
Abstract
Background
HMGCR, SCAP, SREBF1, SREBF2 and TBL2 are well-known genes that are involved in the process of lipid metabolism. However, it is not known whether epigenetic changes of these genes are associated with lipid metabolism. In this study, the methylation levels of the HMGCR, SCAP, SREBF1, SREBF2 and TBL2 genes were analyzed between samples from a hyper-low-density lipoprotein cholesterolemia (hyper-LDL) group and a control group to examine the association between the methylation levels of these genes and the risk of hyper-LDL.
Methods
In this study, a case-control approach was used to explore the association between DNA methylation and hyper-LDL. The DNA methylation levels of HMGCR, SCAP, SREBF1, SREBF2 and TBL2 genes and 231 CpG sites in the promoter regions of these genes were measured in 98 hyper-LDL participants and 89 participants without hypo-LDL.
Results
Compared with participants without hyper-LDL, patients with hyper-LDL TBL2 gene had lower methylation levels (11.93 vs. 12.02, P = 0.004). The methylation haplotypes with significant abundance in the TBL2 gene are tcttttttttt (P = 0.034), ctttttttcct (P = 0.025), ctctttctttt (P = 0.040), ccttttttttt (P = 0.028), and tctttttttttttttt.
Conclusion
The study demonstrates that participants with hyper-LDL have lower methylation of TBL2. The results suggest that DNA methylation of TBL2 can decrease the risk for hyper-LDL in humans.
Similar content being viewed by others

Background
With the development of economy and changes in lifestyle, especially aging population and acceleration of urbanization, the prevalence of Coronary artery disease (CAD) is clearly on the rise which is the main cause of death [1, 2]. Several epidemiological studies have confirmed that hyperlipidemia has a significant association with CAD in the past decades [3]. Clinical studies have revealed that the prevalence of CAD increases as the low-density lipoprotein (LDL) level increases in plasma. In addition, total cholesterol (TC) ranging from 5.2 to 6.2 mmol/L lead to a threefold increase in the morbidity of cardiovascular disease [4]. Many gene mutations have been shown to be responsible for hyperlipidemia, and the main causes of hyperlipidemia are LDL receptor (LDLR) gene defects [5]. In addition, the internalization of the LDLR caused by the proprotein convertase subtilisin kexin type 9 (PCSK9) and ApoB can result in a similar phenotype [6]. Along with genetic mutations, epigenetic changes are also associated with hyperlipidemia. A large number of studies have shown that DNA methylation, as an important epigenetic mode, can regulate gene expression by changing chromatin structure, DNA conformation, DNA stability and interaction between DNA and proteins [7, 8].
Previous studies have revealed that the conversion of HMG-CoA to mevalonate is catalyzed by HMG-CoA reductase (HMGCR), which limits the process of cholesterol biosynthesis. Previous studies also have found that sterol regulatory element-binding transcription factors (SREBFs) control cholesterol homeostasis by regulating the transcription of cholesterol and lipid metabolism genes. SREBF cleavage activator protein (SCAP) and SREBFs can form a complex when the cell sterols are depleted. The complex is transported to the Golgi apparatus and triggers the amino-terminal transcriptional activation domain of SREBFs after being processed by two specific proteases. The activated complex then enters the nucleus, where it combines with the promoter region of the target gene [9, 10]. In a previous study, the transducin (β)-like 2 (TBL2) gene was identified as a new genetic locus affecting lipid concentration [11], which can lead to hypertriglyceridemia disease. However, how TBL2 affects cholesterol has not yet been reported.
The HMGCR, SCAP, SREBF1, SREBF2 and TBL2 genes are well-known genes involved in the lipid metabolism. However, it is not known whether there are any epigenetic associations in the lipid metabolism. Among the hyperlipidemias, high-density lipoproteinemia has the greatest impact on the human body and is most likely to cause various diseases. Low-density lipoprotein can cause changes in other blood lipid indicators. This study sequenced the levels of DNA methylation of HMGCR, SCAP, SREBF1, SREBF2 and TBL2 genes between samples from a hyper-low-density lipoprotein cholesterolemia (hyper-LDL) group and a control group to explore whether the methylation levels of these genes have relevance to risk of hyper-LDL.
Methods
Study population
Before the research started, our team formulated a research plan on August 21, 2012, and followed it (Additional file 1). The Ethical Review Board of First Affiliated Hospital of Xinjiang Medical University approved this study (Additional file 2). The methodology of this study is based on the Declaration of Helsinki, and kept the personal information of the participants confidential. All participants voluntarily participated in this study and signed an informed consent form. The research plan was designed according to STROBE checklists-case-control (Additional file 3). Upon reviewing case-control studies on DNA methylation, the sample sizes of the case and control groups ranged from 80 to 90. Therefore, this study recruited 100 hyper-LDL participants as the hyper-LDL group and randomly enrolled 100 age- and sex-matched participants as the control group. Two hundred samples were tested for DNA methylation. This study recruited 98 participants as the hyper-LDL group and 89 participants as the control group due to sample contamination (Fig. 1). All participants were Han Chinese and enrolled from the First Affiliated Hospital of Xinjiang Medical University from 2012 to 2015.

The CONSORT flow diagram of the study population. CONSORT: CONsolidated Standards of Reporting Trials
The definition of hyper-LDL was a fasting LDL level ≥ 3.1 mmol/L. The inclusion criteria were those with completed inpatient medical records and had no history of taking lipid-lowering medicine. The exclusion criteria were as follows: 1. renal dysfunction, creatinine ≥178 μmol/L; 2. valvular disease and heart failure; 3. chronic inflammatory disease; 4. acute infectious diseases, such as severe pneumonia, cholecystitis, and acute tuberculosis; 5. autoimmune disease; 6. Tumor; and 7. acute cerebral hemorrhage or brain infarction.
DNA isolation and epigenotyping
Fasting venous blood was collected overnight after all participants were informed instructions for attention to take biochemical detection. Blood cells from participants were collected for whole genomic DNA extraction using commercial kits (TIANGEN Biotech, Beijing, China). Whole genomic DNA was diluted with 75% ethanol to final concentration (more than 10 ng/mL) for sequencing and methylation analysis.
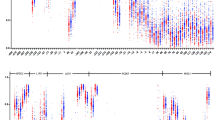
CpG islands distributed in promoters and first exon regions of HMGCR, SCAP, SREBF1, SREBF2 and TBL2 were captured and sequenced for methylation genotyping and analysis. The criteria of eligible CpG islands were as follows: (1) GC content not lower than 50%; (2) 200–1000 bp length; (3) The ratio of observed/expected CpG dinucleotides not lower than 0.60. Ultimately, three CpG regions of HMGCR, one that of SCAP, three that of SREBF1/2 and four that of TBL2 were identified in eligible CpG islands complying with the criteria and analyzed (Fig. 2). BiSulfite Amplicon Sequencing (BSAS) was applied to quantitative analysis of DNA methylation. The principle of the analysis is that sodium bisulfite gives priority to deamination of unmethylated cytosine residues to thymine, while methyl-cytosine remains unmodified. Bisulfite was used to base conversion of 1μg genomic DNA complying with EZ DNA Methylation-GOLD Kit (ZYMO RESEARCH, CA, USA). The samples were sequenced using Illumina MiSeq Benchtop Sequencer (CA, USA) after PCR amplification in target CpG islands (HotStarTaq polymerase kit, TAKARA, Tokyo, Japan) and library construction. The average coverage of all samples was >600X. The CpG regions tested were defined as the distance (in bp) between CpG sites and transcription start site (TSS). The methylated cytosine/total cytosine ratio was defined as the CpG site methylation level. The average methylation level of all detected CpG sites was defined as the gene methylation level.

CpG sites sequenced around the promoters of HMGCR, SCAP, SREBF1/2, TBL2. The eligible CpG sites sequenced in this study are indicated by green lines with arrows. All regions are distributed in the CpG islands of gene promoters and first exon. HMGCR: HMG-CoA reductase; SREBF: Sterol regulatory element binding transcription factors; SCAP: SREBF cleavage activator protein; TBL2: transducin (β)-like 2; TSS: transcription start site
Statistical analysis
The Shapiro–Wilk test assessed the normality of parameters. The measurement data that met the normality assumption are represented as the means±SD. An independent-sample T-test was used to distinguish the hyper-LDL group from the control group. Median (interquartile range) represented the measurement data that did not suit the normality assumption and was tested with the Mann-Whitney U test. Further, the Chi-square test discriminated the categorical data, such as smoking and hypertension. Methylation levels of different genes were assessed between the hyper-LDL group and the control group by the Mann–Whitney U test. The Mann–Whitney U test was used to determine the abundance of each methylated haplotype, to screen methylated haplotypes with significant differences in abundance. The contributions of the major factors for hyper-LDL were assessed by logistic regression analyses with effect ratios (OR and 95% CI). IBM SPSS Statistics Version 22.0 was used to analyze data. A two-tailed value of P < 0.05 was considered statistically significant.
Result
One hundred eighty-seven participants were recruited in this study. The basic characteristics and blood lipid profiles are shown in Table 1. The average age of 187 analyzed participants was 59.93 ± 10.93 years, and 61 (32.6%) were male. The study population have 101 (54%) with hypertension, 144 (77%) with CAD and 46 (24.7%) with diabetes. The hyper-LDL group had a higher prevalence of hypertension (63.3 vs. 43.8%, P = 0.008) and higher TC level (5.21 ± 1.03 vs. 3.57 ± 0.70 mmol/L, P = 0.001), TG level (1.76(0.51–5.32) vs. 1.24(0.35–6.21) mmol/L, P = 0.001), LDL level (3.57(2.44–5.92) vs. 1.98(1–3.42) mmol/L, P = 0.001) and glucose level (5.78(3.41–17.85) vs. 4.84(2.53–13.54) mmol/L, P = 0.001) (Table 1).
A total of 231 CpG sites were identified as methylation sites (44 in HMGCR, 16 in SCAP, 64 in SREBF1, 47 in SREBF2 and 60 in TBL2) according to the measurement results of the target regions (Additional file 4 shows the detailed information of each site). In these 231 CpG islands detected, the statistical differences in methylation sites between the two groups of participants are shown in Table 2. The average methylation levels of CpG sites measured within HMGCR, SCAP, and SREBF1/2 were not significantly correlated with hyper-LDL. However, the methylation level of TBL2 was significantly related to hyper-LDL (Table 3).
The methylation levels of candidate genes compared between two groups are shown in Table 2. Participants in the hyper-LDL group had lower methylation levels (11.93% vs. 12.02%, P = 0.004). The methylation haplotypes with significant abundance in the TBL2 gene were tcttttttttt (P = 0.034), ctttttttcct (P = 0.025), ctctttctttt (P = 0.040), ccttttttttt (P = 0.028), tctttttttttttttt (P = 0.019), tttttttttttttttc (P = 0.031) and tttttttttttttctt (P = 0.015) (Table 4).
This study performed univariate and multivariate logistic regression analyses with effect ratios (OR and 95% CI) to evaluate the contributions of risk factors to hyper-LDL. The DNA methylation of TBL2 is a protective factor for hyper-LDL (Table 5). Participants with high levels of methylation of TBL2 have a 78% lower risk of hyper-LDL (P = 0.017 OR = 0.221 95% CI = 0.064–0.765). In this study, TG (P = 0.003 OR = 1.744 95% CI = 1.211–2.511) and glucose (P = 0.014 OR = 1.26 95% CI = 1.049–1.513) value are treated as continuous variables in the logistic regression model to manifest they are independent risk factors for hyper-LDL. Participants with higher blood glucose or TG levels increase 26 and 74% risk to develop hyper-LDL independently.
Discussion
This study confirms that the increased DNA methylation in TBL2 decreases the susceptibility of hyper-LDL. DNA methylation is a kind of pre-transcriptional modification that is characterized by specifically adding methyl groups to a nucleotide. DNA methylation regulates gene expression and maintains genomic integrity by cooperating with modified nucleosome proteins [12]. CpG islands are distributed unevenly throughout the genome where DNA methylation mainly occurs [13,14,15]. In vivo, approximately 80% of CpGs are methylated in normal healthy cells [16, 17]. The enzymes that catalyze the DNA methylation are named DNA methyltransferases (DNMTs): DNMT1 essentially maintains DNA methylation during cell division by catalyzing the addition of methyl groups to cytosine, generating 5-methylcytosine (5-mC) and cytosine, while DNMT3a and DNMT3b are important for de novo methylation during the methylation process [12, 18,19,20]. CG methylation in promoters close to transcriptional initiation sites usually inhibits gene expression [21,22,23]. There are two ways that promoter methylation inhibits gene transcription: first, by physically blocking the binding of transcription factors to gene promoters, and second, by binding to the methyl-CpG-binding domain, which recruits repressive machinery, such as histone and chromatin modifiers, to the loci that cause chromatin compaction [24]. Biologically, because they act as docking sites for methyl-binding proteins, methylated CpG islands are markers of gene suppression. In fact, they can produce spatial barriers that specifically bind transcription factors to gene promotors, recruit transcription inhibitors or block activation protein binding [8, 25].
In a previous study, the TBL2 gene was identified as a genetic locus affecting lipid concentration, which can lead to hypertriglyceridemia and increase the risk of coronary artery disease [11, 26,27,28,29,30,31]. The TBL2 gene is located in chromosome 7q11.23, and the resulting protein has five WD40 domains [32]. The WD40 repeat protein TBL2 was found to be a candidate transitional epithelial response protein 1 (TERE1)-interacting protein. The TERE1-TBL2 complex may be related to the metabolism of cholesterol [33]. Previous studies have demonstrated that abnormal expression of TERE1 or TBL2 can affect intracellular cholesterol levels [34]. It was also found that TERE1 directly interacts with HMGCR [35]. Schnyder’s Corneal Dystrophy is caused by TERE1 mutations, which have been demonstrated to have an influence on TERE1 interaction with APOE [34, 36] and with HMGCR [35]. In this study, the DNA methylation level of TBL2 decreased in participants with hyper-LDL, which generally inhibits gene expression. The upregulation of TBL2 gene expression, which is positively associated with the TERE1-TBL2 complex, may affect the expression of genes such as APOE and HMGGR and can ultimately lead to downregulation of cholesterol.
Previous studies have found that TBL2 can regulate the PKR-like ER-resident kinase (PERK) pathway. The TBL2 protein is localized in endoplasmic reticulum (ER) and interacts with PERK with the binding of the WD40 domain to the 60S ribosome subunit. Under ER stress, the TBL2-60S ribosome complex mediates the translation of specific proteins that can activate PERK oligomerization and autophosphorylation [37,38,39,40,41]. Activated PERK then phosphorylates eukaryotic initiation factor 2 alpha (eIF2a) of Ser-51 [42, 43]. Under the condition of eIF2a phosphorylation, most RNA transcription was inhibited, while the translation of RNA represented by activating transcription factor 4 (ATF4) was enhanced [44]. Previous studies also have found that TBL2 can directly affect the expression of ATF4 by binding the WD40 domain to ATF4 mRNA [45]. It has been reported that ATF4 is related to lipid metabolism. Kun-Yun Yeh et al. found that overexpression of ATF4 in zebrafish increased lipid accumulation in blood vessels [46]. Chunxia Wang et al. found that in ATF4-knockout mice, fat mobilization increased and fatty acid synthesis decreased. They identified that ATF4 can regulate lipid metabolism [47]. Previous studies have also found that the absence of ATF4 causes an increase in free cholesterol in the liver but a decrease in serum cholesterol [48]. In addition, ATF4 deficiency reduced the accumulation of triglycerides and the expression of SREBP-1c and CHREBP. It is further speculated that ATF4 affects lipid metabolism by affecting SREBP-1c and CHREBP genes [49]. In conclusion, the increased expression of TBL2 caused by the decrease of methylation level leads to the activation of the PERK pathway, which leads to the increase of cholesterol levels (Fig. 3).

Mechanism of the role of TBL2 in hyper-LDL. BL2: Transducin (β)-like 2; TERE1: Transitional epithelial response protein 1; HMGCR: HMG-CoA reductase; SREBP: Sterol regulatory element-binding transcription factor protein; CHREBP: Carbohydrate response element-binding protein; PERK: PKR-like ER-resident kinase; eIF2a: Eukaryotic Initiation Factor 2 alpha; ATF4: Activating transcription factor 4
Study strengths and limitations
There are several strengths in this study. First, this study first found DNA methylation levels and haplotypes at some CPG sites of TBL2 are associated with hyper-LDL. Second, Although the mechanism of candidate genes (HMGCR, SCAP, SREBF1/2) in lipid metabolism have been studied clearly, this study first found that some CpG sites of these genes are related to hyper-LDL. DNA methylation may participate in these genes to regulate lipid metabolism. Third, this is a random case-control study. The subject of the study is people, which has certain clinical application value. This study also has limitations. First, this study is a correlation study. Further functional studies are needed to interpret mechanisms that relate TBL2 methylation to hyper-LDL. Second, this is a single-center study, and all participants in the study were Han Chinese, which may affect the generalizability of the findings.
Conclusion
This study demonstrates that participants with hyper-LDL have lower methylation levels of TBL2. The results suggest that DNA methylation of TBL2 can decrease the risk of hyper-LDL in humans. This study found a variety of DNA methylation haplotypes with statistical differences, which can tell us how the gene is mainly methylated in hyper-LDL. These different haplotypes can help us develop some specific intercalators or activators to act on CpG sites that can replace or activate the DNA methylation of this gene to treat hyper-LDL in the future.
Availability of data and materials
The data sets generated and analyzed during the current study are available from the corresponding author upon reasonable request (Additional file 4).
References
Rosano G. The New ESC Guidelines for the Diagnosis and Management of Chronic Coronary Syndromes: the Good and the Not So Good. Curr Probl Cardio. 2020:100554. Advance online publication.
Ford TJ, Berry C. Angina: contemporary diagnosis and management. Heart. 2020;106:387–98.
Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, et al. Heart disease and stroke Statistics-2017 update: a report from the American Heart Association. Circulation. 2017;135:e146–603.
Stamler J, Daviglus ML, Garside DB, Dyer AR, Greenland P, Neaton JD. Relationship of baseline serum cholesterol levels in 3 large cohorts of younger men to long-term coronary, cardiovascular, and all-cause mortality and to longevity. JAMA. 2000;284:311–8.
Palacios L, Grandoso L, Cuevas N, Olano-Martin E, Martinez A, Tejedor D, Stef M. Molecular characterization of familial hypercholesterolemia in Spain. Atherosclerosis. 2012;221:137–42.
Fazio S, Linton MF. When it looks like familial hypercholesterolemia...but is not. Circ Cardiovasc Genet. 2012;5:599–601.
Ding YN, Tang X, Chen HZ, Liu DP. Epigenetic regulation of vascular aging and age-related vascular diseases. Adv Exp Med Biol. 2018;1086:55–75.
Luo C, Hajkova P, Ecker JR. Dynamic DNA methylation: in the right place at the right time. Science. 2018;361:1336–40.
Brown MS, Radhakrishnan A, Goldstein JL. Retrospective on cholesterol homeostasis: the central role of Scap. Annu Rev Biochem. 2018;87:783–807.
Chen L, Chen XW, Huang X, Song BL, Wang Y, Wang Y. Regulation of glucose and lipid metabolism in health and disease. Sci China Life Sci. 2019;62:1420–58.
Kim YJ, Go MJ, Hu C, Hong CB, Kim YK, Lee JY, Hwang JY, Oh JH, Kim DJ, Kim NH, et al. Large-scale genome-wide association studies in east Asians identify new genetic loci influencing metabolic traits. Nat Genet. 2011;43:990–5.
Jin Z, Liu Y. DNA methylation in human diseases. Genes Dis. 2018;5:1–8.
Field AE, Robertson NA, Wang T, Havas A, Ideker T, Adams PD. DNA methylation clocks in aging: categories, causes, and consequences. Mol Cell. 2018;71:882–95.
Horvath S, Raj K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet. 2018;19:371–84.
Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38:23–38.
Pinnick KE, Karpe F. DNA methylation of genes in adipose tissue. Proc Nutr Soc. 2011;70:57–63.
Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204–20.
Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet. 2010;11:191–203.
Greenberg MVC, Bourc'his D. The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol. 2019;20:590–607.
Michalak EM, Burr ML, Bannister AJ, Dawson MA. The roles of DNA, RNA and histone methylation in ageing and cancer. Nat Rev Mol Cell Biol. 2019;20:573–89.
Grunau C, Hindermann W, Rosenthal A. Large-scale methylation analysis of human genomic DNA reveals tissue-specific differences between the methylation profiles of genes and pseudogenes. Hum Mol Genet. 2000;9:2651–63.
Song F, Smith JF, Kimura MT, Morrow AD, Matsuyama T, Nagase H, Held WA. Association of tissue-specific differentially methylated regions (TDMs) with differential gene expression. Proc Natl Acad Sci U S A. 2005;102:3336–41.
Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, Schubeler D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet. 2005;37:853–62.
Clouaire T, de Las Heras JI, Merusi C, Stancheva I. Recruitment of MBD1 to target genes requires sequence-specific interaction of the MBD domain with methylated DNA. Nucleic Acids Res. 2010;38:4620–34.
Prokhortchouk A, Hendrich B, Jorgensen H, Ruzov A, Wilm M, Georgiev G, Bird A, Prokhortchouk E. The p120 catenin partner Kaiso is a DNA methylation-dependent transcriptional repressor. Genes Dev. 2001;15:1613–8.
Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–13.
Wang J, Ban MR, Zou GY, Cao H, Lin T, Kennedy BA, Anand S, Yusuf S, Huff MW, Pollex RL, Hegele RA. Polygenic determinants of severe hypertriglyceridemia. Hum Mol Genet. 2008;17:2894–9.
Willer CJ, Sanna S, Jackson AU, Scuteri A, Bonnycastle LL, Clarke R, Heath SC, Timpson NJ, Najjar SS, Stringham HM, et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet. 2008;40:161–9.
Blattmann P, Schuberth C, Pepperkok R, Runz H. RNAi-based functional profiling of loci from blood lipid genome-wide association studies identifies genes with cholesterol-regulatory function. PLoS Genet. 2013;9:e1003338.
Hegele RA, Ban MR, Hsueh N, Kennedy BA, Cao H, Zou GY, Anand S, Yusuf S, Huff MW, Wang J. A polygenic basis for four classical Fredrickson hyperlipoproteinemia phenotypes that are characterized by hypertriglyceridemia. Hum Mol Genet. 2009;18:4189–94.
Kraja AT, Chasman DI, North KE, Reiner AP, Yanek LR, Kilpelainen TO, Smith JA, Dehghan A, Dupuis J, Johnson AD, et al. Pleiotropic genes for metabolic syndrome and inflammation. Mol Genet Metab. 2014;112:317–38.
Perez Jurado LA, Wang YK, Francke U, Cruces J. TBL2, a novel transducin family member in the WBS deletion: characterization of the complete sequence, genomic structure, transcriptional variants and the mouse ortholog. Cytogenet Cell Genet. 1999;86:277–84.
Fredericks WJ, McGarvey T, Wang H, Zheng Y, Fredericks NJ, Yin H, Wang LP, Hsiao W, Lee R, Weiss JS, et al. The TERE1 protein interacts with mitochondrial TBL2: regulation of trans-membrane potential, ROS/RNS and SXR target genes. J Cell Biochem. 2013;114:2170–87.
Fredericks WJ, McGarvey T, Wang H, Lal P, Puthiyaveettil R, Tomaszewski J, Sepulveda J, Labelle E, Weiss JS, Nickerson ML, et al. The bladder tumor suppressor protein TERE1 (UBIAD1) modulates cell cholesterol: implications for tumor progression. DNA Cell Biol. 2011;30:851–64.
Nickerson ML, Bosley AD, Weiss JS, Kostiha BN, Hirota Y, Brandt W, Esposito D, Kinoshita S, Wessjohann L, Morham SG, et al. The UBIAD1 prenyltransferase links menaquinone-4 [corrected] synthesis to cholesterol metabolic enzymes. Hum Mutat. 2013;34:317–29.
McGarvey TW, Nguyen TB, Malkowicz SB. An interaction between apolipoprotein E and TERE1 with a possible association with bladder tumor formation. J Cell Biochem. 2005;95:419–28.
Tsukumo Y, Tsukahara S, Furuno A, Iemura S, Natsume T, Tomida A. The endoplasmic reticulum-localized protein TBL2 interacts with the 60S ribosomal subunit. Biochem Biophys Res Commun. 2015;462:383–8.
Tsukumo Y, Tsukahara S, Furuno A, Iemura S, Natsume T, Tomida A. TBL2 is a novel PERK-binding protein that modulates stress-signaling and cell survival during endoplasmic reticulum stress. PLoS One. 2014;9:e112761.
Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–17.
Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110:1389–98.
Ma Y, Hendershot LM. The role of the unfolded protein response in tumour development: friend or foe? Nat Rev Cancer. 2004;4:966–77.
Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–108.
Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904.
Gebauer F, Hentze MW. Molecular mechanisms of translational control. Nat Rev Mol Cell Biol. 2004;5:827–35.
Tsukumo Y, Tsukahara S, Furuno A, Iemura S, Natsume T, Tomida A. TBL2 associates with ATF4 mRNA via its WD40 domain and regulates its translation during ER stress. J Cell Biochem. 2016;117:500–9.
Yeh KY, Lai CY, Lin CY, Hsu CC, Lo CP, Her GM. ATF4 overexpression induces early onset of hyperlipidaemia and hepatic steatosis and enhances adipogenesis in zebrafish. Sci Rep. 2017;7:16362.
Wang C, Huang Z, Du Y, Cheng Y, Chen S, Guo F. ATF4 regulates lipid metabolism and thermogenesis. Cell Res. 2010;20:174–84.
Fusakio ME, Willy JA, Wang Y, Mirek ET, Al Baghdadi RJ, Adams CM, Anthony TG, Wek RC. Transcription factor ATF4 directs basal and stress-induced gene expression in the unfolded protein response and cholesterol metabolism in the liver. Mol Biol Cell. 2016;27:1536–51.
Ren LP, Yu X, Song GY, Zhang P, Sun LN, Chen SC, Hu ZJ, Zhang XM. Impact of activating transcription factor 4 signaling on lipogenesis in HepG2 cells. Mol Med Rep. 2016;14:1649–58.
Acknowledgements
We thank our colleagues for their help. We also thank all the participants for their support.
Funding
This work was supported by NSFC grant number 2017E0269.
Author information
Authors and Affiliations
Contributions
Li Yang and Shuai Liu contributed equally to this work. Li Yang and Shuai Liu were responsible for the statistical analysis and wrote this paper. Yong-Tao Wang and Han Min provided the database. Dilare Adi and Xiao-Mei Li were responsible for collecting clinical data. Zhen yan Fu and Yi-Ning Yang revised the paper critically for important intellectual content. Yi-Tong Ma is accountable for all aspects of the work and funding. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The Ethical Review Board of the First Affiliated Hospital of Xinjiang Medical University approved this study. The IRB approval number is 20181129–10. The study complied with the standards of the Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
None.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 2: Appendix 2
. Approval report of the Medical Ethics Committee of the First Affiliated Hospital of Xinjiang Medical University
Additional file 3: Appendix 3
. STROBE Statement—Checklist of items that should be included in reports of case-control studies
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, Y., Liu, S., Wang, YT. et al. TBL2 methylation is associated with hyper-low-density lipoprotein cholesterolemia: a case-control study. Lipids Health Dis 19, 186 (2020). https://doi.org/10.1186/s12944-020-01359-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12944-020-01359-8