
Abstract
Atherosclerosis is considered as an inflammatory and chronic disorder with an important immunologic component, which underlies the majority of cardiovascular diseases; condition that belongs to a group of noncommunicable diseases that to date and despite of prevention and treatment approaches, they remain as the main cause of death worldwide, with 17.5 million of deaths every year. The impact of lipids in human health and disease is taking center stage in research, due to lipotoxicity explained by elevated concentration of circulating lipids, in addition to altered adipose tissue metabolism, and aberrant intracellular signaling. Immune response and metabolic regulation are highly integrated systems and the proper function of each one is dependent on the other. B lymphocytes express a variety of receptors that can recognize foreign, endogenous or modified self-antigens, among them oxidized low density lipoproteins, which are the main antigens in atherosclerosis. Mechanisms of B cells to recognize, remove and present lipids are not completely clear. However, it has been reported that B cell can recognize/remove lipids through a range of receptors, such as LDLR, CD1d, FcR and SR, which might have an atheroprotector or proatherogenic role during the course of atherosclerotic disease. Pertinent literature related to these receptors was examined to inform the present conclusions.
Similar content being viewed by others

Background
Cardiovascular diseases (CVDs) belong to a group of noncommunicable diseases (NCDs) that to date and despite of prevention and treatment approaches, they remain as the main cause of death worldwide [1, 2]. Atherosclerosis is considered as an inflammatory and chronic disorder with an important immunologic component, which underlies the majority of CVDs [3–5]. This process is favored by inadequate life styles like the use of cigarette, physical inactivity, unhealthy diets, harmful use of alcohol and in some cases even genetic susceptibilities; leading to metabolic changes and promoting the development of other kind of conditions like hypertension, dyslipidemia, overweight and diabetes, which constitute comorbidities of the so called “metabolic syndrome” (MetS) [6–8].
In 1998 the first formal definition of MetS was published by the World Health Organization (WHO) which specifies that besides insulin resistance, that results to be the major underlying risk factor of the MetS, two additional risk factors are necessary for MetS diagnosis, such as obesity, hypertension, high triglyceride levels, reduced high-density lipoprotein (HDL) levels, or microalbuminuria. However, the latest reported definition, communicated by representatives of the International Diabetes Federation; National Heart, Lung and Blood Institute; American Heart Association among others in 2009, considers abdominal obesity as a mandatory requirement for the diagnosis of MetS, including the presence of any of the three risk factors among the five previously described [9–14].
MetS is about dysregulation of a wide range of parameters that easily disrupt the immunological balance, through unhealthy behavioral patterns that lead to physiopathological changes, a similar case to CVD. The impact of lipids in human health and disease is taking center stage in research [15], due to lipotoxicity explained by elevated concentration of circulating lipids in non-adipose tissue, in addition to altered adipose tissue metabolism, and aberrant intracellular signaling [16]. Indeed, unbalanced production of pro and anti-inflammatory adipokines, as well as the participation of many cells of the immune system, contribute to the development of MetS, which also constitute specific features of CVD [17].
Immune response and metabolic regulation are highly integrated systems and the proper function of each one is dependent on the other. Dysfunction of these central homeostatic mechanisms can lead to a cluster of chronic metabolic disorders, characterized by “chronic inflammation”, which drives to a sustained interaction between parenchymal and stromal cells in response to exogenous and endogenous stress, ending in tissue dysfunction and remodeling [17–19]. Among this group of disorders we can find obesity, type 2 diabetes and CVD [17, 20]. In 2014, the WHO reported that NCDs continue to be the global leading cause of death; with a 46.2% associated to CVDs, representing 17.5 million deaths every year. Diabetes caused an additional 1.5 million of deaths, and it is estimated that for 2030, close to 400 million people worldwide will be affected by this disease, with 90% of these being type 2 diabetes [2, 21, 22]. Likewise, the WHO estimates that more than one billion adults worldwide are overweight, from which 300 million are clinically obese [23]. Since their projections show that NCDs will be responsible for a significant increase in the total number of deaths in the next decade, these chronic inflammatory diseases that involve lipid metabolism dysregulation are a matter of public health concern [21].
Studies of tissue morphology and transcriptional profiling in several obesity animal models as well as in obese subjects, have shown that the development of insulin resistance is related to adipose tissue remodeling, which involves cellular infiltration of macrophages inducing inflammatory pathways and ectopic lipid accumulation [24–26]. In this sense it was demonstrated that even modest levels of overweight/obesity elicit modifications in adipose tissue immune function [27].
Recently, it has been reported that early B and T lymphocyte trafficking and infiltration, could promote initiation and perpetuation of adipose tissue inflammation [28–30]. Winer et al. reported that in diet induced obese (DIO) mice, the depletion of early B cells (which currently accumulate in this model), by anti-CD20 treatment, could protect from insulin resistance despite weight gain; while transfer of DIO-IgG exacerbated the metabolic disease. This suggests that B cells contribute to insulin resistance probably by mechanisms such as antigen presentation to T cells, secreting inflammatory cytokines and/or producing pathogenic antibodies [22, 31]. Also, in diabetes mellitus patients, alterations of B cell toll like receptor (TLR) function have been described favoring inflammation, by increased expression of surface TLR4, elevation of pro-inflammatory IL8, and decreased levels of the anti-inflammatory/protective cytokine IL10 [32]. In fact in 2002, the Leiden 85-Plus study found a direct association between the decreased IL10 levels, type 2 diabetes and MetS [33]. In parallel, B cell activation is usually detected in both inherited (e.g., Gaucher disease) [34], and acquired lipid metabolism disorders such as atherosclerosis, in which its participation is contradictory. Some results showed increased atherosclerosis in B cell deficient LDL receptor-deficient (Ldlr −/−) mice, suggesting an atheroprotector role, whereas others have reported that the depletion of B cells through the use of specific monoclonal antibodies against CD20 molecule, reduce the development of atherosclerosis in both, apolipoprotein E single knockout (Apoe −/−) and Ldlr −/− mice, showing an atherogenic effect [35, 36].
B lymphocytes express a variety of receptors that recognize foreign, endogenous or modified self-antigens, among them oxidized low density lipoproteins (oxLDL), which are the main antigens in atherosclerosis. B cell mechanisms to recognize, remove and present lipids are not completely clear. However, it has been reported that B cells can remove lipid antigens through a B cell receptor (BCR) dependent via, but also there is internalization and antigen presentation to invariant natural killer T (iNKT) cells by BCR independent via, associated to low density lipoprotein receptor (LDLR) expression on activated B cells [37–41].
CD1 is included among the receptors expressed in B cells that have the ability to present lipid antigens. They belong to β2 microglobulin family associated polypeptides, which relate with the major histocompatibility complex (MHC) class I and II. Also, B cells express Fc receptors (FcR), which through their immunoreceptor tyrosine-based activation or inhibitory motifs (ITAMs or ITIMs) initiate and propagate early signaling events leading to cell-specific responses [42], and scavenger receptors (SR), which belong to the family of pattern recognition receptors (PRR), that recognize pathogen associated molecular patterns (PAMPs), as well as modified antigens, such as death associated molecular patterns (DAMPs), including modified host derived molecules like oxLDL [43, 44] (Table 1).
The disruption of cellular homeostasis through oxidative stress and contribution to cell death by generation of toxic intermediates during aberrant lipid metabolism, and enhanced pro-inflammatory immunological pathways, could occur in vascular, cardiac and adipose tissue diseases. This opens up the possibility that immune cells often interacting with oxidized products, and more specifically B cells, could participate in the process of lipotoxicity at the injured areas during the tissue remodeling process, leading to development and establishment of the disease or even regulating the inflammatory process. This supports the importance of understanding the receptors that could be involved in lipids recognition and/or removal by B cells.
Receptors involved in lipids recognition-removal by B cells
LDLR
Lymphocytes obtain cholesterol from serum low density lipoproteins (LDL) through its specific LDLR, which is internalized along with LDL; since these cells do not synthetize enough cholesterol to support their membranes [45–47]. The main function of this mechanism is to transfer cholesterol from plasma LDL into the cell in a controlled manner [48]. LDL endocytosis and subsequent lysosomal degradation induces the release of free cholesterol, which suppresses 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMG-CoA) activity, stimulates acyl-CoA cholesterol acyltransferase (ACAT) and regulates the LDLR activity by a feedback mechanism [49, 50].
LDLR is widely expressed in different cell types, including T and B lymphocytes [51]. However there are important differences in cholesterol metabolism among these lymphocyte subpopulations; for example the content of both, free and ester cholesterol is slightly lower in B cells than in T cells. In fact, a conclusion of a study more than two decades ago, pointed out that B cells had deficient LDL catabolism compared with T cells, in terms of internalization of LDLR-LDL complex [52]. However, recently it has been reported that B lymphocytes purified from peripheral blood express LDLR, and are able to internalize LDL with a four-fold increase in the expression of this receptor compared with non-stimulated T and NKT cells. Also, an up regulation of this receptor has been reported after B cell activation through stimulation with different concentrations of IL2 or pokeweed mitogen, which could suggest that LDL internalization could be important for B cells metabolism and maybe even immunoglobulin (Ig) production [51]. Additionally, besides the cell type, some hormones may play a role in the regulation of LDLR activity, such as insulin, which decreases LDL metabolism in lymphocytes by lowering its binding to LDL receptor, with a consequent decreased internalization and degradation of LDL [48]. This is evident in secondary hypercholesterolemias of endocrine origin, and in diabetes, in which the lack of insulin increases the levels of triglycerides, very low density lipoproteins (VLDL) and LDL [53].
It is clear that a defect in the expression or internalization of LDLR leads to an increase in circulating plasma LDL, predisposing it to oxidation, condition that contributes in a great manner to the physiopathology of atherosclerosis [54]. Finally, it is also important to highlight that LDL removal through LDLR might be relevant not only for B cell metabolism, but also for LDL antigen presentation, which could participate in the inflammatory process observed in atherosclerosis [55].
FcR
A critical role for FcR has been described in B cell survival following antigen presentation [56], and in an enhanced antibody response to antigenic challenge [57]. Although FcRs are not directly involved in lipids removal, they could indirectly participate in this process, due to their capability to recognize Ig directed to modified lipids. In this sense, immunological system has the ability to recognize and respond effectively to neoepitopes formed by lipid peroxidation [58]. This includes natural antibodies, which bind to oxidized phospholipids contained in oxLDL. The importance of these antibodies in the context of diseases with a lipid component is evidenced by the observation that in the Apoe −/− atherosclerosis model, mice fed with high fat diet (HFD) present high titers of anti-oxLDL IgM [59].
However, FcμR is not the only FcR expressed on B cells; actually the most widely expressed is the FcγRIIB. FcγR, are important cell surface effector molecules that bind the Fc portion of IgG. In mice, four classes of FcγRs have been identified, FcγRI, FcγRIIB, FcγRIII and FcγRIV, and three of them may interact with immune complexes, such as FcγRI, FcγRIII, and FcγRIIB [60]. This last molecule contains an ITIM in its cytoplasmic tail, leading to negative regulation of BCR signaling affecting B cell activation and differentiation [61–63].
In humans, FcγRIIA and FcγRIIB conform the FcγRII subfamily, and in both cases they bind the Fc portion of IgG with low affinity [64]. FcγRs are differentially expressed in human leukocytes. Both isoforms of CD32, respectively the activating and inhibitory members are expressed in B cells, dependent on its activation status [65]. FcγRI (CD64) and FcγRIIA (CD32) are highly expressed on monocytes, macrophages, and granulocytes, whereas FcγRIIIA (CD16) mainly in macrophages and a small subset of monocytes, as well as in smooth muscle cells and endothelial cells, which also express FcγRIIA [66], and finally FcγRIIIB which is expressed on granulocytes [67, 68].
Apoe −/−mice with a deficiency in the γ chain expression of FcγR, and fed with high fat diet, exhibited a limited development and progression of atherosclerosis, suggesting a proatherogenic effect that could be associated with the loss of FcγRI and FcγRIIIA, and the overexpression of the inhibitory FcγRIIB characteristic of this mouse model. These mice also showed a ∼ 50% less atherosclerotic lesion as well as reduced expression of inflammatory molecules, such as MCP-1 and RANTES and influence over the macrophage phenotypic balance [69, 70]. This agrees with the observation that the absence of FcγRIIB induces increased atherosclerosis development in Ldlr −/− atherosclerosis mouse model, as well as increased activation and expansion of B cells [71]. However, the role of this receptor is not yet clear, since recently it was reported that FcγRIIB deficiency reduces atherosclerosis in hyperlipidemic Apoe −/− mice, as well as induction of T regulatory cells and increased secretion of atheroprotective cytokines such as IL10 and TGFβ, which are partially responsible for the attenuation of the disease [72].
Even though the role of FcR in lipid removal is not clear, their contribution in atherosclerosis development has been previously described either directly in lipids clearance or indirectly during inflammation. The role of FcγRIIB during atherosclerosis is at least partially explained by the recognition of IgG antibodies directed to oxLDL antigens, which could result in protective immunity or down regulation of pre-existing proatherogenic immune responses [71]. This is supported by the observation that human atherosclerosis lesions as well as sera from these patients, presents IgG deposits directed to oxLDL [73, 74]. The inhibitory character of FcγRIIB confers the ability to modulate B cell activity through regulation of BCR-mediated signaling, controlling proliferation, class switching, and B cell differentiation to plasma cells [75, 76], probably regulating the inflammation observed in atherosclerosis. However, activating FcγR are also involved in atherosclerosis, including FcγRIA, FcγRIIA, and FcγRIIIA, which have been described in aorta lesions from patients with atherosclerosis [66, 68].
SR
SRs are distinguished by their wide ability to recognize several ligands, such as microbial, environmental, endogenous and self-modified antigens, either by endocytosis or phagocytosis, contributing to the outcome of the immune system responses. According to their multidomain structure, SR are classified in eight different classes: A, B, C, D, E, F, G and H [77]. In this review, we mainly focus on CD36 and SR class B type I (SR-BI), due to their proved expression on B cells and their relevance in lipid removal involved in atherosclerosis. These receptors belong to class B and D, respectively [43].
Besides macrophages and dendritic cells, other cell types including B, and some epithelial and endothelial cells express SR [78]. Specifically in B cells, a differential expression of some SRs such as CD36 and CD68 has been described, depending on the B cell subset studied. According to the differential analysis of gene expression by DNA microarrays, these receptors are expressed on marginal zone (MZ) but not on follicular (FO) B cells [79], even though for example the function of CD68 on MZ B cells remains to be determined.
CD36
CD36 belongs to the class B SR [80], and is a type III receptor with two transmembrane domains, an extracellular loop with multiple glycosylation sites and two short intracellular tails [43]. CD36 in both murine and human versions, mediates not just the specific uptake of oxLDL, but also its intracellular accumulation and degradation, according to experiments with transfected cells with this molecule [80, 81]. This is also supported by the observation that CD36 null mice present a fast and significant increase in the levels of circulating cholesterol, non-esterified free fatty acids and triacylglycerol [82].
Although, the role of CD36 in B cells is not yet clear, it has been reported to be a specific marker for MZ B cells, due to the absence in its expression on B1 B cells. Also, it was reported that its expression could be rapidly induced on mature B cells, like FO B cells by TLR and CD40 stimulation. However, the lack of CD36 in CD36 −/− mice reveal a minimal effect on mature B cells development, which is evidenced in the transitional B cells stages [83]. Major effect is observed in plasma cell generation and specific antibody responses to infectious diseases in vivo [83, 84]. Just like CD68, differential expression of CD36 in B cells, could be related with the diverse roles of B cells subsets in immune responses mediated by SRs.
In the context of lipid related diseases, it was shown that the absence of CD36 protects against atherosclerotic lesion development in Apoe −/− mice, which approximately developed just a 20% of lesion assessed by en face analysis of whole aortas, compared with control mice [85–87]. Whereas groups of triple knockout CD36 −/− Msr1 −/− Apoe −/− mice, deficient not just for CD36, but also for SR-AI and SR-AII receptors, which also recognize modified LDL such as acetylated LDL (AcLDL) and oxLDL, exhibited increased serum cholesterol levels and larger atherosclerotic lesions located in aortic sinus compared with Apoe −/− mice as controls. This suggests that the lipid uptake mediated through SR, protected against atherosclerosis lesion formation rather than promote it [88]. Finally, no difference was observed between the size of aortic lesion in Ldlr −/− CD36 −/− and Ldlr −/− CD36 +/+ mice fed with western diet. However, bone marrow transplant from Ldlr −/− CD36 −/− into Apoe −/− mice induced 38.4% less lesion area compared with those receiving Ldlr −/− CD36 +/+ transplant [89]. These contradictory results could indicate that the role of this receptor is not only related with aberrant lipid metabolism, but also with persistent inflammatory milieu probably modulating disease progression.
SR-BI
SR-BI, belongs to the class B receptors of SR family, which have a loop structure similar to CD36. This receptor contains two alternative splice variants (SR-BI and SR-BII), and contrary to CD36 receptor, SR-BI recognizes mainly HDL and is responsible for reverse cholesterol transport; however it also recognizes modified lipids, among them oxLDL, as well as native LDL, suggesting that SR-BI could play a role in normal LDL metabolism [90] and could present a selective lipid uptake from HDL [91, 92].
SR-BI is expressed in different tissues such as adrenal glands, steroidogenic tissue and hepatocytes, as well as in different types of cells, like monocytes, macrophages and dendritic cells [93]. According to experiments evaluating total spleen RNA derived from C57BL/6 mice, SR-BI as well as its splice variants, CD36 and macrophage receptor with collagenous structure (MARCO) are expressed in these cells [94]. Another observation that suggests a possible role of SR-BI in B cells, is that the dual engagement of SR-BI and TLR9 by CpG down regulates IL6 and IL10 cytokines, as well as IgM production [94].
Previously, it has been reported that SR-BI expression protects against early onset atherosclerosis development in mouse models of the disease such as Apoe −/− and Ldlr −/− [95, 96]. SR-BI −/− Apoe −/− mice fed with standard chow diet developed occlusive coronary artery atherosclerosis, as well as significant atherosclerotic lesions, compared with control mice [97]. It has furthermore been demonstrated that transplantation of bone marrow from SR-BI −/− mice into female Ldlr −/− mice, induced a twofold reduction of the mean atherosclerotic lesion area after 4 weeks of HFD. According to previous reports, SR-BI in bone marrow derived cells has a dual role in atherosclerotic lesion development, depending on disease stage [98]. Recent data from our collaborators showed that mice with advanced atherosclerosis have a variety of alterations in the frequency and phenotype of B lymphocytes, most of which were associated with dyslipidemia. This was observed in spleen and aortic tissue, suggesting the role of B cells in atherosclerosis both as a systemic and a localized disease [99, 100].
B cells involved in lipid immune responses through CD1 receptors
CD1
Even though CD1 is not directly involved in lipids recognition/removal, it participates in the antigen presentation of lipids, leading to immune responses that contribute with the inflammatory process. This receptors belong to a family of β2 microglobulin associated proteins related to the MHC class I and II, but encoded by genes outside the MHC. Humans have five CD1 isoforms which divide in three groups; the first one comprises CD1a, CD1b and CD1c; the second includes CD1d, and the third one CD1e molecule. In mice, there is a single class of CD1 molecule (mCD1d) which is homologous to human CD1d [101–105].
CD1d
CD1d expression has been described in different cell types, including B cells, which express low levels of this marker [106]. In contrast to classical peptide antigen presentation, CD1 molecules have evolved to present lipid antigens or hydrophobic peptide antigens [107, 108] to T cells, activating a specialized T cell subset called iNKT cells, which express the T cell receptor (TCR) rearrangement Vα14-Jα281 (also known as Jα18 or Jα15) in mice and Vα24-Jα18 in humans [55, 109]. These TCR molecules recognize exogenous glycosphingolipid antigens such as α-galactosylceramide (α-GalCer, isolated from marine sponges), α-glucosylceramides, or diacylglycerol (isolated from the Gram-negative bacteria Sphingomonas and Borrelia burgdorferi spirochete, respectively) [39, 110–115].
In human cells, it has been reported that the interaction between iNKT and B cells trough CD1d even in the absence of α-GalCer, promote proliferation of memory and naive B lymphocytes, as well as Ig production. One of the hypothesis to explain this, suggests that B cells express an endogenous glycolipid different to α-GalCer, associated to CD1d and recognized by iNKT cells [41, 111]. Interestingly, Kain et al. have showed the presence of α-linked contaminants by blocking its stimulatory activity using antagonists, among them anti-CD1-α-GalCer antibody L363. Also confirming that β-glucosylceramides have no stimulatory properties towards NKT cells [116]. However, more recent studies conclude that β-glucosylceramides primarily constitute an NKT type II (CD1d-restricted T cells lacking iTCR) ligand, which display a distinct cytokine profile and provide robust help to B cells [34]. On the other hand, interaction of B cells with lipids is also evidenced by the observation that lipid activated iNKT cells lead to the activation of B cells. Also, it has been suggested that iNKT cells can convert tolerogenic B cells into immunogenic antigen presenting cells (APCs), which can generate long-lasting cytotoxic immunity; mechanism that could depend on the expression of functional CD1d [117]. Direct interaction of B and iNKT cells induce antibody responses during humoral immunity, which is an important mechanism dependent on the expression of CD1d on B cells [38]. This interaction could participate in the inflammatory responses that involve lipids removal and subsequent inflammation, like the one observed in atherosclerosis. In fact, it has been reported that the activation of CD1d-restricted iNKT cells exacerbates atherosclerosis [118, 119]. Additionally, the relevance of CD1d-restricted NKT cells in atherosclerosis is demonstrated by a 40% reduction in fatty streaks formation in CD1d −/− Ldlr −/− mice compared with competent CD1d Ldlr −/− mice. However, the importance of CD1d on lesion progression is just transitory and does not significantly affect the inflammatory cytokine milieu of mature lesions [120].
Also, in other chronic inflammatory disorders such as autoimmune diseases, recent data indicates that CD1d deficiency worsens autoantibody production and nephritis in a genetically susceptible lupus mouse model (BWF1) [121], as well in the hydrocarbon oil-induced model of lupus nephritis [122]. In parallel, iNKT cells participate in the regulation of autoantibody production, since autoreactive B cells are selectively reduced in the presence of activated iNKTs in a CD1d-contact dependent manner [123]. Most recently, it has also been established that inappropriate presentation of CD1d-restricted self-lipids by autoimmune B cells is a crucial mechanism leading to iNKT cell hyperactivation, proliferation, and apoptosis in autoimmune mice [124]. In the same way it has been proposed that healthy B cells are pivotal for iNKT cell homeostasis, related to the fact that in healthy donors, B cells are essential for iNKT cells expansion and activation, conditions that fail in patients with systemic lupus erythematous (SLE) due to altered CD1d recycling [125].
Conlusions
Lipid dysregulation is the result of several inadequate life styles that progress to metabolic changes. This has huge impact during development and establishment of lipid related diseases, especially in atherosclerosis, which underlies the majority of CVDs. In this sense, immune responses and metabolic regulation are highly integrated, even a slight disruption of this interaction could lead to a silent but dangerous systemic imbalance. This is reflected by an elevated concentration of circulating lipids, due to altered metabolism of lipids, aberrant intracellular signaling, as well as unbalanced production of pro and anti-inflammatory cytokines and the participation of various immune cells. This miscommunication can transform into a steady state of inflammation, which could lead to tissue dysfunction and remodeling.
B cells might play specific roles in chronic inflammatory disorders; however it has been hard to establish because of its variability in terms of frequency and functionality among disease models, animal strains and diets. Despite the fact that other cells such as macrophages are the main subject of study in this area, participation of B cells in lipid inflammatory conditions is evidenced by the expression of a variety of receptors that can recognize foreign, endogenous or self-modified antigens such as oxidized lipids, which suggests the participation of B cells in lipotoxicity.
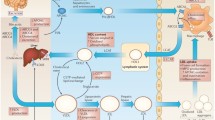
In the early stages of CVDs, hypercholesterolemia induces the diffusion of LDL particles through the activated endothelium, which in turn cause the expression of adhesion molecules which recruit, arrest and finally ends in the entrance of immune system cells to the sub-endothelial space. In mice, it has been reported that cells and soluble factors circulate from aortic tertiary lymphoid organs (ATLOs), which locate in the adventitia of diseased vessels. During the development of atherosclerotic disease, we speculate that B cells have an important participation, since the expression of LDLR contributes with the recognition of LDL molecules and its antigenic presentation trough CD1d to iNKTs exacerbates the disease. The progressive establishment of the inflammatory process and the presence of these molecules in the sub-endothelial space, induce its oxidation generating oxLDL, which are recognized by SR such as CD36, inducing an increased removal of these molecules. In parallel, we can find the up regulation of FcγRIIB receptor that through its inhibitory activity induces anti-inflammatory responses after the interaction with immune complexes of IgG and oxLDL, decreasing inflammation (Fig. 1).

Participation of B cell receptors during atherosclerotic plaque formation. During atherosclerotic plaque formation we speculate that B cells have an important participation. The expression of LDLR contributes with the recognition of LDL molecules and its antigenic presentation trough CD1d to iNKTs exacerbates the disease. Persistence presence of LDL molecule favors its oxidation; oxLDLs are recognized by CD36 and SR-BI, leading to the development of lipid load cells either contributing to atherosclerosis (panel in red) or depending on the disease stage, diminishing the inflammatory process (panel in green). On the contrary, the up regulation of FcγRIIB receptors with inhibitory activity induce anti-inflammatory responses after recognition of immune complexes of IgG and oxLDL associated with an atheroprotector role (panel in green)
All these observations open a number of possibilities regarding to the clinical application of B cells for both the early diagnosis and treatment of atherosclerosis and other lipid metabolism diseases in humans. B cells are clearly involved in lipid mediated diseases as well as in chronic inflammatory conditions, probably by the recognition of lipids through some of the receptors mentioned here, but more work needs to be done in this field.
Abbreviations
- ACAT:
-
Acyl-CoA: cholesterol acyltransferase
- AcLDL:
-
Acetylated LDL
- APCs:
-
Antigen presenting cells
- Apoe −/− :
-
Apolipoprotein E single knockout
- ATLOS:
-
Aortic tertiary lymphoid organs
- BCR:
-
B cell receptor
- CVDs:
-
Cardiovascular diseases
- DAMPs:
-
Death associated molecular patterns
- DIO:
-
Diet induced obese
- FcR:
-
Fc receptors
- FO:
-
Follicular
- HDL:
-
High-density lipoprotein
- HFD:
-
High fat diet
- HMG-CoA:
-
3-hydroxy-3-methyl-glutaryl-CoA
- Ig:
-
Immunoglobulin
- iNKT:
-
invariant Natural killer T
- ITAM:
-
Immunoreceptor tyrosine-based activation motif
- ITIM:
-
Immunoreceptor tyrosine-based inhibitory motif
- LDL:
-
Low density lipoprotein
- LDLR:
-
Low density lipoprotein receptor
- Ldlr −/− :
-
Low density lipoprotein receptor-deficient
- MARCO:
-
Macrophage receptor with collagenous structure
- MetS:
-
Metabolic syndrome
- MHC:
-
Major histocompatibility complex
- MZ:
-
Marginal zone
- NCDs:
-
Noncommunicable diseases
- oxLDL:
-
Oxidized low density lipoproteins
- PAMPs:
-
Pathogen associated molecular patterns
- PRR:
-
Pattern recognition receptors
- SLE:
-
Systemic lupus erythematous
- SR:
-
Scavenger receptors
- SR-BI:
-
SR class B type I
- TCR:
-
T cell receptor
- TLR:
-
Toll like receptor
- VLDL:
-
Very low density lipoproteins
- WHO:
-
World Health Organization
- α-GalCer:
-
α-galactosylceramide
References
Legein B, Temmerman L, Biessen EAL, Lutgens E. Inflammation and immune system interactions in atherosclerosis. Cell Mol Life Sci CMLS. 2013;70:3847–69.
World Health Organization. Global status report on noncommunicable diseases 2014: attaining the nine global noncommunicable diseases targets; a shared responsibility. Geneva: World Health Organization; 2014.
Goldschmidt-Clermont PJ, Dong C, Seo DM, Velazquez OC. Atherosclerosis, inflammation, genetics, and stem cells: 2012 update. Curr Atheroscler Rep. 2012;14(3):201–10.
Tuttolomondo A, Di Raimondo D, Pecoraro R, Arnao V, Pinto A, Licata G. Atherosclerosis as an inflammatory disease. Curr Pharm Des. 2012;18(28):4266–88.
Gu H, Tang C, Yang Y. Psychological stress, immune response, and atherosclerosis. Atherosclerosis. 2012;223(1):69–77.
World Health Organization. Global health risks: mortality and burden of disease attributable to selected major risks. Geneva: World Health Organization; 2009. p. 62.
Kok BPC, Brindley DN. Myocardial fatty acid metabolism and lipotoxicity in the setting of insulin resistance. Heart Fail Clin. 2012;8(4):643–61.
Schilling JD, Mann DL. Diabetic cardiomyopathy: bench to bedside. Heart Fail Clin. 2012;8(4):619–31.
Alberti KG, Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med J Br Diabet Assoc. 1998;15(7):539–53.
Balkau B, Charles MA. Comment on the provisional report from the WHO consultation. European Group for the Study of Insulin Resistance (EGIR). Diabet Med J Br Diabet Assoc. 1999;16(5):442–3.
Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA. 2001;285(19):2486–97.
Grundy SM, Brewer HB, Cleeman JI, Smith SC, Lenfant C. American Heart Association, et al. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation. 2004;109(3):433–8.
Alberti KGMM, Zimmet P, Shaw J. IDF Epidemiology Task Force Consensus Group. The metabolic syndrome--a new worldwide definition. Lancet. 2005;366(9491):1059–62.
Alberti KGMM, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120(16):1640–5.
Aon MA, Bhatt N, Cortassa SC. Mitochondrial and cellular mechanisms for managing lipid excess. Front Physiol. 2014;5:282.
Wende AR, Symons JD, Abel ED. Mechanisms of lipotoxicity in the cardiovascular system. Curr Hypertens Rep. 2012;14(6):517–31.
Suganami T, Tanaka M, Ogawa Y. Adipose tissue inflammation and ectopic lipid accumulation. Endocr J. 2012;59(10):849–57.
Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6(12):1191–7.
Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454(7203):428–35.
Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–7.
Alwan A, World Health Organization. Global status report on noncommunicable diseases 2010. Geneva: World Health Organization; 2011.
Winer DA, Winer S, Chng MHY, Shen L, Engleman EG. B Lymphocytes in obesity-related adipose tissue inflammation and insulin resistance. Cell Mol Life Sci CMLS. 2014;71(6):1033–43.
Guilbert JJ. The world health report 2002-reducing risks, promoting healthy life. Educ Health Abingdon Engl. 2003;16(2):230.
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–808.
Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112(12):1821–30.
Bouloumié A, Casteilla L, Lafontan M. Adipose tissue lymphocytes and macrophages in obesity and insulin resistance: makers or markers, and which comes first? Arterioscler Thromb Vasc Biol. 2008;28(7):1211–3.
Travers RL, Motta AC, Betts JA, Bouloumié A, Thompson D. The impact of adiposity on adipose tissue-resident lymphocyte activation in humans. Int J Obes. 2005;39(5):762–9.
Duffaut C, Galitzky J, Lafontan M, Bouloumié A. Unexpected trafficking of immune cells within the adipose tissue during the onset of obesity. Biochem Biophys Res Commun. 2009;384(4):482–5.
Kintscher U, Hartge M, Hess K, Foryst-Ludwig A, Clemenz M, Wabitsch M, et al. T-lymphocyte infiltration in visceral adipose tissue: a primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. Arterioscler Thromb Vasc Biol. 2008;28(7):1304–10.
DeFuria J, Belkina AC, Jagannathan-Bogdan M, Snyder-Cappione J, Carr JD, Nersesova YR, et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc Natl Acad Sci U S A. 2013;110(13):5133–8.
Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med. 2011;17(5):610–7.
Jagannathan M, McDonnell M, Liang Y, Hasturk H, Hetzel J, Rubin D, et al. Toll-like receptors regulate B cell cytokine production in patients with diabetes. Diabetologia. 2010;53(7):1461–71.
van Exel E, Gussekloo J, de Craen AJM, Frölich M, Bootsma-Van Der Wiel A, Westendorp RGJ, et al. Low production capacity of interleukin-10 associates with the metabolic syndrome and type 2 diabetes : the Leiden 85-Plus Study. Diabetes. 2002;51(4):1088–92.
Nair S, Boddupalli CS, Verma R, Liu J, Yang R, Pastores GM, et al. Type II NKT-TFH cells against Gaucher lipids regulate B-cell immunity and inflammation. Blood. 2015;125(8):1256–71.
Major AS, Fazio S, Linton MF. B-lymphocyte deficiency increases atherosclerosis in LDL receptor-null mice. Arterioscler Thromb Vasc Biol. 2002;22(11):1892–8.
Ait-Oufella H, Herbin O, Bouaziz J-D, Binder CJ, Uyttenhove C, Laurans L, et al. B cell depletion reduces the development of atherosclerosis in mice. J Exp Med. 2010;207(8):1579–87.
Lang GA, Exley MA, Lang ML. The CD1d-binding glycolipid alpha-galactosylceramide enhances humoral immunity to T-dependent and T-independent antigen in a CD1d-dependent manner. Immunology. 2006;119(1):116–25.
Lang GA, Devera TS, Lang ML. Requirement for CD1d expression by B cells to stimulate NKT cell-enhanced antibody production. Blood. 2008;111(4):2158–62.
Barral P, Eckl-Dorna J, Harwood NE, De Santo C, Salio M, Illarionov P, et al. B cell receptor-mediated uptake of CD1d-restricted antigen augments antibody responses by recruiting invariant NKT cell help in vivo. Proc Natl Acad Sci U S A. 2008;105(24):8345–50.
Leadbetter EA, Brigl M, Illarionov P, Cohen N, Luteran MC, Pillai S, et al. NK T cells provide lipid antigen-specific cognate help for B cells. Proc Natl Acad Sci U S A. 2008;105(24):8339–44.
Galli G, Nuti S, Tavarini S, Galli-Stampino L, De Lalla C, Casorati G, et al. CD1d-restricted help to B cells by human invariant natural killer T lymphocytes. J Exp Med. 2003;197(8):1051–7.
Van Laethem F, Leo O. Membrane lipid rafts: new targets for immunoregulation. Curr Mol Med. 2002;2(6):557–70.
Plüddemann A, Neyen C, Gordon S. Macrophage scavenger receptors and host-derived ligands. Methods San Diego Calif. 2007;43(3):207–17.
Moore KJ, Freeman MW. Scavenger receptors in atherosclerosis: beyond lipid uptake. Arterioscler Thromb Vasc Biol. 2006;26(8):1702–11.
Cuthbert JA, Lipsky PE. Immunoregulation by low density lipoproteins in man. Inhibition of mitogen-induced T lymphocyte proliferation by interference with transferrin metabolism. J Clin Invest. 1984;73(4):992–1003.
Ho YK, Smith RG, Brown MS, Goldstein JL. Low-density lipoprotein (LDL) receptor activity in human acute myelogenous leukemia cells. Blood. 1978;52(6):1099–114.
Hiramatsu K, Sakai H, Endoh M, Arimori S. Surface properties of LDL-binding lymphocytes in human peripheral blood. Immunology. 1980;39(3):311–6.
Suresh S, Warty V, Virji M, Sanghvi A. Effect of insulin on low-density-lipoprotein metabolism in human lymphocytes in vitro. Biochem J. 1986;233(2):565–70.
Goldstein JL, Brown MS. The low-density lipoprotein pathway and its relation to atherosclerosis. Annu Rev Biochem. 1977;46:897–930.
Ho YK, Brown S, Bilheimer DW, Goldstein JL. Regulation of low density lipoprotein receptor activity in freshly isolated human lymphocytes. J Clin Invest. 1976;58(6):1465–74.
De Sanctis JB, Blanca I, Rivera H, Bianco NE. Expression of low-density lipoprotein receptors in peripheral blood and tonsil B lymphocytes. Clin Exp Immunol. 1998;113(2):206–12.
Sanghvi A, Warty V. Differences in the binding, internalization and catabolism of low-density lipoprotein between normal human T and B lymphocytes. Biochem J. 1985;227(2):397–404.
Schmitz G, Brüning T, Kovacs E, Barlage S. Fluorescence flow cytometry of human leukocytes in the detection of LDL receptor defects in the differential diagnosis of hypercholesterolemia. Arterioscler Thromb J Vasc Biol Am Heart Assoc. 1993;13(7):1053–65.
Parthasarathy S, Rankin SM. Role of oxidized low density lipoprotein in atherogenesis. Prog Lipid Res. 1992;31(2):127–43.
Allan LL, Hoefl K, Zheng D-J, Chung BK, Kozak FK, Tan R, et al. Apolipoprotein-mediated lipid antigen presentation in B cells provides a pathway for innate help by NKT cells. Blood. 2009;114(12):2411–6.
Kubagawa H, Oka S, Kubagawa Y, Torii I, Takayama E, Kang D-W, et al. Identity of the elusive IgM Fc receptor (FcmuR) in humans. J Exp Med. 2009;206(12):2779–93.
Hjelm F, Carlsson F, Getahun A, Heyman B. Antibody-mediated regulation of the immune response. Scand J Immunol. 2006;64(3):177–84.
Chou M-Y, Fogelstrand L, Hartvigsen K, Hansen LF, Woelkers D, Shaw PX, et al. Oxidation-specific epitopes are dominant targets of innate natural antibodies in mice and humans. J Clin Invest. 2009;119(5):1335–49.
Palinski W, Hörkkö S, Miller E, Steinbrecher UP, Powell HC, Curtiss LK, et al. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J Clin Invest. 1996;98(3):800–14.
Pricop L, Redecha P, Teillaud JL, Frey J, Fridman WH, Sautès-Fridman C, et al. Differential modulation of stimulatory and inhibitory Fc gamma receptors on human monocytes by Th1 and Th2 cytokines. J Immunol Baltim Md 1950. 2001;166(1):531–7.
Aman MJ, Tosello-Trampont AC, Ravichandran K. Fc gamma RIIB1/SHIP-mediated inhibitory signaling in B cells involves lipid rafts. J Biol Chem. 2001;276(49):46371–8.
Ravetch JV, Lanier LL. Immune inhibitory receptors. Science. 2000;290(5489):84–9.
Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol. 2001;19:275–90.
Zhang CY, Booth JW. Differences in endocytosis mediated by FcγRIIA and FcγRIIB2. Mol Immunol. 2011;49(1–2):329–37.
Rabinovitch N, Gelfand EW. Expression of functional activating and inhibitory Fcgamma receptors on human B cells. Int Arch Allergy Immunol. 2004;133(3):285–94.
Ratcliffe NR, Kennedy SM, Morganelli PM. Immunocytochemical detection of Fcgamma receptors in human atherosclerotic lesions. Immunol Lett. 2001;77(3):169–74.
Rothe G, Gabriel H, Kovacs E, Klucken J, Stöhr J, Kindermann W, et al. Peripheral blood mononuclear phagocyte subpopulations as cellular markers in hypercholesterolemia. Arterioscler Thromb Vasc Biol. 1996;16(12):1437–47.
Pfeiffer JR, Howes PS, Waters MA, Hynes ML, Schnurr PP, Demidenko E, et al. Levels of expression of Fcgamma receptor IIA (CD32) are decreased on peripheral blood monocytes in patients with severe atherosclerosis. Atherosclerosis. 2001;155(1):211–8.
Hernández-Vargas P, Ortiz-Muñoz G, López-Franco O, Suzuki Y, Gallego-Delgado J, Sanjuán G, et al. Fcgamma receptor deficiency confers protection against atherosclerosis in apolipoprotein E knockout mice. Circ Res. 2006;99(11):1188–96.
Mallavia B, Oguiza A, Lopez-Franco O, Recio C, Ortiz-Muñoz G, Lazaro I, et al. Gene Deficiency in Activating Fcγ Receptors Influences the Macrophage Phenotypic Balance and Reduces Atherosclerosis in Mice. PLoS One. 2013;8(6):e66754.
Zhao M, Wigren M, Dunér P, Kolbus D, Olofsson KE, Björkbacka H, et al. FcgammaRIIB inhibits the development of atherosclerosis in low-density lipoprotein receptor-deficient mice. J Immunol Baltim Md 1950. 2010;184(5):2253–60.
Ng HP, Zhu X, Harmon EY, Lennartz MR, Nagarajan S. Reduced Atherosclerosis in apoE-inhibitory FcγRIIb-Deficient Mice Is Associated With Increased Anti-Inflammatory Responses by T Cells and Macrophages. Arterioscler Thromb Vasc Biol. 2015;35(5):1101–12.
Virella G, Koskinen S, Krings G, Onorato JM, Thorpe SR, Lopes-Virella M. Immunochemical characterization of purified human oxidized low-density lipoprotein antibodies. Clin Immunol Orlando Fla. 2000;95(2):135–44.
Lehtimäki T, Lehtinen S, Solakivi T, Nikkilä M, Jaakkola O, Jokela H, et al. Autoantibodies against oxidized low density lipoprotein in patients with angiographically verified coronary artery disease. Arterioscler Thromb Vasc Biol. 1999;19(1):23–7.
Nimmerjahn F, Ravetch JV. Fcgamma receptors: old friends and new family members. Immunity. 2006;24(1):19–28.
Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8(1):34–47.
Krieger M. The other side of scavenger receptors: pattern recognition for host defense. Curr Opin Lipidol. 1997;8(5):275–80.
Mukhopadhyay S, Gordon S. The role of scavenger receptors in pathogen recognition and innate immunity. Immunobiology. 2004;209(1–2):39–49.
Zhang P, Li W, Wang Y, Hou L, Xing Y, Qin H, et al. Identification of CD36 as a new surface marker of marginal zone B cells by transcriptomic analysis. Mol Immunol. 2007;44(4):332–7.
Endemann G, Stanton LW, Madden KS, Bryant CM, White RT, Protter AA. CD36 is a receptor for oxidized low density lipoprotein. J Biol Chem. 1993;268(16):11811–6.
Boullier A, Gillotte KL, Hörkkö S, Green SR, Friedman P, Dennis EA, et al. The binding of oxidized low density lipoprotein to mouse CD36 is mediated in part by oxidized phospholipids that are associated with both the lipid and protein moieties of the lipoprotein. J Biol Chem. 2000;275(13):9163–9.
Febbraio M, Abumrad NA, Hajjar DP, Sharma K, Cheng W, Pearce SF, et al. A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J Biol Chem. 1999;274(27):19055–62.
Won W-J, Bachmann MF, Kearney JF. CD36 is differentially expressed on B cell subsets during development and in responses to antigen. J Immunol Baltim Md 1950. 2008;180(1):230–7.
Corcoran L, Vremec D, Febbraio M, Baldwin T, Handman E. Differential regulation of CD36 expression in antigen-presenting cells: Oct-2 dependence in B lymphocytes but not dendritic cells or macrophages. Int Immunol. 2002;14(10):1099–104.
Febbraio M, Podrez EA, Smith JD, Hajjar DP, Hazen SL, Hoff HF, et al. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest. 2000;105(8):1049–56.
Kuchibhotla S, Vanegas D, Kennedy DJ, Guy E, Nimako G, Morton RE, et al. Absence of CD36 protects against atherosclerosis in ApoE knock-out mice with no additional protection provided by absence of scavenger receptor A I/II. Cardiovasc Res. 2008;78(1):185–96.
Febbraio M, Guy E, Silverstein RL. Stem cell transplantation reveals that absence of macrophage CD36 is protective against atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24(12):2333–8.
Moore KJ, Kunjathoor VV, Koehn SL, Manning JJ, Tseng AA, Silver JM, et al. Loss of receptor-mediated lipid uptake via scavenger receptor A or CD36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. J Clin Invest. 2005;115(8):2192–201.
Kennedy DJ, Kuchibhotla SD, Guy E, Park YM, Nimako G, Vanegas D, et al. Dietary cholesterol plays a role in CD36-mediated atherogenesis in LDLR-knockout mice. Arterioscler Thromb Vasc Biol. 2009;29(10):1481–7.
Acton SL, Scherer PE, Lodish HF, Krieger M. Expression cloning of SR-BI, a CD36-related class B scavenger receptor. J Biol Chem. 1994;269(33):21003–9.
Krieger M. Charting the fate of the “good cholesterol”: identification and characterization of the high-density lipoprotein receptor SR-BI. Annu Rev Biochem. 1999;68:523–58.
Rigotti A, Trigatti BL, Penman M, Rayburn H, Herz J, Krieger M. A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc Natl Acad Sci U S A. 1997;94(23):12610–5.
Terpstra V, van Amersfoort ES, van Velzen AG, Kuiper J, van Berkel TJ. Hepatic and extrahepatic scavenger receptors: function in relation to disease. Arterioscler Thromb Vasc Biol. 2000;20(8):1860–72.
Zhu P, Liu X, Treml LS, Cancro MP, Freedman BD. Mechanism and Regulatory Function of CpG Signaling via Scavenger Receptor B1 in Primary B Cells. J Biol Chem. 2009;284(34):22878–87.
Trigatti B, Rayburn H, Viñals M, Braun A, Miettinen H, Penman M, et al. Influence of the high density lipoprotein receptor SR-BI on reproductive and cardiovascular pathophysiology. Proc Natl Acad Sci U S A. 1999;96(16):9322–7.
Covey SD, Krieger M, Wang W, Penman M, Trigatti BL. Scavenger receptor class B type I-mediated protection against atherosclerosis in LDL receptor-negative mice involves its expression in bone marrow-derived cells. Arterioscler Thromb Vasc Biol. 2003;23(9):1589–94.
Braun A, Trigatti BL, Post MJ, Sato K, Simons M, Edelberg JM, et al. Loss of SR-BI expression leads to the early onset of occlusive atherosclerotic coronary artery disease, spontaneous myocardial infarctions, severe cardiac dysfunction, and premature death in apolipoprotein E-deficient mice. Circ Res. 2002;90(3):270–6.
Van Eck M, Bos IST, Hildebrand RB, Van Rij BT, Van Berkel TJC. Dual role for scavenger receptor class B, type I on bone marrow-derived cells in atherosclerotic lesion development. Am J Pathol. 2004;165(3):785–94.
Rincón-Arévalo H, Castaño D, Villa-Pulgarín J, Rojas M, Vásquez G, Ramírez-Pineda JR, et al. Dyslipidemia-associated alterations in B cell subpopulation frequency and phenotype during experimental atherosclerosis. Atherosclerosis. 2016;247:118–26.
Rincón-Arévalo H, Castaño D, Villa-Pulgarín J, Rojas M, Vásquez G, Correa LA, et al. Data in support of dyslipidemia-associated alterations in B cell subpopulations frequency and phenotype during experimental atherosclerosis. Data Brief. 2016;7:958–72.
Hameg A, Apostolou I, Leite-De-Moraes M, Gombert JM, Garcia C, Koezuka Y, et al. A subset of NKT cells that lacks the NK1.1 marker, expresses CD1d molecules, and autopresents the alpha-galactosylceramide antigen. J Immunol Baltim Md 1950. 2000;165(9):4917–26.
Matsuda JL, Kronenberg M. Presentation of self and microbial lipids by CD1 molecules. Curr Opin Immunol. 2001;13(1):19–25.
Calabi F, Jarvis JM, Martin L, Milstein C. Two classes of CD1 genes. Eur J Immunol. 1989;19(2):285–92.
Porcelli SA. The CD1 family: a third lineage of antigen-presenting molecules. Adv Immunol. 1995;59:1–98.
Porcelli SA, Modlin RL. The CD1 system: antigen-presenting molecules for T cell recognition of lipids and glycolipids. Annu Rev Immunol. 1999;17:297–329.
Blumberg RS, Terhorst C, Bleicher P, McDermott FV, Allan CH, Landau SB, et al. Expression of a nonpolymorphic MHC class I-like molecule, CD1D, by human intestinal epithelial cells. J Immunol Baltim Md 1950. 1991;147(8):2518–24.
Beckman EM, Brenner MB. MHC class I-like, class II-like and CD1 molecules: distinct roles in immunity. Immunol Today. 1995;16(7):349–52.
Castaño AR, Tangri S, Miller JE, Holcombe HR, Jackson MR, Huse WD, et al. Peptide binding and presentation by mouse CD1. Science. 1995;269(5221):223–6.
Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol. 2007;25:297–336.
Singh N, Hong S, Scherer DC, Serizawa I, Burdin N, Kronenberg M, et al. Cutting edge: activation of NK T cells by CD1d and alpha-galactosylceramide directs conventional T cells to the acquisition of a Th2 phenotype. J Immunol Baltim Md 1950. 1999;163(5):2373–7.
Galli G, Pittoni P, Tonti E, Malzone C, Uematsu Y, Tortoli M, et al. Invariant NKT cells sustain specific B cell responses and memory. Proc Natl Acad Sci U S A. 2007;104(10):3984–9.
Brossay L, Chioda M, Burdin N, Koezuka Y, Casorati G, Dellabona P, et al. CD1d-mediated recognition of an alpha-galactosylceramide by natural killer T cells is highly conserved through mammalian evolution. J Exp Med. 1998;188(8):1521–8.
Kinjo Y, Wu D, Kim G, Xing G-W, Poles MA, Ho DD, et al. Recognition of bacterial glycosphingolipids by natural killer T cells. Nature. 2005;434(7032):520–5.
Mattner J, Debord KL, Ismail N, Goff RD, Cantu C, Zhou D, et al. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature. 2005;434(7032):525–9.
Kinjo Y, Tupin E, Wu D, Fujio M, Garcia-Navarro R, Benhnia MR-E-I, et al. Natural killer T cells recognize diacylglycerol antigens from pathogenic bacteria. Nat Immunol. 2006;7(9):978–86.
Kain L, Webb B, Anderson BL, Deng S, Holt M, Costanzo A, et al. The identification of the endogenous ligands of natural killer T cells reveals the presence of mammalian α-linked glycosylceramides. Immunity. 2014;41(4):543–54.
Chung Y, Kim B-S, Kim Y-J, Ko H-J, Ko S-Y, Kim D-H, et al. CD1d-restricted T cells license B cells to generate long-lasting cytotoxic antitumor immunity in vivo. Cancer Res. 2006;66(13):6843–50.
Tupin E, Nicoletti A, Elhage R, Rudling M, Ljunggren H-G, Hansson GK, et al. CD1d-dependent activation of NKT cells aggravates atherosclerosis. J Exp Med. 2004;199(3):417–22.
Nakai Y, Iwabuchi K, Fujii S, Ishimori N, Dashtsoodol N, Watano K, et al. Natural killer T cells accelerate atherogenesis in mice. Blood. 2004;104(7):2051–9.
Aslanian AM, Chapman HA, Charo IF. Transient role for CD1d-restricted natural killer T cells in the formation of atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2005;25(3):628–32.
Yang J-Q, Wen X, Liu H, Folayan G, Dong X, Zhou M, et al. Examining the role of CD1d and natural killer T cells in the development of nephritis in a genetically susceptible lupus model. Arthritis Rheum. 2007;56(4):1219–33.
Yang J-Q, Singh AK, Wilson MT, Satoh M, Stanic AK, Park J-J, et al. Immunoregulatory role of CD1d in the hydrocarbon oil-induced model of lupus nephritis. J Immunol Baltim Md 1950. 2003;171(4):2142–53.
Yang J-Q, Wen X, Kim PJ, Singh RR. Invariant NKT cells inhibit autoreactive B cells in a contact- and CD1d-dependent manner. J Immunol Baltim Md 1950. 2011;186(3):1512–20.
Tan AH-M, Chong WP-K, Ng S-W, Basri N, Xu S, Lam K-P. Aberrant presentation of self-lipids by autoimmune B cells depletes peripheral iNKT cells. Cell Rep. 2014;9(1):24–31.
Bosma A, Abdel-Gadir A, Isenberg DA, Jury EC, Mauri C. Lipid-antigen presentation by CD1d(+) B cells is essential for the maintenance of invariant natural killer T cells. Immunity. 2012;36(3):477–90.
Acknowledgements
Not applicable.
Funding
LCE received financial support from the doctoral scholarship from Colciencias.
Availability of data and material
Not applicable.
Authors’ contributions
LCE wrote this paper and performed the suggested changes. LMY made significant changes to all the versions of this paper. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Echeverri Tirado, L.C., Yassin, L.M. B cells interactions in lipid immune responses: implications in atherosclerotic disease. Lipids Health Dis 16, 30 (2017). https://doi.org/10.1186/s12944-016-0390-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12944-016-0390-5