
Abstract
Hematological malignancies are a highly heterogeneous group of diseases with varied molecular and phenotypical characteristics. SWI/SNF (SWItch/Sucrose Non-Fermentable) chromatin remodeling complexes play significant roles in the regulation of gene expression, being essential for processes such as cell maintenance and differentiation in hematopoietic stem cells. Furthermore, alterations in SWI/SNF complex subunits, especially in ARID1A/1B/2, SMARCA2/4, and BCL7A, are highly recurrent across a wide variety of lymphoid and myeloid malignancies. Most genetic alterations cause a loss of function of the subunit, suggesting a tumor suppressor role. However, SWI/SNF subunits can also be required for tumor maintenance or even play an oncogenic role in certain disease contexts. The recurrent alterations of SWI/SNF subunits highlight not only the biological relevance of SWI/SNF complexes in hematological malignancies but also their clinical potential. In particular, increasing evidence has shown that mutations in SWI/SNF complex subunits confer resistance to several antineoplastic agents routinely used for the treatment of hematological malignancies. Furthermore, mutations in SWI/SNF subunits often create synthetic lethality relationships with other SWI/SNF or non-SWI/SNF proteins that could be exploited therapeutically. In conclusion, SWI/SNF complexes are recurrently altered in hematological malignancies and some SWI/SNF subunits may be essential for tumor maintenance. These alterations, as well as their synthetic lethal relationships with SWI/SNF and non-SWI/SNF proteins, may be pharmacologically exploited for the treatment of diverse hematological cancers.
Similar content being viewed by others
Background
Hematological malignancies are cancers that arise in blood-forming tissue. In 2020, over 1.2 million people worldwide were diagnosed with hematological malignancies and over 700 thousand people died from them [1]. Taken together, hematological malignancies accounted for approximately 7.5% of new cancer diagnoses and 7.8% of cancer deaths worldwide in the year 2020, and they ranked fifth among all cancers in terms of prevalence and mortality.
Hematological malignancies are broadly classified as lymphomas, which originate in the lymphatic system; multiple myeloma (MM), which involves plasma cells from the bone marrow; and leukemias, which affect cells from the bone marrow or blood. Moreover, hematological malignancies can affect various types of cells from the blood and from the immune system at various stages of differentiation. In particular, they most commonly affect the B cell lineage (~ 67% of all hematological malignancies), followed by myeloid cells (~ 30%), and T cells or natural killer (NK) cells (~ 3%) [2]. More specific classifications of hematological malignancies can be highly complex, involving over 70 subtypes [3,4,5]. Among lymphomas, the most common subtypes include diffuse large B-cell lymphoma (DLBCL, ~ 15% of all hematological malignancies), marginal zone lymphoma (MZL, ~ 7%), follicular lymphoma (FL, ~ 6%), and Hodgkin lymphoma (HL, ~ 5%), all of which originate in B cells [2]. In addition, MM encompasses ~ 12% of all hematological malignancies. Among leukemias, chronic lymphocytic leukemia (CLL) represents ~ 13% of all hematological malignancies, acute myeloid leukemia (AML) represents ~ 7%, and acute lymphoblastic leukemia (ALL) represents ~ 2%. Other frequent hematological malignancies include chronic myeloproliferative neoplasms (MPN, ~ 10%) and myelodysplastic syndromes (MDS, ~ 6%), both of which affect myeloid cells in the bone marrow. Furthermore, each type of hematological malignancy can be highly heterogeneous at a molecular level and at a clinical level, and genetics-based subclassifications have been gaining popularity in recent years to better explain this heterogeneity [6,7,8,9].
This wide range of hematological malignancies arises from an aberrant regulation of hematopoiesis, a process that requires an exquisite modulation of gene expression. Different mechanisms tightly regulate hematopoiesis, among which epigenetic processes have stood out as crucial players in determining hematopoietic cell fate decisions [10, 11]. As a result, epigenetic abnormalities are frequent in hematological malignancies. Specifically, proteins involved in the modification of chromatin landscapes, such as those from SWI/SNF (SWItch/Sucrose Non-Fermentable) complexes, polycomb repressive complexes, and DNA methyltransferases, are recurrently mutated in a wide variety of hematological neoplasms [12,13,14,15,16,17,18,19,20,21,22]. Moreover, due to the reversible nature of epigenetics, several therapies target different epigenetic regulators to reset the altered transcriptional programs of hematological cancers and hinder tumor progression (reviewed in [23,24,25,26]). Importantly, hematological malignancies are more vulnerable to this type of therapeutic intervention than other tumors [22]. Indeed, there is increasing evidence of dependencies of hematological tumors on certain epigenetic modulators, such as chromatin remodeling complexes, highlighting the attractive potential of epigenetic therapies for the treatment of hematological malignancies.
This review summarizes the biological and therapeutic implications of SWI/SNF chromatin remodeling complexes in hematological malignancies. First, it introduces the role of SWI/SNF complexes in normal hematopoiesis. Next, it discusses the role of individual SWI/SNF subunits in the promotion, suppression, or maintenance of various hematological malignancies. Then, it overviews the recurrence of genetic, epigenetic, and gene expression alterations in SWI/SNF complex subunits in hematological malignancies. Finally, it outlines the clinical implications of SWI/SNF complexes in hematological malignancies in the contexts of SWI/SNF status as a predictor of drug response, SWI/SNF proteins as drug targets, and synthetic lethality involving SWI/SNF proteins.
SWI/SNF complexes and their role in hematopoiesis
SWI/SNF complexes are ATP-dependent chromatin remodelers that use the energy from the hydrolysis of ATP to mobilize nucleosomes along DNA, eject them, or even change their composition [27, 28]. Consequently, these multi-subunit complexes modulate chromatin accessibility to relevant molecular players such as the transcriptional machinery, DNA-binding proteins, cofactors, regulators, and other proteins of the DNA replication and DNA repair processes [29,30,31]. Hence, an adequate SWI/SNF function is critical for several differentiation processes, including hematopoiesis and hematopoietic stem cell (HSC) maintenance [32,33,34]. However, SWI/SNF complexes may play opposing roles in normal and in malignant hematopoiesis, adding a layer of complexity to their function [35,36,37].
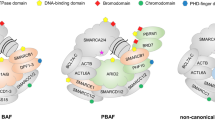
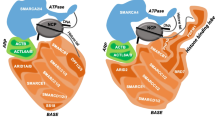
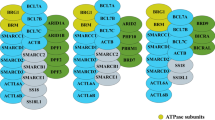
To date, 29 genes have been identified to code for SWI/SNF members, but only 12 to 15 of them coexist in a single SWI/SNF complex. Thus, there is a high variability of SWI/SNF assemblies that include lineage-restricted subunits [28, 38]. Importantly, this combinatorial potential provides a remarkable diversity of SWI/SNF complexes that are tissue-specific and contribute to controlling the transcription of lineage-specific genes [39].
In general, mammalian SWI/SNF complexes belong to three subfamilies: BRG1/BRM-associated factor (BAF), polybromo-associated BAF (PBAF), and a recently discovered subfamily named non-canonical BAF (ncBAF) [40]. The three subfamilies share a core of scaffold subunits (SMARCC1/2, and SMARCD1/D2/D3), and either of the two mutually exclusive ATPase-helicase subunits: SMARCA4 (also known as BRG1) or SMARCA2 (BRM). Additionally, each of those subfamilies contains a specific set of subunits that characterize the resulting assembly, such as the ARID (AT-rich interaction domain) subunits ARID1A/B or ARID2, and the novel non-canonical subunits BRD9, BICRA (GLTSCR1), and BICRAL (GLTSCR1L) [40, 41]. Within each SWI/SNF subfamily, the subunits can be arranged in different combinations. The resulting assemblies are key to determine not only the genomic sites of action of the different SWI/SNF complexes, but also their interaction with other transcription factors [42,43,44,45], DNA repair proteins [31, 46], and DNA replication proteins [47, 48], among many others. Increasing evidence has demonstrated that these changes in the SWI/SNF interactome are crucial for the maintenance of hematological gene expression patterns. Specifically, SWI/SNF complexes can also interact with hematopoietic-specific transcription factors, including EKLF, RUNX1, PU.1, IKAROS, GATA1, and CEBPα [49,50,51,52,53].
Growing evidence has demonstrated the essential role of SWI/SNF complexes in normal hematopoiesis (Table 1). Independent studies have described the important functions of many SWI/SNF subunits, such as ACTL6A, ARID1A, ARID2, PBRM1, PHF10, and SMARCA2 for the maintenance of HSCs [37, 54,55,56,57,58]. In addition, some SWI/SNF subunits play relevant roles in both myeloid and lymphoid differentiation, such as ARID1A [55, 59] and SMARCA4 [60,61,62,63,64,65,66,67]. On the other hand, other subunits are specific to certain processes within one of those lineages. For instance, SMARCE1 is involved in the differentiation of CD4+ and CD8+ T cells [64], while ARID1B and SMARCD2 regulate erythropoiesis [68] and granulocytic maturation [69, 70], respectively.
Contribution of SWI/SNF complexes to hematological malignancies
Because SWI/SNF complexes are involved in the homeostasis of hematological processes, alterations in SWI/SNF subunits can contribute to the onset or progression of hematological malignancies. The roles of different SWI/SNF subunits in different hematological malignancies can be highly diverse, ranging from being tumor suppressive, to being required for tumor maintenance, or even being oncogenic (Table 2). In line with this functional diversity, as will be detailed later, genetic alterations in SWI/SNF subunits are highly recurrent in various hematological malignancies, such as DLBCL [7, 8, 82, 83], whereas they are extremely rare in others, such as AML (excluding the acute promyelocytic leukemia (APL) subtype) [51, 84]. When present, SWI/SNF mutations in hematological malignancies usually cause loss of function, suggesting a tumor suppressor role. On the other hand, in certain contexts, such as in non-APL AML, normal function of at least some SWI/SNF subunits may be required for tumor maintenance [36, 51, 85]. However, although the essentiality of SWI/SNF for non-APL AML maintenance has been extensively demonstrated in vivo, to our knowledge few in vivo studies have explored the functional consequences of SWI/SNF alterations in other hematological malignancies [86].
Importantly, genetic alterations involving different SWI/SNF complex subunits in the same disease can have vastly different functional effects because the remaining residual SWI/SNF complexes may have different biological activities [36]. For instance, the oncogenic fusion protein KMT2A-MLLT1 (also known as MLL1-ENL1), which has been recurrently observed in ALL, AML, and MLL (mixed-lineage leukemia) [103], forms aberrant SWI/SNF complexes named EBAFb (ENL-associated BAF-containing BAF250b) [104]. These EBAFb complexes, which contain SMARCA4 as their catalytic subunit, activate the transcription of genes that are aberrantly expressed in MLL [104]. In the remainder of this section, we summarize the main functional studies on the role of SWI/SNF subunits in hematological malignancies, most of which have been performed in myeloid leukemia.
ATPase-helicases: SMARCA4 and SMARCA2
Although, in many tumor types, SMARCA4 displays loss-of-function mutations characteristic of a tumor suppressor, an additional role as a tumor-supportive gene in certain malignancies is emerging. Indeed, SMARCA4-containing SWI/SNF complexes may be essential for the maintenance of AML, opening new therapeutic possibilities [51, 59, 85, 102]. Mechanistically, SMARCA4-containing SWI/SNF complexes may modulate the well-known oncogene MYC specifically in AML. This modulation has been thoroughly demonstrated in the mouse cell line RN2 (KMT2A-rearranged NRASG12D AML) [51, 102], as well as in the human cell line ME-1 (AML harboring the CBFB-MYH11 fusion gene) [105]. However, another study in a different murine AML model (KMT2A-rearranged NRASWT) failed to reproduce the modulation of Myc by SMARCA4, although it did confirm that SMARCA4, along with SMARCD2 and DPF2, are required for AML maintenance [85].
Regarding SMARCA2, its individual role in hematological malignancies remains unknown. In one study, dual loss of SMARCA2 and SMARCA4 activity either by using allosteric dual inhibitors, such as BRM011 and BRM014, or by knock-down approaches, significantly impaired the viability of a diverse panel of AML cell lines, including those that did not display sensitivity to SMARCA4 knockdown [88].
ARID subunits: ARID1A, ARID1B, ARID2
The ARID subunits of SWI/SNF complexes can show both tumor suppressor and tumor supportive properties depending on the cellular model, disease subtype, and disease stage in which they are evaluated. For example, inhibition of Arid1a abrogated proliferation in RN2 cells, suggesting that ARID1A-containing SWI/SNF complexes promoted AML maintenance in this context [51]. However, another report indicated that ARID1A may act as a barrier to uncontrolled cell division in two human AML cell lines: HL-60 (APL) and THP-1 (KMT2A-rearranged) [89]. In particular, decreasing ARID1A expression in both cell lines suppressed apoptosis and boosted the proliferating ability of AML cells through the TGF-ꞵ1/SMAD3 pathway.
Regarding ARID1B, despite it sharing high homology with ARID1A [106], this subunit has been more linked to a suppressive role in leukemia rather than to a contributing role in tumor maintenance. Specifically, in a recent report by Bluemn et al., knockout of Arid1b promoted both AML initiation and progression in a mouse model of KMT2A-rearranged AML [36]. Interestingly, the authors failed to find phenotypical consequences upon knocking down Arid1b in vitro. In addition, another study showed that knocking down ARID1B in an APL cell line (NB4) under treatment with All-trans retinoic acid (ATRA) blocked blast differentiation [84].
In contrast to ARID1B, the effects of ARID2 may be dependent on the AML stage. In particular, Bluemn et al. found that loss of Arid2 enhances leukemogenesis at the initial stages of AML, but a functioning ARID2 is later required for AML maintenance [36]. Similarly to Arid1b, the phenotypical effects of knocking out Arid2 were observed in vivo but not in vitro. Moreover, the transcriptomic changes were different and, in some cases, opposing when comparing the effects of knocking out Arid1b or Arid2 in the same mouse model. A likely explanation for this discrepancy is that, since ARID1B and ARID2 belong to different SWI/SNF subfamilies (BAF and PBAF, respectively), the biological activity of the residual complexes remaining upon knockout of Arid1b or Arid2 may be different.
Core subunits: SMARCB1, SMARCC1, SMARCD1/2
The core subunits of SWI/SNF complexes have been described to play essential roles in different hematological malignancies because of their crucial function as structural scaffolds. For instance, the knockdown of Smarcc1, Smarcd1, or Smarcd2 impaired the survival of mouse AML cells, confirming their tumor-maintaining role in this tumor type [51, 85]. Moreover, the loss of Smarcb1 can also induce the development of T-cell lymphoma in mouse models [90]. Regarding the latter core subunit, its absence in AML cells generates a residual SWI/SNF complex with SMARCC1 as one of its core subunits, which regulates the expression of oncogenic programs crucial for the survival and migration of AML cells [92].
Accessory subunits
The specific role of some accessory SWI/SNF subunits remains largely unknown. However, their mutational frequency in certain tumor types suggests that they are important contributors to tumorigenesis. This is the case for BCL7A (B-cell CLL/lymphoma 7 protein family member A), which is expressed in the nuclei of germinal center B lymphocytes, but it is lost in mature plasma cells. This subunit is frequently mutated in DLBCL, and reintroduction of wild type BCL7A impaired the proliferation of DLBCL cells both in vitro and in vivo, pointing to a tumor suppressor role [86]. Moreover, recurrent mutations in its first splice donor site cause a deletion of 27 amino acids in the N-terminal portion of the protein, preventing its assembly into the SWI/SNF complex [86].
On the other hand, the accessory subunit BCL11A may have oncogenic functions across multiple hematological malignancies in view of its genetic patterns and expression alterations, which will be detailed in later sections. BCL11A is a zinc-finger transcription factor highly expressed in several hematopoietic lineages. It has been observed that BCL11A may induce the cell cycle progression of hematopoietic cells by directly inhibiting p21 expression [95], and it may deregulate the expression of a plethora of genes involved in apoptosis [107] or those regulated by other myeloid tumor suppressors such as PU.1 [94].
The paralog of BCL11A, BCL11B, is required for T cell development [77]. As a result, it can have either tumor suppressive or oncogenic functions in different hematological malignancies. In T-ALL, BCL11B may be a haploinsufficient tumor suppressor gene, in part because it may help maintain a differentiated T cell state [98]. Interestingly, a haploinsufficient tumor suppressor role of BCL11B has also been proposed in CML [97]. In contrast, in a subset of acute leukemias of ambiguous lineage (ALALs), BCL11B acts as an oncogene by driving specific gene expression programs [99, 100]. Mechanistically, it has been proposed that these leukemias originate when BCL11B becomes aberrantly activated in hematopoietic stem and progenitor cells (HSPCs), which promotes cell self-renewal, blocks myeloid differentiation, and activates the transcription of T lineage genes without proper differentiation into T cells, giving rise to the ambiguous lineage phenotype [99].
Finally, the bromodomain-containing subunit BRD9 may be essential for the maintenance of various hematological malignancies, including ALL, MM, and AML [101, 102]. In the mouse AML cell line RN2, BRD9 associates with SMARCA4-containing SWI/SNF complexes [102]. Knockdown of BRD9 impaired cell growth in RN2 cells and in a panel of human leukemia cell lines, but not in human epithelial cell lines or in mouse embryonic fibroblasts. This lineage-specific dependence highlights BRD9 as a potential target for novel chemotherapeutic agents, as will be detailed in later sections.
Genetic alterations of SWI/SNF subunits in hematological malignancies
Mutations in epigenetic modifiers and chromatin remodeling genes, including SWI/SNF complexes, are a major hallmark of many hematological malignancies, such as various NHLs [108], T cell lymphomas [109, 110], and dendritic cell neoplasms [111]. In addition, even in those hematological malignancies in which SWI/SNF alterations are not a major hallmark, inactivating mutations in SWI/SNF genes, especially ARID1A, are usually present at low frequencies (≤ 5%) [112, 113].
Various SWI/SNF genes, such as ARID1A/1B/2 and SMARCA2/4, are recurrently mutated across a wide variety of hematological and non-hematological cancers, whereas others, such as BCL7A, are specifically mutated in certain hematological malignancies [86, 114, 115]. Although the molecular mechanisms by which SWI/SNF mutations may promote such a wide variety of cancers remain largely unknown, two main models have been proposed (reviewed in [116]). One model proposes that, because SWI/SNF mutations impair DNA repair and genome stability, they create an overall mutagenic environment that may be favorable in different cancer contexts. This model is supported by the observation that SWI/SNF deficient tumors often have a high mutation burden [117], but it is unclear if the high mutation rate of SWI/SNF is the cause or the consequence of the high tumor mutation burden. Alternatively, another model suggests that SWI/SNF mutations affect the genome-wide chromatin accessibility patterns, possibly disrupting the function of lineage-specific transcription factors, which may further alter transcriptional programs in a cell type-specific manner. Indeed, as discussed in previous sections, many SWI/SNF subunits (including the widely mutated ARID1A and SMARCA4) play specific roles in the differentiation of lymphoid and myeloid lineages and in the maintenance of some hematological malignancies. Overall, further research should clarify the relative contribution of each proposed mechanism in different cancer contexts.
Several mechanistic studies support the idea that SMARCA4 mutations can promote cell type-specific oncogenesis by dysregulating lineage-specific transcriptional programs. Across a wide variety of hematological and non-hematological cancers, mutations in SMARCA4 are usually missense and heterozygous, and many of them have been proven to be dominant negative in at least some cellular contexts [118, 119]. SMARCA4 missense mutations frequently disrupt conserved positions involved in either its ATPase activity, its DNA helicase activity, or its overall tridimensional structure. Interestingly, in Smarca4-knockout mouse embryonic stem cells, which also lack expression of Smarca2, overexpression of human SMARCA4 harboring different missense mutations in various conserved positions had converging effects on chromatin accessibility [118]. In particular, the tested mutations mostly decreased the accessibility of a large number of enhancers and superenhancers, many of which were specific to either pro-B cells, T cells, macrophages, bone marrow tissue, or thymus, which could explain why SMARCA4 mutations are ubiquitous across different cancer types while promoting cell type-specific oncogenesis. For example, in a previous study, SMARCA4 had been reported to promote the expression of the MYC oncogene specifically in leukemia, even though both MYC and SMARCA4 are expressed ubiquitously across many cell types [51]. To explain this phenomenon, it was proposed that SWI/SNF binds to a MYC superenhancer only in leukemia cells, and that the superenhancer is bound by lineage-specific transcription factors. Of note, these findings were not reproduced by a later study in a different AML model [85]. In practice, the effect of mutations in SMARCA4 in cancer must be interpreted in the context of the status of its paralog SMARCA2. Indeed, in SMARCA4-deficient lung cancer cell lines, knocking down SMARCA2 decreased cell viability, and subsequently restoring different SMARCA4 mutants had varying abilities to compensate for the phenotype caused by SMARCA2 loss [119]. This phenomenon has critical implications in the context of anti-SMARCA2/4 therapeutic interventions, as the status of one protein may determine the efficacy of inhibiting the other. Finally, intriguingly, some SMARCA4 mutants that had negligible chromatin remodeling activity were able to revert the growth defects caused by SMARCA2 depletion [119].
Regarding ARID1A, about half of its mutations found across different hematological and non-hematological cancers are truncating (mainly nonsense or frameshift) [118]. Whereas truncating mutations in ARID1A are under strong positive selection, missense mutations have unknown significance and they have been proposed to be passengers [120]. In mice, loss of Arid1a impairs the ability of hematopoietic stem cells to differentiate into myeloid and lymphoid lineages by decreasing the accessibility of key loci that regulate hematopoiesis, such as Cebpa, Cd34, Csf1, IL6ra, and Gata2 [55]. Therefore, this observation may explain why loss-of-function mutations in ARID1A are observed in a wide variety of hematological cancers.
In contrast to SMARCA2/4 and ARID1A/1B/2 genes, BCL7A seems to be specifically mutated in lymphomas that are either derived from or phenotypically similar to germinal center B cells, such as germinal center B cell-like (GCB) DLBCL [86], FL [121], Burkitt lymphoma (BL) [122], and MM [123]. In all cases, BCL7A mutations accumulate in its promoter, its 5’-untranslated region (UTR), its first exon, and its first intron, following mutational patterns consistent with aberrant somatic hypermutation (aSHM) caused by off-target activity of the activation-induced cytidine deaminase (AID) enzyme. AID activity occurs during the germinal center reaction, and therefore AID mutational signatures are specific to germinal center-derived malignancies [124]. In DLBCL, certain ‘hotspot’ BCL7A mutations, such as those at its first splice donor site, may be bona fide driver events [86]. In contrast, it is currently unclear if BCL7A mutations are under selection in FL, BL, and MM. Finally, the hypermutated region of BCL7A has been identified as a super-enhancer in DLBCL [125].
Another recurrent alteration involving SWI/SNF genes across various hematological malignancies is the gain or amplification of chromosome 2p16.1, which affects both BCL11A and the nearby REL gene, and which can lead to BCL11A overexpression in some cases [93, 126]. Gain of chromosome 2p16.1 has been recurrently observed in DLBCL [7, 8], FL [108], BL [108], HL [127], and primary mediastinal B-cell lymphoma (PMBL) [128, 129]. However, while some reports state that BCL11A is the main specific target of the copy number gains [108], others have found that the nearby REL gene is amplified at a greater degree than BCL11A [129, 130], and many argue that BCL11A is not a specific target of the recurrent copy number gains [8, 127, 131].
In the remainder of this section, the genetic alterations (point mutations, copy number alterations, and genomic rearrangements) reported in SWI/SNF genes in hematological malignancies of various cell origins will be summarized (Table 3 and Fig. 1). Results from Data Release 33.1 of the International Cancer Genome Consortium (ICGC) and Data Release 12.0 of the Genomics, Evidence, Neoplasia, Information, Exchange (GENIE) Project, as well as individual (often disease-specific) studies, will be included [112, 113]. Whenever appropriate, data across multiple studies will be integrated but, when an individual study differs greatly from other work, it will be reported separately. Such differences between studies may stem from various factors: (i) small sample sizes, which can lead to inaccurate estimates of mutation frequencies; (ii) differences in the composition of the cohorts, for example in terms of disease subtypes; (iii) sequencing strategies (for example, targeted sequencing may not cover all SWI/SNF genes); (iv) differences in data processing and filtering of mutations; and (v) inclusion of matched normal samples. Indeed, studies that report unusually high mutation frequencies of individual genes often lack matched normal samples.

Summary of the main genetic alterations in SWI/SNF subunits in hematological malignancies. Because alteration frequencies can vary greatly between cohorts of the same disease (Table 3), they have been roughly categorized in three discrete groups. Double arrows indicate paralogous subunits that can play equivalent roles in different complexes. ALAL: acute leukemia of ambiguous lineage; ALL: acute lymphoblastic leukemia; APL: acute promyelocytic leukemia; BL: Burkitt lymphoma; BPDCN: blastic plasmacytoid dendritic cell neoplasm; CLL: chronic lymphocytic leukemia; CML: chronic myeloid leukemia; CTCL: cutaneous T cell lymphoma; DLBCL: diffuse large B cell lymphoma; EATL: enteropathy-associated T cell lymphoma; ENKTL: extranodal NK/T cell lymphoma; FL: follicular lymphoma; HSTCL: hepatosplenic T cell lymphoma; LPL: Lymphoplasmacytic lymphoma; MCL: mantle cell lymphoma; MDS: myelodysplastic syndrome; MF: mycosis fungoides; MZL: marginal zone lymphoma; PLL: prolymphocytic leukemia; PMBL: primary mediastinal large B-cell lymphoma; PTCL: peripheral T cell lymphoma, not otherwise specified. *Limited study, see Table 3
B-cell lymphomas
Diffuse large B-cell lymphoma
DLBCL is the most common type of NHL. SWI/SNF subunits are mutated in over a third of DLBCLs (Fig. 2A). According to an integration of whole-exome sequencing (WES) and whole-genome sequencing (WGS) studies in over 2000 patients, the top mutated SWI/SNF genes in DLBCL are ARID1A (8.6%), ARID1B (7.0%), BCL7A (6.1%), SMARCA4 (5.2%), and ACTB (4.8%) (Fig. 2C) [7, 8, 82, 83]. Moreover, targeted sequencing studies in DLBCL, collectively including over 2400 samples, have found the same five SWI/SNF genes as highly mutated at similar frequencies [9, 108, 112, 130, 132]. In addition, gains of chromosome 2p16.1, which contains BCL11A, affect 7-28% DLBCLs [7, 8, 108, 129, 130]. Remarkably, over a third of ARID1A mutations are truncating (nonsense or frameshift). Also of note, the mutation frequency of BCL7A and, consequently, of the SWI/SNF complex, is strongly dependent on the proportion of GCB DLBCLs in the cohort [7, 8, 82, 83].

Mutations in SWI/SNF genes in diffuse large B cell lymphoma (DLBCL). A Proportion of DLBCLs that have at least one mutation (mut.) in one SWI/SNF gene in whole-exome sequencing (WES) or whole-genome sequencing (WGS) studies. WT: wild type. Data sources: [7, 8, 82, 83]. In the datasets of Reddy et al. and Schmitz et al., previously missed splice site mutations from our previous reports have been added [86, 115]. B Distribution of mutations in BCL7A, shown at the protein level. Exon 1 (amino acids 1-31) is highlighted in red. Data sources: [7,8,9, 82, 83, 108]. C Distribution of mutations in SWI/SNF genes in DLBCL in WES or WGS studies. Data sources: [7, 8, 82, 83]. For the cohort of Chapuy et al., only paired samples were included in the plot. ABC: activated B cell-like; GCB: germinal center B cell-like
Over 50%, and up to 70%, of mutations in BCL7A in DLBCL are either truncating or within three mutational hotspots: the start codon, arginine 11, or the first splice donor site (Fig. 2B) [9, 86]. These mutations often affect different alleles from the same sample, suggesting biallelic inactivation patterns that are consistent with a tumor suppressor role of BCL7A in DLBCL [86]. The strongest mutational hotspot in BCL7A in DLBCL is its first splice donor site, which had been overlooked by some large-scale studies [82, 86]. Recently, our group showed that mutations in the first splice donor site of BCL7A cause an in-frame deletion of 27 codons in its first exon, impairing its tumor suppressor function in DLBCL [86].
Follicular lymphoma
Nearly all FLs are mutated in at least one chromatin remodeling gene, and the SWI/SNF complex is among the most highly mutated chromatin remodeling complexes in FL [108, 133]. In particular, various reports in independent FL cohorts have found recurrent point mutations in ARID1A (mutation frequency ~ 6-15%, combined N = 973) and in SMARCA4 (5-8%) [108, 112, 133, 134]. Importantly, mutations in ARID1A have been associated with better disease-free survival in FL [177]. Other SWI/SNF genes harboring recurrent point mutations in FL include BCL7A, ARID1B, ARID2, and BCL11B [112]. Moreover, chromosome 2p16.1 is amplified in ~ 22% FLs [108].
Recurrent BCL7A mutations have been consistently observed in independent FL cohorts, but the mutation frequencies are highly variable, ranging from 4% (N = 138) [133] to 11% (N = 199) [108] and up to 19% (N = 105) [134]. In all cases, the mutations concentrate in the first exon and the first splice donor site of BCL7A, as was observed in DLBCL [86]. Indeed, the mutations are thought to be caused by aSHM in late stages of the disease and they may be associated with FL transformation to DLBCL [121]. On the other hand, this model does not explain the large discrepancies in the mutation rate of BCL7A between cohorts, as the two most discrepant studies both focused on early FL. Overall, more work will be necessary to clarify the role of BCL7A mutations and aSHM in FL progression and transformation.
Burkitt lymphoma
More than 1 in every 4 BLs harbors a mutation in at least one SWI/SNF gene [112, 122, 135,136,137,138]. Three SWI/SNF genes are mutated in BL at frequencies above 15% in a mutually exclusive manner and have been proposed as drivers of the disease: ARID1A, SMARCA4, and BCL7A [135, 137, 138]. Other SWI/SNF genes recurrently mutated in BL include ARID2, SMARCA2, PBRM1, BCL11A, ACTL6A, SMARCC2, SMARCD1, DPF1, BRD7, and BRD9 [112, 137]. However, all of them are mutated at frequencies below 5%, and, to our knowledge, all their mutations reported so far are missense of unknown significance. Moreover, amplification of chromosome 2p16.1 occurs in ~ 11% BLs [108].
ARID1A is mutated in ~ 15-45% of BL patients [112, 122, 135, 136]. Mutations in ARID1A are often truncating (e.g., nonsense or frameshift), causing loss of function. Interestingly, ARID1A has been identified as an essential gene in BL by CRISPR-knockout screening [122]. Moreover, germline mutations in ARID1A may predispose to BL [135].
SMARCA4 is mutated in ~ 14-38% of BLs [108, 112, 122, 135,136,137,138]. In contrast to ARID1A, the vast majority of SMARCA4 mutations in BL are missense, but still they are predicted to have a high functional impact [136]. Among SWI/SNF mutations, those in SMARCA4 are the most enriched in BL compared to other non-Hodgkin lymphomas [108]. Furthermore, SMARCA4 mutations co-occur with MYC translocations in BL [108].
BCL7A was initially reported as a target of a three-way chromosomal rearrangement in a BL cell line [178]. Since then, various studies have confirmed that BCL7A is recurrently mutated in BL at frequencies of ~ 7% in coding regions and up to 43% when also considering non-coding regions, harboring mutational patterns consistent with aSHM [108, 138]. However, it is unclear if the coding or non-coding mutations of BCL7A are under positive selection in BL [135].
Importantly, some SWI/SNF genes are differentially mutated in the different subtypes of BL. For example, mutations in BCL7A are, together with those in BCL6, highly enriched in the endemic subtype of BL, which is associated with Epstein-Barr virus (EBV) infection, whereas mutations in SMARCA4 are enriched in the sporadic subtype, which is usually EBV-negative [122, 138]. In addition, mutations in ARID1A, SMARCA4, and BCL7A are significantly depleted in human immunodeficiency virus (HIV)-associated BL [122].
Hodgkin lymphoma
HL has two major subtypes: classical HL (cHL) and nodular lymphocyte-predominant HL (NLPHL). Two SWI/SNF genes have been proposed as drivers of cHL based on their accumulation of mutations: ARID1A (mutation frequency ~ 9-26%) and ACTB (mutation frequency ~ 26%) [112, 139, 140]. Whereas mutations in ARID1A are mostly deleterious (e.g., nonsense, frameshift, or splice-altering), mutations in ACTB are usually missense. ARID1A-mutant cHLs have an increased burden of driver mutations, which may be associated with a higher sensitivity to PD-1 blockade [179]. On the other hand, mutations in ACTB may be associated with aberrant cytokinesis, which is thought to be a hallmark of cancerous HL cells [140]. Other SWI/SNF genes that may be recurrently mutated in cHL include BCL7A and SMARCA4 [180, 181]. Furthermore, gain of chromosome 2p16.1, which contains BCL11A, is the most recurrent copy number alteration in cHL, affecting ~ 55% of the cases (N = 44) [127]. Finally, to our knowledge, no SWI/SNF mutations have been described in NLPHL, although only a couple dozen samples have been sequenced comprehensively so far [182, 183].
Other B-cell lymphomas
In primary mediastinal large B-cell lymphoma (PMBL), a wide variety of SWI/SNF genes are recurrently mutated, but discrepancies between studies are high [112, 141, 184]. Whereas one study reported that the top mutated SWI/SNF genes in PMBL are ARID1A (21%, N = 33) and SMARCA4 (15%, N = 33) [112], another argued that only ACTB is mutated above the background mutation rate (33%, N = 95) [141]. Moreover, gains of chromosome 2p16.1 affecting BCL11A are frequent in PMBL [128, 129].
In mantle cell lymphoma (MCL), recurrent mutations have been observed in SMARCA4 (mutation frequency ~ 6-10%), ARID2, ARID1A, and ARID1B (mutation frequency ~ 3-6% each) [108, 112, 142, 143]. Other SWI/SNF genes, such as SMARCB1, SMARCD1, PBRM1, and BCL11B, are mutated at lower frequencies [112].
In marginal zone lymphoma (MZL), recurrent mutations have been reported in ARID1A (7%) and ARID1B (4%) [112]. Interestingly, approximately half of the mutations in both genes are frameshift or nonsense. The mutation frequencies are consistently lower in meta-analyses than in individual studies, possibly because not all studies included in meta-analyses may have sequenced both genes [144, 145]. Other SWI/SNF genes recurrently mutated at lower frequencies are SMARCA4, SMARCB1, and PBRM1.
SWI/SNF genes are also frequently altered in lymphoplasmacytic lymphoma (LPL), including its most common subtype, known as Waldenström Macroglobulinemia (WM), and its precursor form, known as IgM monoclonal gammopathy of undetermined significance. In particular, ARID1A harbors recurrent inactivating mutations in ~ 5-17% of WM patients, suffering biallelic inactivation in some cases [112, 147, 148]. Furthermore, ARID1B is deleted in 50% of WMs [148].
T- and NK-cell lymphomas
The most common mature T cell neoplasm is peripheral T cell lymphoma, not otherwise specified (PTCL, NOS). In various PTCL, NOS cohorts, recurrent point mutations have been reported in ARID1A, ARID2, ARID1B, SMARCA2, and SMARCA4 [110, 112, 149,150,151]. Importantly, most mutations in ARID1A and ARID2 are truncating (e.g., nonsense or frameshift), and each gene may be deleted in ~ 5% PTCLs, NOS [110, 149].
Cutaneous T cell lymphomas (CTCLs) include mycosis fungoides and Sézary syndrome. In CTCLs, somatic copy number alterations are an order of magnitude more frequent than point mutations [152,153,154]. Importantly, ARID1A is among the top altered genes in CTCLs, especially in Sézary syndrome [112, 152, 153]. In particular, the frequencies of ARID1A deletions in various CTCL cohorts were 28% (N = 25), 32% (N = 37), 58% (N = 40), and 55% (N = 94) [152,153,154,155]. Furthermore, point mutations in ARID1A have been identified in ~ 8% CTCLs (combined N = 679), and about half of them are truncating [112, 152,153,154,155,156,157]. Interestingly, most ARID1A mutations and deletions are heterozygous. Although it has been proposed that the remaining wild-type allele may be silenced by epigenetic mechanisms, leading to no ARID1A protein expression, further data are needed to confirm this hypothesis [152]. Other SWI/SNF genes recurrently altered in CTCLs are SMARCE1 and SMARCD2 (each amplified in 20% Sézary syndrome patients, N = 25), ARID2 (deleted in 8% Sézary syndrome patients, N = 25), and SMARCB1 (point mutation frequency ~ 3%, combined N = 433) [112, 152,153,154, 156]. Finally, one study reported that BCL7A is recurrently deleted in mycosis fungoides (frequency = 44%, N = 16) [158], but this finding was not reproduced by a later study (N = 22) [185].
In hepatosplenic T cell lymphoma (HSTCL), chromatin remodeling genes are mutated in over 60% of the patients [109]. In particular, ARID1B is mutated in 18% HSTCLs, and SMARCA2 is mutated in 10% HSTCLs (N = 68). However, over a third of the reported ARID1B mutations are synonymous, and therefore their contribution to oncogenesis is unclear.
SWI/SNF mutations are rare (≤5-6%) in other types of T cell lymphomas, including angioimmunoblastic T cell lymphoma [186,187,188,189,190], extranodal NK/T cell lymphoma [159,160,161,162,163], monomorphic epitheliotropic intestinal T cell lymphoma [191,192,193], and anaplastic large-cell lymphoma (ALCL) [112, 194,195,196,197]. The mutations, if any, mostly affect ARID1A, and many are truncating. Interestingly, one ALCL case suffered a fusion between ARID1A and EPB41 [194]. Finally, non-synonymous mutations in BCL11B may affect 12% of enteropathy-associated T-cell lymphomas [164].
Multiple myeloma
Although comprehensive catalogs of MM driver genes are currently available, SWI/SNF genes do not seem to be mutated at high recurrence in their coding sequences [198, 199]. On the other hand, mutations in the 5’-UTR of BCL7A affect up to 76% of MM patients [123, 199, 200]. BCL7A is a tumor suppressor gene that is downregulated in MM compared to normal plasma cells, but it is unclear whether the non-coding mutations observed in MM affect BCL7A expression and whether the mutations are under selection [123]. The patterns of mutations in BCL7A in MM may be consistent with AID activity, whose mutational signature in MM is mostly found in non-coding regions [201]. Future research should explore whether the non-coding mutations in BCL7A in MM are a mere sign of past AID activity or whether they are under selection.
B- and T-cell leukemias
Acute lymphoblastic leukemia (ALL) can affect the hematopoietic precursors of both B and T cells. In B-ALL, although SWI/SNF alterations are rare (≤5%), recent statistically powerful studies have predicted some SWI/SNF genes as putative drivers. In particular, Ma et al. reported that ARID2 (11/218 mutant patients, 5%) and ACTB (1.4%) may be B-ALL drivers [165]. On the other hand, Brady et al. identified as candidate B-ALL drivers ARID2 (2.5% genetic alteration frequency, N = 1428), ARID1A (< 1%), ACTB (< 1%), and BICRAL (< 1%), all of which harbored mostly truncating mutations [166]. In T-ALL, BCL11B is a well-known driver gene that is mutated in 8-10% of the cases (combined N = 1161) [98, 112, 165,166,167]. The mutations are mostly heterozygous and inactivating (e.g., deletions and missense mutations that are predicted to disrupt its DNA-binding domain), suggesting that BCL11B may be a haploinsufficient tumor suppressor in T-ALL. Furthermore, recurrent genomic rearrangements in ~ 2-3% of T-ALLs place TLX3 under the control of BCL11B enhancers, which may in turn inactivate BCL11B expression. Other putative driver genes in T-ALL include ARID1A (~ 3%) and SMARCA4 (~ 3%), the latter of which is mutated in 14% of TLX3-rearranged T-ALLs [165, 166].
In chronic lymphocytic leukemia (CLL), mutations in SWI/SNF genes are rare, affecting < 6% of the patients [112, 113]. The most recurrently mutated SWI/SNF gene is ARID1A, which is mutated in ~ 0.9-1.6% of CLLs (N = 1643). Other recurrently mutated SWI/SNF genes in CLL are ARID1B (1.3%) and PBRM1 (0.9%) [112]. Importantly, over 80% of the ARID1A mutations and over 60% of the ARID1B mutations in CLL are nonsense or frameshift. Finally, recurrent loss of chromosome 22q11 (which encompasses SMARCB1) has been reported in up to 55% of T-cell prolymphocytic leukemias (T-PLL) (combined N = 34), but SMARCB1 may not be significantly downregulated in the affected cases [168, 169].
Myeloid malignancies
In MDS, ARID2 has been identified as a putative driver by some studies (reviewed by [202]). Although the frequency of somatic point mutations in ARID2 is relatively low (~ 1-2%, combined N = 898), the mutations are mostly truncating, and they are thought to be early events [112, 171]. Moreover, ARID2 is deleted in an additional ~ 1% MDSs [171]. Mutations and deletions in ARID2 in MDS may co-occur and act synergistically with those in components of the Polycomb Repressive Complex 2 (PRC2), especially EZH2 and JARID2 [171]. Overall, ARID2-deficient MDS has been proposed as a distinct entity with unique genetic and phenotypical features [171]. Besides ARID2, no other SWI/SNF gene is generally considered to be a driver of MDSs, although rare mutations in ARID1A and ARID1B have also been reported (mutation frequency ~ 0.5% each) [112]. In stark contrast to previous studies, Yao et al. reported that SWI/SNF genes are mutated in 17.8% of MDSs in a single-center cohort (N = 118) [172]. The top 3 mutated SWI/SNF genes were ARID1A, ARID2, and ARID1B. The source of the large discrepancies between Yao et al’s report and previous studies is unclear.
In other myeloid malignancies, mutations in SWI/SNF genes seem to be extremely rare (< 1%), and, if present, they mostly affect ARID1A. This is the case for AML [51], CML [203], and myeloproliferative neoplasms such as juvenile myelomonocytic leukemia [204, 205], chronic myelomonocytic leukemia [206], chronic neutrophilic leukemia [207], chronic eosinophilic leukemia [208], and others [112, 209,210,211,212,213]. However, there are notable exceptions. In APL, recurrent point mutations (mostly truncating) have been found in ARID1A (5%, N = 165) and ARID1B (3%) [84]. Interestingly, ARID1B mutations were enriched in relapsed APL (12%, N = 77) compared to newly diagnosed APL. In addition, recurrent deletions of SMARCB1 in CML patients have been reported at frequencies ranging from 30% (N = 46) [91] to 80% (N = 20) [170].
Other hematological malignancies
About one third of ALALs are characterized by genomic alterations that cause aberrant activation of BCL11B [99, 100]. There are two main mechanisms by which this activation occurs: (i) translocations that place BCL11B under the control of enhancers that are active in HSPCs; or (ii) de novo generation of a superenhancer by tandem amplification of an enhancer located downstream of BCL11B [99, 100]. Interestingly, the most frequent event places BCL11B under regulatory elements located upstream of ARID1B. Furthermore, aberrant activation of BCL11B strongly co-occurs with FLT3 alterations.
In blastic plasmacytoid dendritic cell neoplasm (BPDCN), mutations (mostly loss-of-function) and deletions in ARID1A seem to be recurrent, although cohort sizes have been low (8 altered patients, combined N = 70) [111, 112, 173,174,175,176]. Importantly, loss-of-function ARID1A mutations have been inferred to be early events in at least some BPDCNs [176]. Another WES study in BPDCN, which mostly lacked matched normal samples, found 15/47 (32%) cases mutated in ARID1A [214]. Most of the mutations in ARID1A were missense. Furthermore, the authors defined two molecular subtypes of BPDCN, one of which was enriched in mutations in ARID2, SMARCA4, and PBRM1 [214].
SWI/SNF genes are also altered in extremely rare hematological malignancies, some of which only have one reported case. For example, a recent case report described a highly aggressive pediatric hematopoietic malignancy that was associated with biallelic loss of SMARCB1, a phenomenon that is normally associated with rhabdoid tumors [215].
Finally, recurrent SWI/SNF mutations have been identified at low frequencies in other hematological malignancies, albeit mostly in limited cohorts lacking matched normal samples. These include ARID1B, ARID2, BCL11B, and SMARCA2 mutations in post-transplant lymphoproliferative disorders [189, 216], ARID1A mutations in primary histiocytic sarcoma [217], and ARID1A mutations in aggressive Langerhans cell histiocytosis [218].
Epigenetic and other expression alterations
Epigenetic alterations are defined as heritable modifications of phenotype that do not affect the DNA genomic sequence itself, and they are important regulators of gene expression [219]. In hematological [92] and non-hematological [117] malignancies, expression alteration of SWI/SNF subunits attributable to epigenetic silencing or other mechanisms has been reported (Table 4).
One of the most widely studied mechanisms of epigenetic regulation is promoter hypermethylation. For example, SMARCB1 is downregulated in AML because of repressive methylation of the CpG islands in its promoter, compared to non-tumor hematopoietic cells [92]. Moreover, in CTCL, one study found that the promoter region of BCL7A was hypermethylated, and its expression was successfully restored after treatment with a demethylating agent (decitabine) [221]. However, to our knowledge, little evidence has been found of hypermethylation affecting other SWI/SNF genes in hematological malignancies.
Chromosomal translocations and genomic amplifications can also be a source for expression alterations, such as the ones affecting BCL11A. However, as stated in previous sections, it is unclear if BCL11A is a specific target of these genomic alterations, and, in fact, these amplifications are often not associated with increases in BCL11A mRNA expression [8, 129, 131]. Regardless, many studies have shown that BCL11A is upregulated in several B cell malignancies (CLL, NHL, Hodgkin disease), as well as in ALL, AML, CML, and natural killer/T cell lymphoma (NKTL) [93, 96, 222,223,224,225,226]. Furthermore, aberrant allele-specific expression of BCL11B is a defining feature of a subgroup of ALALs [99].
Finally, another mechanism of gene expression regulation are microRNAs (miRNAs). MiRNAs are small, ~ 18-25 nucleotides long, highly conserved non-coding RNAs that regulate gene expression by binding to target mRNAs, usually impeding their translation and, thus, downregulating their expression [230]. The regulation of SWI/SNF complex subunits by miRNAs has been studied in different tumors. For instance, SMARCA4 has been found to have two alternative 3’UTRs in lung tissue containing active binding sites for miR-155, miR-199, and miR-101 [231]. Because the activity of miRNAs is frequently tissue-specific, it is unknown whether this regulation could be extended to hematological malignancies. At least, that seems to be the case for the regulation of SMARCA4 by miR-155 in some hematological malignancies [227, 228]. In another study in CLL patients, a series of upregulated miRNAs (miR-22, miR-34a, miR-146b, and miR-181b) were found to be responsible for the downregulation of ARID1B [220].
Clinical implications of hematological malignancies with altered SWI/SNF complexes
The recurrent alteration of SWI/SNF complexes during cancer development not only shows the biological relevance of these chromatin remodelers in hematological malignancies, but also highlights their clinical potential. Growing evidence has demonstrated that alterations in SWI/SNF genes result in vulnerabilities and drug resistance phenomena in hematological malignancies, among other cancers (Fig. 3). Some of these susceptibilities are subunit and/or cell-type specific, while others could be more widely applicable.

Therapeutic opportunities and resistances generated by altered SWI/SNF complexes in hematological malignancies. Altered subunits (depicted in red) create dependencies on other subunits or pathways/activities that can be exploited (blunt red arrows). Aberrant SWI/SNF complexes confer resistance to routinely used antineoplastic drugs (black arrows). The rest of the subunits of the SWI/SNF complex are shown in grey. ALL: acute lymphoblastic leukemia; AML: acute myeloid leukemia; CML: chronic myeloid leukemia; MCL: mantle cell lymphoma; MM: multiple myeloma; NHL: non-Hodgkin lymphoma; PRC2: polycomb repressive complex 2
Altered SWI/SNF complex and drug resistance
Increasing evidence has shown that mutations in SWI/SNF complex subunits confer resistance to several antineoplastic agents routinely used for the treatment of hematological malignancies, including ibrutinib, venetoclax, doxorubicin, paclitaxel or vinblastine. Specifically, in MCL, an upregulation of the pro-survival gene BCL2L1 (Bcl-xL) has been observed when SMARCA4 harbors loss-of-function mutations, which results in primary resistance to ibrutinib and venetoclax treatments or, eventually, in relapse after dual exposure to these agents [232]. Likewise, in the haploid CML cell line HAP1, depletion of SMARCB1 or ARID1A increased resistance to doxorubicin, and depletion of SMARCB1 also increased resistance to paclitaxel and vinblastine [233]. In the case of SMARCB1, the resistance was created through the enhancement of the expression of ABCB1 (ATP binding cassette subfamily B member 1), an ATP-dependent promiscuous drug efflux pump, which mediates a well-known mechanism of resistance to several chemotherapeutic drugs [234]. In this scenario, removing SMARCA4 could reverse the effects of the loss of SMARCB1, including ABCB1 overexpression and doxorubicin resistance. Consequently, designing clinical approaches that aim to target these pathogenic residual SWI/SNF complexes may boost sensitivity to chemotherapeutics. Moreover, SWI/SNF complexes are also required for glucocorticoid-dependent transcription. Glucocorticoids, such as prednisolone, asparaginase, or vincristine, are regularly used for the treatment of ALL, and resistance to glucocorticoids has been defined as an important adverse prognostic factor in newly diagnosed ALL patients. Increased expression of several SWI/SNF subunits, including SMARCA4, ARID1A, SMARCB1, and SMARCC2 has been associated with prednisolone sensitivity, whereas the lack of expression correlates with resistance to prednisolone treatment in ALL [235].
Finally, the FDA has approved Vorinostat for the treatment of cutaneous T-cell lymphoma. This drug, also known as SAHA, inhibits all zinc-related HDACs [236], being the first HDAC inhibitor applied to clinical studies. When applied as a single agent, it has exhibited limited results [237], although preclinical studies of a DLBCL therapy combining SAHA and EZH2 inhibitors have shown a clear synergistic effect [238]. In addition, it has been described that lung tumoral cells lacking the SWI/SNF-subunit SMARCA4 are refractory to SAHA treatment [239].
Synthetic lethality-based therapeutic approaches for altered SWI/SNF complex subunits in hematological malignancies
Mutations in SWI/SNF genes often create dependencies either on genes encoding other SWI/SNF subunits or on other pathways or activities that may become therapeutic targets in this context. These phenomena point to a scenario where subunit mutations do not completely halt SWI/SNF activity, resulting in abnormal SWI/SNF complexes with an alternative residual activity that supports cancer progression [240, 241]. Synthetic lethality occurs when two genetic events (i.e., mutations) are combined, resulting in a loss of cell fitness. Several synthetic lethal relationships have been described in altered SWI/SNF-driven cancers whereby cell viability upon the loss of one subunit (e.g., SMARCA4, SMARCB1, ARID1A) uniquely relies on the presence of either other SWI/SNF paralog subunit or a downstream gene or cellular pathway [242]. One of the best-characterized examples of the synthetic lethality between subunit paralogs has been described in non-small cell lung carcinoma cell lines lacking SMARCA4, where SMARCA2 has been identified as the upmost synthetic lethal dependency necessary for cell proliferation [243].
Synthetic lethality between SWI/SNF complex subunits
In hematological malignancies, most of the SWI/SNF subunit-targeting therapies described so far are based on small inhibitors targeting either SMARCA4/SMARCA2 or BRD9. Given that either loss-of-function point mutations or inactivation of SMARCA4 have been outlined in several hematological malignancies, targeting its paralog (SMARCA2) might be a potential therapeutic alternative for the treatment of these cancers. Indeed, it has been shown that several hematopoietic cancer cell lines are exquisitely sensitive to dual SMARCA4/SMARCA2 catalytic inhibitors [244, 245]. A further study in AML revealed that this sensitivity is not driven entirely by SMARCA4 dependence, but also requires the concomitant deprivation of SMARCA2 activity [88]. Hence, SMARCA2/SMARCA4 inhibitors target recurrent leukemic transcriptional programs in cell lines, resulting in a wide range of phenotypic effects, including apoptosis and differentiation. This results in a reduction of tumor growth in an AML xenograft mouse model after the treatment, thus demonstrating the therapeutic potential of SWI/SNF inhibition. This therapeutic avenue can be streamlined with the recent development of proteolysis targeting chimera (PROTAC) degraders of SMARCA2 and SMARCA4 [246], and further optimization of a SMARCA2 selective, orally active VHL-based degrader [247]. Indeed, the clinical use of PROTACs could be of great interest, given the success of several clinical trials where PROTACs are being applied to inhibit key proteins in hematological malignancies, other than SWI/SNF. These clinical trials include targets such as IRAK4 in DLBCL, BTK in B-cell malignancies, IKZF1/3 in MM and CML, and GSPT1 in AML [248]. Likewise, BRD9, a key component of the ncBAF complex, has been proposed as a therapeutic target in a group of leukemias. Particularly in AML, small-molecule inhibitors of BRD9, including BI-7273 and I-BRD9, regulate the cell proliferation rate [102]. Moreover, with the help of PROTACs developed to target BRD9 (dBRD9 and VZ185), it has been possible to verify that anti-cancer activity against AML might be achieved just by blocking the BRD9 bromodomain. However, in those cancers where SMARCB1 is mutated the blockage of the bromodomain is not sufficient, requiring a complete degradation of BRD9, hinting to a need for structural disruption of ncBAF [249,250,251]. More recently, Weisberg and collaborators [101] analysed the dependency on BRD9 in a variety of hematological cancers, including MM, ALL and AML, using novel small molecule inhibitors (EA-89-YM35), degraders (QA-68-ZU81), and RNA interference. They found that after depletion of the BRD9 protein, apoptosis was prominently triggered in ALL and MM, whereas AML cells exhibited terminal differentiation. In addition, these authors observed that the effects of a plethora of chemotherapeutic compounds and targeted therapies against MM, AML and ALL were enhanced upon BRD9 degradation. These findings point to a novel strategy for ALL and MM through the targeting of BRD9, either alone or combined with other compounds.
Synthetic lethality involving non-SWI/SNF complex subunits
Regarding synthetic lethal dependencies displayed by mutant SWI/SNF complexes and other proteins involved in different cellular pathways, Polycomb Repressor Complexes (PRCs) stand out as one of the best-studied examples. PRCs have opposing gene-regulatory activities to those of SWI/SNF [252]. Specifically, the recruitment of SWI/SNF complexes results in a displacement of both PRC1 and PRC2 that is abolished when SMARCB1 is mutated [253]. Thus, the synthetic lethal relationship between PRC2 inhibition and SMARCB1 loss is one of the most actionable dependencies that can be currently targeted with inhibitory compounds against the core subunits of PRC2, EZH2 and EED [254, 255]. Among the different EZH2 inhibitors that have been developed, tazemetostat, also known as EPZ-6438, has shown promising preclinical results [237, 256,257,258]. This low molecular weight compound is a highly specific competitive inhibitor of the cofactor S-adenosyl methionine (SAM) with limited effects in other lysine methyl transferases, including EZH1. This compound reduces trimethylation marks given that SAM is required for H3K27 methylation by EZH2 [257]. Noticeably, there is an active phase II clinical trial (NCT03213665-MATCH) aiming to determine the performance of tazemetostat in pediatric NHL patients harboring SMARCB1 or SMARCA4 gene mutations. Interestingly, the synthetic lethality displayed between SWI/SNF and PRC2 is being assessed not only in hematological malignancies, but in solid tumors as well. Indeed, a phase II clinical trial (NCT05023655) is recruiting patients to analyze the clinical benefit of tazemetostat in ARID1A-mutant solid tumors.
Given that SWI/SNF complexes contribute to the regulation of enhancer function by facilitating the acetylation of H3K27, defective complexes may alter H3K27 acetylation patterns and unleash or support cancerous transcriptional programs. Therefore, further investigation of reagents that modify histone acetylation levels might be of interest regarding hematological malignancies with abnormal SWI/SNF complexes [259]. In this line, it has been shown that loss of SMARCB1 increased recruitment of an endogenous, residual, nuclear SWI/SNF complex and associated histone acetyltransferases (HATs) to target loci, thereby promoting H3K27Ac and thus gene expression that, through Rac activation, enhanced AML cell migration and survival. This finding highlights the tumor suppressor role of SMARCB1 and illustrates the function of a residual SWI/SNF complex in maintaining an oncogenic gene expression program in AML [92].
Conclusion and perspectives
SWI/SNF complexes are ubiquitous epigenetic regulators that play major roles in normal and aberrant hematopoiesis. Indeed, a wide variety of hematological malignancies harbor recurrent alterations in SWI/SNF genes, and the clinical implications of such alterations are just starting to be explored. Recent studies on SWI/SNF-targeting chemotherapeutic agents, as well as on the role of SWI/SNF alterations in drug resistance and the creation of targetable synthetic lethalities in SWI/SNF-defective tumors, are opening new paths for improving cancer treatment that hold a promising future. On the other hand, mechanistic knowledge on the role of SWI/SNF in hematological malignancies is largely limited, and improving this knowledge may unlock novel therapeutic opportunities.
In-depth mechanistic studies of SWI/SNF function and of the consequences of SWI/SNF alterations are mostly lacking in hematological contexts, especially outside of AML. Most knowledge is relatively recent, as advanced epigenomic techniques are necessary to unveil the complex mechanisms of action of SWI/SNF. Importantly, one major question is why similar SWI/SNF alterations are found across such a wide range of hematological and non-hematological cancers. The involvement of SWI/SNF complexes in DNA repair and genome stability may help to explain this observation. In addition, recent work has revealed that SWI/SNF targets lineage-specific regulatory elements across the genome, and that at least some loss-of-function alterations in SWI/SNF genes affect their accessibility. However, most of this work has been performed either in non-hematological models or in AML, which is rarely mutated in SWI/SNF genes. Therefore, future research should explore the role of wild type and mutant SWI/SNF on the accessibility of lineage-specific loci with special emphasis on hematological malignancies that have a high mutation rate of SWI/SNF genes, such as DLBCL.
Another layer of complexity is the fact that the subunit composition of SWI/SNF is dynamic, which has critical implications in mechanistic, phenotypical, and clinical studies. Specifically, when a SWI/SNF subunit is inactivated by either mutation, silencing, or drug treatment, residual SWI/SNF complexes may remain functional, and they may even gain new functions that can be oncogenic, as is the case of SMARCB1-deficient AML. In turn, these phenomena can generate synthetic lethality relationships that can be exploited therapeutically. In this context, future studies should explore further synthetic lethality relationships in SWI/SNF-mutant hematological cancers, which may reveal novel therapeutic opportunities.
Furthermore, novel lines of research could aim to therapeutically rectify alterations in the expression of SWI/SNF genes in hematological malignancies. For example, demethylating agents may restore the expression of hypermethylated SWI/SNF genes to deliver tumor suppressor activity, as has been shown in preclinical studies. Moreover, future studies could explore the therapeutic potential of targeting SWI/SNF subunits that are overexpressed in cancer, such as BCL11A or BCL11B, using approaches such as RNA interference or PROTACs.
Overall, in our view, ongoing preclinical and clinical studies are only scratching the surface of the clinical potential of targeting SWI/SNF or exploiting its synthetic lethality relationships in hematological malignancies, and we expect that future research will reveal new options for improving the treatment of such a diverse group of cancers.
Abbreviations
- ABCB1:
-
ATP binding cassette subfamily B member 1
- AID:
-
Activation-induced cytidine deaminase
- ALAL:
-
Acute leukemia of ambiguous lineage
- ALCL:
-
Anaplastic large-cell lymphoma
- ALL:
-
Acute lymphoblastic leukemia
- AML:
-
Acute myeloid leukemia
- APL:
-
Acute promyelocytic leukemia
- ARID:
-
AT-rich interaction domain
- aSHM:
-
Aberrant somatic hypermutation
- ATRA:
-
All-trans retinoic acid
- BAF:
-
BRG1/BRM-associated factor
- BCL7A :
-
B-cell CLL/lymphoma 7 protein family member A
- BL:
-
Burkitt lymphoma
- BPDCN:
-
Blastic plasmacytoid dendritic cell neoplasm
- cHL:
-
Classical HL
- CLL:
-
Chronic lymphocytic leukemia
- CML:
-
Chronic myeloid leukemia
- CTCLs:
-
Cutaneous T cell lymphomas
- DLBCL:
-
Diffuse large B-cell lymphoma
- EBAFb:
-
ENL-associated BAF-containing BAF250b
- EBV:
-
Epstein-Barr virus
- FL:
-
Follicular lymphoma
- GCB:
-
Germinal center B cell-like
- GENIE:
-
Genomics, Evidence, Neoplasia, Information, Exchange
- HAT:
-
Histone acetyltransferase
- HIV:
-
Human immunodeficiency virus
- HL:
-
Hodgkin lymphoma
- HSC:
-
Hematopoietic stem cell
- HSPC:
-
Hematopoietic stem and progenitor cell
- HSTCL:
-
Hepatosplenic T cell lymphoma
- ICGC:
-
International Cancer Genome Consortium
- LPL:
-
Lymphoplasmacytic lymphoma
- MCL:
-
Mantle cell lymphoma
- MDR1:
-
Multidrug resistant
- MDS:
-
Myelodysplastic syndrome
- MM:
-
Multiple myeloma
- MPN:
-
Myeloproliferative neoplasm
- MZL:
-
Marginal zone lymphoma
- ncBAF:
-
Non-canonical BAF
- NHL:
-
Non-hodgkin lymphoma
- NK:
-
Natural killer
- NKTL:
-
Natural killer/T cell lymphoma
- NLPHL:
-
Nodular lymphocyte-predominant HL
- PBAF:
-
Polybromo-associated BAF
- PgP:
-
P-glycoprotein pump
- PLL:
-
Prolymphocytic leukemia
- PMBL:
-
Primary mediastinal B-cell lymphoma
- PRC:
-
Polycomb repressor complex
- PROTAC:
-
Proteolysis targeting chimera
- PTCL, NOS:
-
Peripheral T cell lymphoma, not otherwise specified
- SS:
-
Sézary syndrome
- SWI/SNF:
-
Switch/Sucrose Non-Fermentable
- UTR:
-
Untranslated region
- WES:
-
Whole-exome sequencing
- WGS:
-
Whole-genome sequencing
- WM:
-
Waldenström macroglobulinemia
References
International Agency for Research on Cancer. Cancer Today. Available from: https://gco.iarc.fr/today. [cited 10 Jan 2023].
Smith A, Howell D, Crouch S, Painter D, Blase J, Wang HI, et al. Cohort Profile: The Haematological Malignancy Research Network (HMRN): a UK population-based patient cohort. Int J Epidemiol. 2018;47:700–700g Oxford Academic. Available from: https://academic.oup.com/ije/article/47/3/700/4958802 [cited 8 Jul 2022].
Swerdlow SH, Campo E, Pileri SA, Lee Harris N, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375–90 American Society of Hematology. Available from: https://ashpublications.org/blood/article/127/20/2375/35286/The-2016-revision-of-the-World-Health-Organization [cited 8 Jul 2022].
Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, de Araujo IBO, Berti E, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia. 2022;36(7):1720–48 Nature Publishing Group. Available from: https://www.nature.com/articles/s41375-022-01620-2[cited 11 Jul 2022].
Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022;36(7):1703–19 Nature Publishing Group. Available from: https://www.nature.com/articles/s41375-022-01613-1 [cited 11 Jul 2022].
Morin RD, Scott DW. DLBCL subclassification: divide and conquer? Blood. 2020;135:1722–4 American Society of Hematology. Available from: https://ashpublications.org/blood/article/135/20/1722/454958/DLBCL-subclassification-divide-and-conquer [cited 11 Nov 2022].
Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med. 2018;378:1396–407 New England Journal of Medicine (NEJM/MMS). Available from: https://www.nejm.org/doi/10.1056/NEJMoa1801445 [cited 8 Jul 2022].
Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med. 2018;24(5):679–90 Nature Publishing Group. Available from: https://www.nature.com/articles/s41591-018-0016-8 [cited 8 Jul 2022].
Lacy SE, Barrans SL, Beer PA, Painter D, Smith AG, Roman E, et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: a Haematological Malignancy Research Network report. Blood. 2020;135:1759–71 American Society of Hematology. Available from: https://ashpublications.org/blood/article/135/20/1759/452715/Targeted-sequencing-in-DLBCL-molecular-subtypes [cited 25 Jul 2022].
Hu D, Shilatifard A. Epigenetics of hematopoiesis and hematological malignancies. Genes Dev. 2016;30:2021–41 Cold Spring Harbor Laboratory Press.
Goyama S, Kitamura T. Epigenetics in normal and malignant hematopoiesis: an overview and update 2017. Cancer Sci. 2017;108:553–62 Blackwell Publishing Ltd.
Huether R, Dong L, Chen X, Wu G, Parker M, Wei L, et al. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat Commun. 2014;5:3630. Nature Publishing Group. Available from: https://www.nature.com/articles/ncomms4630. [cited 17 Jul 2022].
Ntziachristos P, Tsirigos A, van Vlierberghe P, Nedjic J, Trimarchi T, Flaherty MS, et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat Med. 2012;18(2):298–302 Nature Publishing Group. Available from: https://www.nature.com/articles/nm.2651 [cited 17 Jul 2022].
The Cancer Genome Atlas Research Network. Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N Engl J Med. 2013;368:2059–74.
Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42:181–5.
Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303.
Neumann M, Heesch S, Schlee C, Schwartz S, Gökbuget N, Hoelzer D, et al. Whole-exome sequencing in adult ETP-ALL reveals a high rate of DNMT3A mutations. Blood. 2013;121:4749–52 American Society of Hematology.
Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471:189–96.
Abdel-Wahab O, Levine RL. Mutations in epigenetic modifiers in the pathogenesis and therapy of acute myeloid leukemia. Blood. 2013;121:3563–72 American Society of Hematology.
Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones A, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42:722–6.
Nikoloski G, Langemeijer SMC, Kuiper RP, Knops R, Massop M, Tönnissen ERLTM, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010;42:665–7.
Love C, Sun Z, Jima D, Li G, Zhang J, Miles R, et al. The genetic landscape of mutations in Burkitt lymphoma. Nat Genet. 2012;44:1321–5.
Gallipoli P, Huntly BJP. Novel epigenetic therapies in hematological malignancies: current status and beyond. Semin Cancer Biol. 2018;51:198–210 Academic Press.
Cruz-Rodriguez N, Combita AL, Zabaleta J. Epigenetics in Hematological Malignancies. Methods Mol Biol. 2018;1856:87–101 Available from: https://pubmed.ncbi.nlm.nih.gov/30178247/ [cited 17 Jul 2022].
Dimopoulos K, Grønbæk K. Epigenetic therapy in hematological cancers. APMIS. 2019;127:316–28 Available from: https://pubmed.ncbi.nlm.nih.gov/30859683/ [cited 17 Jul 2022].
Jones PA, Ohtani H, Chakravarthy A, de Carvalho DD. Epigenetic therapy in immune-oncology. Nat Rev Cancer. 2019;19(3):151–61 Nature Publishing Group. Available from: https://www.nature.com/articles/s41568-019-0109-9 [cited 17 Jul 2022].
Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304 Available from: https://pubmed.ncbi.nlm.nih.gov/19355820/ [cited 24 Jul 2022].
Wilson BG, Roberts CWM. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11(7):481–92 Nature Publishing Group. Available from: https://www.nature.com/articles/nrc3068 [cited 19 Jul 2022].
Hodges C, Kirkland JG, Crabtree GR. The Many Roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer. Cold Spring Harb Perspect Med. 2016;6:a026930 Cold Spring Harbor Laboratory Press. Available from: http://perspectivesinmedicine.cshlp.org/content/6/8/a026930.full [cited 24 Jul 2022].
Savas S, Skardasi G. The SWI/SNF complex subunit genes: their functions, variations, and links to risk and survival outcomes in human cancers. Crit Rev Oncol Hematol. 2018;123:114–31 Elsevier.
Ribeiro-Silva C, Vermeulen W, Lans H. SWI/SNF: complex complexes in genome stability and cancer. DNA Repair (Amst). 2019;77:87–95 Elsevier.
Kadoch C, Crabtree GR. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci Adv. 2015;1:e1500447 Available from: https://pubmed.ncbi.nlm.nih.gov/26601204/ [cited 17 Jan 2023].
Tu Z, Zheng Y. Role of ATP-dependent chromatin remodelers in hematopoietic stem and progenitor cell maintenance. Curr Opin Hematol. 2022;29:174–80 Available from: https://pubmed.ncbi.nlm.nih.gov/35787545/ [cited 14 Sep 2022].
Wang Z, Wang P, Li Y, Peng H, Zhu Y, Mohandas N, et al. Interplay between cofactors and transcription factors in hematopoiesis and hematological malignancies. Signal Transduct Target Ther. 2021;6(1):24.
Bluemn T, Schmitz J, Chen Y, Zheng Y, Zhang Y, Zheng S, et al. Arid2 regulates hematopoietic stem cell differentiation in normal hematopoiesis. Exp Hematol. 2021;94:37–46 Available from: https://pubmed.ncbi.nlm.nih.gov/33346030/ [cited 4 Aug 2022].
Bluemn T, Schmitz J, Zheng Y, Burns R, Zheng S, DeJong J, et al. Differential roles of BAF and PBAF subunits, Arid1b and Arid2, in MLL-AF9 leukemogenesis. Leukemia. 2022;36:946–55 Available from: https://pubmed.ncbi.nlm.nih.gov/35022500/ [cited 4 Aug 2022].
Liu L, Wan X, Zhou P, Zhou X, Zhang W, Hui X, et al. The chromatin remodeling subunit Baf200 promotes normal hematopoiesis and inhibits leukemogenesis. J Hematol Oncol. 2018;11:27 BioMed Central Ltd.
Pulice JL, Kadoch C. Composition and Function of Mammalian SWI/SNF Chromatin Remodeling Complexes in Human Disease. Cold Spring Harb Symp Quant Biol. 2016;81:53–60 Available from: https://pubmed.ncbi.nlm.nih.gov/28408647/ [cited 19 Jul 2022].
Wu JI, Lessard J, Crabtree GR. Understanding the words of chromatin regulation. Cell. 2009;136:200–6 Available from: https://pubmed.ncbi.nlm.nih.gov/19167321/ [cited 19 Jul 2022].
Mashtalir N, D’Avino AR, Michel BC, Luo J, Pan J, Otto JE, et al. Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell. 2018;175:1272–1288.e20 Available from: https://pubmed.ncbi.nlm.nih.gov/30343899/ [cited 13 Sep 2022].
Alpsoy A, Dykhuizen EC. Glioma tumor suppressor candidate region gene 1 (GLTSCR1) and its paralog GLTSCR1-like form SWI/SNF chromatin remodeling subcomplexes. J Biol Chem. 2018;293:3892–903.
King HW, Klose RJ. The pioneer factor OCT4 requires the chromatin remodeller BRG1 to support gene regulatory element function in mouse embryonic stem cells. Elife. 2017;6:e22631 Available from: https://pubmed.ncbi.nlm.nih.gov/28287392/ [cited 13 Sep 2022].
Sammak S, Allen MD, Hamdani N, Bycroft M, Zinzalla G. The structure of INI1/hSNF5 RPT1 and its interactions with the c-MYC: MAX heterodimer provide insights into the interplay between MYC and the SWI/SNF chromatin remodeling complex. FEBS J. 2018;285:4165–80 Available from: https://pubmed.ncbi.nlm.nih.gov/30222246/ [cited 13 Sep 2022].
Barisic D, Stadler MB, Iurlaro M, Schübeler D. Mammalian ISWI and SWI/SNF selectively mediate binding of distinct transcription factors. Nature. 2019;569:136–40.
Carcamo S, Nguyen CB, Grossi E, Filipescu D, Alpsoy A, Dhiman A, et al. Altered BAF occupancy and transcription factor dynamics in PBAF-deficient melanoma. Cell Rep. 2022;39:110637 Available from: https://pubmed.ncbi.nlm.nih.gov/35385731/ [cited 13 Sep 2022].
Hays E, Nettleton E, Carter C, Morales M, Vo L, Passo M, et al. The SWI/SNF ATPase BRG1 stimulates DNA end resection and homologous recombination by reducing nucleosome density at DNA double strand breaks and by promoting the recruitment of the CtIP nuclease. Cell Cycle. 2020;19:3096–114 Available from: https://pubmed.ncbi.nlm.nih.gov/33044911/ [cited 13 Sep 2022].
Bayona-Feliu A, Barroso S, Muñoz S, Aguilera A. The SWI/SNF chromatin remodeling complex helps resolve R-loop-mediated transcription-replication conflicts. Nat Genet. 2021;53:1050–63 Available from: https://pubmed.ncbi.nlm.nih.gov/33986538/ [cited 13 Sep 2022].
Lo CSY, van Toorn M, Gaggioli V, Dias MP, Zhu Y, Manolika EM, et al. SMARCAD1-mediated active replication fork stability maintains genome integrity. Sci Adv. 2021;7:eabe7804 Available from: https://pubmed.ncbi.nlm.nih.gov/33952518/ [cited 13 Sep 2022].
Armstrong JA, Bieker JJ, Emerson BM. A SWI/SNF-related chromatin remodeling complex, E-RC1, is required for tissue-specific transcriptional regulation by EKLF in vitro. Cell. 1998;95:93–104 Available from: https://pubmed.ncbi.nlm.nih.gov/9778250/ [cited 14 Sep 2022].
Bakshi R, Hassan MQ, Pratap J, Lian JB, Montecino MA, van Wijnen AJ, et al. The human SWI/SNF complex associates with RUNX1 to control transcription of hematopoietic target genes. J Cell Physiol. 2010;225:569–76 Available from: https://pubmed.ncbi.nlm.nih.gov/20506188/ [cited 14 Sep 2022].
Shi J, Whyte WA, Zepeda-Mendoza CJ, Milazzo JP, Shen C, Roe JS, et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013;27:2648–62 Available from: https://pubmed.ncbi.nlm.nih.gov/24285714/ [cited 14 Sep 2022].
Im H, Grass JA, Johnson KD, Kim SI, Boyer ME, Imbalzano AN, et al. Chromatin domain activation via GATA-1 utilization of a small subset of dispersed GATA motifs within a broad chromosomal region. Proc Natl Acad Sci U S A. 2005;102:17065–70.
O’Neill DW, Schoetz SS, Lopez RA, Castle M, Rabinowitz L, Shor E, et al. An Ikaros-containing chromatin-remodeling complex in adult-type Erythroid cells. Mol Cell Biol. 2000;20:7572–82.
Krasteva V, Buscarlet M, Diaz-Tellez A, Bernard MA, Crabtree GR, Lessard JA. The BAF53a subunit of SWI/SNF-like BAF complexes is essential for hemopoietic stem cell function. Blood. 2012;120:4720–32 Available from: https://pubmed.ncbi.nlm.nih.gov/23018638/ [cited 14 Sep 2022].
Han L, Madan V, Mayakonda A, Dakle P, Woon TW, Shyamsunder P, et al. Chromatin remodeling mediated by ARID1A is indispensable for normal hematopoiesis in mice. Leukemia. 2019;33(9):2291–305 Nature Publishing Group. Available from: https://www.nature.com/articles/s41375-019-0438-4 [cited 14 Sep 2022].
Lee H, Dai F, Zhuang L, Xiao ZD, Kim J, Zhang Y, et al. BAF180 regulates cellular senescence and hematopoietic stem cell homeostasis through p21. Oncotarget. 2016;7:19134–46 Available from: https://pubmed.ncbi.nlm.nih.gov/26992241/ [cited 14 Sep 2022].
Krasteva V, Crabtree GR, Lessard JA. The BAF45a/PHF10 subunit of SWI/SNF-like chromatin remodeling complexes is essential for hematopoietic stem cell maintenance. Exp Hematol. 2017;48:58–71.e15 Available from: https://pubmed.ncbi.nlm.nih.gov/27931852/ [cited 14 Sep 2022].
Naidu SR, Capitano M, Ropa J, Cooper S, Huang X, Broxmeyer HE. Chromatin remodeling subunit BRM and valine regulate hematopoietic stem/progenitor cell function and self-renewal via intrinsic and extrinsic effects. Leukemia. 2022;36:821–33 Available from: https://pubmed.ncbi.nlm.nih.gov/34599272/ [cited 15 Sep 2022].
Astori A, Tingvall-Gustafsson J, Kuruvilla J, Coyaud E, Laurent EMN, Sunnerhagen M, et al. ARID1a Associates with Lymphoid-Restricted Transcription Factors and Has an Essential Role in T Cell Development. J Immunol. 2020;205:1419–32 The American Association of Immunologists. Available from: https://www.jimmunol.org/content/205/5/1419 [cited 14 Sep 2022].
Bultman SJ, Gebuhr TC, Magnuson T. A Brg1 mutation that uncouples ATPase activity from chromatin remodeling reveals an essential role for SWI/SNF-related complexes in β-globin expression and erythroid development. Genes Dev. 2005;19:2849–61 Cold Spring Harbor Laboratory Press. Available from: http://genesdev.cshlp.org/content/19/23/2849.full [cited 14 Sep 2022].
Griffin CT, Brennan J, Magnuson T. The chromatin-remodeling enzyme BRG1 plays an essential role in primitive erythropoiesis and vascular development. Development. 2008;135:493–500.
Hu G, Schones DE, Cui K, Ybarra R, Northrup D, Tang Q, et al. Regulation of nucleosome landscape and transcription factor targeting at tissue-specific enhancers by BRG1. Genome Res. 2011;21:1650–8 Available from: https://pubmed.ncbi.nlm.nih.gov/21795385/ [cited 15 Sep 2022].
Vradii D, Wagner S, Doan DN, Nickerson JA, Montecino M, Lian JB, et al. Brg1, the ATPase subunit of the SWI/SNF chromatin remodeling complex, is required for myeloid differentiation to granulocytes. J Cell Physiol. 2006;206:112–8 John Wiley & Sons, Ltd. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/jcp.20432 [cited 14 Sep 2022].
Chi TH, Wan M, Zhao K, Taniuchi I, Chen L, Uttman DR, et al. Reciprocal regulation of CD4/CD8 expression by SWI/SNF-like BAF complexes. Nature. 2002;418(6894):195–9 Nature Publishing Group. Available from: https://www.nature.com/articles/nature00876 [cited 14 Sep 2022].
Chi TH, Wan M, Lee PP, Akashi K, Metzger D, Chambon P, et al. Sequential roles of Brg, the ATPase subunit of BAF chromatin remodeling complexes, in thymocyte development. Immunity. 2003;19:169–82 Available from: https://pubmed.ncbi.nlm.nih.gov/12932351/ [cited 14 Sep 2022].
Gebuhr TC, Kovalev GI, Bultman S, Godfrey V, Su L, Magnuson T. The role of Brg1, a catalytic subunit of mammalian chromatin-remodeling complexes, in T cell development. J Exp Med. 2003;198:1937–49 Available from: https://pubmed.ncbi.nlm.nih.gov/14676303/ [cited 14 Sep 2022].
Choi J, Ko M, Jeon S, Jeon Y, Park K, Lee C, et al. The SWI/SNF-like BAF Complex Is Essential for Early B Cell Development. J Immunol. 2012;188:3791–803 American Association of Immunologists. Available from: https://www.jimmunol.org/content/188/8/3791 [cited 14 Sep 2022].
Azad P, Caldwell AB, Ramachandran S, Spann NJ, Akbari A, Villafuerte FC, et al. ARID1B, a molecular suppressor of erythropoiesis, is essential for the prevention of Monge’s disease. Exp Mol Med. 2022;54:777–87 Available from: https://pubmed.ncbi.nlm.nih.gov/35672450/ [cited 14 Sep 2022].
Priam P, Krasteva V, Rousseau P, D’Angelo G, Gaboury L, Sauvageau G, et al. SMARCD2 subunit of SWI/SNF chromatin-remodeling complexes mediates granulopoiesis through a CEBPɛ dependent mechanism. Nat Genet. 2017;49(5):753–64 Nature Publishing Group. Available from: https://www.nature.com/articles/ng.3812 [cited 15 Sep 2022].
Witzel M, Petersheim D, Fan Y, Bahrami E, Racek T, Rohlfs M, et al. Chromatin-remodeling factor SMARCD2 regulates transcriptional networks controlling differentiation of neutrophil granulocytes. Nat Genet. 2017;49:742–52 Available from: https://pubmed.ncbi.nlm.nih.gov/28369036/ [cited 15 Jan 2023].
Liu P, Keller JR, Ortiz M, Tessarollo L, Rachel RA, Nakamura T, et al. Bcl11a is essential for normal lymphoid development. Nat Immunol. 2003;4:525–32 Available from: https://pubmed.ncbi.nlm.nih.gov/12717432/ [cited 14 Sep 2022].
Sankaran VG, Menne TF, Xu J, Akie TE, Lettre G, van Handel B, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322:1839–42. American Association for the Advancement of Science. Available from: https://www.science.org/doi/10.1126/science.1165409. [cited 14 Sep 2022].
Sankaran VG, Xu J, Ragoczy T, Ippolito GC, Walkley CR, Maika SD, et al. Developmental and species-divergent globin switching are driven by BCL11A. Nature. 2009;460:1093–7 Available from: https://pubmed.ncbi.nlm.nih.gov/19657335/ [cited 14 Sep 2022].
Xu J, Sankaran VG, Ni M, Menne TF, Puram RV, Kim W, et al. Transcriptional silencing of gamma-globin by BCL11A involves long-range interactions and cooperation with SOX6. Genes Dev. 2010;24:783–98.
Liu N, Hargreaves VV, Zhu Q, Kurland JV, Hong J, Kim W, et al. Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch. Cell. 2018;173:430–442.e17 Available from: https://pubmed.ncbi.nlm.nih.gov/29606353/ [cited 14 Sep 2022].
Luc S, Huang J, McEldoon JL, Somuncular E, Li D, Rhodes C, et al. Bcl11a Deficiency Leads to Hematopoietic Stem Cell Defects with an Aging-like Phenotype. Cell Rep. 2016;16:3181–94 Available from: https://pubmed.ncbi.nlm.nih.gov/27653684/ [cited 15 Jan 2023].
Wakabayashi Y, Watanabe H, Inoue J, Takeda N, Sakata J, Mishima Y, et al. Bcl11b is required for differentiation and survival of alphabeta T lymphocytes. Nat Immunol. 2003;4:533–9 Available from: https://pubmed.ncbi.nlm.nih.gov/12717433/ [cited 14 Sep 2022].
Loo CS, Gatchalian J, Liang Y, Leblanc M, Xie M, Ho J, et al. A Genome-wide CRISPR Screen Reveals a Role for the Non-canonical Nucleosome-Remodeling BAF Complex in Foxp3 Expression and Regulatory T Cell Function. Immunity. 2020;53:143–157.e8 Available from: https://pubmed.ncbi.nlm.nih.gov/32640256/ [cited 14 Sep 2022].
Tu J, Liu X, Jia H, Reilly J, Yu S, Cai C, et al. The chromatin remodeler Brg1 is required for formation and maintenance of hematopoietic stem cells. FASEB J. 2020;34:11997–2008 Available from: https://pubmed.ncbi.nlm.nih.gov/32738093/ [cited 15 Sep 2022].
Choi J, Kim YK, Park K, Nah J, Yoon SS, Kim DW, et al. MicroRNA-139-5p regulates proliferation of hematopoietic progenitors and is repressed during BCR-ABL-mediated leukemogenesis. Blood. 2016;128:2117–29 Available from: https://pubmed.ncbi.nlm.nih.gov/27605510/ [cited 15 Sep 2022].
Lee KY, Choi YI, Kim J, Choi JW, Sohn DH, Lee C, et al. Down-Regulation of the SWI/SNF Chromatin Remodeling Activity by TCR Signaling Is Required for Proper Thymocyte Maturation. J Immunol. 2007;178:7088–96 American Association of Immunologists. Available from: https://www.jimmunol.org/content/178/11/7088 [cited 14 Sep 2022].
Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and functional drivers of diffuse large B cell lymphoma. Cell. 2017;171:481–494.e15 Cell Press.
Arthur SE, Jiang A, Grande BM, Alcaide M, Cojocaru R, Rushton CK, et al. Genome-wide discovery of somatic regulatory variants in diffuse large B-cell lymphoma. Nat Commun. 2018;9(1):4001.
Madan V, Shyamsunder P, Han L, Mayakonda A, Nagata Y, Sundaresan J, et al. Comprehensive mutational analysis of primary and relapse acute promyelocytic leukemia. Leukemia. 2016;30(8):1672–81 Nature Publishing Group. Available from: https://www.nature.com/articles/leu201669 [cited 16 Sep 2022].
Cruickshank AV, Sroczynska P, Sankar A, Miyagi S, Rundsten CF, Johansen JV, et al. SWI/SNF Subunits SMARCA4, SMARCD2 and DPF2 Collaborate in MLL-Rearranged Leukaemia Maintenance. PLoS One. 2015;10:e0142806 Available from: https://pubmed.ncbi.nlm.nih.gov/26571505/ [cited 3 Oct 2022].
Baliñas-Gavira C, Rodríguez MI, Andrades A, Cuadros M, Álvarez-Pérez JC, Álvarez-Prado ÁF, et al. Frequent mutations in the amino-terminal domain of BCL7A impair its tumor suppressor role in DLBCL. Leukemia. 2020;34:2722–35 Available from: https://pubmed.ncbi.nlm.nih.gov/32576963/ [cited 16 Sep 2022].
Buscarlet M, Krasteva V, Ho L, Simon C, Hébert J, Wilhelm B, et al. Essential role of BRG, the ATPase subunit of BAF chromatin remodeling complexes, in leukemia maintenance. Blood. 2014;123:1720–8 Available from: https://pubmed.ncbi.nlm.nih.gov/24478402/ [cited 3 Oct 2022].
Rago F, Rodrigues LU, Bonney M, Sprouffske K, Kurth E, Elliott GN, et al. Exquisite Sensitivity to Dual BRG1/BRM ATPase Inhibitors Reveals Broad SWI/SNF Dependencies in Acute Myeloid Leukemia. Mol Cancer Res. 2022;20:361–72 Available from: https://pubmed.ncbi.nlm.nih.gov/34799403/ [cited 14 Sep 2022].
Ren T, Wang J, Tang W, Chen D, Wang S, Zhang X, et al. ARID1A has prognostic value in acute myeloid leukemia and promotes cell proliferation via TGF-β1/SMAD3 signaling. Clin Exp Med. 2022. Available from: https://pubmed.ncbi.nlm.nih.gov/35867200/. [cited 16 Sep 2022].
Roberts CWM, Leroux MM, Fleming MD, Orkin SH. Highly penetrant, rapid tumorigenesis through conditional inversion of the tumor suppressor gene Snf5. Cancer Cell. 2002;2:415–25 Cell Press.
Grand F, Kulkarni S, Chase A, Goldman JM, Gordon M, Cross NC. Frequent Deletion of hSNF5/INI1, a Component of the SWI/SNF Complex, in Chronic Myeloid Leukemia. Cancer Res. 1999;59:3870–4 Available from: https://aacrjournals.org/cancerres/article/59/16/3870/505411/Frequent-Deletion-of-hSNF5-INI1-a-Component-of-the [cited 4 Oct 2022].
Chatterjee SS, Biswas M, Boila LD, Banerjee D, Sengupta A. SMARCB1 Deficiency integrates epigenetic signals to oncogenic gene expression program maintenance in human acute myeloid leukemia. Mol Cancer Res. 2018;16:791–804 American Association for Cancer Research Inc. Available from: https://aacrjournals.org/mcr/article/16/5/791/89912/SMARCB1-Deficiency-Integrates-Epigenetic-Signals [cited 4 Oct 2022].
Satterwhite E, Sonoki T, Willis TG, Harder L, Nowak R, Arriola EL, et al. The BCL11 gene family: involvement of BCL11A in lymphoid malignancies. Blood. 2001;98:3413–20 American Society of Hematology. Available from: https://ashpublications.org/blood/article/98/12/3413/53221/The-BCL11-gene-family-involvement-of-BCL11A-in [cited 16 Sep 2022].
Sunami Y, Yokoyama T, Yoshino S, Takahara T, Yamazaki Y, Harada H, et al. BCL11A promotes myeloid leukemogenesis by repressing PU.1 target genes. Blood Adv. 2022;6:1827–43.
Yin B, Delwel R, Valk PJ, Wallace MR, Loh ML, Shannon KM, et al. A retroviral mutagenesis screen reveals strong cooperation between Bcl11a overexpression and loss of the Nf1 tumor suppressor gene. Blood. 2009;113:1075–85 Available from: https://pubmed.ncbi.nlm.nih.gov/18948576/ [cited 16 Sep 2022].
Shi H, Li C, Feng W, Yue J, Song J, Peng A, et al. BCL11A Is Oncogenic and Predicts Poor Outcomes in Natural Killer/T-Cell Lymphoma. Front Pharmacol. 2020;11:820 Available from: https://pubmed.ncbi.nlm.nih.gov/32625084/ [cited 16 Sep 2022].
Nagamachi A, Yamasaki N, Miyazaki K, Oda H, Miyazaki M, Honda Z, et al. Haploinsufficiency and acquired loss of Bcl11b and H2AX induces blast crisis of chronic myelogenous leukemia in a transgenic mouse model. Cancer Sci. 2009;100:1219–26 Available from: https://onlinelibrary.wiley.com/doi/10.1111/j.1349-7006.2009.01172.x [cited 2 Nov 2022].
Gutierrez A, Kentsis A, Sanda T, Holmfeldt L, Chen S-C, Zhang J, et al. The BCL11B tumor suppressor is mutated across the major molecular subtypes of T-cell acute lymphoblastic leukemia. Blood. 2011;118:4169–73.
Montefiori LE, Bendig S, Gu Z, Chen X, Pölönen P, Ma X, et al. Enhancer hijacking drives oncogenic BCL11B expression in lineage-ambiguous stem cell leukemia. Cancer Discov. 2021;11:2846–67.
di Giacomo D, la Starza R, Gorello P, Pellanera F, Kalender Atak Z, de Keersmaecker K, et al. 14q32 rearrangements deregulating BCL11B mark a distinct subgroup of T and myeloid immature acute leukemia. Blood. 2021;138:773–84.
Weisberg E, Chowdhury B, Meng C, Case AE, Ni W, Garg S, et al. BRD9 degraders as chemosensitizers in acute leukemia and multiple myeloma. Blood Cancer J. 2022;12(7):110.
Hohmann AF, Martin LJ, Minder JL, Roe JS, Shi J, Steurer S, et al. Sensitivity and engineered resistance of myeloid leukemia cells to BRD9 inhibition. Nat Chem Biol. 2016;12:672–9 Nat Publ Group.
Ugale A, Säwén P, Dudenhöffer-Pfeifer M, Wahlestedt M, Norddahl GL, Bryder D. MLL-ENL-mediated leukemia initiation at the interface of lymphoid commitment. Oncogene. 2017;36(22):3207–12 Nature Publishing Group. Available from: https://www.nature.com/articles/onc2016470 [cited 2022 Aug 10].
Nie Z, Yan Z, Chen EH, Sechi S, Ling C, Zhou S, et al. Novel SWI/SNF chromatin-remodeling complexes contain a mixed-lineage leukemia chromosomal translocation partner. Mol Cell Biol. 2003;23:2942–52 Available from: https://pubmed.ncbi.nlm.nih.gov/12665591/ [cited 10 Aug 2022].
Pulikkan JA, Hegde M, Ahmad HM, Belaghzal H, Illendula A, Yu J, et al. CBFβ-SMMHC Inhibition Triggers Apoptosis by Disrupting MYC Chromatin Dynamics in Acute Myeloid Leukemia. Cell. 2018;174:172–186.e21 Cell Press. Available from: http://www.cell.com/article/S0092867418307098/fulltext [cited 23 Jan 2023].
Wilsker D, Probst L, Wain HM, Maltais L, Tucker PW, Moran E. Nomenclature of the ARID family of DNA-binding proteins. Genomics. 2005;86:242–51 Available from: https://pubmed.ncbi.nlm.nih.gov/15922553/ [cited 3 Oct 2022].
Wu H, Gao Y, Ding L, He D, Li Y. Gene expression profile analysis of SUDHL6 cells with siRNA-mediated BCL11A downregulation. Cell Biol Int. 2014;38:1205–14 Available from: https://pubmed.ncbi.nlm.nih.gov/25044937/ [cited 19 Oct 2022].
Ma MCJ, Tadros S, Bouska A, Heavican T, Yang H, Deng Q, et al. Subtype-specific and co-occurring genetic alterations in B-cell non-Hodgkin lymphoma. Haematologica. 2022;107:690–701 Ferrata Storti Foundation. Available from: https://haematologica.org/article/view/haematol.2020.274258 [cited 15 Jul 2022].
McKinney M, Moffitt AB, Gaulard P, Travert M, de Leval L, Raffeld ANM, et al. The genetic basis of hepatosplenic T-cell lymphoma. Cancer Discov. 2017;7:369–79. American Association for Cancer Research Inc. Available from: https://aacrjournals.org/cancerdiscovery/article/7/4/369/5954/The-Genetic-Basis-of-Hepatosplenic-T-cell. [cited 21 Jul 2022].
Watatani Y, Sato Y, Miyoshi H, Sakamoto K, Nishida K, Gion Y, et al. Molecular heterogeneity in peripheral T-cell lymphoma, not otherwise specified revealed by comprehensive genetic profiling. Leukemia. 2019;33(12):2867–83 Nature Publishing Group. Available from: https://www.nature.com/articles/s41375-019-0473-1 [cited 19 Jul 2022].
Sapienza MR, Abate F, Melle F, Orecchioni S, Fuligni F, Etebari M, et al. Blastic plasmacytoid dendritic cell neoplasm: genomics mark epigenetic dysregulation as a primary therapeutic target. Haematologica. 2019;104:729–37 Ferrata Storti Foundation. Available from: https://haematologica.org/article/view/8847 [cited 25 Jul 2022].
Sweeney SM, Cerami E, Baras A, Pugh TJ, Schultz N, Stricker T, et al. AACR project genie: Powering precision medicine through an international consortium. Cancer Discov. 2017;7:818–31 American Association for Cancer Research Inc. Available from: https://aacrjournals.org/cancerdiscovery/article/7/8/818/6485/AACR-Project-GENIE-Powering-Precision-Medicine [cited 12 Jul 2022].
Zhang J, Bajari R, Andric D, Gerthoffert F, Lepsa A, Nahal-Bose H, et al. The International Cancer Genome Consortium Data Portal. Nat Biotechnol. 2019;37(4):367–9 Nature Publishing Group. Available from: https://www.nature.com/articles/s41587-019-0055-9 [cited 18 Jul 2022].
Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45(6):592–601 Nature Publishing Group. Available from: https://www.nature.com/articles/ng.2628 [cited 28 Jul 2022].
Andrades A, Álvarez-Pérez JC, Patiño-Mercau JR, Cuadros M, Baliñas-Gavira C, Medina PP. Recurrent splice site mutations affect key diffuse large B-cell lymphoma genes. Blood. 2022;139:2406–10 American Society of Hematology. Available from: https://ashpublications.org/blood/article/139/15/2406/483332/Recurrent-splice-site-mutations-affect-key-diffuse [cited 28 Jul 2022].
Mittal P, Roberts CWM. The SWI/SNF complex in cancer — biology, biomarkers and therapy. Nat Rev Clin Oncol. 2020;17(7):435–48 Nature Publishing Group. Available from: https://www.nature.com/articles/s41571-020-0357-3 [cited 10 Jan 2023].
Peinado P, Andrades A, Cuadros M, Rodriguez MI, Coira IF, Garcia DJ, et al. Multi-omic alterations of the SWI/SNF complex define a clinical subgroup in lung adenocarcinoma. Clin Epigenetics. 2022;14(1):42.
Hodges HC, Stanton BZ, Cermakova K, Chang C-Y, Miller EL, Kirkland JG, et al. Dominant-negative SMARCA4 mutants alter the accessibility landscape of tissue-unrestricted enhancers. Nat Struct Mol Biol. 2018;25:61–72. https://doi.org/10.1038/s41594-017-0007-3.
Fernando TM, Piskol R, Bainer R, Sokol ES, Trabucco SE, Zhang Q, et al. Functional characterization of SMARCA4 variants identified by targeted exome-sequencing of 131,668 cancer patients. Nat Commun. 2020;11:5551. https://doi.org/10.1038/s41467-020-19402-8.
Martincorena I, Raine KM, Gerstung M, Dawson KJ, Haase K, van Loo P, et al. Universal patterns of selection in Cancer and somatic tissues. Cell. 2017;171:1029–1041.e21. https://doi.org/10.1016/j.cell.2017.09.042 Elsevier.
Pasqualucci L, Khiabanian H, Fangazio M, Vasishtha M, Messina M, Holmes AB, et al. Genetics of Follicular Lymphoma Transformation. Cell Rep. 2014;6:130–40 Elsevier. Available from: http://www.cell.com/article/S2211124713007857/fulltext [cited 14 Jul 2022].
Panea RI, Love CL, Shingleton JR, Reddy A, Bailey JA, Moormann AM, et al. The whole-genome landscape of Burkitt lymphoma subtypes. Blood. 2019;134:1598–607 American Society of Hematology. Available from: https://ashpublications.org/blood/article/134/19/1598/375002/The-whole-genome-landscape-of-Burkitt-lymphoma [cited 11 Jul 2022].
Chakraborty C, Morelli E, Linares M, Anderson KC, Samur MK, Fulciniti M, et al. Recurrent Non-Coding Mutations in BCL7A May Have Significant Functional Consequence in Multiple Myeloma. Blood. 2019;134:857 American Society of Hematology. Available from: https://ashpublications.org/blood/article/134/Supplement_1/857/427069/Recurrent-Non-Coding-Mutations-in-BCL7A-May-Have [cited 15 Jul 2022].
Hübschmann D, Kleinheinz K, Wagener R, Bernhart SH, López C, Toprak UH, et al. Mutational mechanisms shaping the coding and noncoding genome of germinal center derived B-cell lymphomas. Leukemia. 2021;35(7):2002–16 Nature Publishing Group. Available from: https://www.nature.com/articles/s41375-021-01251-z [cited 28 Jul 2022].
Bal E, Kumar R, Hadigol M, Holmes AB, Hilton LK, Loh JW, et al. Super-enhancer hypermutation alters oncogene expression in B cell lymphoma. Nature. 2022;607:808–15. Nature Publishing Group. Available from: https://www.nature.com/articles/s41586-022-04906-8. [cited 8 Jul 2022].
Yin J, Xie X, Ye Y, Wang L, Che F. BCL11A: a potential diagnostic biomarker and therapeutic target in human diseases. Biosci Rep. 2019;39:BSR20190604 Available from: https://pubmed.ncbi.nlm.nih.gov/31654056/ [cited 31 Oct 2022].
Martín-Subero JI, Gesk S, Harder L, Sonoki T, Tucker PW, Schlegelberger B, et al. Recurrent involvement of the REL and BCL11A loci in classical Hodgkin lymphoma. Blood. 2002;99:1474–7 American Society of Hematology. Available from: https://ashpublications.org/blood/article/99/4/1474/110037/Recurrent-involvement-of-the-REL-and-BCL11Aloci-in [cited 26 Jul 2022].
Weniger MA, Pulford K, Gesk S, Ehrlich S, Banham AH, Lyne L, et al. Gains of the proto-oncogene BCL11A and nuclear accumulation of BCL11AXL protein are frequent in primary mediastinal B-cell lymphoma. Leukemia. 2006;20(10):1880–2 Nature Publishing Group. Available from: https://www.nature.com/articles/2404324 [cited 26 Jul 2022].
Bea S, Zettl A, Wright G, Salaverria I, Jehn P, Moreno V, et al. Diffuse large B-cell lymphoma subgroups have distinct genetic profiles that influence tumor biology and improve gene-expression-based survival prediction. Blood. 2005;106:3183–90 American Society of Hematology. Available from: https://ashpublications.org/blood/article/106/9/3183/21944/Diffuse-large-B-cell-lymphoma-subgroups-have [cited 26 Jul 2022].
Karube K, Enjuanes A, Dlouhy I, Jares P, Martin-Garcia D, Nadeu F, et al. Integrating genomic alterations in diffuse large B-cell lymphoma identifies new relevant pathways and potential therapeutic targets. Leukemia. 2018;32(3):675–84 Nature Publishing Group. Available from: https://www.nature.com/articles/leu2017251 [cited 26 Jul 2022].
Kwiecinska A, Ichimura K, Berglund M, Dinets A, Sulaiman L, Collins VP, et al. Amplification of 2p as a genomic marker for transformation in lymphoma. Genes Chromosom Cancer. 2014;53:750–68 John Wiley & Sons, Ltd. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/gcc.22184 [cited 26 Jul 2022].
Bolen CR, Klanova M, Trnény M, Sehn LH, He J, Tong J, et al. Prognostic impact of somatic mutations in diffuse large B-cell lymphoma and relationship to cell-of-origin: data from the phase III GOYA study. Haematologica. 2020;105:2298–307 Ferrata Storti Foundation. Available from: https://haematologica.org/article/view/9561 [cited 25 Jul 2022].
Green MR, Kihira S, Liu CL, Nair RV, Salari R, Gentles AJ, et al. Mutations in early follicular lymphoma progenitors are associated with suppressed antigen presentation. Proc Natl Acad Sci U S A. 2015;112:E1116–25 National Academy of Sciences. Available from: www.pnas.org/cgi/doi/10.1073/pnas.1501199112 [cited 15 Jul 2022].
Krysiak K, Gomez F, White BS, Matlock M, Miller CA, Trani L, et al. Recurrent somatic mutations affecting B-cell receptor signaling pathway genes in follicular lymphoma. Blood. 2017;129:473–83 American Society of Hematology. Available from: https://ashpublications.org/blood/article/129/4/473/36199/Recurrent-somatic-mutations-affecting-B-cell [cited 14 Jul 2022].
López C, Kleinheinz K, Aukema SM, Rohde M, Bernhart SH, Hübschmann D, et al. Genomic and transcriptomic changes complement each other in the pathogenesis of sporadic Burkitt lymphoma. Nat Commun. 2019;10(1):1459.
Love C, Sun Z, Jima D, Li G, Zhang J, Miles R, et al. The genetic landscape of mutations in Burkitt lymphoma. Nat Genet. 2012;44(12):1321–5 Available from: https://www.nature.com/articles/ng.2468. Nature Publishing Group [cited 11 Jul 2022].
Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature. 2012;490(7418):116–20 Nature Publishing Group. Available from: https://www.nature.com/articles/nature11378 [cited 11 Jul 2022].
Kaymaz Y, Oduor CI, Yu H, Otieno JA, Ong’echa JM, Moormann AM, et al. Comprehensive transcriptome and mutational profiling of Endemic Burkitt lymphoma reveals EBV type-specific differences. Mol Cancer Res. 2017;15:563–76 American Association for Cancer Research Inc. Available from: https://aacrjournals.org/mcr/article/15/5/563/281647/Comprehensive-Transcriptome-and-Mutational [cited 15 Jul 2022].
Spina V, Bruscaggin A, Cuccaro A, Martini M, di Trani M, Forestieri G, et al. Circulating tumor DNA reveals genetics, clonal evolution, and residual disease in classical Hodgkin lymphoma. Blood. 2018;131:2413–25 American Society of Hematology. Available from: https://ashpublications.org/blood/article/131/22/2413/37097/Circulating-tumor-DNA-reveals-genetics-clonal [cited 12 Jul 2022].
Wienand K, Chapuy B, Stewart C, Dunford AJ, Wu D, Kim J, et al. Genomic analyses of flow-sorted Hodgkin Reed-Sternberg cells reveal complementary mechanisms of immune evasion. Blood Adv. 2019;3:4065–80 American Society of Hematology. Available from: https://ashpublications.org/bloodadvances/article/3/23/4065/429586/Genomic-analyses-of-flow-sorted-Hodgkin-Reed [cited 12 Jul 2022].
Mottok A, Hung SS, Chavez EA, Woolcock B, Telenius A, Chong LC, et al. Integrative genomic analysis identifies key pathogenic mechanisms in primary mediastinal large B-cell lymphoma. Blood. 2019;134:802–13 American Society of Hematology. Available from: https://ashpublications.org/blood/article/134/10/802/260637/Integrative-genomic-analysis-identifies-key [cited 12 Jul 2022].
Beà S, Valdés-Mas R, Navarro A, Salaverria I, Martín-Garcia D, Jares P, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci U S A. 2013;110:18250–5 PNAS. Available from: www.pnas.org/lookup/suppl/doi:10.1073/pnas.1314608110/-/DCSupplemental.www.pnas.org/cgi/doi/10.1073/pnas.1314608110 [cited 12 Jul 2022].
Zhang J, Jima D, Moffitt AB, Liu Q, Czader M, Hsi ED, et al. The genomic landscape of mantle cell lymphoma is related to the epigenetically determined chromatin state of normal B cells. Blood. 2014;123:2988–96 American Society of Hematology. Available from: https://ashpublications.org/blood/article/123/19/2988/32656/The-genomic-landscape-of-mantle-cell-lymphoma-is [cited 12 Jul 2022].
Jaramillo Oquendo C, Parker H, Oscier D, Ennis S, Gibson J, Strefford JC. Systematic Review of Somatic Mutations in Splenic Marginal Zone Lymphoma. Sci Rep. 2019;9(1):10444.
Vela V, Juskevicius D, Dirnhofer S, Menter T, Tzankov A. Mutational landscape of marginal zone B-cell lymphomas of various origin: organotypic alterations and diagnostic potential for assignment of organ origin. Virchows Arch. 2022;480:403–13 Springer Science and Business Media Deutschland GmbH. Available from: https://link.springer.com/article/10.1007/s00428-021-03186-3 [cited 14 Jul 2022].
Parry M, Rose-Zerilli MJJ, Ljungström V, Gibson J, Wang J, Walewska R, et al. Genetics and prognostication in splenic marginal zone lymphoma: Revelations from deep sequencing. Clin Cancer Res. 2015;21:4174–83 American Association for Cancer Research Inc. Available from: https://aacrjournals.org/clincancerres/article/21/18/4174/117432/Genetics-and-Prognostication-in-Splenic-Marginal [cited 14 Jul 2022].
Varettoni M, Zibellini S, Defrancesco I, Ferretti VV, Rizzo E, Malcovati L, et al. Pattern of somatic mutations in patients with Waldenström macroglobulinemia or IgM monoclonal gammopathy of undetermined significance. Haematologica. 2017;102:2077–85.
Hunter ZR, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. The genomic landscape of Waldenström macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood. 2014;123:1637–46 American Society of Hematology. Available from: https://ashpublications.org/blood/article/123/11/1637/105746/The-genomic-landscape-of-Waldenstrom [cited 2022 Jul 19].
Heavican TB, Bouska A, Yu J, Lone W, Amador C, Gong Q, et al. Genetic drivers of oncogenic pathways in molecular subgroups of peripheral T-cell lymphoma. Blood. 2019;133:1664–76 American Society of Hematology. Available from: https://ashpublications.org/blood/article/133/15/1664/273305/Genetic-drivers-of-oncogenic-pathways-in-molecular [cited 19 Jul 2022].
Schatz JH, Horwitz SM, Teruya-Feldstein J, Lunning MA, Viale A, Huberman K, et al. Targeted mutational profiling of peripheral T-cell lymphoma not otherwise specified highlights new mechanisms in a heterogeneous pathogenesis. Leukemia. 2015;29(1):237–41 Nature Publishing Group. Available from: https://www.nature.com/articles/leu2014261 [cited 21 Jul 2022].
Ji MM, Huang YH, Huang JY, Wang ZF, Fu D, Liu H, et al. Histone modifier gene mutations in peripheral T-cell lymphoma not otherwise specified. Haematologica. 2018;103:679–87.
Choi J, Goh G, Walradt T, Hong BS, Bunick CG, Chen K, et al. Genomic landscape of cutaneous T cell lymphoma. Nat Genet. 2015;47(9):1011–9 Nature Publishing Group. Available from: https://www.nature.com/articles/ng.3356 [cited 20 Jul 2022].
da Silva Almeida AC, Abate F, Khiabanian H, Martinez-Escala E, Guitart J, Tensen CP, et al. The mutational landscape of cutaneous T cell lymphoma and Sézary syndrome. Nat Genet. 2015;47(12):1465–70 Nature Publishing Group. Available from: https://www.nature.com/articles/ng.3442 [cited 20 Jul 2022].
Park J, Daniels J, Wartewig T, Ringbloom KG, Martinez-Escala ME, Choi S, et al. Integrated genomic analyses of cutaneous T-cell lymphomas reveal the molecular bases for disease heterogeneity. Blood. 2021;138:1225–36.
Wang L, Ni X, Covington KR, Yang BY, Shiu J, Zhang X, et al. Genomic profiling of Sézary syndrome identifies alterations of key T cell signaling and differentiation genes. Nat Genet. 2015;47(12):1426–34 Nature Publishing Group. Available from: https://www.nature.com/articles/ng.3444 [cited 20 Jul 2022].
Park J, Yang J, Wenzel AT, Ramachandran A, Lee WJ, Daniels JC, et al. Genomic analysis of 220 CTCLs identifies a novel recurrent gain-of-function alteration in RLTPR (p.Q575E). Blood. 2017;130:1430–40 American Society of Hematology. Available from: https://ashpublications.org/blood/article/130/12/1430/36502/Genomic-analysis-of-220-CTCLs-identifies-a-novel [cited 20 Jul 2022].
Song X, Chang S, Seminario-Vidal L, de Mingo PA, Tordesillas L, Song X, et al. Genomic and Single-Cell Landscape Reveals Novel Drivers and Therapeutic Vulnerabilities of Transformed Cutaneous T-cell Lymphoma. Cancer Discov. 2022;12:1294–313 NLM (Medline). Available from: https://aacrjournals.org/cancerdiscovery/article/12/5/1294/694564/Genomic-and-Single-Cell-Landscape-Reveals-Novel [cited 20 Jul 2022].
Carbone A, Bernardini L, Valenzano F, Bottillo I, de Simone C, Capizzi R, et al. Array-based comparative genomic hybridization in early-stage mycosis fungoides: recurrent deletion of tumor suppressor genes BCL7A, SMAC/DIABLO, and RHOF. Genes Chromosom Cancer. 2008;47:1067–75 John Wiley & Sons, Ltd. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/gcc.20601 [cited 10 Jul 2022].
Choi S, Go JH, Kim EK, Lee H, Lee WM, Cho C-S, et al. Mutational analysis of extranodal NK/T-cell lymphoma using targeted sequencing with a comprehensive cancer panel. Genomics Inform. 2016;14:78–84.
Koo GC, Tan SY, Tang T, Poon SL, Allen GE, Tan L, et al. Janus kinase 3-activating mutations identified in natural killer/T-cell lymphoma. Cancer Discov. 2012;2:591–7. American Association for Cancer Research. Available from: https://aacrjournals.org/cancerdiscovery/article/2/7/591/3362/Janus-Kinase-3-Activating-Mutations-Identified-in. [cited 20 Jul 2022].
Lee S, Park HY, Kang SY, Kim SJ, Hwang J, Lee S, et al. Genetic alterations of JAK/STAT cascade and histone modification in extranodal NK/T-cell lymphoma nasal type. Oncotarget. 2015;6:17764–76 Impact Journals. Available from: https://www.oncotarget.com/article/3776/text/ [cited 20 Jul 2022].
Dobashi A, Tsuyama N, Asaka R, Togashi Y, Ueda K, Sakata S, et al. Frequent BCOR aberrations in extranodal NK/T-cell lymphoma, nasal type. Genes Chromosom Cancer. 2016;55:460–71 John Wiley & Sons, Ltd. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/gcc.22348 [cited 20 Jul 2022].
Jiang L, Gu ZH, Yan ZX, Zhao X, Xie YY, Zhang ZG, et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat Genet. 2015;47(9):1061–6 Nature Publishing Group. Available from: https://www.nature.com/articles/ng.3358 [cited 20 Jul 2022].
Moffitt AB, Ondrejka SL, McKinney M, Rempel RE, Goodlad JR, Teh CH, et al. Enteropathy-associated T cell lymphoma subtypes are characterized by loss of function of SETD2. J Exp Med. 2017;214:1371–86. https://doi.org/10.1084/jem.20160894 The Rockefeller University Press [cited 21 Jul 2022].
Ma X, Liu Y, Liu Y, Alexandrov LB, Edmonson MN, Gawad C, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018;555(7696):371–6 Nature Publishing Group. Available from: https://www.nature.com/articles/nature25795 [cited 18 Jan 2023].
Brady SW, Roberts KG, Gu Z, Shi L, Pounds S, Pei D, et al. The genomic landscape of pediatric acute lymphoblastic leukemia. Nat Genet. 2022;54:1376–89 Available from: https://pubmed.ncbi.nlm.nih.gov/36050548/ [cited 20 Oct 2022].
Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. 2017;49(8):1211–8 Nature Publishing Group. Available from: https://www.nature.com/articles/ng.3909 [cited 18 Jan 2023].
Soulier J, Pierron G, Vecchione D, Garand R, Brizard F, Sigaux F, et al. A complex pattern of recurrent chromosomal losses and gains in T-cell prolymphocytic leukemia. Genes Chromosom Cancer. 2001;31:248–54 John Wiley & Sons, Ltd. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/gcc.1141 [cited 16 Jan 2023].
Bug S, Dürig J, Oyen F, Klein-Hitpass L, Martin-Subero JI, Harder L, et al. Recurrent loss, but lack of mutations, of the SMARCB1 tumor suppressor gene in T-cell prolymphocytic leukemia with TCL1A–TCRAD juxtaposition. Cancer Genet Cytogenet. 2009;192:44–7 Elsevier.
Specchia G, Albano F, Anelli L, Storlazzi CT, Zagaria A, Liso A, et al. Derivative chromosome 9 deletions in chronic myeloid leukemia are associated with loss of tumor suppressor genes. Leuk Lymphoma. 2004;45:689–94.
Sakai H, Hosono N, Nakazawa H, Przychodzen B, Polprasert C, Carraway HE, et al. A novel genetic and morphologic phenotype of ARID2-mediated myelodysplasia. Leukemia. 2018;32(3):839–43 Nature Publishing Group. Available from: https://www.nature.com/articles/leu2017319 [cited 22 Jul 2022].
Yao H, Huo L, Ping N, Liu H, Li H, Ding Z, et al. Recurrent mutations in multiple components of the SWI/SNF complex in myelodysplastic syndromes and acute myeloid leukaemia. Br J Haematol. 2022;196:441–4 John Wiley & Sons, Ltd. Available from: https://onlinelibrary.wiley.com/doi/full/10.1111/bjh.17795 [cited 22 Jul 2022].
Menezes J, Acquadro F, Wiseman M, Gómez-López G, Salgado RN, Talavera-Casañas JG, et al. Exome sequencing reveals novel and recurrent mutations with clinical impact in blastic plasmacytoid dendritic cell neoplasm. Leukemia. 2014;28(4):823–9 Nature Publishing Group. Available from: https://www.nature.com/articles/leu2013283 [cited 25 Jul 2022].
Taylor J, Kim SS, Stevenson KE, Yoda A, Kopp N, Louissaint A, et al. Loss-of-function mutations in the splicing factor ZRSR2 are common in Blastic Plasmacytoid dendritic cell neoplasm and have male predominance. Blood. 2013;122:741.
Togami K, Chung SS, Madan V, Booth CAG, Kenyon CM, Cabal-Hierro L, et al. Sex-Biased ZRSR2 Mutations in Myeloid Malignancies Impair Plasmacytoid Dendritic Cell Activation and Apoptosis. Cancer Discov. 2022;12:522–41 American Association for Cancer Research Inc. Available from: https://aacrjournals.org/cancerdiscovery/article/12/2/522/678508/Sex-Biased-ZRSR2-Mutations-in-Myeloid-Malignancies [cited 25 Jul 2022].
Wang L, Yang M, Zhang X, Yang C, Huang X, Wang Z, et al. ARID1A mutation in blastic plasmacytoid dendritic cell neoplasm. Haematologica. 2017;102:e470–2 Ferrata Storti Foundation. Available from: https://haematologica.org/article/view/8271 [cited 25 Jul 2022].
Pastore A, Jurinovic V, Kridel R, Hoster E, Staiger AM, Szczepanowski M, et al. Integration of gene mutations in risk prognostication for patients receiving first-line immunochemotherapy for follicular lymphoma: a retrospective analysis of a prospective clinical trial and validation in a population-based registry. Lancet Oncol. 2015;16:1111–22 Elsevier. Available from: http://www.thelancet.com/article/S1470204515001692/fulltext [cited 14 Jul 2022].
Zani VJ, Asou N, Jadayel D, Heward JM, Shipley J, Nacheva E, et al. Molecular Cloning of Complex Chromosomal Translocation t(8;14;12)(q24.1;q32.3;q24.1) in a Burkitt Lymphoma Cell Line Defines a New Gene (BCL7A) With Homology to Caldesmon. Blood. 1996;87:3124–34.
Shen J, Ju Z, Zhao W, Wang L, Peng Y, Ge Z, et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med. 2018;24(5):556–62 Nature Publishing Group. Available from: https://www.nature.com/articles/s41591-018-0012-z [cited 12 Jul 2022].
Mata E, Fernández S, Astudillo A, Fernández R, García-Cosío M, Sánchez-Beato M, et al. Genomic analyses of microdissected Hodgkin and Reed-Sternberg cells: mutations in epigenetic regulators and p53 are frequent in refractory classic Hodgkin lymphoma. Blood Cancer J. 2019;9(3):34. Nature Publishing Group. Available from: https://www.nature.com/articles/s41408-019-0195-7. [cited 12 Jul 2022].
Reichel J, Chadburn A, Rubinstein PG, Giulino-Roth L, Tam W, Liu Y, et al. Flow sorting and exome sequencing reveal the oncogenome of primary Hodgkin and Reed-Sternberg cells. Blood. 2015;125:1061–72 American Society of Hematology. Available from: https://ashpublications.org/blood/article/125/7/1061/34180/Flow-sorting-and-exome-sequencing-reveal-the [cited 12 Jul 2022].
Hartmann S, Schuhmacher B, Rausch T, Fuller L, Döring C, Weniger M, et al. Highly recurrent mutations of SGK1, DUSP2 and JUNB in nodular lymphocyte predominant Hodgkin lymphoma. Leukemia. 2016;30(4):844–53 Nature Publishing Group. Available from: https://www.nature.com/articles/leu2015328 [cited 12 Jul 2022].
Song JY, Egan C, Gong Q, Venkataraman G, Herrera AF, Ottesen R, et al. Mutation Spectrum of transformed nodular lymphocyte predominant Hodgkin lymphoma and comparison with De novo diffuse large B-cell lymphoma. Blood. 2017;130:1463.
Chapuy B, Stewart C, Dunford AJ, Kim J, Wienand K, Kamburov A, et al. Genomic analyses of PMBL reveal new drivers and mechanisms of sensitivity to PD-1 blockade. Blood. 2019;134:2369–82 American Society of Hematology. Available from: https://ashpublications.org/blood/article/134/26/2369/422650/Genomic-analyses-of-PMBL-reveal-new-drivers-and [cited 12 Jul 2022].
van Doorn R, van Kester MS, Dijkman R, Vermeer MH, Mulder AA, Szuhai K, et al. Oncogenomic analysis of mycosis fungoides reveals major differences with Sézary syndrome. Blood. 2009;113:127–36 American Society of Hematology. Available from: https://ashpublications.org/blood/article/113/1/127/24416/Oncogenomic-analysis-of-mycosis-fungoides-reveals [cited 20 Jul 2022].
Odejide O, Weigert O, Lane AA, Toscano D, Lunning MA, Kopp N, et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood. 2014;123:1293–6 American Society of Hematology. Available from: https://ashpublications.org/blood/article/123/9/1293/32919/A-targeted-mutational-landscape-of [cited 21 Jul 2022].
Yoo HY, Sung MK, Lee SH, Kim S, Lee H, Park S, et al. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat Genet. 2014;46(4):371–5 Nature Publishing Group. Available from: https://www.nature.com/articles/ng.2916 [cited 21 Jul 2022].
Cheng S, Zhang W, Inghirami G, Tam W. Mutation analysis links angioimmunoblastic T-cell lymphoma to clonal hematopoiesis and smoking. Elife. 2021;10:e66395 eLife Sciences Publications Ltd.
Butzmann A, Sridhar K, Jangam D, Song H, Singh A, Kumar J, et al. Mutations in JAK/STAT and NOTCH1 genes are enriched in post-transplant Lymphoproliferative disorders. Front Oncol. 2022;11:790481.
Vallois D, Dobay MPD, Morin RD, Lemonnier F, Missiaglia E, Juilland M, et al. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T-cell–derived lymphomas. Blood. 2016;128:1490–502 American Society of Hematology. Available from: https://ashpublications.org/blood/article/128/11/1490/35240/Activating-mutations-in-genes-related-to-TCR [cited 22 Jul 2022].
Roberti A, Dobay MP, Bisig B, Vallois D, Boéchat C, Lanitis E, et al. Type II enteropathy-associated T-cell lymphoma features a unique genomic profile with highly recurrent SETD2 alterations. Nat Commun. 2016;7(1):12602. Nature Publishing Group. Available from: https://www.nature.com/articles/ncomms12602. [cited 21 Jul 2022].
Chen C, Gong Y, Yang Y, Xia Q, Rao Q, Shao Y, et al. Clinicopathological and molecular genomic features of monomorphic epitheliotropic intestinal T-cell lymphoma in the Chinese population: a study of 20 cases. Diagn Pathol. 2021;16:114. BioMed Central Ltd. Available from: https://diagnosticpathology.biomedcentral.com/articles/10.1186/s13000-021-01173-5. [cited 21 Jul 2022].
Veloza L, Cavalieri D, Missiaglia E, Ledoux-Pilon A, Bisig B, Pereira B, et al. Monomorphic epitheliotropic intestinal T-cell lymphoma comprises morphologic and genomic heterogeneity impacting outcome. Haematologica. 2023;108:1. Ferrata Storti Foundation (Haematologica). Available from: https://haematologica.org/article/view/haematol.2022.281226. [cited 21 Jul 2022].
Crescenzo R, Abate F, Lasorsa E, Tabbo’ F, Gaudiano M, Chiesa N, et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell. 2015;27:516–32 Cell Press. Available from: http://www.cell.com/article/S153561081500094X/fulltext [cited 21 Jul 2022].
Lobello C, Tichy B, Bystry V, Radova L, Filip D, Mraz M, et al. STAT3 and TP53 mutations associate with poor prognosis in anaplastic large cell lymphoma. Leukemia. 2020;35(5):1500–5 Nature Publishing Group. Available from: https://www.nature.com/articles/s41375-020-01093-1 [cited 21 Jul 2022].
Luchtel RA, Zimmermann MT, Hu G, Dasari S, Jiang M, Oishi N, et al. Recurrent MSCE116K mutations in ALK-negative anaplastic large cell lymphoma. Blood. 2019;133:2776–89 American Society of Hematology. Available from: https://ashpublications.org/blood/article/133/26/2776/272770/Recurrent-MSCE116K-mutations-in-ALK-negative [cited 21 Jul 2022].
Zhong L-H, Wu Z-D, Wang J-C, Wu Z-Z, Chen F-F, Zhu W-F, et al. Molecular profiling of Chinese systemic anaplastic large cell lymphoma patients: novel evidence of genetic heterogeneity. Ann Transl Med. 2021;9:128 AME Publishing Company. Available from: https://atm.amegroups.com/article/view/60489/html [cited 21 Jul 2022].
Bolli N, Biancon G, Moarii M, Gimondi S, Li Y, de Philippis C, et al. Analysis of the genomic landscape of multiple myeloma highlights novel prognostic markers and disease subgroups. Leukemia. 2018;32:2604–16.
Hoang PH, Dobbins SE, Cornish AJ, Chubb D, Law PJ, Kaiser M, et al. Whole-genome sequencing of multiple myeloma reveals oncogenic pathways are targeted somatically through multiple mechanisms. Leukemia. 2018;32(11):2459–70 Nature Publishing Group. Available from: https://www.nature.com/articles/s41375-018-0103-3 [cited 15 Jul 2022].
Hoang PH, Cornish AJ, Sherborne AL, Chubb D, Kimber S, Jackson G, et al. An enhanced genetic model of relapsed IGH-translocated multiple myeloma evolutionary dynamics. Blood Cancer J. 2020;10(10):101. Nature Publishing Group. Available from: https://www.nature.com/articles/s41408-020-00367-2. [cited 15 Jul 2022].
Maura F, Bolli N, Minvielle S, Gloznik D, Szalat R, Fullam A, et al. Analysis of Mutational Signatures Suggest That Aid Has an Early and Driver Role in Multiple Myeloma. Blood. 2016;128:116 American Society of Hematology. Available from: https://ashpublications.org/blood/article/128/22/116/96279/Analysis-of-Mutational-Signatures-Suggest-That-Aid [cited 15 Jul 2022].
Ogawa S. Genetics of MDS. Blood. 2019;133:1049–59 American Society of Hematology. Available from: https://ashpublications.org/blood/article/133/10/1049/272730/Genetics-of-MDS [cited 22 Jul 2022].
Kim T, Tyndel MS, Kim HJ, Ahn J-S, Choi SH, Park HJ, et al. Spectrum of somatic mutation dynamics in chronic myeloid leukemia following tyrosine kinase inhibitor therapy. Blood. 2017;129:38–47.
Behnert A, Meyer J, Parsa JY, Hechmer A, Loh ML, Olshen A, et al. Exploring the genetic and epigenetic origins of juvenile myelomonocytic leukemia using newborn screening samples. Leukemia. 2022;36:279–82 Available from: https://pubmed.ncbi.nlm.nih.gov/34183765/ [cited 21 Oct 2022].
Fiñana C, Gómez-Molina N, Alonso-Moreno S, Belver L. Genomic and Epigenomic Landscape of Juvenile Myelomonocytic Leukemia. Cancers (Basel). 2022;14:1335 Available from: https://pubmed.ncbi.nlm.nih.gov/35267643/ [cited 21 Oct 2022].
Patel AB, Deininger MW. Genetic complexity of chronic myelomonocytic leukemia. Leuk Lymphoma. 2021;62:1031–45 Available from: https://pubmed.ncbi.nlm.nih.gov/33337259/ [cited 2022 Oct 21].
Zhang H, Wilmot B, Bottomly D, Dao KHT, Stevens E, Eide CA, et al. Genomic landscape of neutrophilic leukemias of ambiguous diagnosis. Blood. 2019;134:867–79 Available from: https://pubmed.ncbi.nlm.nih.gov/31366621/ [cited 21 Oct 2022].
Qu SQ, Qin TJ, Xu ZF, Zhang Y, Jia YJ, Ai XF, et al. Targeted sequencing analysis of hyper-eosinophilic syndrome and chronic eosinophilic leukemia. Zhonghua Xue Ye Xue Za Zhi. 2018;39:501–6.
Merker JD, Roskin KM, Ng D, Pan C, Fisk DG, King JJ, et al. Comprehensive whole-genome sequencing of an early-stage primary myelofibrosis patient defines low mutational burden and non-recurrent candidate genes. Haematologica. 2013;98:1689–96 Available from: https://haematologica.org/article/view/6830 [cited 22 Jul 2022].
Sadler B, Chorzalska AD, Bonal DM, Haller G, Oakes A, Petersen M, et al. Whole genome sequencing identifies a recurrent mutation in complement factor I (CFI) in primary Myelofibrosis (PMF). Blood. 2021;138:1472.
Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, et al. Somatic CALR Mutations in Myeloproliferative Neoplasms with Nonmutated JAK2. N Engl J Med. 2013;369:2391–405 Massachusetts Medical Society. Available from: https://www.nejm.org/doi/10.1056/NEJMoa1312542 [cited 2022 Jul 22].
Cabagnols X, Favale F, Pasquier F, Messaoudi K, Defour JP, Ianotto JC, et al. Presence of atypical thrombopoietin receptor (MPL) mutations in triple-negative essential thrombocythemia patients. Blood. 2016;127:333–42 American Society of Hematology. Available from: https://ashpublications.org/blood/article/127/3/333/34889/Presence-of-atypical-thrombopoietin-receptor-MPL [cited 22 Jul 2022].
Milosevic Feenstra JD, Nivarthi H, Gisslinger H, Leroy E, Rumi E, Chachoua I, et al. Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood. 2016;127:325–32 American Society of Hematology. Available from: https://ashpublications.org/blood/article/127/3/325/34897/Whole-exome-sequencing-identifies-novel-MPL-and [cited 22 Jul 2022].
Künstner A, Schwarting J, Witte HM, Bernard V, Stölting S, Kusch K, et al. Integrative molecular profiling identifies two molecularly and clinically distinct subtypes of blastic plasmacytoid dendritic cell neoplasm. Blood Cancer J. 2022;12(7):101. Nature Publishing Group. Available from: https://www.nature.com/articles/s41408-022-00699-1. [cited 25 Jul 2022].
Kinnaman MD, Hamill D, Yabe M, Powell J, Benhamida J, Hasselblatt M, et al. Aggressive hematopoietic malignancy characterized by Biallelic loss of SMARCB1. JCO Precis Oncol. 2020;4:1280–4. https://doi.org/10.1200/PO.20.00215.
Margolskee E, Jobanputra V, Jain P, Chen J, Ganapathi K, Nahum O, et al. Genetic landscape of T- and NK-cell post-transplant lymphoproliferative disorders. Oncotarget. 2016;7:37636–48.
Egan C, Nicolae A, Lack J, Chung HJ, Skarshaug S, Pham TA, et al. Genomic profiling of primary histiocytic sarcoma reveals two molecular subgroups. Haematologica. 2020;105:951–60 Ferrata Storti Foundation. Available from: https://haematologica.org/article/view/9331 [cited 25 Jul 2022].
Jansen C, Dykstra J, Callaway D, Lynch D, Cunningham A, Frohm ML. Aggressive Langerhans cell histiocytosis following T-cell acute lymphoblastic leukemia. Pediatr Blood Cancer. 2020;67:e28704 John Wiley & Sons, Ltd. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/pbc.28704 [cited 25 Jul 2022].
Yao L, Yin H, Hong M, Wang Y, Yu T, Teng Y, et al. RNA methylation in hematological malignancies and its interactions with other epigenetic modifications. Leukemia. 2021;35:1243–57 Available from: https://pubmed.ncbi.nlm.nih.gov/33767371/ [cited 4 Aug 2022].
Saleh LM, Wang W, Herman SEM, Saba NS, Anastas V, Barber E, et al. Ibrutinib downregulates a subset of miRNA leading to upregulation of tumor suppressors and inhibition of cell proliferation in chronic lymphocytic leukemia. Leukemia. 2017;31:340–9.
van Doorn R, Zoutman WH, Dijkman R, de Menezes RX, Commandeur S, Mulder AA, et al. Epigenetic profiling of cutaneous T-cell lymphoma: promoter hypermethylation of multiple tumor suppressor genes including BCL7a, PTPRG, and p73. J Clin Oncol. 2005;23:3886–96 Proc Am Soc Clin Oncol.
Deambrogi C, de Paoli L, Fangazio M, Cresta S, Rasi S, Spina V, et al. Analysis of the REL, BCL11A, and MYCN proto-oncogenes belonging to the 2p amplicon in chronic lymphocytic leukemia. Am J Hematol. 2010;85:541–4. Available from: https://pubmed.ncbi.nlm.nih.gov/20575024/. [cited 31 Oct 2022].
Tosic N, Ugrin M, Marjanovic I, Kostic T, Vukovic V, Tomic K, et al. Expression of BCL11A in chronic lymphocytic leukaemia. Int J Lab Hematol. 2023;45:64–71. Wiley. Available from: https://onlinelibrary.wiley.com/doi/full/10.1111/ijlh.13969. [cited 31 Oct 2022].
Wang LL, Yan D, Tang X, Zhang M, Liu S, Wang Y, et al. High Expression of BCL11A Predicts Poor Prognosis for Childhood MLL-r ALL. Front Oncol. 2021;11:755188 Available from: https://pubmed.ncbi.nlm.nih.gov/34938655/ [cited 31 Oct 2022].
Tao H, Ma X, Su G, Yin J, Xie X, Hu C, et al. BCL11A expression in acute myeloid leukemia. Leuk Res. 2016;41:71–5 Available from: https://pubmed.ncbi.nlm.nih.gov/26707798/ [cited 31 Oct 2022].
Yin J, Zhang F, Tao H, Ma X, Su G, Xie X, et al. BCL11A expression in acute phase chronic myeloid leukemia. Leuk Res. 2016;47:88–92 Available from: https://pubmed.ncbi.nlm.nih.gov/27285855/ [cited 31 Oct 2022].
Cuadros M, Sánchez-Martín V, Herrera A, Baliñas C, Martín-Padrón J, Boyero L, et al. BRG1 regulation by miR-155 in human leukemia and lymphoma cell lines. Clin Transl Oncol. 2017;19:1010–7 Available from: https://pubmed.ncbi.nlm.nih.gov/28251496/ [cited 9 Aug 2022].
Chang Y, Cui M, Fu X, Zhang L, Li X, Li L, et al. MiRNA-155 regulates lymphangiogenesis in natural killer/T-cell lymphoma by targeting BRG1. Cancer Biol Ther. 2019;20:31–41.
Li Y, Wang J, Yu M, Wang Y, Zhang H, Yin J, et al. SNF5 deficiency induces apoptosis resistance by repressing SATB1 expression in Sézary syndrome. Leuk Lymphoma. 2018;59:2405–13 Taylor & Francis. Available from: https://www.tandfonline.com/doi/abs/10.1080/10428194.2017.1422861 [cited 16 Jan 2023].
Gebert LFR, MacRae IJ. Regulation of microRNA function in animals. Nat Rev Mol Cell Biol. 2019;20:21–37.
Coira IF, Rufino-Palomares EE, Romero OA, Peinado P, Metheetrairut C, Boyero-Corral L, et al. Expression inactivation of SMARCA4 by microRNAs in lung tumors. Hum Mol Genet. 2015;24:1400–9 Available from: https://pubmed.ncbi.nlm.nih.gov/25355421/ [cited 11 Nov 2022].
Agarwal R, Chan YC, Tam CS, Hunter T, Vassiliadis D, Teh CE, et al. Dynamic molecular monitoring reveals that SWI–SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat Med. 2018;25(1):119–29 Nature Publishing Group. Available from: https://www.nature.com/articles/s41591-018-0243-z [cited 15 Nov 2022].
Dubey R, Lebensohn AM, Bahrami-Nejad Z, Marceau C, Champion M, Gevaert O, et al. Chromatin-Remodeling Complex SWI/SNF Controls Multidrug Resistance by Transcriptionally Regulating the Drug Efflux Pump ABCB1. Cancer Res. 2016;76:5810–21 Available from: https://pubmed.ncbi.nlm.nih.gov/27503929/ [cited 15 Nov 2022].
Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta. 1976;455:152–62 Available from: https://pubmed.ncbi.nlm.nih.gov/990323/ [cited 15 Nov 2022].
Pottier N, Yang W, Assem M, Panetta JC, Pei D, Paugh SW, et al. The SWI/SNF chromatin-remodeling complex and glucocorticoid resistance in acute lymphoblastic leukemia. J Natl Cancer Inst. 2008;100:1792–803. https://doi.org/10.1093/jnci/djn416.
Kim H-J, Bae S-C. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am J Transl Res. 2011;3:166–79 Available from: www.ajtr.org.
Serganova I, Chakraborty S, Yamshon S, Isshiki Y, Bucktrout R, Melnick A, et al. Epigenetic, metabolic, and immune crosstalk in germinal-center-derived B-cell lymphomas: unveiling new vulnerabilities for rational combination therapies. Front Cell Dev Biol. 2022;9:805195 Available from: https://www.frontiersin.org/articles/10.3389/fcell.2021.805195.
Wang X, Wang D, Ding N, Mi L, Yu H, Wu M, et al. The Synergistic Anti-Tumor Activity of EZH2 Inhibitor SHR2554 and HDAC Inhibitor Chidamide through ORC1 Reduction of DNA Replication Process in Diffuse Large B Cell Lymphoma. Cancers (Basel). 2021;13:4249 Available from: https://www.mdpi.com/2072-6694/13/17/4249.
Romero OA, Vilarrubi A, Alburquerque-Bejar JJ, Gomez A, Andrades A, Trastulli D, et al. SMARCA4 deficient tumours are vulnerable to KDM6A/UTX and KDM6B/JMJD3 blockade. Nat Commun. 2021;12:4319. https://doi.org/10.1038/s41467-021-24618-3.
Helming KC, Wang X, Wilson BG, Vazquez F, Haswell JR, Manchester HE, et al. ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat Med. 2014;20:251–4 Available from: https://pubmed.ncbi.nlm.nih.gov/24562383/ [cited 14 Nov 2022].
Xi W, Sansam CG, Thom CS, Metzger D, Evans JA, Nguyen PTL, et al. Oncogenesis caused by loss of the SNF5 tumor suppressor is dependent on activity of BRG1, the ATPase of the SWI/SNF chromatin remodeling complex. Cancer Res. 2009;69:8094–101 Available from: https://pubmed.ncbi.nlm.nih.gov/19789351/ [cited 14 Nov 2022].
Schiaffino-Ortega S, Balinas C, Cuadros M, Medina PP. SWI/SNF proteins as targets in cancer therapy. J Hematol Oncol. 2014;7:81. BioMed Central Ltd. Available from: https://jhoonline.biomedcentral.com/articles/10.1186/s13045-014-0081-5. [cited 11 Nov 2022].
Hoffman GR, Rahal R, Buxton F, Xiang K, McAllister G, Frias E, et al. Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers. Proc Natl Acad Sci U S A. 2014;111:3128–33 Available from: https://pubmed.ncbi.nlm.nih.gov/24520176/ [cited 6 Sep 2022].
Jagani Z, Chenail G, Xiang K, Bushold G, Bhang H-EC, Li A, et al. In-Depth Characterization and Validation in BRG1-Mutant Lung Cancers Define Novel Catalytic Inhibitors of SWI/SNF Chromatin Remodeling. bioRxiv. 2019:812628. Cold Spring Harbor Laboratory. Available from: https://www.biorxiv.org/content/10.1101/812628v1. [cited 5 Sep 2022].
Papillon JPN, Nakajima K, Adair CD, Hempel J, Jouk AO, Karki RG, et al. Discovery of Orally Active Inhibitors of Brahma Homolog (BRM)/SMARCA2 ATPase Activity for the Treatment of Brahma Related Gene 1 (BRG1)/SMARCA4-Mutant Cancers. J Med Chem. 2018;61:10155–72 American Chemical Society. Available from: https://pubs.acs.org/doi/full/10.1021/acs.jmedchem.8b01318 [cited 5 Sep 2022].
Farnaby W, Koegl M, Roy MJ, Whitworth C, Diers E, Trainor N, et al. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat Chem Biol. 2019;15(7):672–80 Nature Publishing Group. Available from: https://www.nature.com/articles/s41589-019-0294-6 [cited 11 Nov 2022].
Kofink C, Trainor N, Mair B, Wöhrle S, Wurm M, Mischerikow N, et al. A selective and orally bioavailable VHL-recruiting PROTAC achieves SMARCA2 degradation in vivo. Nat Commun. 2022;13(1):5969. Nature Publishing Group. Available from: https://www.nature.com/articles/s41467-022-33430-6. [cited 2 Nov 2022].
Békés M, Langley DR, Crews CM. PROTAC targeted protein degraders: the past is prologue. Nat Rev Drug Discov. 2022;21(3):181–200 Nature Publishing Group. Available from: https://www.nature.com/articles/s41573-021-00371-6 [cited 26 Oct 2022].
Michel BC, D’Avino AR, Cassel SH, Mashtalir N, McKenzie ZM, McBride MJ, et al. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat Cell Biol. 2018;20:1410–20 Available from: https://pubmed.ncbi.nlm.nih.gov/30397315/ [cited 5 Sep 2022].
Zoppi V, Hughes SJ, Maniaci C, Testa A, Gmaschitz T, Wieshofer C, et al. Iterative Design and Optimization of Initially Inactive Proteolysis Targeting Chimeras (PROTACs) Identify VZ185 as a Potent, Fast, and Selective von Hippel-Lindau (VHL) Based Dual Degrader Probe of BRD9 and BRD7. J Med Chem. 2019;62:699–726 American Chemical Society. Available from: https://pubs.acs.org/doi/full/10.1021/acs.jmedchem.8b01413 [cited 8 Sep 2022].
Wang X, Wang S, Troisi EC, Howard TP, Haswell JR, Wolf BK, et al. BRD9 defines a SWI/SNF sub-complex and constitutes a specific vulnerability in malignant rhabdoid tumors. Nat Commun. 2019;10:1881 Available from: https://pubmed.ncbi.nlm.nih.gov/31015438/ [cited 5 Sep 2022].
Tamkun JW, Deuring R, Scott MP, Kissinger M, Pattatucci AM, Kaufman TC, et al. brahma: A regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2/SWI2. Cell. 1992;68:561–72. Elsevier. Available from: http://www.cell.com/article/009286749290191E/fulltext. [cited 18 Jul 2022].
Kadoch C, Williams RT, Calarco JP, Miller EL, Weber CM, Braun SMG, et al. Dynamics of BAF–Polycomb complex opposition on heterochromatin in normal and oncogenic states. Nat Genet. 2016;49(2):213–22 Nature Publishing Group. Available from: https://www.nature.com/articles/ng.3734 [cited 18 Jul 2022].
Kim KH, Roberts CWM. Targeting EZH2 in cancer. Nat Med. 2016;22:128–34 Available from: https://pubmed.ncbi.nlm.nih.gov/26845405/ [cited 7 Sep 2022].
Kim W, Bird GH, Neff T, Guo G, Kerenyi MA, Walensky LD, et al. Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer. Nat Chem Biol. 2013;9:643–50 Available from: https://pubmed.ncbi.nlm.nih.gov/23974116/ [cited 7 Sep 2022].
Gulati N, Béguelin W, Giulino-Roth L. Enhancer of zeste homolog 2 (EZH2) inhibitors. Leuk Lymphoma. 2018;59:1574–85 Taylor and Francis Ltd.
Morin RD, Arthur SE, Assouline S. Treating lymphoma is now a bit EZ-er. Blood Adv. 2021;5:2256–63. https://doi.org/10.1182/bloodadvances.2020002773.
Straining R, Eighmy W. Tazemetostat: EZH2 Inhibitor. J Adv Pract Oncol. 2022;13:158–63 Harborside Press, LLC.
Nakayama RT, Pulice JL, Valencia AM, McBride MJ, McKenzie ZM, Gillespie MA, et al. SMARCB1 is required for widespread BAF complex–mediated activation of enhancers and bivalent promoters. Nat Genet. 2017;49:1613–23.
Acknowledgements
The authors thank the Ph.D. program of Biochemistry and Molecular Biology of the University of Granada and to the Aula de Investigacion sobre la Leucemia infantil: Héroes contra la Leucemia de la Universidad de Granada (https://leucemiainfantil.ugr.es/).
Funding
P.P.M.’s laboratory is funded by Aula de Investigacion sobre la Leucemia infantil: Héroes contra la Leucemia, the Spanish Ministry of Science and Innovation (PID2021-126111OB-I00), Junta de Andalucía (PIGE-0440-2019, PI-0135-2020, P20_00688), University of Granada (B-CTS-480-UGR20), OTRI-UGR (P32_2020_001) and the Spanish Association Against Cancer (LAB-AECC-2018). A.A. was supported by an FPU17/00067 fellowship (Spanish Ministry of Science, Innovation and Universities) and by the Programa de Contratos Puente (Plan Propio 2022) of the University of Granada. P.P. is supported by a contract funded by Programa Operativo de Empleo Juvenil y de la Iniciativa de Empleo Juvenil 2021-2023 n° 8114). J.S-H. is supported by a grant from the Scientific Foundation of the Spanish Association Against Cancer in Granada (#PRDGR21428SANJ). D.J.G. is supported by a “Fundación Benéfica Anticáncer Santa Cándida y San Francisco Javier” predoctoral fellowship. A.M.A. is supported by an FPU17/01258 fellowship. A.M.M.G is supported by a “UGR Plan Propio” contract funded by the Spanish Ministry of Universities and European Union Next Generation.
Author information
Authors and Affiliations
Contributions
A.A., P.P., J.C.Á-P., J.S.-H., D.J.G., A.M.A., A.M.M.-G, and P.P.M. performed the bibliography searches and wrote the initial draft of the manuscript. P.P.M. coordinated the project. All authors read, reviewed, and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors consent to publication.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Andrades, A., Peinado, P., Alvarez-Perez, J.C. et al. SWI/SNF complexes in hematological malignancies: biological implications and therapeutic opportunities. Mol Cancer 22, 39 (2023). https://doi.org/10.1186/s12943-023-01736-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-023-01736-8