
Abstract
Human in vivo models of systemic inflammation are used to study the physiological mechanisms of inflammation and the effect of drugs and nutrition on the immune response. Although in vivo lipopolysaccharide (LPS) challenges have been applied as methodological tool in clinical pharmacology studies, detailed information is desired on dose-response relationships, especially regarding LPS hyporesponsiveness observed after low-dose in vivo LPS administration. A study was performed to assess the in vivo inflammatory effects of low intravenous LPS doses, and to explore the duration of the induced LPS hyporesponsiveness assessed by subsequent ex vivo LPS challenges.
This was a randomized, double-blind, placebo-controlled study with single ascending low doses of LPS (0.5, 1 and 2 ng/kg body weight) administered to healthy male volunteers (3 cohorts of 8 subjects, LPS:placebo 6:2). The in vivo inflammatory response was assessed by measurement of cytokines and CRP. Ex vivo LPS challenges were performed (at −2, 6, 12, 24, 48 and 72 hours relative to in vivo LPS administration) to estimate the duration and magnitude of LPS hyporesponsiveness by assessment of cytokine release (TNF-α, IL-1β, IL-6, IL-8).
LPS administration dose-dependently increased body temperature (+1.5°C for 2 ng/kg LPS), heart rate (+28 bpm for 2 ng/kg LPS), CRP and circulating cytokines which showed clearly distinctive increases from placebo already at the lowest LPS dose level tested (0.5 ng/kg, contrast for timeframe 0–6 hours: TNF-α +413%, IL-6 +288%, IL-8 +254%; all p ≤ 0.0001). In vivo LPS administration dose-dependently induced a period of hyporesponsiveness in the ex vivo LPS-induced cytokine release (IL-1β, IL-6 and TNF-α), with maximal hyporesponsiveness observed at 6 hours, lasting no longer than 12 hours. For IL-6 and IL-8, indications for immune cell priming were observed.
We demonstrated that an in vivo LPS challenge, with LPS doses as low as 0.5 ng/kg, elicits a cytokine response that is clearly distinctive from baseline cytokine levels. This study expanded the knowledge about the dose-effect relationship of LPS-induced hyporesponsiveness. As such, the low-dose LPS challenge has been demonstrated to be a feasible methodological tool for future clinical studies exploring pharmacological or nutritional immune-modulating effects.
Similar content being viewed by others


Introduction
Human models of systemic inflammation have been developed with the purpose to explore the molecular mechanisms and physiological significance of the systemic inflammatory response encountered in acute as well as chronic inflammatory conditions, such as sepsis, trauma, type 2 diabetes, atherosclerosis, and Alzheimer s disease, in a controlled, standardized experimental setting. A better understanding of the underlying molecular and pathophysiological mechanisms could lead to optimized prevention and treatment of these disorders, associated with morbidity and mortality [1]. In addition, human models of systemic inflammation can be applied in clinical pharmacology studies to assess the effects of specific interventions (medicinal or non-medicinal) on the inflammatory response in non-diseased populations.
Human endotoxemia is often used as a model of systemic inflammation. In this experimental setting, purified lipopolysaccharide (LPS, also referred to as endotoxin) from the cell membrane of Escherichia coli (E. coli) or other Gram-negative bacteria is administered intravenously to healthy volunteers resulting in flu-like symptoms, increased production of C-reactive protein (CRP) and increased concentrations of pro- and anti-inflammatory cytokines. Since the effects of E. coli are highly reproducible, this is the predominant bacterial source used [1]. LPS induces an inflammatory response via stimulation of Toll-like receptors (TLRs), basic signaling receptors of the innate immune system activated by tissue damage or by molecules associated with pathogen-associated molecular patterns (PAMPs) on invading microorganisms. LPS is known to activate multiple intracellular pathways (e.g. the MyD88-dependent and TRIF-dependent pathways) [2],[3].
The human endotoxemia model has been studied extensively and commonly applies relatively high LPS doses (2–4 ng/kg body weight) [1],[4]-[8]. However, an endotoxemia model applying such relatively high LPS doses is not preferred as methodological tool in clinical pharmacology studies since the elicited immune response is so strong that potential effects of immune-modulating interventions may not be observed, other homeostatic mechanisms may be temporarily impaired, and, importantly, the elicited immune response at these LPS doses is not free of risk for the volunteer. Studies applying lower LPS doses have been performed [9],[10], but thorough characterization of a human endotoxemia model at lower LPS dose levels is desired. In the current study, 0.5 ng/kg was selected as the lowest LPS dose to be administered intravenously because an LPS dose of 0.2 ng/kg was shown previously to elicit no cytokine response in vivo[11]. We performed a study to characterize the LPS dose relationship of the human inflammatory response at low LPS doses (0.5, 1 and 2 ng/kg) administered to healthy volunteers. Furthermore, we explored the effects of such an in vivo LPS challenge on the inflammatory response induced by subsequent ex vivo LPS challenges. It has been described that an in vivo LPS challenge induces hyporesponsiveness to following in vivo or ex vivo LPS challenges. The biochemical mechanisms accounting for this hyporesponsiveness have been demonstrated to involve negative regulators such as IRAK-M, SOCS-1, SHIP, ST2 and IL-10 [2],[3],[12]-[18] and downregulation of CD14 [19].
It has been reported that ex vivo LPS hyporesponsiveness following an in vivo LPS challenge was resolved after 1 week [4]. However, the exact time course of this phenomenon and relation to LPS dose level is unclear. Since such information could be important for (repeated) application of in vivo and ex vivo LPS challenges in clinical pharmacology studies, characterization of this hyporesponsiveness was an objective of our study.
Methods
Subjects
Twenty-four healthy male volunteers, aged 18–28 years (inclusive) with a BMI of 18 to 25 kg/m2 and a body weight ≥ 56 kg, participated in this study. After providing informed consent, subjects were medically screened within 3 weeks prior to participation. Exclusion criteria included history of sepsis, cardiovascular disease, previous syncope or malignancy, haemorrhagic diathesis, any active inflammatory or infectious disease, renal impairment, diabetes mellitus, thyroid dysfunction, and prior exposure to endotoxin in an experimental setting within 4 weeks of the anticipated exposure. Any use of medication that in the opinion of the investigator would complicate or compromise the study or interfere with the study objectives was not permitted during the study. The study was conducted in accordance with the Declaration of Helsinki and Guideline for Good Clinical Practice, and was approved by the Medical Ethics Review Board of the Academic Medical Center, Amsterdam, The Netherlands.
Study design
This was a randomized, blinded, placebo controlled study of ascending single doses of 0.5, 1 and 2 ng/kg LPS (U.S. Reference Escherichia Coli (E. Coli) endotoxin CC-RE-Lot 3 (O113:H, 10:K negative, National Institute of Health, Bethesda, MD, US, approximately 10 EU/ng); or placebo), administered to healthy male subjects as an intravenous bolus over 2 minutes. Each cohort included eight healthy subjects, of which six subjects received LPS and two placebo (sodium chloride 0.9%). Subjects were prehydrated with 1500 mL glucose/saline (2.5% glucose/0.45% sodium chloride) 2 hours prior to LPS(/placebo) administration, followed by an intravenous drip of 150 mL/hr for a period of 6 hours. After LPS/placebo administration, subjects were confined to the clinical research unit for 24 hours.
Safety monitoring
Safety monitoring was performed by adverse events monitoring, physical examination, assessment of electrocardiogram (ECG) and vital signs, and laboratory evaluations (routine hematology, chemistry, coagulation, and semi-quantitative dipstick urinalysis). In case of clinically significant findings in dipstick analysis, a microscopic investigation of the urine was performed. For subject safety, maximally two subjects were treated within one day, with a lag time of at least 40 minutes between subjects. All blinded safety data collected up to at least 24 hours after LPS/placebo administration were reviewed before the decision was made to escalate the LPS dose level and proceed with the next cohort.
Inflammatory markers
The systemic inflammatory response was assessed by frequent measurement of C-reactive protein (CRP) and a panel of cytokines (IL-1β, IL-6, IL-8 and TNF-α) using a human ultra-sensitive 4-plex (MSD). CRP levels were measured as part of the standard chemistry panel. Samples for cytokine analysis were collected in sodium heparin (Greiner) tubes. In addition, the effect of an in vivo LPS challenge on cytokine release (IL-1β, IL-6, IL-8, TNF-α) induced by an ex vivo LPS challenge was studied. Blood samples were collected in sodium heparin tubes (Greiner) before and 6, 12, 24, 48 and 72 hours after the in vivo LPS administration. Whole blood cultures were prepared with a 1:1 dilution with RPMI 1640 medium and incubated with LPS (E. Coli O111:B4, manufactured by Sigma-Aldrich, Saint Louis, MO, US, catalog number L-3012, approximately 10 EU/ng) for 24 hours at 37°C, 5% CO2. Cultures were centrifuged and supernatants were used for cytokine assessment using the earlier mentioned cytokine 4-plex with a 20-fold dilution. Whole blood cultures were performed by Good Biomarker Sciences, Leiden, The Netherlands.
Statistical analysis
Statistical analysis was performed for circulating inflammatory markers and ex vivo-induced cytokines, which were log-transformed prior to analysis. These repeatedly measured parameters were analyzed with a mixed model of variance with treatment, time, and treatment by time as fixed factors and subject as random factor and the baseline measurement as covariate. A variance components (co)variance structure was used to model the within-subject errors, the Kenward-Roger approximation to estimate denominator degrees of freedom and the restricted maximum likelihood method to estimate model parameters. Contrasts were calculated within the model for each parameter over the following time profiles: baseline to 6 hours post-dose for circulating cytokines; baseline to 24 hours post-dose for CRP; and at 6 hours post-dose for ex vivo-induced cytokines. The general treatment effect and specific contrasts were reported with the estimated difference, the 95% confidence interval (CI), the least square mean (LSM) estimates and the p-value. Graphs of the LSMs estimates over time by treatment present 95% confidence intervals as error bars and change from baseline LSMs estimates. All analyses were performed using SAS for Windows Version 9.1.3 (SAS Institute, Inc., Cary, NC, USA).
Results
Safety monitoring
Single intravenous low doses of LPS were well tolerated in healthy male subjects. Observed adverse events (AEs) were of mild severity and self-limiting without therapeutic intervention. The most frequent occurring AEs, probably or possibly related to treatment, were headache, observed in 66.7% of the LPS-treated subjects and 33.3% of the placebo-treated subjects and feeling cold, observed in 44.4% of the LPS-treated subjects and none of the placebo-treated subjects. No clinically relevant changes or unexpected treatment-related trends were observed in supine systolic and diastolic blood pressure, body temperature, or ECG-derived parameters following administration of LPS (Figure 1). LPS dose-dependently increased body temperature and heart rate, with a maximal increase amounting approximately 1.5°C and 28 ± 13.2 bpm for the highest LPS dose tested, observed at 3–4 hours after LPS administration.

Vital signs: temperature (°C, panel A ), heart rate (bpm, panel B ), systolic blood pressure (mmHg, panel C ), diastolic blood pressure (mmHg, panel D ), change from baseline with standard deviation as error bars.
LPS administration resulted in a dose-dependent decrease in monocyte count (maximal change from baseline −1.9 ± 2.3, −4.8 ± 3.6 and −7.7 ± 1.5% at 6 hours post-LPS for doses of 0.5, 1 and 2 ng/kg, respectively), returning to baseline levels within 12 to 24 hours post-LPS (data not shown). In addition, LPS administration resulted in decreased blood platelet count levels (minimal change from baseline of −15 ± 9.4, −28 ± 14.4, and −31 ± 9.1*10^9/L at 4 hours post-dose, data not shown) and an increase in neutrophil count (maximum change from baseline 25.8 ± 3.5, 40.4 ± 9.5, and 42.9 ± 6.2% at 4 hours post-LPS for doses of 0.5, 1 and 2 ng/kg, respectively) and leukocyte count (maximum change from baseline 4.0 ± 1.3, 4.4 ± 1.2, 6.3 ± 1.4*10^9/L at 4–6 hours post-LPS), returning to baseline at 12–24 hours post-dose (data not shown). Eosinophil, erythrocyte, lymphocyte, and basophil counts and hematocrit and hemoglobin slightly decreased after LPS administration, with maximal changes observed 4 hours after LPS administration (data not shown).
Activated partial thromboplastin time (APTT) was variable over the day, ranging from −0.7 to +0.9 s around a baseline concentration of 29.3 ± 1.8 s. LPS administration resulted in a decrease in APTT, with an estimated difference of −2.0 s (p = 0.0151) at 1 ng/kg LPS and a maximal mean decrease from baseline of −3.9 s at 4 hours post-LPS (Figure 2). Although the decrease in APTT upon administration of 2 ng/kg LPS was comparable in size, this difference did not reach a level of statistical significance (p = 0.1233). An effect of LPS administration on Prothrombin Time (PT) was not observed (data not shown).

Activated Partial Thromboplastin Time (APTT) LSMs change from baseline profile, with 95% CI as error bars.
Circulating inflammatory markers
CRP levels were low in the placebo-treated group (data not shown; baseline concentration 0.85 ± 1.08 mg/L, with a minimal variability over time of maximally 0.30 ± 0.55 mg/L. LPS administration dose-dependently increased CRP, maximal levels observed 24 hours post-dose (11.31 ± 6.73, 15.15 ± 3.93, and 18.42 ± 5.15 mg/L for LPS doses of 0.5, 1 and 2 ng/kg, respectively, Figure 3A; all contrasts presented for the complete time profile up to 24 hours post-dose, versus placebo, p < 0.0001). In the placebo-treated group, circulating cytokine levels were minimal (Figure 3B-D). LPS administration resulted in a dose-dependent increase in TNF-α, IL-6, and IL-8, with maximal levels amounting 221.9 ± 61.2 pg/mL, 314.8 ± 130.9 and 329.4 ± 84.4 pg/mL, respectively. Maximal concentrations were reached at 1.5–3 hours after LPS administration. For all LPS dose levels tested, contrasts for cytokine release versus placebo (time interval 0–6 hours post-dose) reached a distinct level of significance (p < 0.0001). In a considerable number of samples, IL-1β levels were below the limit of quantification (LOQ, 0.6 pg/mL, data not shown). In general, higher levels of IL-1β were observed with increasing LPS doses. For all subjects in the 2 ng/kg dose group, IL-1β levels above LOQ could be detected 3–6 hours post-LPS, ranging from 0.7 to 2.6 pg/mL. No statistical analysis was performed for IL-1β.

CRP (A) time profile graph, with standard deviation as error bars; TNF-α (B) , IL-6 (C) , IL-8 (D) time profile graphs up to 6 hours, with standard deviation as error bars.
Ex-vivo LPS-induced cytokine release
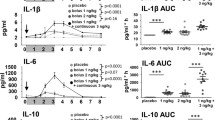
Ex vivo LPS-induced IL-1β, IL-6, IL-8 and TNF-α release was variable over time, as observed in the placebo-treated subjects (Figure 4). In vivo LPS administration dose-dependently decreased ex vivo LPS-induced TNF-α release at the highest two dose levels tested in the first hours after the in vivo LPS challenge (Figure 4A). Maximal mean reduction was observed at 6 hours post-dose with an estimated difference (95% CI) of −66.4% (−81.4 to −39.0%) and −74.7% (−86.0 to −54.3%) for 1 and 2 ng/kg, respectively, which differed significantly from placebo (Table 1, p = 0.0005 and p < 0.0001 for 1 and 2 ng/kg, respectively). Subsequently, TNF-α release increased and exceeded levels as observed for the placebo group at 12 hours post-LPS. Ex vivo LPS-induced IL-1β release basically mirrored the patterns observed for TNF-α release, with a maximal mean reduction at 6 hours post-LPS with an estimated difference (95% CI) of −65.8% (−79.5 to −43.1%) and −84.7% (−90.8 to −74.5%; Figure 4B and Table 1; p < 0.0001 and p < 0.0001 versus placebo for 1 and 2 ng/kg, respectively), and a return to placebo levels at approximately 12 hours post-dose. An in vivo LPS challenge at 1 and 2 ng/kg significantly inhibited the ex vivo LPS-induced release of IL-6 lasting for approximately 12 hours post-LPS and a maximal effect at 6 hours with an estimated difference (95% CI) of −31.3% (−50.8 to −4.0%) and −41.3% (−58.1 to −17.8%; Figure 4C and Table 1; p = 0.0283 and p = 0.0024 versus placebo for 1 and 2 ng/kg, respectively). Remarkably, ex vivo IL-6 release increased after the in vivo administration of 0.5 ng/kg LPS, peaking at 6 hours post-LPS and almost significantly exceeding cytokine levels observed for placebo-treated subjects with an estimated difference (95% CI) of 34.8% (−3.1 to 87.5% (Figure 4C and Table 1; p = 0.0754 versus placebo). A same response was observed for ex vivo IL-8 release: in vivo administration of LPS resulted in an increased IL-8 response to an ex vivo LPS challenge for the two lowest LPS doses tested with an estimated difference (95% CI) of 55.1% (−6.6 to 157.5%) and 19.2% (−28.8 to 99.5%) (Figure 4D and Table 1; at 6 hours, p = 0.0879 and p = 0.4961 versus placebo, for 0.5 and 1 ng/kg, respectively), but not for the 2 ng/kg dose. The observed increases in IL-8 release were followed by a strong decrease up to 12 hours post-dose, which was consistent for all LPS doses tested (Figure 4D).

TNF-α (A) , IL-1β (B) , IL-6 (C) , IL-8 (D) after ex vivo LPS challenge (10 EU/ng, 24 hour incubation) LSMs change from baseline profile, with 95% CI as error bars, in vivo LPS challenge on t = 0.
Discussion
Human endotoxemia has been applied frequently as a controlled and standardized model of systemic inflammation providing mechanistic insight in molecular and physiological inflammatory pathways. The in vivo LPS challenge can also be applied as methodological tool in clinical pharmacology studies to assess the effects of specific interventions (medicinal or non-medicinal) on the inflammatory response in healthy volunteers. This experimental model has been studied extensively, and commonly applies relatively high LPS doses (2–4 ng/kg bodyweight) [1],[4]-[8]. However, such relatively high LPS doses are not preferred for reasons mentioned, and characterization of a human endotoxemia model applying lower LPS dose levels is desired. Therefore, we performed a study to characterize the human inflammatory response induced by low LPS doses administered to healthy volunteers. In addition, we explored the effects of an in vivo LPS challenge on the inflammatory response induced by subsequent ex vivo LPS challenges. Although it is known from literature that in vivo LPS challenge induces hyporesponsiveness to subsequent in vivo or ex vivo LPS challenges [2],[3],[12]-[18], the exact time course of this phenomenon and relation to LPS dose level is unclear.
Administration of low LPS doses (0.5–2 ng/kg) to healthy volunteers was well-tolerated and safe; all reported AEs were of mild severity and self-limiting, and no unexpected treatment-related trends in vital signs or ECG recordings nor in urinary or blood laboratory parameters were measured. LPS administration dose-dependently increased body temperature and heart rate with maximum levels observed at 3–4 hours post-dose (change to baseline of approximately 1.5°C and 28 ± 13.2 bpm). Observed changes in hematology parameters were expected as a result of LPS treatment [5],[8],[20],[21] and subject hydration from 2 hours pre-dose till 6 hours post-dose. Furthermore, LPS administration temporarily inhibited APTT, with a maximal decrease from baseline of approximately 3–4 s at 4 hours post-LPS, in line with previously reported LPS effects on coagulation [22],[23]. There is a close interaction between coagulation and inflammation pathways [24],[25]. Stimulation of monocytes with endotoxin results in an increased expression of tissue factor, the main initiator of coagulation [23],[26],[27]. Based on this observation an increase in PT levels was expected via the extrinsic pathway, however, this could not be confirmed by the results from our study due to a high variability over the 24 hours time profile. Cytokines such as IL-6 and TNF-α are the main mediators of inflammation-induced coagulation [28],[29]. Whereas changes in temperature and heart rate after an in vivo LPS challenge could serve as a pharmacodynamic readout measure for pharmacological or dietary interventions, APTT effect size and contrast versus placebo of an in vivo LPS challenge were limited.
In vivo LPS administration dose-dependently increased circulating CRP and cytokine levels (TNF-α, IL-6 and IL-8). Maximal CRP levels were observed 24 hours post-LPS and maximal cytokine levels were observed 1.5–3 hours post-LPS. A single intravenous dose of LPS as low as 0.5 ng/kg induced a distinct inflammatory response in the healthy volunteers.
A power calculation was performed which showed that in a parallel study design, at an LPS dose level of 0.5 ng/kg, a sample size of 8 subjects per treatment group would provide 80% power to detect an 28% inhibition in the LPS-induced TNF-α response, at a two-sided significance level of 0.05. Under the same conditions, it would be possible to demonstrate an inhibition of the LPS-induced cytokine response of 53% and 49% for IL-6 and CRP, respectively. Given the fact that the inter subject variability on log scale is well comparable between different LPS doses, this power calculation also applies for LPS doses of 1 and 2 ng/kg.
Circulating IL-1β levels were low and for the majority of the samples tested the level was below LOQ (0.6 pg/mL). However, in the highest LPS dose group tested, an increase in circulating IL-1β levels could be demonstrated at 3–6 hours post-LPS, with observed IL-1β levels ranging from 0.7 to 2.6 pg/mL. This is in contrast with IL-1β release following an ex vivo LPS challenge of whole blood cultures, which caused the release of substantial amounts of IL-1β. Reports from other human endotoxemia experiments also note IL-1β responses are very low or lacking, despite high circulating levels of IL-6 and TNFα [30]-[35]. Interestingly, even in cases of severe sepsis, IL-1β can be detected in only a small fraction of patients and corresponds weakly with disease severity [36]. However it can be acutely induced in response to certain surgical procedures, and is implicated in many chronic inflammatory conditions, such as diabetes, cardiovascular disease, and rheumatoid arthritis. Since IL-1β expression is limited to inflammasome activation and requires multiple signals [37],[38], this suggests that low-dose human endotoxemia may be insufficient to induce systemic IL-1β. LPS stimulation in whole blood cultures also induces cell death, which may facilitate inflammasome activation and induce substantial IL-1β release as seen in our ex vivo LPS experiment.
In vivo LPS administration induced LPS hyporesponsiveness as evidenced by ex vivo cytokine release of IL-1β, IL-6 and TNF-α. This hyporesponsiveness was LPS dose-dependent. Although the kinetics of endotoxin hyporesponsiveness have been described previously, the exact time course of the hyporesponsiveness is not well documented [4]. Here we demonstrate that LPS-induced hyporesponsiveness of specific cytokines reached a maximum at 6 hours after the in vivo LPS challenge, and lasted no longer than 12 hours. Interestingly, it has been reported that attenuated cytokine responses in vivo persisted for at least 2 weeks [4]. This indicates that there is a significant discrepancy between the LPS hyporesponsiveness measured after an in vivo LPS challenge, for which the tissue-resident macrophages, migrating leukocytes and endothelial cells are implicated to be the main sources of cytokine production, and an ex vivo LPS challenge, for which only the circulating leukocytes are the source of cytokine release and there is no active clearance of endotoxin since the system is closed [4],[39]. The fact that the estimated duration of the derangement of the immune system induced by an in vivo LPS challenge is dependent on the selected methodology (assessment by ex vivo LPS challenge or in vivo LPS challenge) should be carefully taken into account when designing future clinical pharmacology studies applying in vivo/ex vivo LPS challenges, and this process should be driven by the nature and mechanism of action of the investigational product.
Interestingly, patterns for ex vivo LPS-induced IL-8 release (at all in vivo LPS doses tested) and IL-6 release (at the lowest in vivo LPS dose tested) differed from the patterns observed for IL-1β and TNF-α: a preceding in vivo LPS challenge caused an increased cytokine release after an ex vivo LPS challenge, rather than an inhibition of cytokine release. It may be well possible that immune cells were primed by the low-dose in vivo LPS challenge, resulting in an augmented IL-8 and IL-6 responses after ex vivo LPS stimulation. Priming of innate immune cells by low endotoxin levels has been described before, and allows the immune system to elicit a strong inflammatory response against potential pathogens [2]. Although priming of the murine immune system has been explored rather extensively, the underlying mechanisms in human immunology are poorly understood. Pretreatment of murine macrophage cells with very low doses of LPS results in an augmented cytokine production after subsequent LPS stimulation, which is LPS concentration-dependent [40]-[42]. In general it should be noted that humans are much more sensitive to LPS than mice, indicating the relative poor feasibility of murine models to support human endotoxin responses [43]. The fact that, dependent on the in vivo LPS dose applied and specific cytokine measured, either LPS hyporesponsiveness or LPS priming is observed in a relatively narrow LPS dose range (0.5–2 ng/kg) indicates that a delicate balance exists between endotoxin hyporesponsiveness and endotoxin priming, which is still to be characterized in more detail.
It should be noted that sample collection tubes used for ex vivo LPS challenges contained an endotoxin-like contamination. Although the exact level of contamination could not be expressed in relative endotoxin units, additional experiments indicated that the contamination was TLR4-specific. As a consequence of this contamination, ex vivo LPS challenges were performed at an endotoxin level resulting in a maximal TLR4-mediated response (EC100) rather than the anticipated sub-maximal response level (EC80), which was believed not to affect study outcomes.
Conclusion
Overall, our experiments demonstrate that human endotoxemia induced by commonly applied relatively high LPS doses (exceeding 2 ng/kg) can be avoided: application of LPS doses as low as 0.5 ng/kg result in significant responses in routine safety markers (e.g. temperature, blood pressure and heart rate) and circulating cytokine levels that can function as pharmacodynamic markers. As such, the low-dose LPS challenge has been demonstrated to be a feasible methodological tool for future clinical studies exploring pharmacological or nutritional immune-modulating effects. An in vivo LPS challenge induced immune cell hyporesponsiveness or immune cell priming (dependent on in vivo LPS dose and cytokine readout), determined by repeated ex vivo LPS challenges, but the duration of these effects was limited. These results indicate that a combination of in vivo LPS administration and repeated ex vivo LPS challenges can be applied in clinical pharmacology studies.
Authors’ contributions
MM and KB conceptualized the study and supervised the complete study. MRD, EPP, MM and KB designed the study, KEM was involved in the design of the laboratory methods and carried out the laboratory experiments. MRD, EPP, MM and KB were responsible for the clinical execution of the study and interpretation of the results. All authors were involved in writing the manuscript and approved the final manuscript.
Abbreviations
- AE:
-
Adverse Event
- APTT:
-
Activated Partial Thromboplastin Time
- BMI:
-
Body Mass Index
- bpm:
-
beats per minute
- CD14:
-
Cluster of differentiation 14
- CI:
-
Confidence Interval
- CRP:
-
C-Reactive Protein
- ECG:
-
Electrocardiogram
- E. Coli:
-
Escherichia Coli
- IL-1β:
-
Interleukin-1β
- IL-6:
-
Interleukin-6
- IL-8:
-
Interleukin-8
- IL-10:
-
Interleukin-10
- IRAK-M:
-
Interleukin-1 receptor-associated kinase-M
- LPS:
-
Lipopolysaccharide
- LOQ:
-
Limit Of Quantification
- LSMs:
-
Least Square Means
- PAMP:
-
Pathogen-associated molecular patterns
- PT:
-
Prothrombin Time
- SHIP:
-
SH2 domain-containing inositol 5′-phosphatase
- SOCS-1:
-
Suppressor of Cytokine Signaling-1
- TLR:
-
Toll-like receptor
- TNF-α:
-
Tumor Necrosis Factor-α
- TRIF:
-
TIR domain-containing adapter inducing IFN-β
References
Andreasen AS, Krabbe KS, Krogh-Madsen R, Taudorf S, Pedersen BK, Moller K: Human endotoxemia as a model of systemic inflammation. Curr Med Chem. 2008, 15: 1697-1705. 10.2174/092986708784872393.
Fu Y, Glaros T, Zhu M, Wang P, Wu Z, Tyson JJ, Li L, Xing J: Network topologies and dynamics leading to endotoxin tolerance and priming in innate immune cells. PLoS Comput Biol. 2012, 8: e1002526-10.1371/journal.pcbi.1002526.
Fujihara M, Muroi M, Tanamoto K, Suzuki T, Azuma H, Ikeda H: Molecular mechanisms of macrophage activation and deactivation by lipopolysaccharide: roles of the receptor complex. Pharmacol Ther. 2003, 100: 171-194. 10.1016/j.pharmthera.2003.08.003.
Kox M, De Kleijn KS, Pompe JC, Ramakers BP, Netea MG, Van der Hoeven JG, Hoedemaekers CW, Pickkers P: Differential ex vivo and in vivo endotoxin tolerance kinetics following human endotoxemia. Crit Care Med. 2011, 39: 1866-1870. 10.1097/CCM.0b013e3182190d5d.
Draisma A, Pickkers P, Bouw MP, van der Hoeven JG: Development of endotoxin tolerance in humans in vivo. Crit Care Med. 2009, 37: 1261-1267. 10.1097/CCM.0b013e31819c3c67.
van der Poll T, Coyle SM, Moldawer LL, Lowry SF: Changes in endotoxin-induced cytokine production by whole blood after in vivo exposure of normal humans to endotoxin. J Infect Dis. 1996, 174: 1356-1360. 10.1093/infdis/174.6.1356.
de Vos AF, Pater JM, Van den Pangaart PS, De Kruif MD, Van 't Veer C, Van der Poll T: In vivo lipopolysaccharide exposure of human blood leukocytes induces cross-tolerance to multiple TLR ligands. J Immunol. 2009, 183: 533-542. 10.4049/jimmunol.0802189.
Van Eijk LT, Van der Pluijm RW, Ramakers BP, Dorresteijn MJ, Van der Hoeven JG, Kox M, Pickkers P: Body mass index is not associated with cytokine induction during experimental human endotoxemia. Innate Immun. 2013, 20: 61-67. 10.1177/1753425913481821.
Ferguson JF, Mulvey CK, Patel PN, Shah RY, Doveikis J, Zhang W, Tabita-Martinez J, Terembula K, Eiden M, Koulman A, Griffin JL, Mehta NN, Shah R, Propert KJ, Song WL, Reilly MP: Omega-3 PUFA supplementation and the response to evoked endotoxemia in healthy volunteers. Mol Nutr Food Res. 2013, 58: 601-613. 10.1002/mnfr.201300368.
Nieuwdorp M, Meuwese MC, Mooij HL, van Lieshout MH, Hayden A, Levi M, Meijers JC, Ince C, Kastelein JJ, Vink H, Stroes ES: Tumor necrosis factor-alpha inhibition protects against endotoxin-induced endothelial glycocalyx perturbation. Atherosclerosis. 2009, 202: 296-303. 10.1016/j.atherosclerosis.2008.03.024.
Draisma A, De Goeij M, Wouters CW, Riksen NP, Oyen WJ, Rongen GA, Boerman OC, van Deuren M, van der Hoeven JG, Pickkers P: Endotoxin tolerance does not limit mild ischemia-reperfusion injury in humans in vivo. Innate Immun. 2009, 15: 360-367. 10.1177/1753425909105548.
Morris M, Li L: Molecular mechanisms and pathological consequences of endotoxin tolerance and priming. Arch Immunol Ther Exp (Warsz). 2012, 60: 13-18. 10.1007/s00005-011-0155-9.
Kobayashi K, Hernandez LD, Galan JE, Janeway CA, Medzhitov R, Flavell RA: IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002, 110: 191-202. 10.1016/S0092-8674(02)00827-9.
Sly LM, Rauh MJ, Kalesnikoff J, Song CH, Krystal G: LPS-induced upregulation of SHIP is essential for endotoxin tolerance. Immunity. 2004, 21: 227-239. 10.1016/j.immuni.2004.07.010.
Chang J, Kunkel SL, Chang CH: Negative regulation of MyD88-dependent signaling by IL-10 in dendritic cells. Proc Natl Acad Sci USA. 2009, 106: 18327-18332. 10.1073/pnas.0905815106.
Brint EK, Xu D, Liu H, Dunne A, McKenzie AN, O’Neill LA, Liew FY: ST2 is an inhibitor of interleukin 1 receptor and Toll-like receptor 4 signaling and maintains endotoxin tolerance. Nat Immunol. 2004, 5: 373-379. 10.1038/ni1050.
Nakagawa R, Naka T, Tsutsui H, Fujimoto M, Kimura A, Abe T, Seki E, Sato S, Takeuchi O, Takeda K, Akira S, Yamanishi K, Kawase I, Nakanishi K, Kishimoto T: SOCS-1 participates in negative regulation of LPS responses. Immunity. 2002, 17: 677-687. 10.1016/S1074-7613(02)00449-1.
Kinjyo I, Hanada T, Inagaki-Ohara K, Mori H, Aki D, Ohishi M, Yoshida H, Kubo M, Yoshimura A: SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity. 2002, 17: 583-591. 10.1016/S1074-7613(02)00446-6.
Schaaf B, Luitjens K, Goldmann T, van Bremen T, Sayk F, Dodt C, Dalhoff K, Droemann D: Mortality in human sepsis is associated with downregulation of Toll-like receptor 2 and CD14 expression on blood monocytes. Diagn Pathol. 2009, 4: 12-10.1186/1746-1596-4-12.
Van 't Veer C, Van den Pangaart PS, Van Zoelen MA, De Kruif M, Birjmohun RS, Stroes ES, de Vos AF, Van der Poll T: Induction of IRAK-M is associated with lipopolysaccharide tolerance in a human endotoxemia model. J Immunol. 2007, 179: 7110-7120. 10.4049/jimmunol.179.10.7110.
Sivapalaratnam S, Farrugia R, Nieuwdorp M, Langford CF, van Beem RT, Maiwald S, Zwaginga JJ, Gusnanto A, Watkins NA, Trip MD, Ouwehand WH: Identification of candidate genes linking systemic inflammation to atherosclerosis; results of a human in vivo LPS infusion study. BMC Med Genomics. 2011, 4: 64-10.1186/1755-8794-4-64.
Ungerstedt JS, Soop A, Sollevi A, Blomback M: Bedside monitoring of coagulation activation after challenging healthy volunteers with intravenous endotoxin. Thromb Res. 2003, 111: 329-334. 10.1016/j.thromres.2003.09.028.
De Jonge E, Dekkers PE, Creasey AA, Hack CE, Paulson SK, Karim A, Kesecioglu J, Levi M, Van Deventer SJ, Van der Poll T: Tissue factor pathway inhibitor dose-dependently inhibits coagulation activation without influencing the fibrinolytic and cytokine response during human endotoxemia. Blood. 2000, 95: 1124-1129.
Levi M, van der Poll T, Buller HR: Bidirectional relation between inflammation and coagulation. Circulation. 2004, 109: 2698-2704. 10.1161/01.CIR.0000131660.51520.9A.
Esmon CT: The interactions between inflammation and coagulation. Br J Haematol. 2005, 131: 417-430. 10.1111/j.1365-2141.2005.05753.x.
Levi M, van der Poll T, Ten Cate H: Tissue factor in infection and severe inflammation. Semin Thromb Hemost. 2006, 32: 33-39. 10.1055/s-2006-933338.
Franco RF, De Jonge E, Dekkers PE, Timmerman JJ, Spek CA, Van Deventer SJ, Van Deursen P, Van Kerkhoff L, Van Gemen B, Ten Cate H, Reitsma PH: The in vivo kinetics of tissue factor messenger RNA expression during human endotoxemia: relationship with activation of coagulation. Blood. 2000, 96: 554-559.
Stouthard JM, Levi M, Hack CE, Veenhof CH, Romijn HA, Sauerwein HP, van der Poll T: Interleukin-6 stimulates coagulation, not fibrinolysis, in humans. Thromb Haemost. 1996, 76: 738-742.
van der Poll T, Buller HR, ten Cate H, Wortel CH, Bauer KA, van Deventer SJ, Hack CE, Sauerwein HP, Rosenberg RD, ten Cate JW: Activation of coagulation after administration of tumor necrosis factor to normal subjects. N Engl J Med. 1990, 322: 1622-1627. 10.1056/NEJM199006073222302.
van Deventer SJ, Buller HR, ten Cate JW, Aarden LA, Hack CE, Sturk A: Experimental endotoxemia in humans: analysis of cytokine release and coagulation, fibrinolytic, and complement pathways. Blood. 1990, 76: 2520-2526.
van Eijk LT, Dorresteijn MJ, Smits P, van der Hoeven JG, Netea MG, Pickkers P: Gender differences in the innate immune response and vascular reactivity following the administration of endotoxin to human volunteers. Crit Care Med. 2007, 35: 1464-1469. 10.1097/01.CCM.0000266534.14262.E8.
Coyle SM, Calvano SE, Lowry SF: Gender influences in vivo human responses to endotoxin. Shock. 2006, 26: 538-543. 10.1097/01.shk.0000232589.39001.4d.
Michie HR, Manogue KR, Spriggs DR, Revhaug A, O’Dwyer S, Dinarello CA, Cerami A, Wolff SM, Wilmore DW: Detection of circulating tumor necrosis factor after endotoxin administration. N Engl J Med. 1988, 318: 1481-1486. 10.1056/NEJM198806093182301.
Van Zee KJ, Coyle SM, Calvano SE, Oldenburg HS, Stiles DM, Pribble J, Catalano M, Moldawer LL, Lowry SF: Influence of IL-1 receptor blockade on the human response to endotoxemia. J Immunol. 1995, 154: 1499-1507.
Dorresteijn MJ, van Eijk LT, Netea MG, Smits P, van der Hoeven JG, Pickkers P: Iso-osmolar prehydration shifts the cytokine response towards a more anti-inflammatory balance in human endotoxemia. J Endotoxin Res. 2005, 11: 287-293. 10.1177/09680519050110050501.
Blackwell TS, Christman JW: Sepsis and cytokines: current status. Br J Anaesth. 1996, 77: 110-117. 10.1093/bja/77.1.110.
Netea MG, Nold-Petry CA, Nold MF, Joosten LA, Opitz B, van der Meer JH, van de Veerdonk FL, Ferwerda G, Heinhuis B, Devesa I, Funk CJ, Mason RJ, Kullberg BJ, Rubartelli A, Van der Meer JW, Dinarello CA: Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood. 2009, 113: 2324-2335. 10.1182/blood-2008-03-146720.
Martinon F, Burns K, Tschopp J: The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002, 10: 417-426. 10.1016/S1097-2765(02)00599-3.
Dorresteijn MJ, Draisma A, van der Hoeven JG, Pickkers P: Lipopolysaccharide-stimulated whole blood cytokine production does not predict the inflammatory response in human endotoxemia. Innate Immun. 2010, 16: 248-253. 10.1177/1753425909339923.
West MA, Koons A: Endotoxin tolerance in sepsis: concentration-dependent augmentation or inhibition of LPS-stimulated macrophage TNF secretion by LPS pretreatment. J Trauma. 2008, 65: 893-898. 10.1097/TA.0b013e3181877fde.
Hirohashi N, Morrison DC: Low-dose lipopolysaccharide (LPS) pretreatment of mouse macrophages modulates LPS-dependent interleukin-6 production in vitro. Infect Immun. 1996, 64: 1011-1015.
Deng H, Maitra U, Morris M, Li L: Molecular mechanism responsible for the priming of macrophage activation. J Biol Chem. 2013, 288: 3897-3906. 10.1074/jbc.M112.424390.
Munford RS: Murine responses to endotoxin: another dirty little secret?. J Infect Dis. 2010, 201: 175-177. 10.1086/649558.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Dillingh, M.R., van Poelgeest, E.P., Malone, K.E. et al. Characterization of inflammation and immune cell modulation induced by low-dose LPS administration to healthy volunteers. J Inflamm 11, 28 (2014). https://doi.org/10.1186/s12950-014-0028-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12950-014-0028-1