
Abstract
Background
Cigarette smoke exposure (SE) during pregnancy is the largest modifiable risk factor for the development of lung disorders in offspring. We have previously shown that maternal L-Carnitine treatment can reduce the adverse impacts of maternal SE on renal and brain disorders in offspring. Here, we investigated the effect of maternal L-Carnitine supplementation on lung inflammatory pathways, autophagy, and mitophagy markers in the offspring in response to maternal SE.
Methods
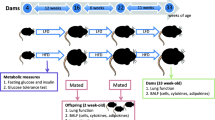
Female BALB/c mice (8 weeks) were exposed to cigarette smoke for 6 weeks prior to mating, during gestation and lactation. Some of the SE dams were given L-Carnitine supplementation (1.5 mM in drinking water, SE + LC) during gestation and lactation. Lungs from the offspring were studied at birth and adulthood (13 weeks).
Results
At birth, in male offspring, there were increased levels of inflammatory markers (phosphorylated(p)-ERK1,2, p-P38 MAPK, p- NF-κB), and inflammasome marker (NLRP3), as well as mitophagy fission marker Drp-1 and autophagosome marker (LC3A/B-II) in the lung. Maternal L-Carnitine supplementation significantly reduced NLRP3 level. In contrast, maternal SE only increased IL1-β in female offspring, which was reversed by maternal L-Carnitine supplementation. At 13 weeks, there was an increase in LC3A/B-II and p- NF-κB in the male SE offspring with reduced p-JNK1,2, which were partially normalised by maternal L-Carnitine treatment. Female offspring were not affected by maternal SE at this age.
Conclusion
Maternal SE had adverse impacts on the male offspring’s lung, which were partially alleviated by maternal L-Carnitine supplementation. Females seem to be less affected by the adverse effects of maternal SE.
Similar content being viewed by others


Background
Smoking during pregnancy is a major cause of maternal and newborn morbidity and mortality [1], with pulmonary diseases being a major adverse outcome [2, 3]. In-utero smoke exposure (SE) reduces lung function in human newborns [4, 5]. Animal models have shown a decreased number of saccules, septal crests, and decreased elastin fibres in foetuses [6] and suckling pups [7], as well as increased airway thickness, collagen deposition, inflammation, and airway hyper-responsiveness due to intrauterine SE [8,9,10].
In humans, certain respiratory diseases, including chronic obstructive pulmonary disease (COPD), occur disproportionately in males and females [11]. The common pathophysiological process includes increased inflammation, oxidative stress, impaired mitochondrial renewal (mitophagy), and cellular self-cleaning mechanism (autophagy) [12]. In keeping with this, our previous murine studies found that the changes in inflammation, oxidative stress, mitophagy, and autophagy have a marked sex difference in the offspring’s brain and kidney following in-utero SE, wherein female offspring are less affected from such adverse effects [13, 14].
Previously, it has only been demonstrated that maternal SE causes lung inflammation in male offspring [15]. Other studies show that prenatal SE can differentially affect DNA methylation in cord blood or lungs in males and females in both human and animal studies. For example, the gene encoding insulin-like growth factor (IGF)-1, a growth factor involved in lung development, was found to be methylated in the babies from smokers, and methylation was most pronounced in male children [16, 17]. In an animal study, CpG site-specific hypo- and hypermethylation of Igf1 exons was observed in the lungs from male and female offspring, respectively. However, protein transcription of IGF-1 is reduced in male offspring, even without in utero SE, which may partially explain sex differences in the susceptibility of developing certain respiratory disorders [18]. The mechanism by which in-utero causes differential DNA methylation between males and females is not known, but a recent study in T cells has demonstrated that the parental source of the X chromosome (male versus female) affects the methylation of the X chromosome in offspring [19], suggesting that the sex chromosomes may be involved.
Given these known differences, we hypothesised that sex differences also exist regarding the effects of in-utero SE on other pulmonary changes.
The regulation of inflammation involves several signalling pathways, such as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and mitogen-activated protein kinase (MAPK) pathways [20]. Three well-characterised subfamilies of MAPK include the extracellular signal-regulated kinase (ERK)1/2, Jun N-terminal kinase (JNK) stress-activated protein kinase, and p38 [21]. NF-κB is often regarded as the master controller of inflammation [22]. Inflammatory response requires a considerable amount of energy derived from the mitochondria [23], whereas mitochondrial function is often compromised during this process. There is a close relationship between the activation of the nucleotide-binding domain and leucine-rich repeat-containing family pyrin domain containing 3 (NLRP3) inflammasome (increasing IL-1β activity) and mitochondrial dysfunction [24]. Thus, the inflammasome is regarded as the bridge between inflammatory response and subsequent mitochondrial damage [24], including oxidative stress [24] and mitochondrial DNA impairment [25]. This has been observed in conditions like COPD but has not been investigated in the setting of maternal SE [26].
Mitophagy is the autophagic elimination of injured mitochondria, which is regulated by fusion and fission [27]. The balance between fusion and fission is essential to mitochondrial integrity. Fission is to separate damaged mitochondrial fragments from the healthy part, while fusion is to generate a new mitochondrion from two healthy mitochondrial fragments [28, 29]. We have observed dysregulated mitophagy in the brain and kidney caused by maternal SE which was associated with organ pathology [13, 30]; however, whether this also occurs in the lung is unknown.
In-utero SE results in considerable foetal oxidative stress and inhibits the endogenous antioxidant activity [31]. Therefore, improving antioxidant ability may alleviate the adverse effects of maternal SE. L-Carnitine has been shown to attenuate age-related disorders by reducing oxidative stress and increasing antioxidant capacity in rats [32, 33]. A clinical study also showed that L-Carnitine supplementation can suppress serum levels of inflammatory cytokines in humans [34]. We have shown that maternal L-Carnitine supplementation during pregnancy and lactation can alleviate brain [13] and renal dysfunction [35] in offspring from the SE mothers. As such, this approach may also ameliorate the adverse impact of maternal SE on lung health in the offspring.
Given the known differences in the susceptibility of developing lung diseases between males and females [36], we hypothesised that in-utero smoke exposure would result in chronic hyperactivation of inflammatory markers and dysregulated autophagy and mitophagy in male offspring, but not in female offspring. Maternal L-carnitine may ameliorate the adverse impact of maternal SE on the offspring’s lung.
Results
Effect of maternal SE on body weight
At postnatal(P)1, both male and female offspring from the SE dams appeared smaller than the SHAM offspring (Table 1). Maternal L-Carnitine supplement during gestation and lactation increased the birth weight of both male and female offspring (P < 0.05 vs SE). There were no differences in body weight among the 3 groups at 13 weeks for both males and females (Table 1). There was a non-significant decrease in the litter size from the SE dams, and the male-female ratio was unaffected Table 2).
Effect of maternal SE on lung p-ERK, p-p38, p-JNK, and p- NF-κB in the offspring
At P1, maternal SE significantly increased the levels of p-ERK1,2 (P < 0.01 vs SHAM, Fig. 1a), p-P38 (P < 0.01 vs SHAM, Fig. 1e) and p-NF-κB (P < 0.01 vs, SHAM, Fig. 1g) in the male offspring. Only p- NF-κB appeared to be partially reversed by maternal L-Carnitine treatment without statistical significance (Fig. 1g). In the female offspring, maternal SE did not significantly affect p-ERK1,2, p-JNK1,2, p-p38, or NF-κB levels, whereas maternal L-Carnitine supplementation significantly reduced p-ERK1,2 (P < 0.05 vs SHAM, P < 0.01 vs SE, Fig. 1b) and p-P38 (P < 0.05 vs SHAM, Fig. 1f) levels.

Lung p-ERK1,2, p-JNK1,2, p-p38 and p- NF-κB in the offspring at P1. Protein levels of p-ERK1,2 a, b, p-JNK1,2 c, d, p-p38 e, f and p- NF-κB g, h in the lung of the male and female offspring at P1. Results are expressed as means ± SE, (male n = 8, female, n = 6–8). Data were analysed by one-way ANOVA followed by Tukey’s post hoc tests. *P < 0.05, **P < 0.01, *** P < 0.001, ****P < 0.0001. ERK, extracellular signal-regulated kinase; JNK, c-JUN N-terminal kinase; p38, p38 Mitogen-activated protein kinase; NF-κB: Nuclear factor-kB. SE, maternal smoke exposure; SE + LC, maternal smoke exposure with L-Carnitine supplement
At 13 weeks, p-JNK1,2 level was lower, and p- NF-κB was higher in the male offspring (P < 0.05 vs SHAM offspring, Fig. 2c, g), which was not affected by maternal L-Carnitine supplementation. In the adult females, neither maternal SE nor maternal L-Carnitine supplementation had any effect on the abovementioned proteins (Fig. 2).

Lung p-ERK1,2, p-JNK1,2, p-p38 and p- NF-κB in the offspring at 13 weeks. Protein levels of p-ERK1,2 a, b, p-JNK1,2 (C, D), p-p38 e, f and p- NF-κB g, h in the lung of the male and female offspring at 13 weeks. Results are expressed as means ± SE, (male n = 7–8, female n = 8). Data were analysed by one-way ANOVA followed by Tukey’s post hoc tests. *P < 0.05, **P < 0.01. ERK, extracellular signal-regulated kinase; JNK, c-JUN N-terminal kinase; p38, p38 Mitogen-activated protein kinase; NF-κB: Nuclear factor-kB. SE, maternal smoke exposure; SE + LC, maternal smoke exposure with L-Carnitine supplement
Effect of maternal SE on lung NLRP3 and IL1-β levels in the offspring
In P1 offspring, an increased NLRP3 and IL-1β was observed in male and female offspring, however only NLRP3 in the SE male (P < 0.01 vs SHAM, Fig. 3a) and IL-1β in SE female (P < 0.05 vs SHAM, Fig. 3d) were significantly higher than the SHAM offspring. Maternal L-Carnitine treatment normalised both markers (P < 0.05 vs SE, Fig. 3a).

Lung inflammasome markers NLRP3 and IL-1 β in the offspring at P1 and 13 weeks. Protein levels of NLRP3 a, b and IL-1β c, d in the lung of male and female offspring at P1. Protein expression of NLRP3 e, g and IL-1β f, h in the lung of male and female offspring at 13 weeks. Results are expressed as means ± SE (male n = 8, female n = 6–8). Data were analysed by one-way ANOVA followed by Tukey’s post hoc tests. *P < 0.05, **P < 0.01, ****P < 0.0001. NLRP3, nucleotide-binding domain and leucine-rich repeat-containing (NLR) family pyrin domain containing 3; SE; maternal smoke exposure; SE + LC, maternal smoke exposure with L-Carnitine supplement
At 13 weeks, maternal cigarette SE significantly increased NLRP3 expression in female offspring (P < 0.01 vs SHAM, Fig. 3f); maternal L-Carnitine supplementation did not have any effect (P < 0.01 vs SE, Fig. 3f). Maternal smoke exposure significantly increased IL-1β level (P < 0.01 vs SHAM, Fig. 3g) in the male offspring, which was further increased after maternal L-Carnitine treatment (P < 0.01 vs SE, Fig. 3g).
Effect of maternal SE on lung mitophagy markers in the offspring
At P1, total cell autophagy marker LC3A/B-II and mitochondrial fission marker Drp-1 protein levels were significantly increased in the male SE offspring (P < 0.05, Fig. 4a, c). Maternal L-carnitine supplementation further increased LC3A/B-II level, but normalised Drp-1 levels in the SE + LC offspring (P < 0.001 vs SHAM, Fig. 4a, c). No changes in autophagy and mitophagy markers were found in P1 female offspring among the 3 groups (Fig. 4b, d, e).

Lung LC3A/B-II, Drp-1 and Opa-1 in the offspring at P1. Protein levels of LC3A/B-II a, b, Drp-1 c, d and Opa-1 e, f in the lung of male and female offspring at P1. Results are expressed as means ± SE, (male n = 8, female n = 6–8). Data were analysed by one-way ANOVA followed by Tukey’s post hoc tests. * P < 0.05, **P < 0.01, ***P < 0.001. LC3A/B, light chain 3 microtubule-associated protein; Drp-1, dynamin-related protein; Opa-1, optic atrophy-1; SE, maternal smoke exposure; SE + LC, maternal smoke exposure with L-Carnitine supplement
At 13 weeks, LC3A/B-II protein was significantly increased by maternal SE in the male offspring (P < 0.01, Fig. 5a) which was not affected by maternal L-Carnitine supplementation. In the female offspring, no difference in autophagy and mitophagy markers was observed among the 3 groups (Fig. 5b, d, e).

Lung LC3A/B-II, Drp-1 and Opa-1 in the offspring at 13 weeks. Protein levels of LC3A/B-II a, b, Drp-1 c, d and Opa-1 e, f in the lung of male and female offspring at 13 weeks. Results are expressed as means ±SE, (male n = 7–8, female n = 8). Data were analysed by one-way ANOVA followed by Tukey’s post hoc tests. **P < 0.01. Drp-1. LC3A/B, light chain 3 microtubule-associated protein; dynamin-related protein; Opa-1, optic atrophy-1; SE, maternal smoke exposure; SE + LC, maternal smoke exposure with L-Carnitine supplement
Discussion
Maternal smoking during pregnancy is well-documented to cause long-term adverse effects on the health outcomes in multiple organs in the offspring, including respiratory, neurological, and renal systems [37]. However, the sex difference in respiratory disorders has not been broadly studied, perhaps due to the preference of using one gender to model asthma or COPD.
In this study, male offspring from the SE dams had smaller body weight from birth to adulthood, consistent with previous animal studies and humans suggesting the reproducibility and human relevance of our model [38, 39]. Maternal SE activated inflammatory NF-κB and MAPK pathways, which were more prominent in the male offspring at P1. It is well known that cigarette smoking can induce inflammation via the MAPK signalling cascade [40], reflected by increased phosphorylation of ERK and P38 [41, 42]. MAPK pathway activation can also lead to increased activation of certain transcription factors, such as NF-κB [43]. In the current study, these effects in P1 male SE offspring are likely due to the chemicals in cigarette smoke including free radicals reaching the foetus via the placenta. NLRP3 inflammasome activation in the male offspring at P1 is in accordance with other inflammatory pathways, especially NF-κB. However, only NF-κB hyperactivation was maintained at adulthood. This may be due to a lack of a second insult after birth. As NF-κB regulates acute responses to external stimuli, its innate hyperactivation may enhance the response to postnatal environmental factors, such as allergens to increase the risk of asthma or tobacco smoking to increase the risk of COPD [44]. This requires further investigation with additional modelling in the offspring.
It is not surprising to observe that female offspring are less affected by the adverse effect of maternal SE compared with the male littermates. Such a lack of response in the females is consistent with our previous observations in the brain and kidney [14, 45, 46]. In adults, there are known differences in innate and adaptive immune responses between males and females which might be related to immune related genes encoded upon the X chromosome being differentially expressed in males and females. Furthermore the sex hormone oestrogen can protect females from developing several diseases [46, 47].
A recent study found that inflammasomes can be the bridge between inflammation and mitochondrial function [47]. There is increasing recognition that mitochondrial dysfunction plays a key role in the development of various diseases including COPD and asthma [27, 48, 49]. Maternal smoking can induce a high level of oxidative stress in the developing foetus [50] which is persistent until adulthood to directly damage the mitochondria [14, 51]. Injured mitochondria can also induce more oxidative stress and inflammation. As such, mitophagy and autophagy are key to recycle mitochondrial fragments and eliminate defective mitochondria to maintain cellular homeostasis [52]. Increased fission maker Drp-1 and autophagosome marker LC3A/B-II in the male SE offspring at birth suggests an increased number of damaged mitochondria due to maternal SE. The fusion marker Opa-1 was not increased suggesting less mitochondrial biogenesis. At adulthood, only LC3A/B-II remained elevated, suggesting higher demand to eliminate injured cellular elements resulting from maternal SE. This may drive the development of lung disorders in the SE offspring [53]. Interestingly, mitophagy markers in the lung were not changed in the female offspring at any age, suggesting a gender-specific response to maternal SE. These results are consistent with our previous research in other organs [13].
In vivo and in vitro studies have demonstrated that L-Carnitine can prevent oxidative stress-induced injury [54,55,56]. In this study, maternal L-Carnitine supplementation increased birth weight in both male and female SE offspring. This suggests that L-Carnitine can ameliorate in-utero underdevelopment caused by maternal SE. Additionally, maternal L-Carnitine supplementation exhibited some anti-inflammatory effects in newborns from the SE dams, by partially suppressing NF-κB activation and NLRP3 inflammasome formation in the males, as well as MAPK pathway and IL-1β in the females. This may be due to its ability to inhibit oxidative stress induced by maternal SE in utero. However, the protection of maternal L-Carnitine supplementation during gestation at birth did not persist until adulthood, especially in the male offspring. The result of increased IL-1β in the male offspring lung, especially in the group with maternal L-Carnitine supplementation is intriguing as there was no increase in inflammasome activation. IL-1β is encoded by its own gene and produced as an in-active protein which is converted into an active protein by the inflammasome. Without a concomitant increase in inflammasome levels our results suggest at 13 weeks that either there is an increase in inactive IL-1β, not active IL-1β or the IL-1β measured by western blotting is produced external to the lungs.
The protective effects of maternal L-Carnitine supplementation have been observed in the other organs, including the brain, kidney, and liver [13, 30, 35, 57], however not in the lung as shown in this study. This is surprising, but may be explained by the limitation of how we assessed the lung in this study. Firstly, we did not collect bronchoalveolar lavage (BAL) fluid, which can provide information on inflammatory cytokine changes and inflammatory cell counts which are more direct ways to assess lung inflammation. We only used one dose of L-carnitine during pregnancy and lactation, which cannot provide the correlation between L-carnitine dose and inflammation. The multiple-dose regime needs to be tested in our future study. The hyperactivation of inflammatory signalling cascades may represent an increased ability to respond to external stimuli such as allergens or cigarette smoke, but itself may not necessarily cause lung diseases. We did not measure reactive oxygen species levels, and as such whilst it is likely that L-Carnitine supplementation acts via scavenging reactive oxygen species, we cannot definitively say this was the case.
Conclusions
In conclusion, there are gender differences in the susceptibility to lung disorders in response to maternal smoking, with male offspring more susceptible to increased inflammatory changes. Maternal L-Carnitine supplementation during pregnancy may partially alleviate the adverse effects of maternal SE on lung health outcomes however only marked in the newborn offspring.
Materials and methods
Animals
The animal experiments were approved by the Animal Care and Ethics Committee at the University of Technology Sydney (ACEC#2011-313A). All protocols were performed according to the Australian National Health and Medical Research Council Guide for the Care and Use of Laboratory Animals. Female BALB/c mice (8 weeks, Animal Resources Centre, Perth, WA, Australia) were housed at 20 ± 2 °C and maintained on a 12 h light, 12 h dark cycle (lights on at 06:00 h) with ad libitum access to standard rodent chow and water. Female BALB/c mice were divided into 3 groups. The SHAM group (n = 11) was exposed to air in a 15 L Perspex chamber for 6 weeks prior to mating, during gestation and lactation, SE group (n = 21) was exposed to cigarette smoke generated from 2 cigarettes (Winfield Red, 1.2 mg nicotine; VIC, Australia) per session (5-min interval between), twice daily during the same period of time as we have previously described [46]. A sub-group of the SE dams (n = 12) was provided with L-Carnitine (Sapphire Bioscience Pty. Ltd., NSW, Australia) directly dissolved in drinking water (1.5 mM, SE + LC) during gestation and lactation as we have previously described [13]. This was available ad libatum. L-Carnitine dose was determined according to a previous publication [58]. Pregnancy was confirmed by significant weight gain 10 days after mateing. P1 mice were sacrificed by decapitation, while adult animals were sacrificed after anaesthetic overdose (Pentothal®, 0.1 mg/g, i.p., Abbott Australasia Pty. Ltd., NSW, Australia) between 9:00–12:00 h. The lungs from the offspring were collected at birth (P1) and adulthood (13 weeks) and stored at − 80 °C for later analysis.
Western blotting
The protein levels of the markers of interest were measured in the lung, including inflammatory markers, phosphate(p)-ERK1,2 (1:2000; Cell Signalling Technology), p-JNK1,2 (1:2000; Cell Signalling Technology), p-p38 MAPK (1: 2000; Cell Signalling Technology), p- NF-κB (1:2000; Cell Signalling Technology), and autophagy markers light chain 3 microtubule-associated protein A/B (LC3A/B)-II (1:2000; Cell Signalling Technology), mitophagy fission marker dynamin-related protein (Drp)-1 (1:2000; Cell Signalling Technology) and mitophagy fusion marker optic atrophy (OPA)-1 (1:2000; Cell Signalling Technology), inflammasome marker NLRP3 (1:2000; Abcam), IL-1β (1:2000; Cell Signalling Technology), and housekeeping protein β-actin (1:10000; Cell Signalling Technology).
The lung was homogenised using cell lysis buffers for total protein and mitochondrial protein extraction through differential centrifugation as previously described [35]. Protein concentrations were measured using DC Protein assay (Bio-Rad, Hercules, CA); 15μg of proteins were separated on Ctiterion™TGX Stain Free Precast Gel (BIO-RAD, USA) and then transferred to PVDF membranes (BIO-RAD, USA), which was then blocked with TBST. The membranes were incubated with the primary antibodies, followed by horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology). Protein expression was detected by SuperSignal West Pico Chemiluminescent substrate (Thermo, MA, USA) by exposing the membrane in ChemiDoc (BIO-RAD, USA). The density of the protein band was determined using Image J (National Institute of Health, Bethesda, Maryland, USA).
Statistical methods
The results are expressed as mean ± SEM. Normality was tested by Shapiro-Wilk test prior to the statistical analysis. If the data were not normally distributed, they were log-transformed to research normality. The differences between groups were analysed by one-way ANOVA followed by Tukey’s post hoc tests (GraphPad Prism 8, GraphPad Software, CA, USA). P < 0.05 was considered statistically significant.
Availability of data and materials
The datasets herein used and analyzed are available from the corresponding author on reasonable request.
Abbreviations
- SE:
-
Smoke exposure
- COPD:
-
Chronic obstructive pulmonary disease
- ERK:
-
Extracellular signal-regulated kinase
- JNK:
-
Jun N-terminal kinase
- NLRP3:
-
Nucleotide-binding domain and leucine-rich repeat-containing family pyrin domain containing 3
- p38:
-
p38 Mitogen-activated protein kinase
- NF-κB:
-
Nuclear factor-κB;
- SE + LC:
-
Maternal smoke exposure with L-Carnitine supplement
References
Warren GW, Alberg AJ, Kraft AS, Cummings KM. The 2014 Surgeon General’s report:the health consequences of smoking–50 years of Progress: a paradigm shift in cancer care. Cancer. 2014;(120):1914–6.
Songül Şahinli A, Marakoğlu K, Kiyici A. Evaluation of the levels of oxidative stress factors and ischemia modified albumin in the cord blood of smoker and non-smoker pregnant women. J Matern Fetal Neonatal Med. 2012;25:1064–8.
Lee LJ, Lupo PJ. Maternal smoking during pregnancy and the risk of congenital heart defects in offspring: a systematic review and metaanalysis. Pediatr Cardiol. 2013;34:398–407.
Hoo A-F, Henschen M, Dezateux C, Costeloe K, Stocks J. Respiratory function among preterm infants whose mothers smoked during pregnancy. Am J Respir Crit Care Med. 1998;158:700–5.
Hanrahan JP, Tager IB, Segal MR, Tosteson TD, Castile RG, Van Vunakis H, Weiss ST, Speizer FE. The effect of maternal smoking during pregnancy on early infant lung function. Am Rev Respir Dis. 1992;145:1129–35.
Collins MH, Moessinger AC, Kleinerman J, Bassi J, Rosso P, Collins AM, James L, Blanc WA. Fetal lung hypoplasia associated with maternal smoking: a morphometric analysis. Pediatr Res. 1985;19:408–12.
Maritz G. Maternal nicotine exposure during gestation and lactation of rats induce microscopic emphysema in the offspring. Exp Lung Res. 2002;28:391–403.
Sekhon H, Proskocil B, Clark J, Spindel E. Prenatal nicotine exposure increases connective tissue expression in foetal monkey pulmonary vessels. Eur Respir J. 2004;23:906–15.
Wongtrakool C, Wang N, Hyde DM, Roman J, Spindel ER. Prenatal nicotine exposure alters lung function and airway geometry through α7 nicotinic receptors. Am J Respir Cell Mol Biol. 2012;46:695–702.
Sandberg KL, Pinkerton KE, Poole SD, Minton PA, Sundell HW. Fetal nicotine exposure increases airway responsiveness and alters airway wall composition in young lambs. Respir Physiol Neurobiol. 2011;176:57–67.
Aryal S, Diaz-Guzman E, Mannino DM. COPD and gender differences: an update. Transl Res. 2013;162:208–18.
Mizumura K, Maruoka S, Shimizu T, Gon Y. Autophagy, selective autophagy, and necroptosis in COPD. Int J Chronic Obstruct Pulmon Dis. 2018;13:3165.
Chan YL, Saad S, Al-Odat I, Oliver BG, Pollock C, Jones NM, Chen H. Maternal L-carnitine supplementation improves brain health in offspring from cigarette smoke exposed mothers. Front Mol Neurosci. 2017;10:33.
Chan YL, Saad S, Pollock C, Oliver B, Al-Odat I, Zaky AA, Jones N, Chen H. Impact of maternal cigarette smoke exposure on brain inflammation and oxidative stress in male mice offspring. Sci Rep. 2016;6:25881.
Chen H, Li G, Chan YL, Chapman DG, Sukjamnong S, Nguyen T, Annissa T, McGrath KC, Sharma P, Oliver BG. Maternal e-cigarette exposure in mice alters DNA methylation and lung cytokine expression in offspring. Am J Respir Cell Mol Biol. 2018;58:366–77.
Wiklund P, Karhunen V, Richmond RC, Parmar P, Rodriguez A, De Silva M, Wielscher M, Rezwan FI, Richardson TG, Veijola J, et al. DNA methylation links prenatal smoking exposure to later life health outcomes in offspring. Clin Epigenetics. 2019;11:97.
Zhang B, Hong X, Ji H, Tang W-Y, Kimmel M, Ji Y, Pearson C, Zuckerman B, Surkan PJ, Wang X. Maternal smoking during pregnancy and cord blood DNA methylation: new insight on sex differences and effect modification by maternal folate levels. Epigenetics. 2018;13:505–18.
Meyer KF, Krauss-Etschmann S, Kooistra W, Reinders-Luinge M, Timens W, Kobzik L, Plosch T, Hylkema MN. Prenatal exposure to tobacco smoke sex dependently influences methylation and mRNA levels of the Igf axis in lungs of mouse offspring. Am J Physiol Lung Cell Mol Physiol. 2017;312:L542–l555.
Golden LC, Itoh Y, Itoh N, Iyengar S, Coit P, Salama Y, Arnold AP, Sawalha AH, Voskuhl RR. Parent-of-origin differences in DNA methylation of X chromosome genes in T lymphocytes. Proc Natl Acad Sci U S A. 2019;116(52)26779-87. https://doi.org/10.1073/pnas.1910072116.
Armstrong L, Hughes O, Yung S, Hyslop L, Stewart R, Wappler I, Peters H, Walter T, Stojkovic P, Evans J. The role of PI3K/AKT, MAPK/ERK and NFκβ signalling in the maintenance of human embryonic stem cell pluripotency and viability highlighted by transcriptional profiling and functional analysis. Hum Mol Genet. 2006;15:1894–913.
Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75:50–83.
Kumar A, Takada Y, Boriek AM, Aggarwal BB. Nuclear factor-κB: its role in health and disease. J Mol Med. 2004;82:434–48.
McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol. 2006;16:R551–60.
Zhou R, Yazdi AS, Menu P, Tschopp JJN. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221-5.
Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DMJI. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36(3):401-14.
Colarusso C, Terlizzi M, Molino A, Pinto A, Sorrentino R. Role of the inflammasome in chronic obstructive pulmonary disease (COPD). Oncotarget. 2017;8:81813–24.
Nam H-S, Izumchenko E, Dasgupta S, Hoque MO. Mitochondria in chronic obstructive pulmonary disease and lung cancer: where are we now? Biomark Med. 2017;11:475–89.
Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11:872.
Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9.
Stangenberg S, Nguyen LT, Chan YL, Zaky A, Pollock CA, Chen H, Saad S. Maternal L-carnitine supplementation ameliorates renal underdevelopment and epigenetic changes in male mice offspring due to maternal smoking. Clin Exp Pharmacol Physiol. 2019;46:183–93.
Isik B, Ceylan A, Isik R. Oxidative stress in smokers and non-smokers. Inhal Toxicol. 2007;19:767–9.
Hagen TM, Ingersoll RT, Wehr CM, Lykkesfeldt J, Vinarsky V, Bartholomew JC, Song MH, Ames BN. Acetyl-L-Carnitine fed to old rats partially restores mitochondrial function and ambulatory activity. Proc Natl Acad Sci U S A. 1998;95:9562.
Liu J, Head E, Gharib AM, Yuan W, Ingersoll RT, Hagen TM, Cotman CW, Ames BN. Memory loss in old rats is associated with brain mitochondrial decay and RNA/DNA oxidation: partial reversal by feeding acetyl-L-carnitine and/or R-alpha -lipoic acid. Proc Natl Acad Sci U S A. 2002;99:2356–61.
Shakeri A, Tabibi H, MJHi H. Effects of l-carnitine supplement on serum inflammatory cytokines, C-reactive protein, lipoprotein (a), and oxidative stress in hemodialysis patients with Lp (a) hyperlipoproteinemia. Hemodial Int. 2010;14(4):498-504.
Nguyen LT, Stangenberg S, Chen H, Al-Odat I, Chan YL, Gosnell ME, Anwer AG, Goldys EM, Pollock CA, Saad S. L-Carnitine reverses maternal cigarette smoke exposure-induced renal oxidative stress and mitochondrial dysfunction in mouse offspring. Am J Physiol Renal Physiol. 2015;308:F689–96.
Wang B, Chen H, Chan YL, Wang G, Oliver BG. Why do intrauterine exposure to air pollution and cigarette smoke increase the risk of asthma? Front Cell Dev Biol. 2020;8:38.
Banderali G, Martelli A, Landi M, Moretti F, Betti F, Radaelli G, Lassandro C, Verduci E. Short and long term health effects of parental tobacco smoking during pregnancy and lactation: a descriptive review. J Transl Med. 2015;13:327.
Isaksen CV, Austgulen R, Chedwick L, Romundstad P, Vatten L, Craven C. Maternal smoking, intrauterine growth restriction, and placental apoptosis. Pediatr Dev Pathol. 2004;7:433–42.
Horta BL, Victora CG, Menezes AM, Halpern R, Barros FC. Low birthweight, preterm births and intrauterine growth retardation in relation to maternal smoking. Paediatr Perinat Epidemiol. 1997;11:140–51.
Zhao J, Harper R, Barchowsky A, YPJAJoP-LC D, Physiology M. Identification of multiple MAPK-mediated transcription factors regulated by tobacco smoke in airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2007;293(2):L480-90.
Li T, Song T, Ni L, Yang G, Song X, Wu L, Liu B, Liu CJB, Communications Br. The p-ERK–pc-Jun–cyclinD1 pathway is involved in proliferation of smooth muscle cells after exposure to cigarette smoke extract. Biochem Biophys Res Commun. 2014;453(3):316-20.
Marumo S, Hoshino Y, Kiyokawa H, Tanabe N, Sato A, Ogawa E, Muro S, Hirai T, MJBpm M. P38 mitogen-activated protein kinase determines the susceptibility to cigarette smoke-induced emphysema in mice. BMC Pulm Med. 2014;14:79.
Aggarwal BB, Takada Y, Shishodia S, Gutierrez AM, Oommen OV, Ichikawa H, Baba Y, Kumar A: Nuclear transcription factor NF-kappa B: role in biology and medicine. 2004.
Hylkema MN, Blacquière MJ. Intrauterine effects of maternal smoking on sensitization, asthma, and chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2009;6:660–2.
Chan YL, Saad S, Al-Odat I, Zaky AA, Oliver B, Pollock C, Li W, Jones NM, Chen H. Impact of maternal cigarette smoke exposure on brain and kidney health outcomes in female offspring. Clin Exp Pharmacol Physiol. 2016;43:1168–76.
Al-Odat I, Chen H, Chan YL, Amgad S, Wong MG, Gill A, Pollock C, Saad S. The impact of maternal cigarette smoke exposure in a rodent model on renal development in the offspring. PLoS One. 2014;9:e103443.
Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221.
Prakash Y, Pabelick CM, Sieck GC. Mitochondrial dysfunction in airway disease. Chest. 2017;152:618–26.
Mabalirajan U, Dinda AK, Kumar S, Roshan R, Gupta P, Sharma SK, Ghosh B. Mitochondrial structural changes and dysfunction are associated with experimental allergic asthma. J Immunol. 2008;181:3540–8.
Noakes PS, Thomas R, Lane C, Mori TA, Barden AE, Devadason SG, Prescott SLJT. Association of maternal smoking with increased infant oxidative stress at 3 months of age. 2007;62:714–7.
Chan Y, Oliver B, Sharma P, Zakarya R, Chen H, Saad S, Santiyanont R, Sukjamnong S: Effect of long-term maternal smoking on the offspring’s lung health. 2017.
Narendra D, Tanaka A, Suen D-F, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803.
Cloonan SM, Choi AMK. Mitochondria in lung disease. J Clin Invest. 2016;126:809–20.
Kolodziejczyk J, Saluk-Juszczak J, Wachowicz B. L-Carnitine protects plasma components against oxidative alterations. Nutrition. 2011;27:693–9.
Mingorance C, Rodríguez-Rodríguez R, Justo ML, de Sotomayor MÁ, Herrera MD. Critical update for the clinical use of L-carnitine analogs in cardiometabolic disorders. Vasc Health Risk Manag. 2011;7:169.
Nguyen LT, Stangenberg S, Chen H, Al-Odat I, Chan YL, Gosnell ME, Anwer AG, Goldys EM, Pollock CA, SJAJoP-RP S. L-Carnitine reverses maternal cigarette smoke exposure-induced renal oxidative stress and mitochondrial dysfunction in mouse offspring. Am J Physiol Renal Physiol. 2015;308(7):F689-96.
Saad S, Al-Odat I, Chan Y, McGrath K, Pollock C, Oliver B, Chen H. Maternal L-carnitine supplementation improves glucose and lipid profiles in female offspring of dams exposed to cigarette smoke. Clin Exp Pharmacol Physiol. 2018;45:694–703.
Ratnakumari L, Qureshi IA, Maysinger D, Butterworth RF. Developmental deficiency of the cholinergic system in congenitally hyperammonemic spf mice: effect of acetyl-L-carnitine. J Pharmacol Exp Ther. 1995;274:437–43.
Funding
Mr. Baoming Wang is supported by the China Scholarship Council. Dr. Yik Lung Chan is supported by an iCare Fellowship. Shengyu Zhou was supported by the China Scholarship Council. Associate Professor Hui Chen was supported by a Research Fellowship for International Young Scientists (81750110554) awarded by the National Natural Science Foundation of China. Professor Brian G Oliver was supported by a Career Development Fellowship II (APP1110368) by National Health & Medical Research Council Australia.
Author information
Authors and Affiliations
Contributions
HC (University of Technology Sydney), SS (University of Sydney), and BGO (University of Technology Sydney) designed the study. BW (University of Technology Sydney), SZ (Shandong University, China) and YLC (University of Technology Sydney) performed all the experiments. BW, HC, SS, BGO, and YLC contributed to the writing of the manuscript. BW and YLC prepared all the figures. All the authors reviewed the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The animal experiments were approved by the Animal Care and Ethics Committee at the University of Technology Sydney (ACEC#2011-313A).
Consent for publication
N/A
Competing interests
There is no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, B., Chan, Y.L., Zhou, S. et al. Offspring sex affects the susceptibility to maternal smoking-induced lung inflammation and the effect of maternal antioxidant supplementation in mice. J Inflamm 17, 24 (2020). https://doi.org/10.1186/s12950-020-00253-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12950-020-00253-5