
Abstract
Cellular senescence (CS), a permanent and irreversible arrest of the cell cycle and proliferation leading to the degeneration of cellular structure and function, has been implicated in various key physiological and pathological processes, particularly in cancer. Initially, CS was recognized as a barrier to tumorigenesis, serving as an intrinsic defense mechanism to protect cells from malignant transformation. However, increasing evidence suggests that senescent cells can promote tumor progression to overt malignancy, primarily through a set of factors known as senescence-associated secretory phenotypes (SASPs), including chemokines, growth factors, cytokines, and stromal metalloproteinases. These factors significantly reshape the tumor microenvironment (TME), enabling tumors to evade immune destruction. Interestingly, some studies have also suggested that SASPs may impede tumor development by enhancing immunosurveillance. These opposing roles highlight the complexity and heterogeneity of CS and SASPs in diverse cancers. Consequently, there has been growing interest in pharmacological interventions targeting CS or SASPs in cancer therapy, such as senolytics and senomorphics, to either promote the clearance of senescent cells or mitigate the harmful effects of SASPs. In this review, we will interpret the concept of CS, delve into the role of SASPs in reshaping the TME, and summarize recent advances in anti-tumor strategies targeting CS or SASPs.
Similar content being viewed by others


Introduction
Cellular senescence (CS) was initially introduced in 1961 [1]. In 1965, it was further described as a manifestation of the finite replicative capacity of diploid cell lines in vivo, characterized by the Hayflick limit, which represents the maximum number of cell divisions achievable before cellular growth arrest [2]. This phenomenon was subsequently defined as replicative senescence, which was considered irreversible and permanent due to the cell's inability to physiologically reverse this cycle arrest [3]. Recent studies have shown that CS can result from exposure to various internal or external stressors, such as replication stress, telomere damage, metabolic disorders, and carcinogenic factors [4], leading to different types of senescence. In addition to arrested growth, another key feature of CS is the senescence-associated secretory phenotype (SASP), which includes a range of proinflammatory and proteolytic factors. SASP is generally diverse and dynamic, varying according to the type of senescent cells and the cellular environment [5]. However, SASP can also influence the surrounding environment, making CS and SASP not only cellular phenomena but also closely related to the development of various diseases, particularly tumors. For instance, SASP can either promote or inhibit tumor progression by remodeling the tumor microenvironment (TME), while the TME, in turn, affects SASP production. This intricate interaction significantly impacts tumor development. Although the mechanisms underlying this complex relationship are not yet fully understood, extensive preclinical studies have demonstrated that targeting senescence and/or SASP can benefit cancer patients.
In this review, we provide an overview of the characteristics and markers of CS, discuss the current mechanistic understanding of CS, and explore the impact of SASP components on the TME. Specifically, we highlight the "double-edged sword" role of SASP in cancer through its remodeling of the TME. Finally, we summarize recent advancements in anti-senescent therapies and propose their potential applications in future cancer treatments.
Types of CS
Replicative senescence
In addition to occurring under physiological conditions, CS can also be induced by various factors through different mechanisms (Fig. 1). Consequently, CS is categorized into different types based on its inducers. Replicative senescence (RS) was first proposed by Hayflick in 1961 and later defined as cell cycle arrest caused by continuous cell culture [1]. A hallmark of RS is the presence of short telomeres, which result from repeated cycles of DNA replication. Mechanistically, when telomeres reach a critical length, they are recognized as DNA double-strand breaks (DSB), which activate a DNA damage response (DDR). The earliest checkpoint kinases, ataxia-telangiectasia mutated (ATM) and ataxia-telangiectasia and Rad 3-related (ATR), are then activated to phosphorylate various proteins, including checkpoint kinase 2 (CHK2) [6]. CHK2 transmits DDR signals by phosphorylating the tumor suppressor protein p53, which is involved in cell cycle arrest, apoptosis, and DNA repair. Phosphorylated p53 subsequently activates the downstream protein p21 [7]. p21 inhibits the phosphorylation of retinoblastoma protein (RB) by restraining the function of cyclin-dependent kinase 2 (CDK2). Finally, the transcription of E2F, an essential protein in the S-phase of the cell cycle, is halted by the downregulation of RB phosphorylation, causing the cell cycle to arrest at the G1 stage and enter senescence [8, 9]. Notably, cancer cells can also induce p21 through retroviral-mediated CHK2 activation without the involvement of p53 [10]. In addition to the p53/p21CIP1 mechanism associated with cell cycle arrest, the p16INK4a/RB pathway can also induce CS. p16, also known as cyclin-dependent kinase inhibitor 2A (CDK2A), binds to the kinases CDK4 and CDK6, forming complexes that block RB phosphorylation, resulting in CS [11]. Currently, an increasing number of factors have been found to play important roles in the p53/p21CIP1 and p16INK4a/RB pathways. For example, a complex has been identified that regulates the cell cycle through the p53/p16-RB-E2F-DREAM pathway [12], and the knockout of YPEL2 reduces cell proliferation in S-phase and promotes endothelial cell senescence by activating the p53/p21 signaling pathway (Fig. 2) [13].

Factors inducing cellular senescence (CS). CS can be triggered by various factors, including oncogenes, telomere shortening, mitochondrial dysfunction, DNA damage response (DDR), protein homeostasis disorders, and chemoradiotherapy

Signaling pathways involved in CS. During DNA replication, telomeres progressively shorten, triggering a DDR and DSB, which activate ATM and ATR, leading to the activation of CHK2. This activation blocks E2F transcription through the p53/p21 and p16/RB pathways, causing the cell cycle to arrest in the G1 phase. Additionally, OIS and TIS can also initiate DDR and DSB, ultimately leading to cell cycle arrest
Premature Senescence
Various stressors can lead to premature senescence, which can be categorized into different types depending on the stimulus. Below are several types of cancer-related premature senescence:
Oncogene-induced senescence (OIS)
OIS is primarily triggered by the expression of certain oncogenes. It was initially observed as a result of the heterotopic expression of HrasV12, an oncogenic form of Ras, in human lung fibroblasts [14]. The mechanisms underlying OIS have since been elucidated: oncogene activation induces the production of reactive oxygen species (ROS), which leads to DSBs and DDR, thereby initiating CS [15]. The TP53/CDKN1A and p16/RB pathways are also involved in OIS. TP53 acts to inhibit cancer cell growth, and its loss can lead to increased invasiveness [16]. Meanwhile, RB plays an important role in maintaining OIS by inhibiting the expression of the DNA transcription factor E2F [17]. OIS is also associated with signs of DNA replication stress, which can cause DSBs and genomic instability in human precancerous lesions [18, 19]. Bartkova et al. demonstrated that CS acts as a barrier to tumor formation in precancerous lesions by activating DNA damage checkpoints in response to DNA replication stress [18]. Furthermore, research has shown that oncogene expression alone does not trigger DDR in the absence of DNA replication. Thus, OIS arises from DDR activation driven by oncogene-induced DNA hyper-replication [20].
Therapy-induced senescence (TIS)
TIS occurs when senescence is induced by various types of chemotherapy or radiation therapy, which cause DSBs and activate DDR [21]. Chemotherapeutic agents such as cisplatin, paclitaxel, bleomycin, and cyclophosphamide are more likely to induce TIS than radiation therapy [22]. The mechanisms by which different chemotherapeutics induce TIS vary. For instance, busulfan induces CS through the p38 pathway, while cisplatin primarily triggers senescence via the p53 pathway [23]. TIS is an effective strategy for suppressing cancer growth and has been shown to benefit cancer patients [24].
SARS-CoV-2 (viral)—induced senescence
The coronavirus disease 2019 (COVID-19) pandemic has brought severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) into the spotlight [25]. Recent studies have revealed that SARS-CoV-2 is closely linked to CS. For example, Yang et al. found that SARS-CoV-2 infection can induce senescence in dopaminergic neurons [26]. Similarly, Meyer et al. reported that SARS-CoV-2 infection triggers epithelial cell senescence and increases the levels of SASPs [27]. Moreover, recent research by Gioia et al. discovered that SARS-CoV-2 infection can cause DNA damage and disrupt the DDR due to the depletion of CHK1. Additionally, the SARS-CoV-2 N protein can bind to damage-induced long non-coding RNAs (lncRNAs), impairing the recruitment of 53BP1 and thereby hindering DNA repair [28].
Rare types of senescence
Rare forms of senescence that do not depend on DNA damage include sodium butyrate- and nuclear barrier-induced senescence, as well as senescence caused by mitochondrial dysfunction, aberrant epigenetic modifications, protein homeostasis disorders, and paracrine signaling [29]. Although these types of senescence are uncommon, further exploration of these different forms and their underlying mechanisms will provide a deeper understanding of age-related diseases and may ultimately benefit anti-senescence strategies for cancer treatment.
Biomarkers of CS
The biological markers of CS vary among different types of senescent cells. For example, lapatinib induces p15- and p27-dependent senescence without detectable upregulation of p16 and p21 in HER2-positive breast cancer cells [30]. Similarly, in prostate cancer cells, androgen primarily upregulates p15 to induce CS [31]. Notably, the expression of these markers can vary even within the same senescent cells. For example, senescence induced by glyoxal is initially mediated predominantly by the protein kinase B/FOXO3a/p27KIP1 signaling pathway but gradually shifts to being driven by the p16INK4/Rb pathway [32]. Furthermore, nonsenescent cells can also express some of these markers, which complicates the identification of CS, particularly in early cultures where a mix of nonsenescent and senescent cells may be present. This complexity may be due to the premature senescence of some primary cells. Additionally, prematurely senescent cells can influence surrounding nonsenescent cells to elevate the levels of certain markers, a phenomenon known as paracrine senescence [33].
SASP components, which include proteins conserved across cell types, are valuable phenotypic markers. However, SASP alone is insufficient to definitively identify senescent cells because several SASP components are also involved in inflammatory processes. Given the complexity of CS markers, a systematic, multiparameter approach has been employed to identify senescence. Recently, a method known as senescence-associated morphological profiles was developed, which uses multidimensional, phenotype-driven assessments to define senescence [34].
Although many markers have been identified (Table 1), none are specific for CS. Typically, a combination of factors, such as cellular morphology and nuclear characteristics, is used to determine whether cells are in a senescent state. The discovery of a specific marker would greatly enhance our understanding of the underlying mechanisms and unique pathology of CS. Since no single marker is currently sufficient to identify senescence, a combination of multiple cellular senescence markers should be considered. For example, Estela et al. proposed a detailed multifactorial guide for evaluating senescence both in vitro and in vivo [35]. As such, this review will not further elaborate on these markers.
Senescence-associated secretory phenotype (SASP)
SASP is a key pathological feature of CS. Despite the arrest in cell cycle and proliferation, senescent cells maintain an active metabolic state and secrete a wide range of factors that influence both themselves and their surrounding environment. These secretory factors collectively form what is known as the SASP. The concept of SASP was first introduced in 2008 as a phenotype in which genotoxic stress-induced aging in human cells results in the secretion of substances associated with inflammation and malignancy [5]. SASP is now recognized as a complex secretory process composed of hundreds of different proteins and nonprotein signaling molecules, including pro-inflammatory factors, proteases, and growth factors. While numerous studies have attempted to characterize the composition of SASP, its exact composition remains unknown [71]. The components and levels of SASP can vary significantly depending on the stimuli that drive senescence, the types of senescent cells involved, and the duration of senescence [5, 72]. Several studies have demonstrated that SASP plays an important role in cancer, with its diverse cytokines exerting differential effects on tumor cells and the TME, as summarized in Table 2. Below, we introduce several key SASP components:
Interleukin-1 (IL-1)
The IL-1 pathway is essential for the expression of most SASP factors, although it is not necessary for the induction of senescence itself. IL-1α can activate SASP through IL-1R signaling, while IL-1β, which shares the same signaling pathway as IL-1α, is also crucial for SASP expression. Both IL-1α and IL-1β can independently affect SASP without a hierarchical relationship between them [73]. Buhl et al. found in their study on hair cell astrocytoma that IL-1β can induce growth arrest and senescence in proliferating hair cell astrocytoma cells, as well as upregulate SASP factors [101]. The release of IL-1α depends on caspase-5 and caspase-11 [102]. Kelly et al. discovered that the expression of DOT1L is necessary for SASP gene expression, possibly through the upregulation of H3K79me2/3 at the IL-1A locus [103].
Interleukin-6 (IL-6)
Wang et al. found that atorvastatin can induce senescence in hepatocellular carcinoma (HCC) cells by inhibiting the IL-6/STAT pathway. Previous studies have shown that high levels of IL-6 are closely associated to HCC [104]. Interestingly, Shriki et al. found that IL-6 deficiency can lead to the deterioration of liver cancer in mice, which is related to severe damage and senescence of SASP [105]. The contradictory effects of IL-6 may be related to the specific environment; for example, in chronic liver injury, IL-6 can inhibit liver injury, fibrosis, and the occurrence of liver cancer, while inhibiting IL-6 in acute liver injury can reduce the risk of liver cancer [106].
Galectin-9
Tarallo et al. first reported that senescent cells secrete Galectin-9 in melanoma [107]. Previous studies have shown that Galectin-9 has immunosuppressive effects in the TME, promoting the apoptosis of T cells and monocytes, increasing the regulation of T cells, helper T cells, and M2 macrophages, and impairing anti-tumor immune responses [108]. On the other hand, some reports suggest that Galectin-9 can inhibit melanoma metastasis [109].
Extracellular vesicles (EVs)
Lehmann et al. were among the first to report an increase in senescence-associated exosome secretion [110]. Suppression of small extracellular vesicle release induces the accumulation of DNA damage and apoptosis-like death in senescent cells [111]. EVs have recently emerged as crucial intermediaries within SASP and have been shown to play various roles in senescence. Similar to the more conventional "soluble" SASP, this vesicular secretome also induces a diverse array of effects that can be beneficial or detrimental depending on the specific cellular environment. These effects are driven by a varied set of EV cargos, including proteins, nucleic acids, and lipids, which support the functional roles of EVs in senescence [112]. Misawa et al. found that senescent cell-derived EVs function as SASP factors by regulating cancer growth. Since numerous EVs are found in all bodily fluids, including blood, saliva, and urine, they are considered valuable targets for liquid biopsy, which is simpler and less invasive than traditional diagnostic techniques. Therefore, EV release might be a potential target for the treatment or prevention of age-related diseases.
CS and SASP on the TME
The impact of SASP on the TME varies significantly depending on the type of cells undergoing senescence and the specific triggers of senescence. For example, SASP can promote the neuroendocrine transdifferentiation of breast cancer cells through the NF-κB pathway [113]. In melanoma, treatment with SASP-associated cytokines supports the immune system's self-sustained surveillance of senescent cells [114]. Furthermore, senescent cells can enhance melanoma metastasis by increasing the production of soluble E-cadherin [115]. In HCC, hepatic SASP facilitates HCC progression by polarizing macrophages, a process closely linked to Bcl3 expression in hepatocytes [116]. SASP components such as Coactosin-like protein 1, Alpha-enolase, and Peroxiredoxin 2 contribute to the proliferation and behavioral changes of HCC cells [117]. Additionally, acute SASP derived from mesenchymal stromal cells induce senescence in immortal prostate cells, but not in prostate cancer cells, suggesting that SASPs from acutely senescent cells may be more effective at preventing cancer initiation within the TME rather than eradicating established cancer cells [118]. Senescent fibroblasts secrete growth differentiation factor 15 (GDF15), a component of the TME that not only promotes the formation of colon cancer but also induces the proliferation, migration, and invasion of colon cancer cells [88]. SASP also has the capacity to recruit various cell types to the tumor periphery. For example, CXCR2 can attract and enhance the protumor properties of tumor-associated macrophages, facilitating epithelial-mesenchymal transition (EMT) [80]. The knockout of BTG1 has been shown to induce senescence and create a microenvironment conducive to angiogenesis and tumor growth, thereby promoting tumor metastasis [119]. Moreover, senescent cells interact with platelets through SASP, increasing platelet aggregation and promoting platelet activation. In vivo, senescent cells recruit platelets to sites of senescence-induced inflammation, altering the TME and leading to endothelial dysfunction, proliferation of premalignant cells, and enhanced tumor invasion and metastasis [120].
Deng et al. discovered that SASP factor IL-6, secreted by senescent tumor cells, elevates adenosine levels in the TME, which, in turn, promotes CD73 expression in macrophages via the JAK/STAT3 signaling pathway, leading to further adenosine accumulation. Their research also showed that even after the clearance of senescent cells, the infiltration of T cells in the TME was impaired. Targeted inhibition of CD73 not only suppresses tumors but also enhances the efficacy of anti-PD-1 monoclonal antibody immunotherapy. They proposed that early remodeling of the TME by senescent macrophages may play a crucial anti-tumor role, which is significant for cancer patient prognosis [121]. Additionally, adenosine has been shown to upregulate PD-L1 expression in human macrophages, further influencing the immune landscape of the TME [122].
Double roles of SASP in tumors
The SASP plays a dual role in tumor biology due to its heterogeneity. SASP can act as both a tumor suppressor and a promoter of tumor initiation and progression. On one hand, SASP is known to enforce cell cycle arrest and recruit immune cells to eliminate damaged or oncogene-expressing cells, acting as a protective mechanism against tumorigenesis. On the other hand, SASP can create an immunosuppressive environment that supports tumor progression and recurrence.
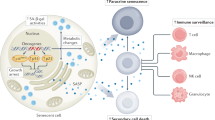
In this review, we summarize the key biological functions of SASP, focusing on the cytokines and intercellular interactions that shape the TME and influence the response to immunotherapy in geriatric oncology. We also discuss current clinical strategies targeting TME components and explore potential therapeutic targets within the senescent TME. Here, we highlight recent advances that underscore the conflicting roles for SASP in tumor development and treatment response (Fig. 3).

The double-edged sword effect of SASP in tumors. The SASP exhibits dual roles in tumor dynamics. On one side, it enforces cell cycle arrest, thereby inhibiting tumor cell proliferation. Additionally, SASP recruits immune cells to clear senescent cells, bolstering immunosurveillance and enhancing antigen presentation. On the opposite side, SASP can promote tumor cell proliferation, facilitate the emergence of cancer stem cells and EMT, and suppress immune responses, contributing to tumor progression and immune evasion
Antitumor effects of SASP
Preserving cell cycle arrest
One of the critical antitumor mechanisms of the SASP is its role in maintaining cell cycle arrest. For example, RNAi inhibition of PAI-1, a p53 target gene, can induce fibroblasts from primary embryonic mice, as well as primary human BJ fibroblasts, to exit from replicative senescence. Conversely, p53 knockout and PAI-1 overexpression induce CS [123]. SASP factors such as IL-6 and CXCR2 maintain cell cycle arrest via p53, thereby inhibiting tumor cell growth and proliferation [124,125,126]. Although the precise mechanism by which SASP maintains cell cycle arrest is not fully understood, it is generally agreed that SASP does not directly cause cell cycle arrest but makes the arrest irreversible once the cell enters this state. Interestingly, rapamycin has been found to inhibit SASP factor expression and reduce SASP-induced inflammation in human fibroblasts without reversing CS, suggesting that targeting SASP does not disrupt age-related cell cycle arrest [127]. This finding implies that cell cycle re-entry might be possible by inhibiting specific SASP factors or targeting downstream molecules.
Immunosurveillance
Although the full mechanisms by which SASP affects immune cells are not completely understood, SASP is theoretically capable of significantly impacting immune responses. In fact, senescent cells can generate a SASP that regulates the microenvironment of surrounding tissue, particularly affecting nearby normal endothelial cells, which in turn mediate NF-κB factors in the SASP. These factors activate CD4 + T cells via STAT1 and inducible costimulator/its ligand signaling, recruiting neighboring T cells and driving immune-mediated senescence surveillance and clearance of senescent cells [128]. For instance, in a mouse model of pancreatic ductal adenocarcinoma, tumor cell senescence following MEK and CDK4/6 inhibition induces SASP, leading to T-cell infiltration [129]. Similarly, in a mouse model of hepatic fibrosis induced by CCl4, activated hepatic astrocytes induce hepatic CS, which is inhibited by the knockout of p53 and p16INK4a [130]. Moreove, senescent hematopoietic stem cells (HSCs) attract NK cells and other immune cells to fibrotic lesions, thereby limiting immune-mediated fibrosis [130]. In mouse liver cancer models, p53-induced CS suppresses macrophages, neutrophils, and NK cells [16], leading to the clearance of tumor cells through phagocytosis and NK cell activation [16]. Interestingly, tumor regression is not observed when p53 is activated in mice lacking both B and T lymphocytes, suggesting that adaptive immunity may not needed for the clearance of senescent tumor cells within the SASP-recruited immune microenvironment [131]. Recent studies have also shown that tumor cell senescence can shift from immune evasion to immune surveillance, with IFN-γ synergizing with SASP to enhance antigen presentation and immune surveillance, ultimately leading to tumor cell rejection [132]. Thus, SASP effects can be leveraged for therapeutic purposes, such as forcing tumor cells into CS or exploiting SASP- mediated immune surveillance in cancer therapy. However, the dual nature of SASP in the TME— possessing both antitumor and tumor-promoting properties— complicates its use as an antitumor strategy.
Pro-tumor effects of SASP
Promotion of tumor cell proliferation
The initial protumor mechanism of SASP is its role in promoting tumor cell proliferation. For example, senescent human fibroblasts can stimulate hyperproliferation and progression of preneoplastic epithelial cells, accelerating tumorigenesis in neoplastic epithelial cells [133]. Compared to ‘young’ mesenchymal stem cells (MSCs), senescent human umbilical cord MSCs (s-UCMSCs) significantly accelerate the proliferation and migration of breast cancer cells via IL-6/STAT-3-dependent signaling [134]. Additionally, senescent breast luminal cells secrete SASP factors, including IL-6 and IL-8, which activate stromal fibroblasts through the STAT3 pathway, leading to tumor development in a paracrine manner [135]. Similarly, human senescent fibroblasts induced by bleomycin foster cancer cell proliferation and promote preneoplastic cells through the secretion of matrix metalloproteinases (MMPs) [136]. Senescent macrophages also contribute to tumor transformation by reshaping the TME [137], and eliminating these senescent macrophages has been shown to ameliorate KRAS-driven lung tumors [138]. Recently, small extracellular vesicles (sEVs) from senescent cells were identified as crucial mediators of protumor functions, with senescent cells increasing EphA2 in sEVs, thereby promoting cancer cell proliferation through activation of the MAPK pathway [139]. Furthermore, the extracellular matrix (ECM) secreted by SASP enhances tumor cells proliferation and provides a conducive environment for tumor progression [140]. For example, ECM production of senescent fibroblasts induced by irradiation boosts the proliferation of premalignant epidermal cells via PI3K and MAPK signaling [141]. Collectively, SASP not only induces CS but also directly promotes tumorigenesis and the proliferation of precancerous cells.
Induce epithelial mesenchymal transition (EMT)
Increasing evidence indicates that SASP can induce EMT, thereby favoring tumor progression. In breast cancer, for example, senescent fibroblasts secrete IL-6 and IL-8, key components of SASP. These cytokines increase vimentin expression, decrease E-cadherin levels, and reduce cytokeratin in noninvasive breast cancer cells [5]. Senescent fibroblasts can also inhibit the differentiation of epithelial cells and reduce the expression of differentiation markers by secreting MMP-3 [93]. Additionally, spindle cells have been observed in pancreatic epithelial cells exposed to senescence-conditioned media, which are poorly tumorigenic [142]. SASP can also induce EMT in nonsenescent cells; for instance, conditioned media from senescent malignant pleural mesothelioma cells trigger the emergence of EMT-like, clonogenic, and chemoresistant cell subpopulations [143]. In addition, senescent fibroblasts induce the morphological and functional differentiation of nonmalignant epithelial cells [93]. Through coculture systems and xenograft models, SASP, primarily from senescent fibroblasts, has been shown to induce EMT, promote angiogenesis, and enhance the progression of premalignant epithelial tumors [144]. Currently, it is well-established that SASP can induce EMT, influencing the differentiation status of tumor cells.
Induction of stemness in tumor cells
Initially, genotoxic-induced SASP was found to endow a subset of irradiated or doxorubicin-treated multiple myeloma cells with a stemness-like, highly tumorigenic state. CS induced by genotoxic stress produces SASP, leading to the release of IP-10 and RANTES in the TME, driving the formation of cancer stem cells (CSCs) [145]. In addition to promoting further differentiation of cells with unstable genomes, the senescent microenvironment grants premalignant cells a stem phenotype [146]. For instance, MCF-7 breast cancer cells in senescent conditioned medium were induced to express EMT programs, while treatment with IL-6 and IL-8 was also effective in inducing breast cancer cells to form mammosphere and exhibit stem-like properties, underscoring the crucial role of SASP in the induction of tumor cell stemness [77]. Currently, SASP is known to promote tumor cell stemness in both tumor and nontumor environment. For example, keratinocytes exposed to SASP upregulate stem cell markers [147], and injury-induced senescence enables in vivo reprogramming in skeletal muscle, suggesting a paracrine effect of senescence on cellular plasticity [148]. Overall, SASP plays a significant role in conferring a stemness-like, highly tumorigenic state to tumors, highlighting the potential of targeting senescent cells to combat tumors.
Immunosuppression and anti-inflammatory effects
Immune cells are a critical component of the TME, and SASP can recruit suppressor immune cells to prevent immune clearance of tumor cells while also inducing immune cell-mediated inflammatory responses that further promote tumor progression. For example, genetic knockout of Sin3B in the mouse genome inhibits pancreatic cancer progression by preventing CS induced by KRAS and reducing IL-1α in the SASP [149]. Notably, some SASP factors have been shown to both inhibit tumor initiation and promote tumor growth, depending on the tumor cell status. For example, chemokines secreted by senescent hepatic cells can inhibit HCC at an early stage but accelerate the growth of fully developed HCC cells, likely due to plasticity in myeloid CCR2 cells [150]. Elevated levels of MMPs in senescent fibroblast SASP from nonmelanoma skin cancer activate membrane PAR-1/thrombin receptors, leading to tumor escape following the senescence of tumor cells [151]. Additionally, Lau et al. found that IL-1 in SASP recruited macrophages in the mouse pancreas, with tissue-resident macrophages promoting pancreatic cancer progression, potentially explaining the role of SASP in promoting pancreatic cancer [122].
Beyond these protumor mechanisms, SASP also protects tumors in other ways. For example, in squamous cell carcinoma of the head and neck, early CS and SASP generation—where chemokine receptor ligands for CXCR2 play an important role— may lead to radioresistance [152]. Through these mechanisms, SASP creates a TME that not only fosters tumor progression but also shields tumors from immune system attacks and external drug elimination once fully developed, posing challenges for clinical anticancer strategies.
In summary, the impact of SASP on tumorigenesis and tumor development is a double-edged sword, with its effects depending largely on the composition of SASP. This duality presents opportunities for therapeutic interventions, such as enhancing immune system-mediated tumor cell killing, inducing tumor CS, and inhibiting tumor immune evasion. Therefore, it is crucial to identify which SASP factors have antitumor or pro-tumor properties to develop strategies that specifically inhibit tumors.
Therapy targeting CS and SASP in tumors
Although CS was once considered an irreversible state of cell cycle arrest, recent evidence suggests that senescent cells can occasionally escape and re-enter the cell cycle under certain conditions, potentially contributing to tumor recurrence [153]. For instance, Zampetides et al. discovered that oncogene-induced senescent cells can occasionally bypass the barrier of irreversible cell cycle arrest and resume proliferation [154]. Similarly, Yu et al. found that the loss of the H3K9me3 marker can enable cells to exit the senescent state and re-enter the cell cycle [155, 156]. Additionally, tumor cells may escape from senescence following anti-tumor treatments, potentially due to accumulated genomic instability [157, 158]. These findings highlight the potential of targeting senescent cells as a promising strategy for treating cancer recurrence.
On the other hand, CS may contribute to the progression of senescence-related diseases, particularly cancer, through the SASP. Indeed, the removal of senescent cells has been shown to slow the progression of senescence-associated diseases [159]. Importantly, senescent cells possess unique defenses against apoptotic stimuli, known as senescent cell antiapoptotic pathways (SCAP), which include the BCL-2/BCL-XL, PI3K/AKT, p53/p21/PAI-1&2, HIF-1α, and tyrosine kinase signaling pathways [160]. Therefore, the development of SCAP-targeting drugs may effectively eliminate senescent cells without harming normal cells. Currently, drugs that specifically target and kill senescent cells are collectively referred to as senolytics [160], and multiple reviews have already detailed their mechanisms and potential [161, 162]. Here, we summarize the current antitumor studies of senolytics, categorizing them according to their therapeutic mechanisms (Table 3).
Senolytics
To date, various senolytic drugs have been developed, including first-generation senolytics like dasatinib and quercetin (D + Q) [160], BCL-2 family inhibitors (such as ABT-263 and ABT-737) [163], and cardiac glycosides like ouabain and digitoxin [170]. Several of these senolytic agents have demonstrated efficacy in 'one-two punch' combination therapies against cancer. For instance, lung and breast cancer cells exposed to chemotherapy agents such as etoposide and doxorubicin typically enter a senescent state. Sequential treatment with ABT-263 can further suppress tumor progression by eliminating these senescent cells [180]. Beyond these drugs, other types of senolytics have emerged. For instance, the plant-derived compound piperlongumine (PL) has been found to selectively kill senescent cells by increasing reactive oxygen species (ROS) levels and inhibiting the PI3K/AKT/mTOR pathway. PL has also been shown to synergize with ABT-263 to enhance its senolytic effects [172, 181]. Additionally, a chimeric antigen receptor (CRT) -T cell targeting urokinase-type plasminogen activator receptor (uPAR) has been developed by Amor et al. for the treatment of liver cancer and hepatic fibrosis. Given that uPAR is less expressed in vital tissues, these CAR-T cells are more disease-specific and safer [182]. Another promising senolytic is the bromodomain and extraterminal domain (BET) family protein degrader (BETd). BETd has been shown to be effective in inhibiting HCC in mice [178]. The recently developed BETd ARV825 exhibits strong senolytic activity in vivo by reducing BRD4 levels, negatively regulating XRCC4 expression in senescent cells, exacerbating DSBs, and positively regulating autophagy gene expression. This results in the apoptosis of senescent cells by blocking non-homologous end joining repair [178].
Given the lack of specificity of general senolytics, more targeted drugs have been developed in recent years. For example, Poblocka et al. are investigating the use of beta-2-microglobulin (B2M), a membrane protein marker on senescent cells, to deliver toxic drugs to these cells via antibody–drug coupling (ADC) [128]. Kento et al. developed an ADC drug that targets senescent dermal fibroblasts using apolipoprotein D (Apo D), which specifically kills senescent human dermal fibroblasts when combination with pyrrolobenzodiazepine, without significant side effects [183].
Additionally, galacto-oligosaccharide nanoparticle delivery systems have been explored for targeting senescent cells [184]. These nanoparticles release their encapsulated senolytics upon digestion by elevated SA-β-gal activity in senescent cells, effectively killing them [184]. Ana et al. developed a galactose-modified doxorubicin prodrug designed to target senescent cells with high SA-β-gal activity, providing a single-molecule approach for senescence-targeted antitumor therapies [185].
While senolytics hold great promise, they are not yet fully developed. The heterogeneity of senescent cells means that different senolytics have their own limitations in clinical practice. Therefore, it is crucial to optimize the clinical use of senolytics to maximize the clearance of senescent cells. Current thinking suggests that senolytics are best administered intermittently rather than continuously, as it takes time for senescent cells to form and generate SASPs. However, whether this approach might have detrimental effects on the organism remains to be explored. Further preclinical animal studies are needed to evaluate the potential adverse effects of intermittent drug administration. Additionally, experimental studies are required to determine the optimal timing, dosage, and other parameters for senolytic therapies. Moreover, senolytics may have side effects; for example, ABT-263 can lead to thrombocytopenia. Therefore, there is a need for more effective and safer senolytics that minimize damage to normal tissues. Alternatively, combining multiple senolytics, such as dasatinib with quercetin or PL with ABT-263, may improve efficacy while reducing adverse effects like hemotoxicity through combination therapy.
In conclusion, while senescent cell clearance is a promising therapeutic approach, it may also negatively impact normal tissue function. For instance, wounds healing may be impaired [41], and the blood-tissue barrier may be damaged, promoting fibrosis [186]. These findings suggest that targeting the detrimental aspects of senescence in the TME without killing senescent cells might be a more refined approach. This has led to the proposal of a novel therapy: senomorphics (Fig. 4).

Mechanism of senolytics and senomorphics. Senolytics induce apoptosis in cells undergoing CS by targeting various SCAPs. These pathways include MDM2, Bcl-2 family members, HSP90, SA-β-gal, PI3K/AKT, Na+-K+ pumps, FOXO4, and ADC. On the other hand, senomorphics inhibit pathways like NF-κB, MAPK, mTOR, and Bcl-2, thereby preventing senescent cells from releasing SASP factors such as IL-6, IL-8, MMP, and others
Senomorphics
Senomorphics are drugs that modify the phenotype of senescent cells, restoring them to a more youthful state without inducing apoptosis. They achieve this by interfering with the inflammatory response of senescent cells and disrupting signaling pathways related to senescence and SASP expression [187]. Compared to senolytics, senomorphics may present fewer side effects since they do not directly kill senescent cells but instead inhibit the development of senescence. Senomorphics primarily target signaling pathways associated with SASP expression, such as NF-κB, JAK/STAT, C/EBPβ, and GATA4, offering promising targets for drug development. Another strategy involves targeting specific components of the SASP using antibodies, such as those against IL-6/8. However, this approach is limited by the heterogeneity of SASP expression across different stages and types of senescent cells, making precise targeting challenging. We have summarized the current research on senomorphics in cancer in the table below (Table 4):
Despite the development of numerous senomorphics, most studies have concentrated on a limited range of common SASP factors. As illustrated in the table above, many senomorphics primarily target IL-6, IL-8, and similar factors, without considering the comprehensive changes within the SASP profile. Consequently, we cannot dismiss the possibility that senomorphics may inadvertently increase the secretion of harmful substances. Thus, while senomorphics represent a promising cancer therapy, they may not offer a complete solution but rather serve as a more beneficial approach within the complex, double-edged nature of SASP. For this reason, future studies must explore the effects of senomorphics on a broader range of SASP components, beyond the commonly studied factors. This includes investigating their impact on ECM composition, microvesicles, and other elements of the TME. In other words, more extensive and detailed research is required to fully understand the role of senomorphics in modulating senescence and SASP in cancer.
Future perspectives and conclusion
CS is a crucial process in growth, development, and cancer. Although significant progress has been made in understanding the mechanisms of CS and SASP, further studies are essential due to the heterogeneity of CS and SASP, which vary by cell type and stage. Additionally, the role of CS and SASP in vivo, particularly within the TME, requires more evidence. Such research could lead to the identification of simpler, more accurate, and more specific biomarkers. Furthermore, an in-depth exploration of the dual effects of SASP might provide new avenues for developing antitumor therapies. However, more specific drugs targeting CS are necessary to ensure their clinical efficacy. As our understanding of the roles and mechanisms of CS and SASP deepens, new tumor-targeting strategies are likely to emerge, ultimately improving clinical outcomes for cancer patients and advancing the field of precision medicine.
Despite the increasing number of studies on senescence characteristics and SASP, numerous challenges remain. Key issues include how to identify CS more simply and specifically, and how to determine the status of different senescent cells at various time points. Given that CS is dynamic in both space and time, early and late factors that activate and sustain CS may differ. While 'one-two punch' strategies hold promise for enhancing cancer therapies, several critical questions persist. For instance, determining the optimal timing for administering senolytics during anticancer regimens, identifying the best sequence of administration, and evaluating whether sequential treatment (initiating senescence followed by senolytics) offers the greatest benefits are crucial considerations. It is also important to recognize that when senescence is activated, it affects the surrounding microenvironment through SASP, creating a bidirectional communication between CS and the microenvironment. The heterogeneity of SASP means that different SASP profiles may contribute to either a favorable or detrimental microenvironment. This review aimed to highlight the dual nature of SASP in the microenvironment and cancer, as SASP has been shown to have both positive and negative effects in cancer, influencing the induction and elimination of CS. A better understanding of the mechanisms underlying CS and SASP could lead to more effective cancer prevention and treatment strategies. In the case of senolytics, attention must be given to their long-term adverse effects, as the elimination of senescent cells could increase the body's burden and trigger dysfunction. For senomorphics, it is important to consider their broader therapeutic impacts. Identifying specific methods for CS detection, elucidating the mechanisms of CS and SASP, and developing more effective anti-senescence drugs will be critical steps forward in cancer therapy.
Availability of data and materials
Not applicable.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- CS:
-
Cellular senescence
- SASP:
-
Senescence-associated secretory phenotype
- TME:
-
Tumor microenvironment
- RS:
-
Replicative senescence
- DSB:
-
DNA double-strand breaks
- DDR:
-
DNA damage response
- RS:
-
Replicative senescence
- ATR:
-
Ataxia telangiectasia and Rad3-related
- ATM:
-
Ataxia telangiectasia mutated
- Rb:
-
Retinoblastoma protein
- CDK2:
-
Cyclin dependent kinase 2
- CDK2A:
-
Cyclin dependent kinase inhibitor 2A
- OIS:
-
Oncogene-induced senescence
References
Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621.
Hayflick L. THE LIMITED IN VITRO LIFETIME OF HUMAN DIPLOID CELL STRAINS. Exp Cell Res. 1965;37:614–36.
Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705.
Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8(9):729–40.
Coppé JP, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6(12):2853–68.
Nagao H, et al. Unique ligand and kinase-independent roles of the insulin receptor in regulation of cell cycle, senescence and apoptosis. Nat Commun. 2023;14(1):57.
De Cecco M, et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature. 2019;566(7742):73–8.
Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9(6):400–14.
Zhu W, Abbas T, Dutta A. DNA replication and genomic instability. Adv Exp Med Biol. 2005;570:249–79.
Aliouat-Denis CM, et al. p53-independent regulation of p21Waf1/Cip1 expression and senescence by Chk2. Mol Cancer Res. 2005;3(11):627–34.
Alcorta DA, et al. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci U S A. 1996;93(24):13742–7.
Kandhaya-Pillai R, et al. Key elements of cellular senescence involve transcriptional repression of mitotic and DNA repair genes through the p53–p16/RB-E2F-DREAM complex. Aging (Albany NY). 2023;15(10):4012–34.
Xu JX, et al. A novel role for YPEL2 in mediating endothelial cellular senescence via the p53/p21 pathway. Mech Ageing Dev. 2023;211: 111803.
Rattanavirotkul N, Kirschner K, Chandra T. Induction and transmission of oncogene-induced senescence. Cell Mol Life Sci. 2021;78(3):843–52.
Vickridge E, et al. The DNA repair function of BCL11A suppresses senescence and promotes continued proliferation of triple-negative breast cancer cells. NAR Cancer. 2022;4(4):zcac028.
Xue W, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445(7128):656–60.
Khalil RM, Diab-Assaf JM. Lemaitre, Emerging Therapeutic Approaches to Target the Dark Side of Senescent Cells: New Hopes to Treat Aging as a Disease and to Delay Age-Related Pathologies. Cells. 2023;12(6):915.
Bartkova J, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444(7119):633–7.
Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319(5868):1352–5.
Di Micco R, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444(7119):638–42.
Prasanna PG, et al. Therapy-Induced Senescence: Opportunities to Improve Anticancer Therapy. J Natl Cancer Inst. 2021;113(10):1285–98.
Demaria M, et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017;7(2):165–76.
Murray, D. and R. Mirzayans, Cellular Responses to Platinum-Based Anticancer Drugs and UVC: Role of p53 and Implications for Cancer Therapy. Int J Mol Sci. 2020;21(16):5766.
Schmitt CA, et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109(3):335–46.
V’Kovski P, et al. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol. 2021;19(3):155–70.
Yang L, et al. SARS-CoV-2 infection causes dopaminergic neuron senescence. Cell Stem Cell. 2024;31(2):196–211.e6.
Meyer K, et al. SARS-CoV-2 Spike Protein Induces Paracrine Senescence and Leukocyte Adhesion in Endothelial Cells. J Virol. 2021;95(17): e0079421.
Gioia U, et al. SARS-CoV-2 infection induces DNA damage, through CHK1 degradation and impaired 53BP1 recruitment, and cellular senescence. Nat Cell Biol. 2023;25(4):550–64.
Park JH, et al. Disruption of nucleocytoplasmic trafficking as a cellular senescence driver. Exp Mol Med. 2021;53(6):1092–108.
McDermott MSJ, et al. HER2-Targeted Tyrosine Kinase Inhibitors Cause Therapy-Induced-Senescence in Breast Cancer Cells. Cancers (Basel). 2019;11(2):197.
Mirzakhani K, et al. The androgen receptor-lncRNASAT1-AKT-p15 axis mediates androgen-induced cellular senescence in prostate cancer cells. Oncogene. 2022;41(7):943–59.
Halkoum R, et al. Glyoxal Induces Senescence in Human Keratinocytes through Oxidative Stress and Activation of the Protein Kinase B/FOXO3a/p27(KIP1) Pathway. J Invest Dermatol. 2022;142(8):2068–2078.e7.
Ogrodnik M. Cellular aging beyond cellular senescence: Markers of senescence prior to cell cycle arrest in vitro and in vivo. Aging Cell. 2021;20(4): e13338.
Wallis R, et al. Senescence-associated morphological profiles (SAMPs): an image-based phenotypic profiling method for evaluating the inter and intra model heterogeneity of senescence. Aging (Albany NY). 2022;14(10):4220–46.
González-Gualda E, et al. A guide to assessing cellular senescence in vitro and in vivo. Febs j. 2021;288(1):56–80.
Yang YK, et al. Changes in phenotype and differentiation potential of human mesenchymal stem cells aging in vitro. Stem Cell Res Ther. 2018;9(1):131.
Oja S, et al. Automated image analysis detects aging in clinical-grade mesenchymal stromal cell cultures. Stem Cell Res Ther. 2018;9(1):6.
Wu F, et al. β-Galactosidase-Activatable Fluorescent and Photoacoustic Imaging of Tumor Senescence. Anal Chem. 2023;95(28):10481–5.
Hu, L., et al., A rationally designed fluorescence probe achieves highly specific and long-term detection of senescence in vitro and in vivo. Aging Cell, 2023: p. e13896.
Hildebrand DG, et al. α-Fucosidase as a novel convenient biomarker for cellular senescence. Cell Cycle. 2013;12(12):1922–7.
Baker DJ, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479(7372):232–6.
Burd CE, et al. Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell. 2013;152(1–2):340–51.
Demaria M, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. 2014;31(6):722–33.
Calcinotto A, Alimonti A. Aging tumour cells to cure cancer: “pro-senescence” therapy for cancer. Swiss Med Wkly. 2017;147: w14367.
López-Domínguez JA, et al. Cdkn1a transcript variant 2 is a marker of aging and cellular senescence. Aging (Albany NY). 2021;13(10):13380–92.
Tyler EJ, et al. Early growth response 2 (EGR2) is a novel regulator of the senescence programme. Aging Cell. 2021;20(3): e13318.
Kim KM, et al. SCAMP4 enhances the senescent cell secretome. Genes Dev. 2018;32(13–14):909–14.
Amor C, et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature. 2020;583(7814):127–32.
Takaya K, Asou T, Kishi K. Selective elimination of senescent fibroblasts by targeting the cell surface protein ACKR3. Int J Mol Sci. 2022;23(12):6531.
Abbadie C, Pluquet O. Unfolded Protein Response (UPR) Controls Major Senescence Hallmarks. Trends Biochem Sci. 2020;45(5):371–4.
Misawa T, et al. Identification of Novel Senescent Markers in Small Extracellular Vesicles. Int J Mol Sci. 2023;24(3):2421.
Boichuck M, et al. c-Met as a new marker of cellular senescence. Aging (Albany NY). 2019;11(9):2889–97.
Thorin-Trescases N, et al. Angptl2 is a Marker of Cellular Senescence: The Physiological and Pathophysiological Impact of Angptl2-Related Senescence. Int J Mol Sci. 2021;22(22):12232.
Wei Z, et al. Pan-senescence transcriptome analysis identified RRAD as a marker and negative regulator of cellular senescence. Free Radic Biol Med. 2019;130:267–77.
Georgakopoulou EA, et al. Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging (Albany NY). 2013;5(1):37–50.
Davan-Wetton CSA, Montero-Melendez T. An optimised protocol for the detection of lipofuscin, a versatile and quantifiable marker of cellular senescence. PLoS ONE. 2024;19(7): e0306275.
Kim SY, et al. Global transcriptional downregulation of TREX and nuclear trafficking machinery as pan-senescence phenomena: evidence from human cells and tissues. Exp Mol Med. 2020;52(8):1351–9.
Pathak RU, Soujanya M, Mishra RK. Deterioration of nuclear morphology and architecture: A hallmark of senescence and aging. Ageing Res Rev. 2021;67: 101264.
Heckenbach I, et al. Nuclear morphology is a deep learning biomarker of cellular senescence. Nat Aging. 2022;2(8):742–55.
Kim Y, et al. nc886, a Non-Coding RNA, Is a New Biomarker and Epigenetic Mediator of Cellular Senescence in Fibroblasts. Int J Mol Sci. 2021;22(24):13673.
Matias I, et al. Loss of lamin-B1 and defective nuclear morphology are hallmarks of astrocyte senescence in vitro and in the aging human hippocampus. Aging Cell. 2022;21(1): e13521.
Curnock R, et al. TFEB-dependent lysosome biogenesis is required for senescence. Embo j. 2023;42(9): e111241.
Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015;15(7):397–408.
Bertolo A, et al. Autofluorescence is a Reliable in vitro Marker of Cellular Senescence in Human Mesenchymal Stromal Cells. Sci Rep. 2019;9(1):2074.
Chen X, et al. Dynamic ultrasound molecular-targeted imaging of senescence in evaluation of lapatinib resistance in HER2-positive breast cancer. Cancer Med. 2023;12(19):19904–20.
He L, et al. Morphology-based deep learning enables accurate detection of senescence in mesenchymal stem cell cultures. BMC Biol. 2024;22(1):1.
Thamarath SS, et al. Rapid and Live-Cell Detection of Senescence in Mesenchymal Stem Cells by Micro Magnetic Resonance Relaxometry. Stem Cells Transl Med. 2023;12(5):266–80.
Psaroudis RT, et al. CD26 is a senescence marker associated with reduced immunopotency of human adipose tissue-derived multipotent mesenchymal stromal cells. Stem Cell Res Ther. 2022;13(1):358.
Adewoye AB, et al. Multiparameter flow cytometric detection and quantification of senescent cells in vitro. Biogerontology. 2020;21(6):773–86.
Galvis D, et al. A dynamical systems model for the measurement of cellular senescence. J R Soc Interface. 2019;16(159):20190311.
Herbstein F, et al. The SASP factor IL-6 sustains cell-autonomous senescent cells via a cGAS-STING-NFκB intracrine senescent noncanonical pathway. Aging Cell. 2024:e14258.
Özcan S, et al. Unbiased analysis of senescence associated secretory phenotype (SASP) to identify common components following different genotoxic stresses. Aging (Albany NY). 2016;8(7):1316–29.
Lau L, et al. Uncoupling the Senescence-Associated Secretory Phenotype from Cell Cycle Exit via Interleukin-1 Inactivation Unveils Its Protumorigenic Role. Mol Cell Biol. 2019;39(12):e00586–18.
Bhaumik D, et al. MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging (Albany NY). 2009;1(4):402–11.
Su Y, et al. S100A13 promotes senescence-associated secretory phenotype and cellular senescence via modulation of non-classical secretion of IL-1α. Aging (Albany NY). 2019;11(2):549–72.
Dong M, et al. Autocrine IL-1β mediates the promotion of corneal neovascularization by senescent fibroblasts. Am J Physiol Cell Physiol. 2018;315(5):C734–c743.
Ortiz-Montero P, Londoño-Vallejo A, Vernot JP. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun Signal. 2017;15(1):17.
Yu YC, et al. Radiation-induced senescence in securin-deficient cancer cells promotes cell invasion involving the IL-6/STAT3 and PDGF-BB/PDGFR pathways. Sci Rep. 2013;3:1675.
Acosta JC, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133(6):1006–18.
Di Mitri D, et al. Re-education of Tumor-Associated Macrophages by CXCR2 Blockade Drives Senescence and Tumor Inhibition in Advanced Prostate Cancer. Cell Rep. 2019;28(8):2156–2168.e5.
Liu Y, et al. Klotho-mediated targeting of CCL2 suppresses the induction of colorectal cancer progression by stromal cell senescent microenvironments. Mol Oncol. 2019;13(11):2460–75.
Coppé JP, et al. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS ONE. 2010;5(2): e9188.
Hwang HJ, et al. Endothelial cells under therapy-induced senescence secrete CXCL11, which increases aggressiveness of breast cancer cells. Cancer Lett. 2020;490:100–10.
Severino V, et al. Insulin-like growth factor binding proteins 4 and 7 released by senescent cells promote premature senescence in mesenchymal stem cells. Cell Death Dis. 2013;4(11): e911.
Xu Q, et al. Targeting amphiregulin (AREG) derived from senescent stromal cells diminishes cancer resistance and averts programmed cell death 1 ligand (PD-L1)-mediated immunosuppression. Aging Cell. 2019;18(6): e13027.
Alessio N, et al. Increase of circulating IGFBP-4 following genotoxic stress and its implication for senescence. Elife. 2020;9:e54523.
Vassilieva I, et al. Paracrine senescence of human endometrial mesenchymal stem cells: a role for the insulin-like growth factor binding protein 3. Aging (Albany NY). 2020;12(2):1987–2004.
Guo Y, et al. Senescence-associated tissue microenvironment promotes colon cancer formation through the secretory factor GDF15. Aging Cell. 2019;18(6): e13013.
Gungor MZ, Uysal M, Senturk S. The Bright and the Dark Side of TGF-β Signaling in Hepatocellular Carcinoma: Mechanisms, Dysregulation, and Therapeutic Implications. Cancers (Basel). 2022;14(4):940.
Rana T, et al. PAI-1 Regulation of TGF-β1-induced Alveolar Type II Cell Senescence, SASP Secretion, and SASP-mediated Activation of Alveolar Macrophages. Am J Respir Cell Mol Biol. 2020;62(3):319–30.
Davalos AR, et al. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J Cell Biol. 2013;201(4):613–29.
Ohkouchi S, et al. Myriad Functions of Stanniocalcin-1 (STC1) Cover Multiple Therapeutic Targets in the Complicated Pathogenesis of Idiopathic Pulmonary Fibrosis (IPF). Clin Med Insights Circ Respir Pulm Med. 2015;9(Suppl 1):91–6.
Parrinello S, et al. Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J Cell Sci. 2005;118(Pt 3):485–96.
Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141(1):52–67.
Zheng X, et al. Downregulation of HINFP induces senescence-associated secretory phenotype to promote metastasis in a non-cell-autonomous manner in bladder cancer. Oncogene. 2022;41(28):3587–98.
Gabasa M, et al. MMP1 drives tumor progression in large cell carcinoma of the lung through fibroblast senescence. Cancer Lett. 2021;507:1–12.
Littlepage LE, et al. Matrix metalloproteinases contribute distinct roles in neuroendocrine prostate carcinogenesis, metastasis, and angiogenesis progression. Cancer Res. 2010;70(6):2224–34.
Rossi M, et al. Pleiotropic effects of BAFF on the senescence-associated secretome and growth arrest. Elife. 2023;12:e84238.
Kuehnemann C, et al. Extracellular Nicotinamide Phosphoribosyltransferase Is a Component of the Senescence-Associated Secretory Phenotype. Front Endocrinol (Lausanne). 2022;13: 935106.
Ezure T, Sugahara M, Amano S. Senescent dermal fibroblasts negatively influence fibroblast extracellular matrix-related gene expression partly via secretion of complement factor D. BioFactors. 2019;45(4):556–62.
Buhl JL, et al. The Senescence-associated Secretory Phenotype Mediates Oncogene-induced Senescence in Pediatric Pilocytic Astrocytoma. Clin Cancer Res. 2019;25(6):1851–66.
Wiggins KA, et al. IL-1α cleavage by inflammatory caspases of the noncanonical inflammasome controls the senescence-associated secretory phenotype. Aging Cell. 2019;18(3): e12946.
Leon KE et al., DOT1L modulates the senescence-associated secretory phenotype through epigenetic regulation of IL1A. J Cell Biol, 2021;20(8).
Wang ST, et al. Atorvastatin-induced senescence of hepatocellular carcinoma is mediated by downregulation of hTERT through the suppression of the IL-6/STAT3 pathway. Cell Death Discov. 2020;6:17.
Shriki A, et al. Multiple Roles of IL6 in Hepatic Injury, Steatosis, and Senescence Aggregate to Suppress Tumorigenesis. Cancer Res. 2021;81(18):4766–77.
Naugler WE, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317(5834):121–4.
Tarallo D, et al. Mitofusin 1 silencing decreases the senescent associated secretory phenotype, promotes immune cell recruitment and delays melanoma tumor growth after chemotherapy. Sci Rep. 2024;14(1):909.
Daley D, et al. Dectin 1 activation on macrophages by galectin 9 promotes pancreatic carcinoma and peritumoral immune tolerance. Nat Med. 2017;23(5):556–67.
Nobumoto A, et al. Galectin-9 suppresses tumor metastasis by blocking adhesion to endothelium and extracellular matrices. Glycobiology. 2008;18(9):735–44.
Lehmann BD, et al. Senescence-associated exosome release from human prostate cancer cells. Cancer Res. 2008;68(19):7864–71.
Misawa T, et al. Biology of extracellular vesicles secreted from senescent cells as senescence-associated secretory phenotype factors. Geriatr Gerontol Int. 2020;20(6):539–46.
Wallis R, Mizen H, Bishop CL. The bright and dark side of extracellular vesicles in the senescence-associated secretory phenotype. Mech Ageing Dev. 2020;189: 111263.
Raynard C, et al. NF-κB-dependent secretome of senescent cells can trigger neuroendocrine transdifferentiation of breast cancer cells. Aging Cell. 2022;21(7): e13632.
Homann L, et al. IFN-γ and TNF Induce Senescence and a Distinct Senescence-Associated Secretory Phenotype in Melanoma. Cells. 2022;11(9):1514.
Kawaguchi K, et al. Cellular senescence promotes cancer metastasis by enhancing soluble E-cadherin production. iScience. 2021;24(9):103022.
Huang Y, et al. The hepatic senescence-associated secretory phenotype promotes hepatocarcinogenesis through Bcl3-dependent activation of macrophages. Cell Biosci. 2021;11(1):173.
Ma Y, et al. Identification and functional analysis of senescence-associated secretory phenotype of premature senescent hepatocytes induced by hexavalent chromium. Ecotoxicol Environ Saf. 2021;211: 111908.
Alessio N, et al. The senescence-associated secretory phenotype (SASP) from mesenchymal stromal cells impairs growth of immortalized prostate cells but has no effect on metastatic prostatic cancer cells. Aging (Albany NY). 2019;11(15):5817–28.
Cheng YC, et al. Loss of the tumor suppressor BTG3 drives a pro-angiogenic tumor microenvironment through HIF-1 activation. Cell Death Dis. 2020;11(12):1046.
Valenzuela CA, et al. SASP-Dependent Interactions between Senescent Cells and Platelets Modulate Migration and Invasion of Cancer Cells. Int J Mol Sci. 2019;20(21):5292.
Deng Y, et al. Tumor cell senescence-induced macrophage CD73 expression is a critical metabolic immune checkpoint in the aging tumor microenvironment. Theranostics. 2024;14(3):1224–40.
Noh JY, et al. Additive Effect of CD73 Inhibitor in Colorectal Cancer Treatment With CDK4/6 Inhibitor Through Regulation of PD-L1. Cell Mol Gastroenterol Hepatol. 2022;14(4):769–88.
Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006;8(8):877–84.
Orjalo AV, et al. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci U S A. 2009;106(40):17031–6.
Guo H, et al. Chemokine receptor CXCR2 is transactivated by p53 and induces p38-mediated cellular senescence in response to DNA damage. Aging Cell. 2013;12(6):1110–21.
Kuilman T, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133(6):1019–31.
Laberge RM, et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2015;17(8):1049–61.
Yin K, et al. Senescence-induced endothelial phenotypes underpin immune-mediated senescence surveillance. Genes Dev. 2022;36(9–10):533–49.
Ruscetti M, et al. Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell. 2020;181(2):424–441.e21.
Krizhanovsky V, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134(4):657–67.
Iannello A, et al. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J Exp Med. 2013;210(10):2057–69.
Chen HA, et al. Senescence Rewires Microenvironment Sensing to Facilitate Antitumor Immunity. Cancer Discov. 2023;13(2):432–53.
Krtolica A, et al. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A. 2001;98(21):12072–7.
Di GH, et al. IL-6 secreted from senescent mesenchymal stem cells promotes proliferation and migration of breast cancer cells. PLoS ONE. 2014;9(11): e113572.
Al-Khalaf HH, et al. Senescent Breast Luminal Cells Promote Carcinogenesis through Interleukin-8-Dependent Activation of Stromal Fibroblasts. Mol Cell Biol. 2019;39(2):e00359–18.
Liu D, Hornsby PJ. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 2007;67(7):3117–26.
Prieto LI, et al. Senescent alveolar macrophages promote early-stage lung tumorigenesis. Cancer Cell. 2023;41(7):1261–1275.e6.
Haston S, et al. Clearance of senescent macrophages ameliorates tumorigenesis in KRAS-driven lung cancer. Cancer Cell. 2023;41(7):1242–1260.e6.
Takasugi M, et al. Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat Commun. 2017;8:15729.
Sprenger CC, Plymate SR, Reed MJ. Aging-related alterations in the extracellular matrix modulate the microenvironment and influence tumor progression. Int J Cancer. 2010;127(12):2739–48.
Kang J, et al. Extracellular matrix secreted by senescent fibroblasts induced by UVB promotes cell proliferation in HaCaT cells through PI3K/AKT and ERK signaling pathways. Int J Mol Med. 2008;21(6):777–84.
Ohuchida K, et al. Radiation to stromal fibroblasts increases invasiveness of pancreatic cancer cells through tumor-stromal interactions. Cancer Res. 2004;64(9):3215–22.
Canino C, et al. SASP mediates chemoresistance and tumor-initiating-activity of mesothelioma cells. Oncogene. 2012;31(26):3148–63.
Krtolica A, et al. Quantification of epithelial cells in coculture with fibroblasts by fluorescence image analysis. Cytometry. 2002;49(2):73–82.
Cahu J, Bustany S, Sola B. Senescence-associated secretory phenotype favors the emergence of cancer stem-like cells. Cell Death Dis. 2012;3(12): e446.
Castro-Vega LJ, et al. The senescent microenvironment promotes the emergence of heterogeneous cancer stem-like cells. Carcinogenesis. 2015;36(10):1180–92.
Ritschka B, et al. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017;31(2):172–83.
Chiche A, et al. Injury-Induced Senescence Enables In Vivo Reprogramming in Skeletal Muscle. Cell Stem Cell. 2017;20(3):407–414.e4.
Rielland M, et al. Senescence-associated SIN3B promotes inflammation and pancreatic cancer progression. J Clin Invest. 2014;124(5):2125–35.
Eggert T, et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell. 2016;30(4):533–47.
Malaquin N, et al. Senescent fibroblasts enhance early skin carcinogenic events via a paracrine MMP-PAR-1 axis. PLoS ONE. 2013;8(5): e63607.
Schoetz U, et al. Early senescence and production of senescence-associated cytokines are major determinants of radioresistance in head-and-neck squamous cell carcinoma. Cell Death Dis. 2021;12(12):1162.
Galanos P, et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat Cell Biol. 2016;18(7):777–89.
Zampetidis CP, et al. A recurrent chromosomal inversion suffices for driving escape from oncogene-induced senescence via subTAD reorganization. Mol Cell. 2021;81(23):4907–4923.e8.
Yu Y, et al. Targeting the Senescence-Overriding Cooperative Activity of Structurally Unrelated H3K9 Demethylases in Melanoma. Cancer Cell. 2018;33(2):32–336.e8.
Milanovic M, et al. Senescence-associated reprogramming promotes cancer stemness. Nature. 2018;553(7686):96–100.
Saleh T, Tyutyunyk-Massey L, Gewirtz DA. Tumor Cell Escape from Therapy-Induced Senescence as a Model of Disease Recurrence after Dormancy. Cancer Res. 2019;79(6):1044–6.
Angelopoulou A, et al. Loss of the tumour suppressor LKB1/STK11 uncovers a leptin-mediated sensitivity mechanism to mitochondrial uncouplers for targeted cancer therapy. Mol Cancer. 2024;23(1):147.
Baker DJ, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530(7589):184–9.
Zhu Y, et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015;14(4):644–58.
D’Ambrosio M, Gil J. Reshaping of the tumor microenvironment by cellular senescence: An opportunity for senotherapies. Dev Cell. 2023;58(12):1007–21.
Schmitt CA, Wang B, Demaria M. Senescence and cancer - role and therapeutic opportunities. Nat Rev Clin Oncol. 2022;19(10):619–36.
van Deursen JM. Senolytic therapies for healthy longevity. Science. 2019;364(6441):636–7.
Jia Y et al. Co-targeting BCL-XL and BCL-2 by PROTAC 753B eliminates leukemia cells and enhances efficacy of chemotherapy by targeting senescent cells. Haematologica. 2023;108(10):2626–38.
Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11(7):515–28.
Saber S, et al. Innovative challenge for the inhibition of hepatocellular carcinoma progression by combined targeting of HSP90 and STAT3/HIF-1α signaling. Biomed Pharmacother. 2023;158: 114196.
Baar MP, et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell. 2017;169(1):132–147.e16.
He Y, et al. Inhibition of USP7 activity selectively eliminates senescent cells in part via restoration of p53 activity. Aging Cell. 2020;19(3): e13117.
Ray-Coquard I, et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol. 2012;13(11):1133–40.
Guerrero A, et al. Cardiac glycosides are broad-spectrum senolytics. Nat Metab. 2019;1(11):1074–88.
Triana-Martínez F, et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat Commun. 2019;10(1):4731.
Zhang X, et al. Oxidation resistance 1 is a novel senolytic target. Aging Cell. 2018;17(4): e12780.
Lin TH et al. Piperlongumine Induces Cellular Apoptosis and Autophagy via the ROS/Akt Signaling Pathway in Human Follicular Thyroid Cancer Cells. Int J Mol Sci. 2023;24(9):8048.
Xu Q, et al. The flavonoid procyanidin C1 has senotherapeutic activity and increases lifespan in mice. Nat Metab. 2021;3(12):1706–26.
Li W, et al. The curcumin analog EF24 is a novel senolytic agent. Aging (Albany NY). 2019;11(2):771–82.
Troiani M, et al. Single-cell transcriptomics identifies Mcl-1 as a target for senolytic therapy in cancer. Nat Commun. 2022;13(1):2177.
Cho HJ, et al. Identification of SYK inhibitor, R406 as a novel senolytic agent. Aging (Albany NY). 2020;12(9):8221–40.
Wakita M, et al. A BET family protein degrader provokes senolysis by targeting NHEJ and autophagy in senescent cells. Nat Commun. 2020;11(1):1935.
Cho HJ, et al. Nintedanib induces senolytic effect via STAT3 inhibition. Cell Death Dis. 2022;13(9):760.
Saleh T, et al. Clearance of therapy-induced senescent tumor cells by the senolytic ABT-263 via interference with BCL-X(L) -BAX interaction. Mol Oncol. 2020;14(10):2504–19.
Wang Y, et al. Discovery of piperlongumine as a potential novel lead for the development of senolytic agents. Aging (Albany NY). 2016;8(11):2915–26.
Li JH, Chen YY. A Fresh Approach to Targeting Aging Cells: CAR-T Cells Enhance Senolytic Specificity. Cell Stem Cell. 2020;27(2):192–4.
Takaya K, Asou T, Kishi K. New Senolysis Approach via Antibody-Drug Conjugate Targeting of the Senescent Cell Marker Apolipoprotein D for Skin Rejuvenation. Int J Mol Sci. 2023;24(6):5857.
Muñoz-Espín D et al. A versatile drug delivery system targeting senescent cells. EMBO Mol Med. 2018;10(9):e9355.
Guerrero A, et al. Galactose-modified duocarmycin prodrugs as senolytics. Aging Cell. 2020;19(4): e13133.
Grosse L, et al. Defined p16(High) Senescent Cell Types Are Indispensable for Mouse Healthspan. Cell Metab. 2020;32(1):87–99.e6.
Kim EC, Kim JR. Senotherapeutics: emerging strategy for healthy aging and age-related disease. BMB Rep. 2019;52(1):47–55.
Lim H, Park H, Kim HP. Effects of flavonoids on senescence-associated secretory phenotype formation from bleomycin-induced senescence in BJ fibroblasts. Biochem Pharmacol. 2015;96(4):337–48.
Menicacci B, et al. Modulation of the Senescence-Associated Inflammatory Phenotype in Human Fibroblasts by Olive Phenols. Int J Mol Sci. 2017;18(11):2275.
Matacchione G, et al. Anti-SASP and anti-inflammatory activity of resveratrol, curcumin and β-caryophyllene association on human endothelial and monocytic cells. Biogerontology. 2021;22(3):297–313.
Woo J, et al. Senotherapeutic-like effect of Silybum marianum flower extract revealed on human skin cells. PLoS ONE. 2021;16(12): e0260545.
Frediani E, et al. Olive phenols preserve lamin B1 expression reducing cGAS/STING/NFκB-mediated SASP in ionizing radiation-induced senescence. J Cell Mol Med. 2022;26(8):2337–50.
Lim JS, et al. Identification of a novel senomorphic agent, avenanthramide C, via the suppression of the senescence-associated secretory phenotype. Mech Ageing Dev. 2020;192: 111355.
Alimbetov D, et al. Suppression of the senescence-associated secretory phenotype (SASP) in human fibroblasts using small molecule inhibitors of p38 MAP kinase and MK2. Biogerontology. 2016;17(2):305–15.
Lee DY, et al. Dietary Curcumin Attenuates Hepatic Cellular Senescence by Suppressing the MAPK/NF-κB Signaling Pathway in Aged Mice. Antioxidants (Basel). 2023;12(6):1165.
Hu Q, et al. Metformin as a senostatic drug enhances the anticancer efficacy of CDK4/6 inhibitor in head and neck squamous cell carcinoma. Cell Death Dis. 2020;11(10):925.
Wang R, et al. Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging Cell. 2017;16(3):564–74.
Jia HJ, et al. Artesunate ameliorates irinotecan-induced intestinal injury by suppressing cellular senescence and significantly enhances anti-tumor activity. Int Immunopharmacol. 2023;119: 110205.
Yang KE, et al. Ginsenoside Rb2 suppresses cellular senescence of human dermal fibroblasts by inducing autophagy. J Ginseng Res. 2023;47(2):337–46.
Zhang Y, et al. Cycloastragenol: A Novel Senolytic Agent That Induces Senescent Cell Apoptosis and Restores Physical Function in TBI-Aged Mice. Int J Mol Sci. 2023;24(7):6554.
Mullen M, et al. Fisetin Attenuates Cellular Senescence Accumulation During Culture Expansion of Human Adipose-Derived Stem Cells. Stem Cells. 2023;41(7):698–710.
Chen DD, et al. HSP90 acts as a senomorphic target in senescent retinal pigmental epithelial cells. Aging (Albany NY). 2021;13(17):21547–70.
Liu S, et al. Simvastatin suppresses breast cancer cell proliferation induced by senescent cells. Sci Rep. 2015;5:17895.
Xu M, et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci U S A. 2015;112(46):E6301–10.
Gao LB, et al. Identification of a small molecule SR9009 that activates NRF2 to counteract cellular senescence. Aging Cell. 2021;20(10): e13483.
Park M, et al. Zileuton Alleviates Radiation-Induced Cutaneous Ulcers via Inhibition of Senescence-Associated Secretory Phenotype in Rodents. Int J Mol Sci. 2022;23(15):8390.
Samakkarnthai P, et al. In vitro and in vivo effects of zoledronic acid on senescence and senescence-associated secretory phenotype markers. Aging (Albany NY). 2023;15(9):3331–55.
Shrestha N, et al. Immunotherapeutic approach to reduce senescent cells and alleviate senescence-associated secretory phenotype in mice. Aging Cell. 2023;22(5): e13806.
Liu H, et al. Rutin is a potent senomorphic agent to target senescent cells and can improve chemotherapeutic efficacy. Aging Cell. 2024;23(1):e13921.
Prattichizzo F, et al. Anti-TNF-α treatment modulates SASP and SASP-related microRNAs in endothelial cells and in circulating angiogenic cells. Oncotarget. 2016;7(11):11945–58.
Acknowledgements
The figures in this article were supported and licensed by BioRender.
Funding
This study was supported by the Outstanding Scientific Fund of Shengjing Hospital and the 345 Talent Project of Shengjing Hospital (No.30C).
Author information
Authors and Affiliations
Contributions
Z.D. and Y.L. wrote the main manuscript text, and Z.Y. and Y.T. revised the manuscript. T.J. and F.X. proofread and supervised during the writing of the original manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics Approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dong, Z., Luo, Y., Yuan, Z. et al. Cellular senescence and SASP in tumor progression and therapeutic opportunities. Mol Cancer 23, 181 (2024). https://doi.org/10.1186/s12943-024-02096-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-024-02096-7