systems reached the stage of clinical research [188].

(this figure is adopted from Yu et al. [189], Wang et al. [190], Liu et al. [191], Phillips et al. [192], Hoffman et al. [193] with required copyright permission)
Schematic illustration of multimodal imaging of radiolabeled inorganic NPs: plasmonic NPs for X-ray visualization, magnetic NPs for MRI visualization, C-based for optical imaging. Si-based NPs requires additional functionalization by contrast agents to be visualized
Plasmonic and magnetic NPs (Au, Fe3O4)
Au NPs are widely used in biology and medicine as drug delivery carries due to their unique optical properties [194]. Indeed, pronounced localized surface plasmon resonance allows their visualization possibility in vitro and in vivo using photoacoustic [195] and X-ray imaging [196]. Optical properties of Au NPs strongly depend on their physicochemical composition. The modern chemical synthetic techniques allow fabrication of monodispersed Au NPs with well-defined size and shape [197]. Recently, Au NPs were also employed as core-particles for radionuclides in radiotherapy and radiodiagnostics.
Similar to the organic NPs, in order to achieve targeting properties of Au NPs-based radionuclide delivery systems, different moieties such as peptides, antibodies, aptamers, nucleic acids are employed. These multifunctional platforms allow targeted delivery of radionuclides with visualization of radionuclides biodistribution by PET/CT or SPECT. In the recent study, 45 nm Au NPs were integrated with radioactive 125I. Navigation of Au NPs-based radionuclide delivery platform to the tumor site was provided by conjugation of Au NPs surface with cyclic Arg-Gly-Asp peptide. In vivo investigation of the developed delivery system revealed acute apoptosis and tumor growth suppression of NCI-H446 tumor-bearing mice after 2 days post treatment [198]. In another in vitro comparative study authors employed 30 nm Au NPs conjugated with mAbs (panitumumab), which target breast cancer (BC) cells overexpressing EGFR. 177Lu was used as radioactive compound in Au-based delivery system. The obtained data showed that the EGFR overexpressing cells demonstrated lower viability than BC cells with lower EGFR expression [199]. McLaughlin et al. used so-called α generator based on Au NPs to target biologically relevant receptors. In particular, female BALB/c mouse was used as a model for the targeted delivery of radionuclides. In this case, multiple α radiations were emitted by the 225Ac daughters while targeting the receptors in vivo. Such radioactive generators can potentially enhance the efficiency of tumor therapy [200]. Au NPs can appear not only as a core element for the radionuclide delivery platform, but as radioactive part of the system as well. Katti et al. demonstrated a novel approach to treat prostate cancer with 198Au NPs, which were modified with glucoside molecules mangiferin. In vivo experiments on severe combined immunodeficient (SCID) mice revealed over 85% reduction of bearing prostate tumor volume as compared to the control groups [201].
Superparamagnetic iron oxide NPs (SPIONs) are also widely used as a core element for the delivery of radionuclides. SPIONs are biocompatible and clinically approved NPs [202], which enable real-time visualization of drug biodistribution with MRI. Similar to Au NPs, SPIONs can be synthesized of various shape and size that affect their MRI contrast [203], as well as interaction with cells and tissues [204]. Such different behavior was demonstrated by Radovic et al. in vivo on example of 90Y labeled differently sized and coated SPIONs. The biodistribution of magnetic NPs was probed on Wistar rats. The data revealed that uncoated SPIONs end up in liver in a lower rate compared to PEGylated NPs [205]. Targeting features of Fe3O4-based radionuclide delivery systems can be achieved either by surface functionalization or by applying magnet in the close proximity to cells. The latter possibility was discussed in the recent work, where magnetic NPs were modified with 165Ho and cytostatic drug, and navigated into tumor area using external magnet. In vitro and in vivo results suggested that the external magnetic field navigation was achieved, since higher amounts of 165Ho were observed in tumor than in liver, which led to the inhibition of tumor growth [206]. In another study, radioisotope 125I was integrated into Fe3O4-Ag heterodimer to achieve dual-modality imaging with MRI and CT. The obtained delivery platform showed high radiolabeling efficiency as well as reduced T2-MRI signal intensity [207]. Recent work demonstrated radiolabeling of Fe3O4 NPs of 20 nm size with sodium pertechnetate (99mTc- pertechnetate) and the developed system was probed on male Wistar rats. The obtained results showed that this type of NPs could be introduced in the laboratory animals via intravenous injection and biodistribution could be controlled by external magnetic field [208]. Isotope 59Fe can be also used as radioactive element incorporated into the crystal lattice of Fe3O4 NPs. An advantage of this approach was the fact that radioactive 59Fe did not alter MRI contrast properties of Fe3O4 NPs. Therefore, the developed carriers enabled bimodal visualization (MRI/CT) as demonstrated in vivo (Fig. 8) [193].

(this figure is reproduced from Hoffman et al. [193] with required copyright permission)
Schematic illustration of SPION-based NPs fabrication (top), sagittal MR images of mouse animal model (bottom). The blue arrows indicate the brain, the red arrows indicate the liver and the yellow arrows indicate the bladder
Silica-based NPs
Silica NPs are already widely used as delivery carriers of various bioactive compounds due to the tailored physicochemical properties [209]. One of the great advantages of silica NPs is its porosity and, thus, high surface area, what makes Si NPs good candidates for employment as drug carriers. Silica NPs are usually prepared using chemical synthesis with a good possibility of size, shape and pore-size variation [210]. It is worth noting that some of silica-based materials are approved by Food and Drug Administration and generally recognized as safe [211]. Therefore, unlike plasmonic NPs, ultrasmall 124I modified silica-based NPs (‘C dots’) were already employed in clinical studies in 2014 on humans. Phillips et al. [192] developed radionuclide Si-based carriers, which possessed dual imaging properties: PET/SPECT and optical (due to the additional labeling with fluorescent markers). The obtained results on metastatic melanoma patients revealed good in vivo particle stability and their distinct, reproducible pharmacokinetics without any adverse effects. The development of Si-based NPs ‘C dots’ was continued in the work by Chen et al. [212]. They were able to optimize the synthesis of carriers in water and fabricated a novel PEGylated ultrasmall silica particles labeled with Cy5 dye. Si-based NPs were further conjugated with chelators and anti-HER2 scFv fragments. This allowed to achieve more efficient tumor-targeting with bulk renal clearance, which surpasses already existing characteristics of scFv molecules and scFv-conjugated NPs larger than 10 nm. The experiments were conducted in vivo on BT-474 tumor-bearing mice and revealed a favorable pharmacokinetics and clearance with decreased hepatic and reticuloendothelial system uptake (Fig. 9).

(this figure was reproduced from Chen et al. [212] with the required copyright permission)
In vivo HER2-targeted PET imaging in xenograft breast cancer models. Serial coronal and axial tomographic PET images acquired at 2, 24, 48, and 72 h post i.v. injection of radiolabeled particle immunoconjugates in groups of tumor-bearing mice as follows: a Targeted group: 89ZrDFO-scFv-PEG-Cy5-C’ dots in BT-474 mice. b Non-targeted group: 89Zr-DFO-Ctr/scFv-PEG-Cy5-C’ dots in BT-474 mice. For each group, maximum intensity projection (MIP) images were also acquired at 48 h p.i. H: heart, B: bladder, L: liver. c Representative MIP PET, CT, and PET/CT fusion images of 89Zr-DFO-scFv-PEG-Cy5-C’ dots in a BT-474 tumor-bearing mouse. BT474 tumors are marked with yellow arrows
Prior first clinical trials of Si-based radionuclide-labeled carriers, series of pre-clinical studies were conducted. For example, mesoporous silica NPs of 400 nm size were used to transport stable isotope 166Ho in mice with orthotopic non-small cell lung cancer A549-luciferase expressing tumor. The administration of the delivery systems were intravenous and biodistribution was monitored using luciferase bioluminescence [213]. Pascual et al. improved targeting properties of developed Si-based systems by functionalization of NPs with Mucin 1 aptamer that is specific to breast cancer. Pronounced targeted delivery of 99mTc into tumor of MDA-MB-231 tumor-bearing Balb/c mice was shown and monitored with SPECT and bioluminescence options [214]. In another study silica NPs were modified with anti-HER2 and labeled with 99mTc. Antibody conjugation allowed to target breast carcinoma cells. Radionuclides delivery efficiency was probed in vivo on tumor xenograft models in mice. Biodistribution of the developed radioactive formulation was monitored with optical fluorescence microscopy due to the additional labeling of Si-based carriers with indocyanine green (ICG) [215].
It is worth mentioning that mesoporous silica NPs can be simultaneously loaded with various cargoes. For example, in the recent study, mesoporous silica NPs were loaded with 89Zr and photosensitizer Chlorin e6, which can be activated by Cherenkov radiation from 89Zr. Therefore, the developed radionuclide delivery systems overcame the limitations of conventional photodynamic therapy, which requires light source to activate photosensitizer [216].
Carbon-based NPs
Carbon-based nanomaterials, which include fullerenes, carbon nanotubes (CNTs), carbon dots (CDs), nanodiamonds, graphene and its derivatives, are of significant interest nowadays due to unique optical, thermal and mechanical properties [217]. These materials can be used as drug delivery carriers, which can be simultaneously visualized by fluorescence microscopy [218]. However, there are some concerns about toxicity and biocompatibility of carbon-based materials. For example, due to the high hydrophobicity and non-biodegradability of CNTs, they possess reduced biocompatibility, what can limit their applications in biology and medicine [219]. It has significant importance in the field of nuclear medicine due to the reduced clearance of the radionuclides, what can increase the toxic effect of fabricated radioformulation. In order to overcome above mentioned drawbacks, the surface of CNTs can be functionalized with biocompatible polymers to improve their solubility and biocompatibility, thus, reduce their toxicity [220]. Therefore, recently single-walled CNTs were successfully employed to deliver radionuclides into cells. In this study, the surface of CNTs was simultaneously modified with tumor-specific mAbs, chelating agent (for radiolabeling with 111In) and fluorescence markers. The biodistribution 111In labeled CNTs was possible to monitor with fluorescence microscopy. Such multifunctional CNT-based delivery platform demonstrated effective targeting features with simultaneous visualization due to fluorescent moieties [221]. In another study, surface of single-walled CNTs was modified with biantennary carbohydrates in order to increase biocompatibility and solubility of CNTs. The cavities of CNTs were filled with Na125I and the developed CNT-based radiocarriers were intravenously administered in mice and tracked with CT. It has been demonstrated that the tunability of CNT surface modification offers versatility of organ specific delivery of radionuclides [222]. Ruggiero et al. developed CNT-based carriers to deliver 225Ac into the tumor angiogenic vessels, which express the monomeric vascular endothelial-cadherin. Antibody and chelating molecules coating significantly decreased toxicity of the delivery platform as shown in the murine model [223].
Likewise CNTs, pristine graphene and graphene oxide (GO) can induce certain toxicity, however, surface coating of graphene and GO by biocompatible polymers reduces their toxic effects [224, 225]. Therefore, graphene-based materials have been also successfully employed to deliver radionuclides in vitro and in vivo. Indeed, in the recent study Chen and co-workers developed graphene-based radionuclide carriers (131I) coated with PEG to reduce their toxicity. Synthesized delivery platforms demonstrated increased efficiency against cancer cells compared with the free 131I isotopes [226]. Surface functionalization can not only reduce toxic effects of graphene materials but also increase affinity of radionuclide labeling. Indeed, non-covalently PEGylated GO nanoplatelets demonstrated increased labeling with 166Ho [227]. In another work, Zhao et al. proved that polydopamine surface functionalization of GO resulted in enhanced adsorption of U(VI) [228].
Other carbon-based material, nanodiamonds, is generally considered to be non-toxic and biocompatible in various cell types [229]. Therefore, this material can potentially be a good candidate to deliver radiotherapeutics into cells and tissues. Rojas et al. labeled nanodiamonds with functionalized amino groups with 18F and monitored the biodistribution of developed systems in rats and mice. Authors observed changes in the biodistribution of the developed formulations by the addition of surfactant agents in suspension of nanodiamonds, [230]. In another study, aminated nanodiamonds were modified with 223Ra and 211Pb radionuclides with the 93% and 94% of labeling efficiency respectively [231].
Inorganic NPs labeling approaches
While radiolabeling of antibodies and organic nanoparticles is mostly limited to the use of chelating agents, modification of inorganic NPs with radionuclides does not always require them. For the chelator-based labeling the suitability of the chelator agent for the radionuclide is essential. Prior the conjugation with the chelator, the surface of NPs should be modified with addition of carboxyl, thiols and amino groups. After that the further radiolabeling process is relatively straightforward. Since most of the chelators, such as DOTA, NOTA, BMEDA, and so forth are reactive to carboxyl, thiol and amine groups, they can be incubated with the modified NPs until the conjugation is complete. Afterwards, using the same strategy, NPs can be simply shaken with radionuclides in an aqueous solution. The main drawback of the chelator-based functionalization is that there is no universal chelating agent suitable for every isotope, which means a proper chelator should be selected prior labeling to achieve an efficient modification [232]. However, such surface modification changes the pharmacokinetics of NPs, which can reduce the overall effectiveness of radionuclide delivery. Another setback of the chelator-based labeling is the possible detachment of radioisotope, which decreases specificity of delivery and therefore hampers the potential therapeutic and diagnostic effect of a delivery system [233].
Therefore, scientific community is also focused on the development of chelator-free labeling approaches. Chelator-free radionuclide labeling is subdivided on “hot-plus-cold” strategy, specific trapping and cation exchange (Fig. 10) [234, 235]. In the “hot-plus-cold” labeling approach radioactive compound is incorporated into the NPs during the synthesis procedure. Such method allows to obtain exceptionally stable labeling with high radiochemical yields. For example, Zhao et al. reported the 64Cu-alloyed AuNPs obtained via “hot-plus-cold” synthesis, showed increased radiolabeling stability [236]. However, the main drawback of this technique it that it can be used only for certain combinations of radionuclide and NPs. Additionally, the synthesis should be shorter than the half-life of chosen radionuclide in order to preserve its functionality [237]. The specific trapping method is based on the ability of some metal ions (in case of metallic nuclides) to form stable bonds with oxygen atoms, which are usually expressed on the surface of some inorganic NPs. Like for “hot-plus-cold” approach, specific trapping requires appropriate combinations of radionuclides and NPs. Moreover, for the in vivo applications, the stability of formed bond should be previously checked. Cation exchange labeling technique replaces an ion in a NP by another ion from the solution. This mechanism is driven by the relative thermodynamic stability of the reactants compared to the products. For this the appropriate solvent should be considered. As for other labeling methods, a specific combination of NP-cation pairing is of great importance for the cation exchange labeling.

(this figure was reproduced from Goel et al. [234] with the required copyright permission)
Schematic illustration of five major strategies for radiolabeling inorganic NPs. a Chelator-mediated complexation, b Specific trapping (b1) and ion-exchange (b2). c Hot-plus-cold precursor synthesis, d Proton beam activation. NP: nanoparticle
As an example of chelator-free radionuclide labeling, Chen et al. developed Si-based delivery systems labeled with 89Zr using deprotonated silanol groups inside mesopores or on the surface of silica NPs as inherent hard oxygen donors for radiolabeling. The resulted systems demonstrated enhanced stability of 89Zr in mice over 3 weeks [238]. Another group showed efficient chelator-free labeling of amorphous Si NPs with clinically relevant radioisotopes (89Zr, 68Ga, 111In, 90Y, 177Lu, 64Cu) by simple adsorption of radionuclides into the NP’s surface. In vivo stability of developed systems was demonstrated and the binding stability was found to be correlated with the hardness of the radioisotope [239]. The same group developed the radiolabeling methods of softer isotopes like 64Cu by functionalization of silica NPs with thiol groups. The labeling efficiency was improved from 74.4% for uncoated silica NPs to 94.5% for sulfur coated silica NPs due to the formation of thermodynamically stable bond [240].
Outlooks
The inorganic NPs can possess unique physicochemical properties, which can be tuned and optimized by choosing appropriate core material. Therefore, it is possible to find an optimal type of NPs, which will provide required functionalities of radionuclide delivery systems. In other words, apart from radionuclide delivery, NPs with magnetic and plasmonic properties make possible simultaneous visualization of developed radiopharmaceuticals by means of MRI or X-ray imaging. Majority of carbon-based delivery carriers can be visualized with optical fluorescence microscopy, however, to reduce toxic effects of these materials an additional surface modification is required. Silica-based carriers enables high loading capacity due to its porosity and wide functionalization possibilities, nevertheless, extra labeling of these carriers with, for example, fluorescence moieties make it possible optical biodistribution monitoring of such formulations.
The ability to perform chelator-free radiolabeling of inorganic NPs allows to achieve more stable incorporation of radionuclides, which reduces off target effects and overall increases amount of delivered cargo.
However, the toxicity of inorganic NPs is still a topic of research. Therefore, despite variety of inorganic NPs and possibilities of their functionalization, nowadays, they are mostly employed in pre-clinical studies with only a few types of inorganic NPs are clinically approved (ultrasmall Si NPs and Fe3O4 NPs as contrast agent).
Additionally, inorganic NPs much less effective for targeting of single tumor cells or tumors with lack of vasculature comparing to the mAbs and their fragments. This means that NPs cannot be employed for treatment of metastatic tumors, due to overall inefficiency [237].
The first steps toward clinical applications of radiolabeled inorganic NPs have been made in order to understand the biodistribution of these carriers inside the human body. There are a number FDA-approved inorganic platforms labeled with radionuclides, which were used for medical imaging. Despite the abovementioned ultrasmall Si NPs, 99mTc-labeled sulfur and stannous fluoride colloids were reported to be used as imaging agents in humans [241]. Therefore, we believe that the clinical employment of the inorganic NPs in the radiomedicine will seriously increase, due to the multimodal imaging, which these particles can offer, as well as fine tuning of their physicochemical properties, which helps to predict the biological cell response and the possible clinical outcomes. The recent studies on radionuclide delivery using inorganic NPs were presented in Table 3.
Microspheres
Another radionuclide delivery systems that have been successfully employed in pre-clinical and clinical studies are microspheres with size distribution in the range of 20–60 µm. Microspheres can be functionalized with active ligands to impart them properties of selectivity. Nowadays, there are three types of radioactive microspheres approved for clinical use—TheraSpheres (BTG, London, UK), SIR-Spheres (Sirtex Medical Limited, North Sydney NSW, Australia) and QuiremSpheres (Quirem Medical BV, Deventer, the Netherlands) [242].
These microspheres are usually used in radioembolization—a procedure, where the microspheres are injected via a catheter into the hepatic arteries. The efficiency of such microspheres is ensured by the fact that healthy hepatic tissue receives its supply mostly from the portal vasculature, while the malignant hepatic tumors are fed by arterial blood. Thus, administration of large microspheres through the hepatic artery leads to the clogging of small tumor capillaries that have a diameter of approximately 8–10 µm. As a result of higher concentration of blood vessels in tumor nodules compared to the normal tissue, the major amount of spheres becomes lodged and emit radiation in the tumor site, leaving the healthy parenchyma relatively unaffected [242,243,244,245].
All three clinically approved types of microspheres share a common mechanism of tumor targeting, each of them differs in the means of fabrication, radioactive labeling and choice of materials. These materials possess different physical traits and are applied in slightly different medical conditions.
TheraSpheres are insoluble glass beads with 90Y as an integral component and with diameter ranging between 20 and 30 µm. The synthesis of glass microspheres can be divided into two main stages: the particle production and radionuclide activation. The beads are obtained by melting at a high temperature a mixture of 89Y oxide (89Y2O3), aluminum oxide (Al2O3) and silicon dioxide (SiO2). The resulting glass is crushed, processed with a flame sprayer to even out the particles and passed through sieves to pick out particles with required distribution. The second stage consists of converting the non-radioactive 89Y to 90Y by neutron bombardment in a nuclear reactor. The production results in the formation of particles with 90Y as an integral component [246]. Such method allows incorporating relatively high amounts of yttrium. The combination of high activity and the particular size distribution allows to minimize the embolic effect, reducing the risk of reflux or stasis/retrograde flow [245, 247, 248].
The SIR-Spheres are microspheres based on biocompatible resin with 90Y impregnated on the particle surface. Each bead has a diameter ranging from 20 to 60 µm, and specific activity of 50 Bq at calibration. The activity of each particle, which is relatively small compared to the TheraSphere, is explained by the means of radionuclide incorporation: the 90Y is bound exclusively to the resin surface in the form of 90Y sulphate.
QuiremSpheres are poly-l-lactic acid based microspheres, containing 165Ho. The size of the particles varies from 15 to 60 µm and the use of poly-l-lactic acid allows to achieve the particle density of 1.4 g/cm3, which is closer to the density of blood (1.06 g/cm3). The 165Ho is activated to 165Ho with neutron activation in a nuclear reactor. Due to the short half-life period of 165Ho, each patient dose of QuiremSpheres needs to be prepared separately, thus, a specific activity can differ in each dose and depends on the needs of a patient [243, 249].
Another difference between yttrium-based microspheres and QuiremSpheres is the biodegradability of the latter. While TheraSpheres and SIR-Spheres remain in the patient liver as a permanent implant, the QuiremSpheres disintegrate leaving only insoluble holmium lactate [250]. Such degradation of microspheres positively influences the outcome of the treatment and enhances the efficiency of consecutive procedures of radioembolization whether they would be required in the future.
Choice of radionuclide
The choice of 90Y as a radionuclide for TheraSpheres and SIR-Spheres was based on its pure β-emitting properties with tissue penetration range of 90Y β-radiation of 2.5 mm (max. 11 mm), which in combination with targeting mechanism majorly restrict irradiation mostly to malignant tissues. In addition to that, 90Y has a physical half-life suitable for the radiotherapy (64.1 h) and 94% of radiation is delivered within 11 days after the drug admission [251].
The post-procedural visualization of microsphere distribution is commonly performed by analyzing the bremsstrahlung radiation produced by the decelerating β−, but such imaging lacks required precision in dose calculation and evaluation. However, the 90Y decay is accompanied by positron emission, which is although relatively weak (0.003%), but still can be detected with highly sensitive enough tomographs, thus, making PET possible with specific equipment (Fig. 11) [252].

(this figure is reproduced from Sangro et al. [253] with required copyright permission)
Superselective segmental radioembolization. a CT scan before treatment showing multinodular tumor. b PET scan showing intense radiation after administration of 90Y resin microspheres. c Significant atrophy of liver segment and lack of tumor activity 1 year after treatment
Although 90Y-based microspheres have proved to be overall effective for hepatic tumor treatment, two major drawbacks lead to the development of 166Ho microspheres. Firstly, in order to achieve a therapeutic amount of 90Y, long neutron activation times (more than 2 weeks) are required. Secondly, it was proved difficult to track the biodistribution of yttrium microspheres, due to the fact 90Y being an almost pure β− emitter [254].
166Ho emits low energy γ rays, which accompany electron radiation, when it reaches its steady state as daughter product 166Er. This makes possible the visualization of microsphere distribution by SPECT/CT. In addition, both holmium and its decay product erbium are lanthanides, which makes possible the visualization of microsphere by the means MRI of the paramagnetic metal [255].
Outlooks
Microspheres have already been successfully implemented in clinical practice long time ago and the recent developments of QuiremSpheres shows that the scientific community is currently developing new approaches in microspheres employment as radionuclide carriers. However, it is important to mention that implementation of microspheres is strictly limited to the radioembolization procedure, which heavily relies on the peculiar blood supply of liver cancer, and, therefore, cannot be used for the treatment of other oncological diseases. Further improvements of the microspheres can be focused around the enhancement of microsphere visualization, which would allow more precise estimation of the delivered radiation dose. In addition, modifications of sphere composition can be made in order to ensure the microsphere degradation and subsequent excretion.
Conclusion and future prospects
The radionuclides and their capacity to emit ionizing radiation have been employed in clinical practice not only for diagnostic, but also for therapy, with relevant contribution in the several fields of medicine. Given the nature of radioisotopes, as well as the desired goal (e.g. biochemical pathway investigation of radionuclide, bioimaging and anomaly detection, curative or palliative treatment or for theranostic purpose), an appropriate carrier platform should be attentively evaluated and suggested. Due to the advances in chemistry and cellular biology, many radionuclide delivery systems have been developed and demonstrated its efficiency compared to individual alternatives in vitro and in vivo. In this review article, we consider the most trendy radionuclide delivery systems, which can be categorized into four groups: (i) antibodies and their fragments, (ii) organic and (iii) inorganic nanoparticles, and (iv) microspheres. The translation of most of them into clinics has not progressed as rapidly as expected, since each of these platforms possess advantages and disadvantages.
Antibody-based delivery systems are widely employed in therapy and diagnostics due to their distinct pharmacokinetics: highly specific binding to the antigens, easy clearance because of relatively small size. The use of intact antibodies is restricted by poor penetration into solid tumors, whereas the immune system undergoes Fc-mediated bystander activation. However, this limitation can be overcome with antibody fragments. Nevertheless, due to the ultrasmall size of antibody fragments it is not always possible to perform effective labeling with radiometals, since pharmacokinetics of antibodies are affected by incorporating of chelators.
The use of NPs as radionuclide delivery platforms is rapidly developing field due to their unique physicochemical and biological characteristics. To date, many efforts are focused on the translation of developed radiopharmaceuticals based on NPs into clinical practice. However, their fully introduction in clinics is postponed due to technical and biological limitations. There is need for the development of more facile procedures for the surface modification and radiolabeling of NPs. At the same time, biological barriers can induce the different response to the same nanomaterials from patient to patient. The additional stabilization of NPs are highly required, since after the first contact of NPs with the biological fluids organic compounds tend to bind to the NPs surface forming so-called “corona” around the particles. This corona can induce NPs aggregation and significantly change pharmacokinetic profile of NP-based radiopharmaceuticals.
Compared to the inorganic NPs, there are some example of successful clinical trials of organic NPs, especially liposomes ([64Cu]MM-302 and 188Re-BMEDA liposomes). The clinical implementation of inorganic NPs is limited by concerns over their toxicity, non-optimal utilization and in vivo behavior. As an advantage, inorganic NPs provide additional possibilities of multimodal imaging, for examples, magnetic NPs as contrast agent for MRI, Au NPs as contrast agent for X-ray imaging, C-based NPs as contrast agent for fluorescence imaging. Inorganic and organic NPs enable high radionuclides payloads, flexibility of radiolabeling options and controlled physicochemical parameters (size, shape, chemical composition, stiffness), what results in increased amount of delivered radionuclides in the site of interest and improved tissue penetration and, thus, the efficiency of radiotherapy.
It seems that the scientific community is now at the stage of the developing the advanced hybrid radionuclide carriers with complex architecture taking advantages from each individual delivery system. The integration of various organic and inorganic NPs (e.g. liposomes, micelles, metal NPs, polymer conjugates etc.) in single smart platform allows to achieve multifunctionality and enable multimodal imaging. It also leads to further increase the loading capacity, enhance the biomembrane-crossing rate, and the tissue-penetration efficiency. At the same time, the functionalization of this hybrid platform with targeting moieties may enable the increased targeting delivery of radionuclides.
Availability of data and materials
Not applicable.
Change history
02 January 2020
After publication of this article, an error was found in the description of the holmium isotopes. 165Ho is a stable isotope a fraction of which is activated to 166Ho by neutron activation in a nuclear reactor [2]. In one paragraph of the published article, describing holmium containing QuiremSpheres, 165Ho should be replaced with 166Ho. The correct description is given below.
Abbreviations
- NIS:
-
expressing sodium/iodide symporter
- DNA:
-
deoxyribonucleic acid
- LET:
-
linear energy transfer
- SPECT:
-
single-photon emission computed tomography
- PET:
-
positron emission tomography
- PET/CT:
-
positron emission tomography/computed tomography
- Fab:
-
antigen-binding fragment
- Fc:
-
fragment crystallizable
- mAbs:
-
monoclonal antibodies
- RIT:
-
radioimmunotherapy
- AUC:
-
area under curve
- FcRn:
-
neonatal Fc receptor
- scFv:
-
single-chain variable fragments
- sdAbs:
-
single domain antibodies
- WT:
-
wild type
- DM:
-
double mutant
- ID/g:
-
injected dose per gram
- VH :
-
heavy chain variable
- VL :
-
light chain variable
- CEA:
-
carcinoembryonic antigen
- PSCA:
-
prostate stem-cell antigen
- PSMA:
-
prostate-specific membrane antigen
- FDG:
-
fluorodeoxyglucose
- TAG-72:
-
tumor-associated glycoprotein 72
- IgG:
-
immunoglobulin G
- CDR3:
-
complementarity-determining region 3
- EGFR:
-
epidermal growth factor receptor
- HER2:
-
human epidermal growth factor receptor 2
- DTPA:
-
diethylenetriaminepentaacetic acid
- 131I-SGMIB:
-
N-succinimidyl 3-guanidinomethyl-5-[131I]iodobenzoate
- bsAbs:
-
bispecific antibodies
- NHS:
-
N-hydroxysuccinimide esters
- SCN:
-
isothiocyanates
- TROP2:
-
trophoblast antigen 2
- HSG:
-
histaminesuccinyl-glycine
- DFO:
-
desferrioxamine
- CPTA:
-
4-(1,4,8,11-tetraazacyclotetradec-1-yl)-methyl benzole acid tetrahydrochloride
- FDA:
-
Food and Drug Administration
- NPs:
-
nanoparticles
- PEG:
-
poly(ethylene glycol)
- NODAGA:
-
2-(4,7-bis(carboxymethyl)-1,4,7-triazonan-1-yl)pentanedioic acid
- DOTA:
-
1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid
- NOTA:
-
1,4,7-triazacyclononane-1,4,7-triacetic acid
- BMEDA:
-
N,N-bis(2-mercaptoethyl)-N’,N’-diethylethylenediamine
- HMPAO:
-
hexamethylpropyleneamine oxime
- MRI:
-
magnetic resonance imaging
- EPR:
-
enhanced permeation and retention
- HSA:
-
human serum albumin
- BSA:
-
bovine serum albumin
- SLN:
-
sentinel lymph node
- DTPA:
-
diethylenetriaminepentaacetic acid
- HSA:
-
human serum albumin
- PAMAM:
-
polyamidoamine
- PPI:
-
polypropyleneimine
- PLL:
-
poly-l-lysine
- MMP-2:
-
matrix metalloproteinase-2
- CTX:
-
chlorotoxin
- HPAO:
-
3-(4′-hydroxyphenyl)propionic acid-OSu
- H4do3a-pyNO‑C :
-
10-[(4-carboxy-1-oxidopyridin-2-yl)methyl]-1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid
- CT:
-
computer tomography
- NIR:
-
near infrared
- DTPA-PE:
-
DTPA-phosphatidylethanolamine
- DTPA-SA:
-
DTPA-stearylamine
- CB-TE2A:
-
2,2′‐(1,4,8,11‐tetraazabicyclo(6.6.2)hexadecane‐4,11‐diyl)diacetic acid
- pHEMA:
-
poly(2-hydroxyethyl methacrylate)
- PCMA:
-
poly(4-methylcoumarin methacrylate)
- DMF:
-
dimethylformamide
- PBLG:
-
poly(γ-benzyl l-glutamate)
- PLA:
-
polylactic acid
- PGA:
-
polyglycolic acid
- PLGA:
-
poly(lactic-co-glycolic acid)
- PEO:
-
polyethylene oxide
- Au NPs:
-
gold nanoparticles
- SCID:
-
severe combined immunodeficient
- SPIONs:
-
superparamagnetic iron oxide nanoparticles
- ICG:
-
indocyanine green
- CNTs:
-
carbon nanotubes
- Cy5.5:
-
cyanine5.5
- CDs:
-
carbon dots
- GO:
-
graphene oxide
References
Yeong C-H, Cheng M, Ng K-H. Therapeutic radionuclides in nuclear medicine: current and future prospects. J Zhejiang Univ Sci B. 2014;15(10):845–63.
Srivastava SC, Mausner LF. Therapeutic radionuclides: production, physical characteristics, and applications. In: Baum RP, editor. Therapeutic nuclear medicine. Berlin: Springer; 2014. p. 11–50.
Oh J-R, Ahn B-C. False-positive uptake on radioiodine whole-body scintigraphy: physiologic and pathologic variants unrelated to thyroid cancer. Am J Nucl Med Mol Imaging. 2012;2(3):362–85.
Liu Z, Xing M. Induction of sodium/iodide symporter (NIS) expression and radioiodine uptake in non-thyroid cancer cells. PLoS ONE. 2012;7(2):e31729.
Huang R, Zhao Z, Ma X, Li S, Gong R, Kuang A. Targeting of tumor radioiodine therapy by expression of the sodium iodide symporter under control of the survivin promoter. Cancer Gene Ther. 2011;18(2):144–52.
Iyer AK, Duan Z, Amiji MM. Nanodelivery systems for nucleic acid therapeutics in drug resistant tumors. Mol Pharm. 2014;11(8):2511–26.
Bazak R, Houri M, Achy SE, Hussein W, Refaat T. Passive targeting of nanoparticles to cancer: a comprehensive review of the literature. Mol Clin Oncol. 2014;2(6):904–8.
Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12):6387–92.
Iyer AK, Khaled G, Fang J, Maeda H. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov Today. 2006;11(17–18):812–8.
Kesharwani P, Ghanghoria R, Jain NK. Carbon nanotube exploration in cancer cell lines. Drug Discov Today. 2012;17(17–18):1023–30.
Islam T, Josephson L. Current state and future applications of active targeting in malignancies using superparamagnetic iron oxide nanoparticles. Cancer Biomark Sect Dis Markers. 2009;5(2):99–107.
Teow Y, Valiyaveettil S. Active targeting of cancer cells using folic acid-conjugated platinum nanoparticles. Nanoscale. 2010;2(12):2607–13.
Patra JK, Das G, Fraceto LF, Campos EVR, Rodriguez-Torres M, Acosta-Torres LS, et al. Nano based drug delivery systems: recent developments and future prospects. J Nanobiotechnology. 2018;16(1):71.
Duvall WL, Croft LB, Ginsberg ES, Einstein AJ, Guma KA, George T, et al. Reduced isotope dose and imaging time with a high-efficiency CZT SPECT camera. J Nucl Cardiol Off Publ Am Soc Nucl Cardiol. 2011;18(5):847–57.
Galea R, Ross C, Wells RG. Reduce, reuse and recycle: a green solution to Canada’s medical isotope shortage. Appl Radiat Isot Data Instrum Methods Use Agric Ind Med. 2014;87:148–51.
Hoedl SA, Updegraff WD. The production of medical isotopes without nuclear reactors or uranium enrichment. Sci Glob Secur. 2015;23(2):121–53.
Yong KJ, Milenic DE, Baidoo KE, Brechbiel MW. Mechanisms of cell killing response from low linear energy transfer (LET) radiation originating from (177)Lu radioimmunotherapy targeting disseminated intraperitoneal tumor xenografts. Int J Mol Sci. 2016;17(5):736.
Kassis AI. Therapeutic radionuclides: biophysical and radiobiologic principles. Semin Nucl Med. 2008;38(5):358–66.
Wright CL, Zhang J, Tweedle MF, Knopp MV, Hall NC. Theranostic imaging of Yttrium-90. Biomed Res Int. 2015;2015:481279.
Graves SA, Hernandez R, Fonslet J, England CG, Valdovinos HF, Ellison PA, et al. Novel preparation methods of 52Mn for immunoPET imaging. Bioconjug Chem. 2015;26(10):2118–24.
McDevitt MR, Sgouros G, Finn RD, Humm JL, Jurcic JG, Larson SM, et al. Radioimmunotherapy with alpha-emitting nuclides. Eur J Nucl Med. 1998;25(9):1341–51.
McDevitt MR, Ma D, Lai LT, Simon J, Borchardt P, Frank RK, et al. Tumor therapy with targeted atomic nanogenerators. Science. 2001;294(5546):1537–40.
Müller C, van der Meulen NP, Benešová M, Schibli R. Therapeutic radiometals beyond 177Lu and 90Y: production and application of promising α-particle, β–particle, and auger electron emitters. J Nucl Med Off Publ Soc Nucl Med. 2017;58(Suppl 2):91S–6S.
Kratochwil C, Bruchertseifer F, Giesel FL, Weis M, Verburg FA, Mottaghy F, et al. 225Ac-PSMA-617 for PSMA-targeted α-radiation therapy of metastatic castration-resistant prostate cancer. J Nucl Med Off Publ Soc Nucl Med. 2016;57(12):1941–4.
Piron B, Paillas S, Boudousq V, Pèlegrin A, Bascoul-Mollevi C, Chouin N, et al. DNA damage-centered signaling pathways are effectively activated during low dose-rate Auger radioimmunotherapy. Nucl Med Biol. 2014;41(Suppl):e75–83.
Behr TM, Béhé M, Löhr M, Sgouros G, Angerstein C, Wehrmann E, et al. Therapeutic advantages of Auger electron- over β-emitting radiometals or radioiodine when conjugated to internalizing antibodies. Eur J Nucl Med Mol Imaging. 2000;27(7):753–65.
Plomp R, Dekkers G, Rombouts Y, Visser R, Koeleman CAM, Kammeijer GSM, et al. Hinge-region o-glycosylation of human immunoglobulin G3 (IgG3). Mol Cell Proteomics MCP. 2015;14(5):1373–84.
Morais M, Ma MT. Site-specific chelator-antibody conjugation for PET and SPECT imaging with radiometals. Drug Discov Today Technol. 2018;30:91–104.
Knowles SM, Wu AM. Advances in immuno-positron emission tomography: antibodies for molecular imaging in oncology. J Clin Oncol Off J Am Soc Clin Oncol. 2012;30(31):3884–92.
Wei W, Ni D, Ehlerding EB, Luo Q-Y, Cai W. PET Imaging of receptor tyrosine kinases in cancer. Mol Cancer Ther. 2018;17(8):1625–36.
Wei W, Jiang D, Ehlerding EB, Luo Q, Cai W. Noninvasive PET imaging of T cells. Trends Cancer. 2018;4(5):359–73.
Freise AC, Wu AM. In vivo imaging with antibodies and engineered fragments. Mol Immunol. 2015;67(2 Pt A):142–52.
Tsai W-TK, Wu AM. Aligning physics and physiology: engineering antibodies for radionuclide delivery. J Label Compd Radiopharm. 2018;61(9):693–714.
Wilder RB, DeNardo GL, DeNardo SJ. Radioimmunotherapy: recent results and future directions. J Clin Oncol. 1996;14(4):1383–400.
Kodaira S, Li HK, Konishi T, Kitamura H, Kurano M, Hasegawa S. Validating α-particle emission from 211At-labeled antibodies in single cells for cancer radioimmunotherapy using CR-39 plastic nuclear track detectors. PLoS ONE. 2017;12(6):e0178472.
Kraeber-Bodere F, Barbet J, Chatal J-F. Radioimmunotherapy: from current clinical success to future industrial breakthrough. J Nucl Med. 2016;57(3):329–31.
Larson SM, Carrasquillo JA, Cheung N-KV, Press OW. Radioimmunotherapy of human tumours. Nat Rev Cancer. 2015;15(6):347–60.
Rösch F, Herzog H, Qaim SM. The beginning and development of the theranostic approach in nuclear medicine, as exemplified by the radionuclide pair 86Y and 90Y. Pharmaceuticals. 2017;10(2):56.
Aluicio-Sarduy E, Ellison PA, Barnhart TE, Cai W, Nickles RJ, Engle JW. PET radiometals for antibody labeling. J Label Compd Radiopharm. 2018;61(9):636–51.
Stern LA, Case BA, Hackel BJ. Alternative non-antibody protein scaffolds for molecular imaging of cancer. Curr Opin Chem Eng. 2013;2(4):425–32.
Hu S, Shively L, Raubitschek A, Sherman M, Williams LE, Wong JYC, et al. Minibody: a novel engineered anti-carcinoembryonic antigen antibody fragment (single-chain Fv-CH3) which exhibits rapid, high-level targeting of xenografts. Cancer Res. 1996;56(13):3055–61.
Sundaresan G, Yazaki PJ, Shively JE, Finn RD, Larson SM, Raubitschek AA, et al. 124I-labeled engineered anti-CEA minibodies and diabodies allow high-contrast, antigen-specific small-animal PET imaging of xenografts in athymic mice. J Nucl Med Off Publ Soc Nucl Med. 2003;44(12):1962–9.
Yazaki PJ, Wu AM, Tsai SW, Williams LE, Ikler DN, Wong JY, et al. Tumor targeting of radiometal labeled anti-CEA recombinant T84.66 diabody and t84.66 minibody: comparison to radioiodinated fragments. Bioconjug Chem. 2001;12(2):220–8.
Tavaré R, McCracken MN, Zettlitz KA, Knowles SM, Salazar FB, Olafsen T, et al. Engineered antibody fragments for immuno-PET imaging of endogenous CD8+ T cells in vivo. Proc Natl Acad Sci USA. 2014;111(3):1108–13.
van Duijnhoven SMJ, Rossin R, van den Bosch SM, Wheatcroft MP, Hudson PJ, Robillard MS. Diabody pretargeting with click chemistry in vivo. J Nucl Med. 2015;56(9):1422–8.
Freise AC, Zettlitz KA, Salazar FB, Lu X, Tavaré R, Wu AM. ImmunoPET imaging of murine CD4+ T cells using anti-CD4 cys-diabody: effects of protein dose on T cell function and imaging. Mol Imaging Biol MIB Off Publ Acad Mol Imaging. 2017;19(4):599–609.
Tavaré R, Escuin-Ordinas H, Mok S, McCracken MN, Zettlitz KA, Salazar FB, et al. An effective immuno-PET imaging method to monitor CD8-dependent responses to immunotherapy. Cancer Res. 2016;76(1):73–82.
Viola-Villegas NT, Sevak KK, Carlin SD, Doran MG, Evans HW, Bartlett DW, et al. Noninvasive imaging of PSMA in prostate tumors with (89)Zr-Labeled huJ591 engineered antibody fragments: the faster alternatives. Mol Pharm. 2014;11(11):3965–73.
Hong H, Sun J, Cai W. Radionuclide-based cancer imaging targeting the carcinoembryonic antigen. Biomark Insights. 2008;3:435–51.
Debie P, Devoogdt N, Hernot S. Targeted nanobody-based molecular tracers for nuclear imaging and image-guided surgery. Antibodies. 2019;8(1):12.
Muyldermans S. Nanobodies: natural single-domain antibodies. Annu Rev Biochem. 2013;82:775–97.
Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol. 2005;23(9):1126–36.
Chatalic KLS, Veldhoven-Zweistra J, Bolkestein M, Hoeben S, Koning GA, Boerman OC, et al. A novel 111In-labeled anti-prostate-specific membrane antigen nanobody for targeted SPECT/CT imaging of prostate cancer. J Nucl Med Off Publ Soc Nucl Med. 2015;56(7):1094–9.
Vaneycken I, Govaert J, Vincke C, Caveliers V, Lahoutte T, De Baetselier P, et al. In vitro analysis and in vivo tumor targeting of a humanized, grafted nanobody in mice using pinhole SPECT/micro-CT. J Nucl Med Off Publ Soc Nucl Med. 2010;51(7):1099–106.
Gainkam LOT, Huang L, Caveliers V, Keyaerts M, Hernot S, Vaneycken I, et al. Comparison of the biodistribution and tumor targeting of two 99mTc-labeled anti-EGFR nanobodies in mice, using pinhole SPECT/Micro-CT. J Nucl Med. 2008;49(5):788–95.
Zhou Z, Vaidyanathan G, McDougald D, Kang CM, Balyasnikova I, Devoogdt N, et al. Fluorine-18 labeling of the HER2-targeting single-domain antibody 2Rs15d using a residualizing label and preclinical evaluation. Mol Imaging Biol MIB Off Publ Acad Mol Imaging. 2017;19(6):867–77.
Rashidian M, Ingram JR, Dougan M, Dongre A, Whang KA, LeGall C, et al. Predicting the response to CTLA-4 blockade by longitudinal noninvasive monitoring of CD8 T cells. J Exp Med. 2017;214(8):2243–55.
D’Huyvetter M, Vincke C, Xavier C, Aerts A, Impens N, Baatout S, et al. Targeted radionuclide therapy with A 177Lu-labeled Anti-HER2 nanobody. Theranostics. 2014;4(7):708–20.
Xavier C, Vaneycken I, D’huyvetter M, Heemskerk J, Keyaerts M, Vincke C, et al. Synthesis, preclinical validation, dosimetry, and toxicity of 68 Ga-NOTA-Anti-HER2 nanobodies for iPET imaging of HER2 receptor expression in cancer. J Nucl Med. 2013;54(5):776–84.
Keyaerts M, Xavier C, Heemskerk J, Devoogdt N, Everaert H, Ackaert C, et al. Phase I study of 68 Ga-HER2-nanobody for PET/CT assessment of HER2 expression in breast carcinoma. J Nucl Med Off Publ Soc Nucl Med. 2016;57(1):27–33.
Strohl WR. Fusion proteins for half-life extension of biologics as a strategy to make biobetters. BioDrugs Clin Immunother Biopharm Gene Ther. 2015;29(4):215–39.
Löfblom J, Feldwisch J, Tolmachev V, Carlsson J, Ståhl S, Frejd FY. Affibody molecules: engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Lett. 2010;584(12):2670–80.
Frejd FY, Kim K-T. Affibody molecules as engineered protein drugs. Exp Mol Med. 2017;49(3):e306.
Orlova A, Wållberg H, Stone-Elander S, Tolmachev V. On the selection of a tracer for PET imaging of HER2-expressing tumors: direct comparison of a 124I-labeled affibody molecule and trastuzumab in a murine xenograft model. J Nucl Med Off Publ Soc Nucl Med. 2009;50(3):417–25.
Altai M, Perols A, Tsourma M, Mitran B, Honarvar H, Robillard M, et al. Feasibility of affibody-based bioorthogonal chemistry-mediated radionuclide pretargeting. J Nucl Med Off Publ Soc Nucl Med. 2016;57(3):431–6.
Tolmachev V, Grönroos T, Yim C-B, Garousi J, Yue Y, Grimm S, et al. Molecular design of radiocopper-labelled affibody molecules. Sci Rep. 2018;8:6542.
Orlova A, Tran TA, Ekblad T, Karlström AE, Tolmachev V. (186)Re-maSGS-Z (HER2:342), a potential affibody conjugate for systemic therapy of HER2-expressing tumours. Eur J Nucl Med Mol Imaging. 2010;37(2):260–9.
Sedykh SE, Prinz VV, Buneva VN, Nevinsky GA. Bispecific antibodies: design, therapy, perspectives. Drug Des Dev Ther. 2018;12:195–208.
Trivedi A, Stienen S, Zhu M, Li H, Yuraszeck T, Gibbs J, et al. Clinical pharmacology and translational aspects of bispecific antibodies. Clin Transl Sci. 2017;10(3):147–62.
Brinkmann U, Kontermann RE. The making of bispecific antibodies. mAbs. 2017;9(2):182–212.
Altai M, Membreno R, Cook B, Tolmachev V, Zeglis BM. Pretargeted imaging and therapy. J Nucl Med Off Publ Soc Nucl Med. 2017;58(10):1553–9.
Luo H, Hernandez R, Hong H, Graves SA, Yang Y, England CG, et al. Noninvasive brain cancer imaging with a bispecific antibody fragment, generated via click chemistry. Proc Natl Acad Sci USA. 2015;112(41):12806–11.
Schoffelen R, Boerman OC, Goldenberg DM, Sharkey RM, van Herpen CML, Franssen GM, et al. Development of an imaging-guided CEA-pretargeted radionuclide treatment of advanced colorectal cancer: first clinical results. Br J Cancer. 2013;109(4):934–42.
van Rij CM, Lütje S, Frielink C, Sharkey RM, Goldenberg DM, Franssen GM, et al. Pretargeted immuno-PET and radioimmunotherapy of prostate cancer with an anti-TROP-2 x anti-HSG bispecific antibody. Eur J Nucl Med Mol Imaging. 2013;40(9):1377–83.
Heskamp S, Hernandez R, Molkenboer-Kuenen JDM, Essler M, Bruchertseifer F, Morgenstern A, et al. α-Versus β-emitting radionuclides for pretargeted radioimmunotherapy of carcinoembryonic antigen-expressing human colon cancer xenografts. J Nucl Med Off Publ Soc Nucl Med. 2017;58(6):926–33.
Ma MT, Blower PJ. CHAPTER 8. Chelators for diagnostic molecular imaging with radioisotopes of copper, gallium and zirconium. In: Crichton RR, Ward RJ, Hider RC, editors. Metal chelation in medicine. Cambridge: Royal Society of Chemistry; 2016. p. 260–312.
Ma MT, Donnelly PS. Peptide targeted copper-64 radiopharmaceuticals. Curr Top Med Chem. 2011;11(5):500–20.
Cusnir R, Imberti C, Hider RC, Blower PJ, Ma MT. Hydroxypyridinone chelators: from iron scavenging to radiopharmaceuticals for PET imaging with gallium-68. Int J Mol Sci. 2017;18(1):116.
Blower PJ. A nuclear chocolate box: the periodic table of nuclear medicine. Dalton Trans. 2015;44(11):4819–44.
Price EW, Orvig C. Matching chelators to radiometals for radiopharmaceuticals. Chem Soc Rev. 2014;43(1):260–90.
Wadas TJ, Wong EH, Weisman GR, Anderson CJ. Coordinating radiometals of copper, gallium, indium, yttrium, and zirconium for PET and SPECT imaging of disease. Chem Rev. 2010;110(5):2858–902.
Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol. 2008;26(8):925–32.
Boswell CA, Mundo EE, Zhang C, Bumbaca D, Valle NR, Kozak KR, et al. Impact of drug conjugation on pharmacokinetics and tissue distribution of anti-STEAP1 antibody-drug conjugates in rats. Bioconjug Chem. 2011;22(10):1994–2004.
Giersing BK, Rae MT, CarballidoBrea M, Williamson RA, Blower PJ. Synthesis and characterization of 111In–DTPA–N-TIMP-2: a radiopharmaceutical for imaging matrix metalloproteinase expression. Bioconjug Chem. 2001;12(6):964–71.
Zeglis BM, Davis CB, Aggeler R, Kang HC, Chen A, Agnew BJ, et al. Enzyme-mediated methodology for the site-specific radiolabeling of antibodies based on catalyst-free click chemistry. Bioconjug Chem. 2013;24(6):1057–67.
Oller-Salvia B, Kym G, Chin JW. Rapid and efficient generation of stable antibody-drug conjugates via an encoded cyclopropene and an inverse-electron-demand diels-alder reaction. Angew Chem Int Ed Engl. 2018;57(11):2831–4.
Adumeau P, Sharma SK, Brent C, Zeglis BM. Site-specifically labeled immunoconjugates for molecular imaging–part 1: cysteine residues and glycans. Mol Imaging Biol MIB Off Publ Acad Mol Imaging. 2016;18(1):1–17.
Nunes JPM, Vassileva V, Robinson E, Morais M, Smith MEB, Pedley RB, et al. Use of a next generation maleimide in combination with THIOMAB™ antibody technology delivers a highly stable, potent and near homogeneous THIOMAB™ antibody-drug conjugate (TDC). RSC Adv. 2017;7(40):24828–32.
O’Shannessy DJ, Dobersen MJ, Quarles RH. A novel procedure for labeling immunoglobulins by conjugation to oligosaccharide moieties. Immunol Lett. 1984;8(5):273–7.
Schwarz U, Wunderlich G, Brossmer R. Transfer of 131I and fluoresceinyl sialic acid derivatives into the oligosaccharide chains of IgG: a new method for site-specific labeling of antibodies. Nucl Med Biol. 1999;26(4):383–8.
Hu M, Chen P, Wang J, Chan C, Scollard DA, Reilly RM. Site-specific conjugation of HIV-1 tat peptides to IgG: a potential route to construct radioimmunoconjugates for targeting intracellular and nuclear epitopes in cancer. Eur J Nucl Med Mol Imaging. 2006;33(3):301–10.
Zeglis BM, Davis CB, Abdel-Atti D, Carlin SD, Chen A, Aggeler R, et al. Chemoenzymatic strategy for the synthesis of site-specifically labeled immunoconjugates for multimodal PET and optical imaging. Bioconjug Chem. 2014;25(12):2123–8.
Paterson BM, Alt K, Jeffery CM, Price RI, Jagdale S, Rigby S, et al. Enzyme-mediated site-specific bioconjugation of metal complexes to proteins: sortase-mediated coupling of copper-64 to a single-chain antibody. Angew Chem Int Ed. 2014;53(24):6115–9.
Rashidian M, Wang L, Edens JG, Jacobsen JT, Hossain I, Wang Q, et al. Enzyme-mediated modification of single-domain antibodies for imaging modalities with different characteristics. Angew Chem Int Ed. 2016;55(2):528–33.
Sugiura G, Kühn H, Sauter M, Haberkorn U, Mier W. Radiolabeling strategies for tumor-targeting proteinaceous drugs. Mol Basel Switz. 2014;19(2):2135–65.
Salacinski PRP, McLean C, Sykes JEC, Clement-Jones VV, Lowry PJ. Iodination of proteins, glycoproteins, and peptides using a solid-phase oxidizing agent, 1,3,4,6-tetrachloro-3α,6α-diphenyl glycoluril (Iodogen). Anal Biochem. 1981;117(1):136–46.
Dillman RO. Radiolabeled anti-CD20 monoclonal antibodies for the treatment of B-cell lymphoma. J Clin Oncol Off J Am Soc Clin Oncol. 2002;20(16):3545–57.
Miller PW, Long NJ, Vilar R, Gee AD. Synthesis of 11C, 18F, 15O, and 13N radiolabels for positron emission tomography. Angew Chem Int Ed Engl. 2008;47(47):8998–9033.
Hou S, Phung DL, Lin W-Y, Wang M, Liu K, Shen CK-F. Microwave-assisted one-pot synthesis of n-succinimidyl-4-[18F]fluorobenzoate ([18F]SFB). JoVE J Vis Exp. 2011;52:e2755.
Eichenberger LS, Patra M, Holland JP. Photoactive chelates for radiolabelling proteins. Chem Commun. 2019;55(16):2257–60.
Patra M, Eichenberger LS, Fischer G, Holland JP. Photochemical conjugation and one-pot radiolabelling of antibodies for immuno-PET. Angew Chem Int Ed. 2019;58(7):1928–33.
Pattni BS, Chupin VV, Torchilin VP. New developments in liposomal drug delivery. Chem Rev. 2015;115(19):10938–66.
Shargh VH, Hondermarck H, Liang M. Antibody-targeted biodegradable nanoparticles for cancer therapy. Nanomedicine. 2015;11(1):63–79.
Greish K. Enhanced permeability and retention (EPR) effect for anticancer nanomedicine drug targeting. In: Grobmyer SR, Moudgil BM, editors. Cancer nanotechnology. Totowa: Humana Press; 2010. p. 25–37.
Riaz MK, Riaz MA, Zhang X, Lin C, Wong KH, Chen X, et al. Surface functionalization and targeting strategies of liposomes in solid tumor therapy: a review. Int J Mol Sci. 2018;19(1):195.
Koren E, Torchilin VP. Cell-penetrating peptides: breaking through to the other side. Trends Mol Med. 2012;18(7):385–93.
Zhang X, Lin C, Lu A, Lin G, Chen H, Liu Q, et al. Liposomes equipped with cell penetrating peptide BR2 enhances chemotherapeutic effects of cantharidin against hepatocellular carcinoma. Drug Deliv. 2017;24(1):986–98.
Petersen AL, Hansen AE, Gabizon A, Andresen TL. Liposome imaging agents in personalized medicine. Adv Drug Deliv Rev. 2012;64(13):1417–35.
Goins B, Bao A, Phillips WT. Techniques for loading technetium-99m and rhenium-186/188 radionuclides into preformed liposomes for diagnostic Imaging and radionuclide therapy. In: D’Souza GGM, editor. liposomes. New York: Springer; 2017. p. 155–78.
Edmonds S, Volpe A, Shmeeda H, Parente-Pereira AC, Radia R, Baguña-Torres J, et al. Exploiting the metal-chelating properties of the drug cargo for in vivo positron emission tomography imaging of liposomal nanomedicines. ACS Nano. 2016;10(11):10294–307.
Kleynhans J, Grobler AF, Ebenhan T, Sathekge MM, Zeevaart J-R. Radiopharmaceutical enhancement by drug delivery systems: a review. J Control Release. 2018;287:177–93.
Kim J, Pandya DN, Lee W, Park JW, Kim YJ, Kwak W, et al. Vivid tumor imaging utilizing liposome-carried bimodal radiotracer. ACS Med Chem Lett. 2014;5(4):390–4.
Kaul A, Chaturvedi S, Attri A, Kalra M, Mishra AK. Targeted theranostic liposomes: rifampicin and ofloxacin loaded pegylated liposomes for theranostic application in mycobacterial infections. RSC Adv. 2016;6(34):28919–26.
Soundararajan A, Bao A, Phillips WT, McManus LM, Goins BA. Chemoradionuclide therapy with 186Re-labeled liposomal doxorubicin: toxicity, dosimetry, and therapeutic response. Cancer Biother Radiopharm. 2011;26(5):603–14.
Zhu C, Bandekar A, Ray S, Pomper M, Bruchertseifer F, Morgenstern A, et al. Anti-PSMA labeled liposomes loaded with Actinium-225 for potential antivascular alpha-radiotherapy. J Nucl Med. 2014;55(supplement 1):640.
Sofou S, Kappel BJ, Jaggi JS, McDevitt MR, Scheinberg DA, Sgouros G. Enhanced retention of the α-particle-emitting daughters of actinium-225 by liposome carriers. Bioconjug Chem. 2007;18(6):2061–7.
Henriksen G, Schoultz BW, Michaelsen TE, Bruland ØS, Larsen RH. Sterically stabilized liposomes as a carrier for α-emitting radium and actinium radionuclides. Nucl Med Biol. 2004;31(4):441–9.
Sgouros G. Alpha-particles for targeted therapy. Adv Drug Deliv Rev. 2008;60(12):1402–6.
Ferreira SMZMD, Domingos GP, dos Santos Ferreira D, Rocha TGR, Serakides R, de Faria Rezende CM, et al. Technetium-99m-labeled ceftizoxime loaded long-circulating and pH-sensitive liposomes used to identify osteomyelitis. Bioorg Med Chem Lett. 2012;22(14):4605–8.
Goins B, Phillips WT, Klipper R. Blood-pool imaging using technetium-99m-labeled liposomes. J Nucl Med Off Publ Soc Nucl Med. 1996;37(8):1374–9.
Azarian V, Gangloff A, Seimbille Y, Delaloye S, Czernin J, Phelps ME, et al. Synthesis and liposome encapsulation of a novel 18F-conjugate of ω-conotoxin GVIA for the potential imaging of N-type Ca2+ channels in the brain by positron emission tomography. J Label Compd Radiopharm. 2006;49(3):269–83.
Man F, Lammers T, de Rosales RT. Imaging nanomedicine-based drug delivery: a review of clinical studies. Mol Imaging Biol. 2018;20(5):683–95.
Lamichhane N, Udayakumar TS, D’Souza WD, Simone CB II, Raghavan SR, Polf J, et al. Liposomes: clinical applications and potential for image-guided drug delivery. Molecules. 2018;23(2):288.
Lopez-Berestein G, Kasi L, Rosenblum MG, Haynie T, Jahns M, Glenn H, et al. Clinical pharmacology of 99mTc-labeled liposomes in patients with cancer. Cancer Res. 1984;44(1):375–8.
Presant CA, Proffitt RT, Turner AF, Williams LE, Winsor D, Werner JL, et al. Successful imaging of human cancer with indium-111-labeled phospholipid vesicles. Cancer. 1988;62(5):905–11.
Koukourakis MI, Koukouraki S, Giatromanolaki A, Kakolyris S, Georgoulias V, Velidaki A, Karkavitsas NN. High intratumoral accumulation of stealth liposomal doxorubicin in sarcomas: rationale for combination with radiotherapy. Acta Oncol. 2000;39(2):207–11.
Lee H, Shields AF, Siegel BA, Miller KD, Krop I, Ma CX, et al. 64Cu-MM-302 positron emission tomography quantifies variability of enhanced permeability and retention of nanoparticles in relation to treatment response in patients with metastatic breast cancer. Clin Cancer Res. 2017;23(15):4190–202.
Brandt M, Cardinale J, Giammei C, Guarrochena X, Happl B, Jouini N, et al. Mini-review: targeted radiopharmaceuticals incorporating reversible, low molecular weight albumin binders. Nucl Med Biol. 2019;70:46–52.
Basuli F, Li C, Xu B, Williams M, Wong K, Coble VL, et al. Synthesis of fluorine-18 radio-labeled serum albumins for PET blood pool imaging. Nucl Med Biol. 2015;42(3):219–25.
Yang M, Hoppmann S, Chen L, Cheng Z. Human serum albumin conjugated biomolecules for cancer molecular imaging. Curr Pharm Des. 2012;18(8):1023–31.
Rhodes BA. Direct labeling of proteins with 99mTc. Int J Radiat Appl Instrum B. 1991;18(7):667–76.
Schibli R, Schubiger AP. Current use and future potential of organometallic radiopharmaceuticals. Eur J Nucl Med Mol Imaging. 2002;29(11):1529–42.
Chakravarty R, Chakraborty S, Guleria A, Kunwar A, Sarma HD, Dash A. Facile one-pot synthesis of intrinsically radiolabeled 64Cu-human serum albumin nanocomposite for cancer targeting. ChemistrySelect. 2017;2(26):8043–51.
Tian L, Chen Q, Yi X, Chen J, Liang C, Chao Y, et al. Albumin-templated manganese dioxide nanoparticles for enhanced radioisotope therapy. Small. 2017;13(25):1700640.
Tomalia DA, Fréchet JMJ. Discovery of dendrimers and dendritic polymers: a brief historical perspective. J Polym Sci Part Polym Chem. 2002;40(16):2719–28.
Liko F, Hindré F, Fernandez-Megia E. Dendrimers as innovative radiopharmaceuticals in cancer radionanotherapy. Biomacromolecules. 2016;17(10):3103–14.
Crampton HL, Simanek EE. Dendrimers as drug delivery vehicles: non-covalent interactions of bioactive compounds with dendrimers. Polym Int. 2007;56(4):489–96.
Tomalia DA, Reyna LA, Svenson S. Dendrimers as multi-purpose nanodevices for oncology drug delivery and diagnostic imaging. Biochem Soc Trans. 2007;35(1):61–7.
Na M, Yiyun C, Tongwen X, Yang D, Xiaomin W, Zhenwei L, et al. Dendrimers as potential drug carriers. Part II. Prolonged delivery of ketoprofen by in vitro and in vivo studies. Eur J Med Chem. 2006;41(5):670–4.
Patri AK, Kukowska-Latallo JF, Baker JR. Targeted drug delivery with dendrimers: comparison of the release kinetics of covalently conjugated drug and non-covalent drug inclusion complex. Adv Drug Deliv Rev. 2005;57(15):2203–14.
Kannan RM, Nance E, Kannan S, Tomalia DA. Emerging concepts in dendrimer-based nanomedicine: from design principles to clinical applications. J Intern Med. 2014;276(6):579–617.
Malik N, Wiwattanapatapee R, Klopsch R, Lorenz K, Frey H, Weener JW, et al. Dendrimers: relationship between structure and biocompatibility in vitro, and preliminary studies on the biodistribution of 125I-labelled polyamidoamine dendrimers in vivo. J Control Release. 2000;65(1):133–48.
Jain K, Kesharwani P, Gupta U, Jain NK. Dendrimer toxicity: let’s meet the challenge. Int J Pharm. 2010;394(1):122–42.
Zhao L, Zhu M, Li Y, Xing Y, Zhao J. Radiolabeled dendrimers for nuclear medicine applications. Molecules. 2017;22(9):1350.
Laznickova A, Biricova V, Laznicek M, Hermann P. Mono(pyridine-N-oxide) DOTA analog and its G1/G4-PAMAM dendrimer conjugates labeled with 177Lu: radiolabeling and biodistribution studies. Appl Radiat Isot. 2014;84:70–7.
Subbarayan M, Shetty SJ, Srivastava TS, Noronha OPD, Samuel AM, Mukhtar H. Water-soluble 99mTc-labeled dendritic novel porphyrins tumor imaging and diagnosis. Biochem Biophys Res Commun. 2001;281(1):32–6.
Agashe HB, Babbar AK, Jain S, Sharma RK, Mishra AK, Asthana A, et al. Investigations on biodistribution of technetium-99m-labeled carbohydrate-coated poly(propylene imine) dendrimers. Nanomed Nanotechnol Biol Med. 2007;3(2):120–7.
Almutairi A, Rossin R, Shokeen M, Hagooly A, Ananth A, Capoccia B, et al. Biodegradable dendritic positron-emitting nanoprobes for the noninvasive imaging of angiogenesis. Proc Natl Acad Sci. 2009;106(3):685–90.
Trembleau L, Simpson M, Cheyne RW, Escofet I, Appleyard MVCAL, Murray K, et al. Development of 18F-fluorinatable dendrons and their application to cancer cell targeting. New J Chem. 2011;35(11):2496–502.
Garrigue P, Tang J, Ding L, Bouhlel A, Tintaru A, Laurini E, et al. Self-assembling supramolecular dendrimer nanosystem for PET imaging of tumors. Proc Natl Acad Sci. 2018;115(45):11454–9.
Kovacs L, Tassano M, Cabrera M, Zamboni CB, Fernández M, Anjos RM, et al. Development of 177Lu-DOTA-dendrimer and determination of Its effect on metal and ion levels in tumor tissue. Cancer Biother Radiopharm. 2015;30(10):405–10.
Zhao L, Zhu J, Cheng Y, Xiong Z, Tang Y, Guo L, et al. Chlorotoxin-conjugated multifunctional dendrimers labeled with radionuclide 131I for single photon emission computed tomography imaging and radiotherapy of gliomas. ACS Appl Mater Interfaces. 2015;7(35):19798–808.
Yang G, Sadeg N, Belhadj-Tahar H. New potential in situ anticancer agent derived from [188Re]rhenium nitro-imidazole ligand loaded 5th generation poly-l-lysine dendrimer for treatment of transplanted human liver carcinoma in nude mice. Drug Des Open Access. 2017;6(1):1–7.
Pinto Reis C, Neufeld RJ, Ribeiro António J, Veiga F. Nanoencapsulation I. Methods for preparation of drug-loaded polymeric nanoparticles. Nanomed Nanotechnol Biol Med. 2006;2(1):8–21.
Kobayashi H, Koyama Y, Barrett T, Hama Y, Regino CAS, Shin IS, et al. Multimodal nanoprobes for radionuclide and five-color near-infrared optical lymphatic imaging. ACS Nano. 2007;1(4):258–64.
Wang Y, Miao Z, Ren G, Xu Y, Cheng Z. A novel affibody bioconjugate for dual-modality imaging of ovarian cancer. Chem Commun. 2014;50(85):12832–5.
Talelli M, Barz M, Rijcken CJF, Kiessling F, Hennink WE, Lammers T. Core-crosslinked polymeric micelles: principles, preparation, biomedical applications and clinical translation. Nano Today. 2015;10(1):93–117.
Movassaghian S, Merkel OM, Torchilin VP. Applications of polymer micelles for imaging and drug delivery. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2015;7(5):691–707.
Allmeroth M, Moderegger D, Gündel D, Buchholz H-G, Mohr N, Koynov K, et al. PEGylation of HPMA-based block copolymers enhances tumor accumulation in vivo: a quantitative study using radiolabeling and positron emission tomography. J Control Release. 2013;172(1):77–85.
Yamamoto F, Yamahara R, Makino A, Kurihara K, Tsukada H, Hara E, et al. Radiosynthesis and initial evaluation of 18F labeled nanocarrier composed of poly(l-lactic acid)-block-poly(sarcosine) amphiphilic polydepsipeptide. Nucl Med Biol. 2013;40(3):387–94.
Yang Z, Zheng S, Harrison WJ, Harder J, Wen X, Gelovani JG, et al. Long-circulating near-infrared fluorescence core-cross-linked polymeric micelles: synthesis, characterization, and dual nuclear/optical imaging. Biomacromolecules. 2007;8(11):3422–8.
Hoang B, Lee H, Reilly RM, Allen C. Noninvasive monitoring of the fate of 111In-labeled block copolymer micelles by high resolution and high sensitivity microSPECT/CT imaging. Mol Pharm. 2009;6(2):581–92.
Cheng C-C, Huang C-F, Ho A-S, Peng C-L, Chang C-C, Mai F-D, et al. Novel targeted nuclear imaging agent for gastric cancer diagnosis: glucose-regulated protein 78 binding peptide-guided 111In-labeled polymeric micelles. Int J Nanomed. 2013;8:1385–91.
Arranja A, Ivashchenko O, Denkova AG, Morawska K, van Vlierberghe S, Dubruel P, et al. SPECT/CT imaging of pluronic nanocarriers with varying poly(ethylene oxide) block length and aggregation state. Mol Pharm. 2016;13(3):1158–65.
Jensen ATI. Radiolabeling of liposomes and polymeric micelles with PET-isotopes. Technical University of Denmark (DTU); 2012.
Pressly ED, Rossin R, Hagooly A, Fukukawa K, Messmore BW, Welch MJ, et al. Structural effects on the biodistribution and positron emission tomography (PET) imaging of well-defined 64Cu-labeled nanoparticles comprised of amphiphilic block graft copolymers. Biomacromolecules. 2007;8(10):3126–34.
Sun G, Hagooly A, Xu J, Nyström AM, Li Z, Rossin R, et al. Facile, Efficient approach to accomplish tunable chemistries and variable biodistributions for shell cross-linked nanoparticles. Biomacromolecules. 2008;9(7):1997–2006.
Hruby M, Konak C, Kucka J, Vetrik M, Filippov SK, Vetvicka D, et al. Thermoresponsive, hydrolytically degradable polymer micelles intended for radionuclide delivery. Macromol Biosci. 2009;9(10):1016–27.
Hara E, Makino A, Kurihara K, Ueda M, Hara I, Kawabe T, et al. Radionuclide therapy using nanoparticle of 131I-Lactosome in combination with percutaneous ethanol injection therapy. J Nanoparticle Res. 2013;15(12):2131.
Lee H, Hoang B, Fonge H, Reilly RM, Allen C. In vivo distribution of polymeric nanoparticles at the whole-body, tumor, and cellular levels. Pharm Res. 2010;27(11):2343–55.
Miura Y, Tsuji AB, Sugyo A, Sudo H, Aoki I, Inubushi M, et al. Polymeric micelle platform for multimodal tomographic imaging to detect scirrhous gastric cancer. ACS Biomater Sci Eng. 2015;1(11):1067–76.
Jensen AI, Binderup T, Ek PK, Grandjean CE, Rasmussen PH, Kjær A, et al. PET imaging with copper-64 as a tool for real-time in vivo investigations of the necessity for cross-linking of polymeric micelles in nanomedicine. J Label Compd Radiopharm. 2017;60(8):366–74.
Ulbrich K, Holá K, Šubr V, Bakandritsos A, Tuček J, Zbořil R. Targeted drug delivery with polymers and magnetic nanoparticles: covalent and noncovalent approaches, release control, and clinical studies. Chem Rev. 2016;116(9):5338–431.
Du JZ, Du XJ, Mao CQ, Wang J. Tailor-made dual pH-sensitive polymer–doxorubicin nanoparticles for efficient anticancer drug delivery. J Am Chem Soc. 2011;133(44):17560–3.
Tang C, Edelstein J, Mikitsh J, Xiao E, Hemphill A, Pagels R, et al. Biodistribution and fate of core-labeled 125 I polymeric nanocarriers prepared by Flash NanoPrecipitation (FNP). J Mater Chem B. 2016;4(14):2428–34.
Allmeroth M, Moderegger D, Gündel D, Koynov K, Buchholz HG, Mohr K, et al. HPMA-LMA copolymer drug carriers in oncology: an in vivo PET study to assess the tumor line-specific polymer uptake and body distribution. Biomacromolecules. 2013;14(9):3091–101.
Subramanian S, Dandekar P, Jain R, Pandey U, Samuel G, Hassan PA, et al. Technetium-99m–labeled poly(dl-Lactide-co-Glycolide) nanoparticles as an alternative for sentinel lymph node imaging. Cancer Biother Radiopharm. 2010;25(6):637–44.
Delgado A, Soriano I, Sánchez E, Oliva M, Évora C. Radiolabelled biodegradable microspheres for lung imaging. Eur J Pharm Biopharm. 2000;50(2):227–36.
Yadav KS, Chuttani K, Mishra AK, Sawant KK. Long circulating nanoparticles of etoposide using PLGA-MPEG and PLGA-pluronic block copolymers: characterization, drug-release, blood-clearance, and biodistribution studies. Drug Dev Res. 2010;71(4):228–39.
Sirianni RW, Zheng M-Q, Patel TR, Shafbauer T, Zhou J, Saltzman WM, et al. Radiolabeling of poly(lactic-co-glycolic acid) (PLGA) nanoparticles with biotinylated F-18 prosthetic groups and imaging of their delivery to the brain with positron emission tomography. Bioconjug Chem. 2014;25(12):2157–65.
Banerjee SR, Foss CA, Horhota A, Pullambhatla M, McDonnell K, Zale S, et al. 111In- and IRDye800CW-labeled PLA–PEG nanoparticle for imaging prostate-specific membrane antigen-expressing tissues. Biomacromolecules. 2017;18(1):201–9.
Pant K, Sedláček O, Nadar RA, Hrubý M, Stephan H. Radiolabelled polymeric materials for imaging and treatment of cancer: quo vadis? Adv Healthc Mater. 2017;6(6):1601115.
Akhlaghi M, Pourjavadi A. Preparation and primary evaluation of 66Ga-DTPA-chitosan in fibrosarcoma bearing mice. Nukleonika. 2011;56(1):41–7.
Wallace AM, Hoh CK, Darrah DD, Schulteis G, Vera DR. Sentinel lymph node mapping of breast cancer via intradermal administration of Lymphoseek. Nucl Med Biol. 2007;34(7):849–53.
Sant SB. Nanoparticles: from theory to applications: Günter Schmid, 2nd ed., Wiley-VCH Verlag, Weinheim, 2010, ISBN: 978-3-527-32589-4. Mater Manuf Process. 2012;27(12):1462–3.
Yang M, Cheng K, Qi S, Liu H, Jiang Y, Jiang H, et al. Affibody modified and radiolabeled gold–Iron oxide hetero-nanostructures for tumor PET, optical and MR imaging. Biomaterials. 2013;34(11):2796–806.
Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33(9):941–51.
Bobo D, Robinson KJ, Islam J, Thurecht KJ, Corrie SR. Nanoparticle-based medicines: a review of FDA-approved materials and clinical trials to date. Pharm Res. 2016;33(10):2373–87.
Yu M, Xu J, Zheng J. Renal clearable luminescent gold nanoparticles: from the bench to the clinic. Angew Chem. 2019;131(13):4156–72.
Wang Z, Qiao R, Tang N, Lu Z, Wang H, Zhang Z, et al. Active targeting theranostic iron oxide nanoparticles for MRI and magnetic resonance-guided focused ultrasound ablation of lung cancer. Biomaterials. 2017;127:25–35.
Liu Z, Robinson JT, Tabakman SM, Yang K, Dai H. Carbon materials for drug delivery & cancer therapy. Mater Today. 2011;14(7–8):316–23.
Phillips E, Penate-Medina O, Zanzonico PB, Carvajal RD, Mohan P, Ye Y, et al. Clinical translation of an ultrasmall inorganic optical-PET imaging nanoparticle probe. Sci Transl Med. 2014;6(260):260ra149.
Hoffman D, Sun M, Yang L, McDonagh PR, Corwin F, Sundaresan G, et al. Intrinsically radiolabelled [59Fe]-SPIONs for dual MRI/radionuclide detection. Am J Nucl Med Mol Imaging. 2014;4(6):548–60.
Shukla R, Bansal V, Chaudhary M, Basu A, Bhonde RR, Sastry M. Biocompatibility of gold nanoparticles and their endocytotic fate inside the cellular compartment: a microscopic overview. Langmuir. 2005;21(23):10644–54.
Mallidi S, Larson T, Tam J, Joshi PP, Karpiouk A, Sokolov K, et al. Multiwavelength photoacoustic imaging and plasmon resonance coupling of gold nanoparticles for selective detection of cancer. Nano Lett. 2009;9(8):2825–31.
Polte J, Erler R, Thünemann AF, Sokolov S, Ahner TT, Rademann K, et al. Nucleation and growth of gold nanoparticles studied via in situ small angle X-ray scattering at millisecond time resolution. ACS Nano. 2010;4(2):1076–82.
Hühn J, Carrillo-Carrion C, Soliman MG, Pfeiffer C, Valdeperez D, Masood A, et al. Selected standard protocols for the synthesis, phase transfer, and characterization of inorganic colloidal nanoparticles. Chem Mater. 2017;29(1):399–461.
Su N, Dang Y, Liang G, Liu G. Iodine-125-labeled cRGD-gold nanoparticles as tumor-targeted radiosensitizer and imaging agent. Nanoscale Res Lett. 2015;10(1):160.
Yook S, Cai Z, Lu Y, Winnik MA, Pignol J-P, Reilly RM. Radiation nanomedicine for EGFR-positive breast cancer: panitumumab-modified gold nanoparticles complexed to the β-particle-emitter, 177Lu. Mol Pharm. 2015;12(11):3963–72.
McLaughlin MF, Woodward J, Boll RA, Wall JS, Rondinone AJ, Kennel SJ, et al. Gold coated lanthanide phosphate nanoparticles for targeted alpha generator radiotherapy. PLoS ONE. 2013;8(1):e54531.
Katti KV, Khoobchandani M, Thipe VC, Al-Yasiri AY, Katti KK, Loyalka SK, et al. Prostate tumor therapy advances in nuclear medicine: green nanotechnology toward the design of tumor specific radioactive gold nanoparticles. J Radioanal Nucl Chem. 2018;318(3):1737–47.
Guardia P, Riedinger A, Kakwere H, Gazeau F, Pellegrino T. Magnetic nanoparticles for magnetic hyperthermia and controlled drug delivery. In: Ruiz-Molina D, Novio F, Roscini C, editors. Bio- and bioinspired nanomaterials. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA; 2014. p. 139–72.
Guardia P, Labarta A, Batlle X. Tuning the size, the shape, and the magnetic properties of iron oxide nanoparticles. J Phys Chem C. 2011;115(2):390–6.
Di Corato R, Espinosa A, Lartigue L, Tharaud M, Chat S, Pellegrino T, et al. Magnetic hyperthermia efficiency in the cellular environment for different nanoparticle designs. Biomaterials. 2014;35(24):6400–11.
Radović M, Calatayud MP, Goya GF, Ibarra MR, Antić B, Spasojević V, et al. Preparation and in vivo evaluation of multifunctional 90Y-labeled magnetic nanoparticles designed for cancer therapy. J Biomed Mater Res A. 2015;103(1):126–34.
Munaweera I, Shi Y, Koneru B, Saez R, Aliev A, Di Pasqua AJ, et al. Chemoradiotherapeutic magnetic nanoparticles for targeted treatment of nonsmall cell lung cancer. Mol Pharm. 2015;12(10):3588–96.
Zhu J, Zhang B, Tian J, Wang J, Chong Y, Wang X, et al. Synthesis of heterodimer radionuclide nanoparticles for magnetic resonance and single-photon emission computed tomography dual-modality imaging. Nanoscale. 2015;7(8):3392–5.
Wang Y-F, Fu C-M, Chuang M-H, Cham T-M, Chung M-I. Magnetically directed targeting aggregation of radiolabelled ferrite nanoparticles. J Nanomater. 2011;2011:1–5.
Trofimov A, Ivanova A, Zyuzin M, Timin A. Porous inorganic carriers based on silica, calcium carbonate and calcium phosphate for controlled/modulated drug delivery: fresh outlook and future perspectives. Pharmaceutics. 2018;10(4):167.
Nooney RI, Thirunavukkarasu D, Chen Y, Josephs R, Ostafin AE. Synthesis of nanoscale mesoporous silica spheres with controlled particle size. Chem Mater. 2002;14(11):4721–8.
Vivero-Escoto JL, Huxford-Phillips RC, Lin W. Silica-based nanoprobes for biomedical imaging and theranostic applications. Chem Soc Rev. 2012;41(7):2673–85.
Chen F, Ma K, Madajewski B, Zhuang L, Zhang L, Rickert K, et al. Ultrasmall targeted nanoparticles with engineered antibody fragments for imaging detection of HER2-overexpressing breast cancer. Nat Commun. 2018;9(1):4141.
Di Pasqua AJ, Miller ML, Lu X, Peng L, Jay M. Tumor accumulation of neutron-activatable holmium-containing mesoporous silica nanoparticles in an orthotopic non-small cell lung cancer mouse model. Inorganica Chim Acta. 2012;393:334–6.
Pascual L, Cerqueira-Coutinho C, García-Fernández A, de Luis B, Bernardes ES, Albernaz MS, et al. MUC1 aptamer-capped mesoporous silica nanoparticles for controlled drug delivery and radio-imaging applications. Nanomed Nanotechnol Biol Med. 2017;13(8):2495–505.
Yamaguchi H, Tsuchimochi M, Hayama K, Kawase T, Tsubokawa N. Dual-labeled near-infrared/99mTc imaging probes using PAMAM-coated silica nanoparticles for the imaging of HER2-expressing cancer cells. Int J Mol Sci. 2016;17(7):1086.
Kamkaew A, Cheng L, Goel S, Valdovinos HF, Barnhart TE, Liu Z, et al. Cerenkov radiation induced photodynamic therapy using chlorin e6-loaded hollow mesoporous silica nanoparticles. ACS Appl Mater Interfaces. 2016;8(40):26630–7.
Chen D, Dougherty CA, Zhu K, Hong H. Theranostic applications of carbon nanomaterials in cancer: focus on imaging and cargo delivery. J Control Release. 2015;210:230–45.
Mundra RV, Wu X, Sauer J, Dordick JS, Kane RS. Nanotubes in biological applications. Curr Opin Biotechnol. 2014;28:25–32.
Yan L, Zhao F, Li S, Hu Z, Zhao Y. Low-toxic and safe nanomaterials by surface-chemical design, carbon nanotubes, fullerenes, metallofullerenes, and graphenes. Nanoscale. 2011;3(2):362–82.
Singh S, Vardharajula S, Tiwari P, Eroğlu E, Vig K, Dennis V, et al. Functionalized carbon nanotubes: biomedical applications. Int J Nanomed. 2012;7:5361–74.
McDevitt MR, Chattopadhyay D, Kappel BJ, Jaggi JS, Schiffman SR, Antczak C, et al. Tumor targeting with antibody-functionalized, radiolabeled carbon nanotubes. J Nucl Med. 2007;48(7):1180–9.
Hong SY, Tobias G, Al-Jamal KT, Ballesteros B, Ali-Boucetta H, Lozano-Perez S, et al. Filled and glycosylated carbon nanotubes for in vivo radioemitter localization and imaging. Nat Mater. 2010;9(6):485–90.
Ruggiero A, Villa HC, Holland JP, Sprinkle SR, et al. Imaging and treating tumor vasculature with targeted radiolabeled carbon nanotubes. Int J Nanomed. 2010;5:783–802.
Duch MC, Budinger GRS, Liang YT, Soberanes S, Urich D, Chiarella SE, et al. Minimizing oxidation and stable nanoscale dispersion improves the biocompatibility of graphene in the lung. Nano Lett. 2011;11(12):5201–7.
Zhang S, Yang K, Feng L, Liu Z. In vitro and in vivo behaviors of dextran functionalized graphene. Carbon. 2011;49(12):4040–9.
Chen L, Zhong X, Yi X, Huang M, Ning P, Liu T, et al. Radionuclide 131I labeled reduced graphene oxide for nuclear imaging guided combined radio- and photothermal therapy of cancer. Biomaterials. 2015;66:21–8.
Kim J, Jay M. Neutron-activatable radionuclide cancer therapy using graphene oxide nanoplatelets. Nucl Med Biol. 2017;52:42–8.
Zhao Z, Li J, Wen T, Shen C, Wang X, Xu A. Surface functionalization graphene oxide by polydopamine for high affinity of radionuclides. Colloids Surf Physicochem Eng Asp. 2015;482:258–66.
Schrand AM, Huang H, Carlson C, Schlager JJ, Ōsawa E, Hussain SM, et al. Are diamond nanoparticles cytotoxic? J Phys Chem B. 2007;111(1):2–7.
Rojas S, Gispert JD, Martín R, Abad S, Menchón C, Pareto D, et al. Biodistribution of amino-functionalized diamond nanoparticles. ACS Nano. 2011;5(7):5552–9.
Garashchenko BL, Dogadkin NN, Borisova NE, Yakovlev RY. Sorption of 223Ra and 211Pb on modified nanodiamonds for potential application in radiotherapy. J Radioanal Nucl Chem. 2018;318(3):2415–23.
Liu S. Bifunctional coupling agents for radiolabeling of biomolecules and target-specific delivery of metallic radionuclides. Adv Drug Deliv Rev. 2008;60(12):1347–70.
Goel S, Chen F, Ehlerding EB, Cai W. Intrinsically radiolabeled nanoparticles: an emerging paradigm. Small Weinh Bergstr Ger. 2014;10(19):3825–30.
Goel S, England CG, Chen F, Cai W. Positron emission tomography and nanotechnology: a dynamic duo for cancer theranostics. Adv Drug Deliv Rev. 2017;113:157–76.
Kamkaew A, Ehlerding EB, Cai W. Nanoparticles as radiopharmaceutical cectors. In: Lewis JS, Windhorst AD, Zeglis BM, editors. Radiopharmaceutical chemistry. Cham: Springer International Publishing; 2019. p. 181–203.
Zhao Y, Sultan D, Detering L, Cho S, Sun G, Pierce R, et al. Copper-64-alloyed gold nanoparticles for cancer imaging: improved radiolabel stability and diagnostic accuracy. Angew Chem Int Ed Engl. 2014;53(1):156–9.
Pratt EC, Shaffer TM, Grimm J. Nanoparticles and radiotracers: advances toward radionanomedicine. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2016;8(6):872–90.
Chen F, Goel S, Valdovinos HF, Luo H, Hernandez R, Barnhart TE, et al. In vivo integrity and biological fate of chelator-free zirconium-89-labeled mesoporous silica nanoparticles. ACS Nano. 2015;9(8):7950–9.
Shaffer TM, Wall MA, Harmsen S, Longo VA, Drain CM, Kircher MF, et al. Silica nanoparticles as substrates for chelator-free labeling of oxophilic radioisotopes. Nano Lett. 2015;15(2):864–8.
Shaffer TM, Harmsen S, Khwaja E, Kircher MF, Drain CM, Grimm J. Stable radiolabeling of sulfur-functionalized silica nanoparticles with copper-64. Nano Lett. 2016;16(9):5601–4.
Thakor AS, Jokerst JV, Ghanouni P, Campbell JL, Mittra E, Gambhir SS. Clinically approved nanoparticle imaging agents. J Nucl Med. 2016;57(12):1833–7.
Reinders MTM, Smits MLJ, van Roekel C, Braat AJAT. Holmium-166 microsphere radioembolization of hepatic malignancies. Semin Nucl Med. 2019;49(3):237–43.
Bretcanu O, Evans I. Glasses for treatment of liver cancer by radioembolization. In: Marchi J, editor. Biocompatible glasses. Cham: Springer International Publishing; 2016. p. 267–83.
Kennedy AS. Radioactive microspheres for liver cancers. US Oncol Rev. 2005;1(1):25–8.
Lewandowski R, Salem R. Yttrium-90 radioembolization of hepatocellular carcinoma and metastatic disease to the liver. Semin Interv Radiol. 2006;23(1):064–72.
Bogni A, Pascali C. Medical Devices for Radioembolization. In: Bombardieri E, Seregni E, Evangelista L, Chiesa C, Chiti A, editors. Clinical applications of nuclear medicine targeted therapy. Cham: Springer International Publishing; 2018. p. 107–18.
Maccauro M, Aliberti G, Chiesa C, Spreafico C. HCC Radioembolization with yttrium-90 glass microspheres (TheraSphere). In: Bombardieri E, Seregni E, Evangelista L, Chiesa C, Chiti A, editors. Clinical Applications of nuclear medicine targeted therapy. Cham: Springer International Publishing; 2018. p. 119–25.
Kulik LM, Atassi B, van Holsbeeck L, Souman T, Lewandowski RJ, Mulcahy MF, et al. Yttrium-90 microspheres (TheraSphere®) treatment of unresectable hepatocellular carcinoma: downstaging to resection, RFA and bridge to transplantation. J Surg Oncol. 2006;94(7):572–86.
Radosa CG, Radosa JC, Grosche-Schlee S, Zöphel K, Plodeck V, Kühn JP, et al. Holmium-166 radioembolization in hepatocellular carcinoma: feasibility and safety of a new treatment option in clinical practice. Cardiovasc Intervent Radiol. 2019;42(3):405–12.
Zielhuis SW, Nijsen JFW, Krijger GC, van het Schip AD, Hennink WE. Holmium-loaded poly(l-lactic acid) microspheres: in vitro degradation study. Biomacromol. 2006;7(7):2217–23.
Wu T-J, Chiu H-Y, Yu J, Cautela MP, Sarmento B, das Neves J, et al. Nanotechnologies for early diagnosis, in situ disease monitoring, and prevention. In: Uskoković V, Uskoković DP, editors. Nanotechnologies in preventive and regenerative medicine. Amsterdam: Elsevier; 2018. p. 1–92.
Carlier T, Willowson KP, Fourkal E, Bailey DL, Doss M, Conti M. 90Y-PET imaging: exploring limitations and accuracy under conditions of low counts and high random fraction. Med Phys. 2015;42(7):4295–309.
Sangro B, Gardini AC. Radioembolisation in hepatocellular carcinoma: principles of management. In: Cross T, Palmer DH, editors. Liver cancers: from mechanisms to management. Cham: Springer International Publishing; 2019. p. 139–52.
Atassi B, Bangash AK, Bahrani A, Pizzi G, Lewandowski RJ, Ryu RK, et al. Multimodality imaging following 90Y radioembolization: a comprehensive review and pictorial essay. RadioGraphics. 2008;28(1):81–99.
van de Maat GH, Seevinck PR, Elschot M, Smits MLJ, de Leeuw H, van het Schip AD, et al. MRI-based biodistribution assessment of holmium-166 poly(l-lactic acid) microspheres after radioembolisation. Eur Radiol. 2013;23(3):827–35.
Acknowledgements
This work was supported by the grant of the Russian Science Foundation, No. 19-75-10010 (A.S.T.). The part of this work related to the delivery of radiotherapeutics with inorganic nanoparticles was founded with Russian Science Foundation, Grant N 19-75-00039 (MVZ). The research is funded from Tomsk Polytechnic University Competitiveness Enhancement Program.
Funding
This work was supported by the grant of the Russian Science Foundation, No. 19-75-10010 (A.S.T.). The part of this work related to the delivery of radionuclides radiotherapeutics with inorganic nanoparticles was founded with Russian Science Foundation, Grant N 19-75-00039 (MVZ).
Author information
Authors and Affiliations
Contributions
OOP, ARM, MVZ and AST gathered the research data and made substantial contributions to interpretation of literature. All made substantial contribution to draft article and revised it critically, emphasizing important intellectual content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
We have included and adapted for this publication 7 Figures (Figs. 3, 5, 7, 8, 9, 10 and 11) from the previously published articles with required copyright permission from the copyright holder. We have mentioned this in the manuscript with the proper citation.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Peltek, O.O., Muslimov, A.R., Zyuzin, M.V. et al. Current outlook on radionuclide delivery systems: from design consideration to translation into clinics. J Nanobiotechnol 17, 90 (2019). https://doi.org/10.1186/s12951-019-0524-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12951-019-0524-9